Cas9 Protein vs. mRNA Delivery: A Strategic Guide to Optimizing Editing Efficiency for Therapeutics
This article provides a comprehensive analysis for researchers and drug development professionals on the critical choice between delivering the CRISPR-Cas9 system as a pre-complexed protein (RNP) or as mRNA.
Cas9 Protein vs. mRNA Delivery: A Strategic Guide to Optimizing Editing Efficiency for Therapeutics
Abstract
This article provides a comprehensive analysis for researchers and drug development professionals on the critical choice between delivering the CRISPR-Cas9 system as a pre-complexed protein (RNP) or as mRNA. We explore the foundational principles of each cargo type, detailing advanced delivery methodologies like lipid nanoparticles (LNPs) and virus-like particles (VLPs). The content delves into troubleshooting key challenges such as off-target effects, immunogenicity, and editing kinetics, while presenting the latest optimization strategies from AI-guided LNP design to novel nanostructures. Finally, we offer a comparative validation of therapeutic applications, synthesizing data from recent clinical trials and preclinical studies to guide the selection and refinement of CRISPR-based therapies for enhanced efficacy and safety.
Cas9 Cargo Fundamentals: Understanding Protein RNP and mRNA Delivery Mechanisms
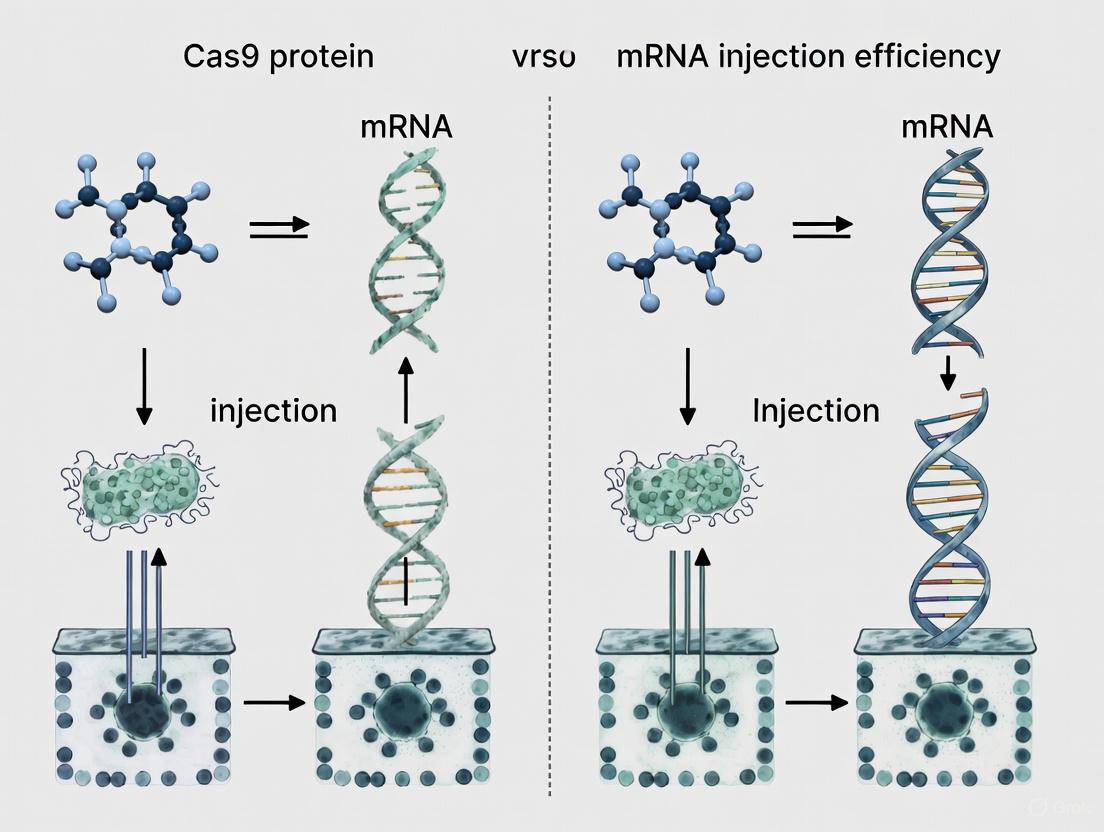
For researchers optimizing the efficiency of Cas9 protein versus mRNA delivery, the choice of cargo form is a fundamental experimental design decision. The CRISPR-Cas9 system can be delivered as plasmid DNA (pDNA), messenger RNA (mRNA), or a pre-assembled ribonucleoprotein (RNP) complex. Each form has distinct implications for editing kinetics, specificity, biosafety, and delivery requirements [1]. This guide provides a technical breakdown of these cargo types, supported by experimental data and protocols, to help you troubleshoot common issues and select the optimal strategy for your application.
Cargo Comparison and Selection Guide
The table below summarizes the key characteristics of the three primary CRISPR cargo forms to inform your experimental design.
| Cargo Form | Key Advantages | Key Disadvantages | Ideal Use Cases |
|---|---|---|---|
| Plasmid DNA (pDNA) | Cost-effective for production; stable and easy to handle [2]. | Requires nuclear entry; prolonged Cas9 expression increases off-target effects and immune responses [2] [1]. | Large-scale, low-cost screening where high specificity is not the primary concern. |
| mRNA | Faster editing onset than pDNA; transient expression reduces off-target risks [2]. | Requires in-cell translation; can trigger innate immune responses [2]. | Applications requiring faster results than pDNA but where RNP delivery is inefficient. |
| Ribonucleoprotein (RNP) | Fastest editing kinetics (immediately active); highest specificity; minimal off-target effects; low immunogenicity; transient activity [3] [2] [1]. | More complex production; limited shelf-life; challenging delivery in some systems [4]. | Therapeutic applications [3], sensitive cells (e.g., stem cells [4]), and experiments demanding the highest fidelity. |
Frequently Asked Questions (FAQs) and Troubleshooting
FAQ 1: Why is RNP often considered the gold standard for therapeutic development?
RNP complexes are favored for therapeutics due to their superior safety and specificity profile. Because the complex is active immediately upon delivery and degrades rapidly inside the cell, the window for off-target editing is significantly reduced compared to pDNA and mRNA, which require transcription and/or translation and lead to prolonged Cas9 expression [3] [2] [1]. Furthermore, RNP delivery avoids the risk of unintended integration of foreign DNA into the host genome, and it elicits a lower immune response than nucleic acid-based delivery methods [3].
FAQ 2: My gene editing efficiency is low with RNPs. What could be the cause?
Low efficiency with RNPs is often a delivery issue. Consider the following troubleshooting steps:
- Verify Delivery Method: RNPs are large, charged complexes. Ensure your delivery method (e.g., electroporation, lipofection with specialized reagents) is optimized for large macromolecules [4]. Standard plasmid transfection reagents may not be effective.
- Check RNP Quality and Stability: Use fresh, properly assembled RNPs. The complex can be unstable, and improper assembly or storage can lead to dissociation and loss of activity.
- Optimize N/P Ratio: When using polymeric or lipid-based nanoparticles, the charge ratio between cationic polymers (N) and anionic phosphates of the nucleic acid/protein (P) is critical. An optimal N/P ratio ensures efficient complex formation, cellular uptake, and endosomal escape [3] [2].
FAQ 3: Can I improve HDR efficiency when using RNP complexes?
Yes. Homology-Directed Repair (HDR) is generally less efficient than error-prone Non-Homologous End Joining (NHEJ). To enhance HDR with RNPs:
- Use the TILD-CRISPR Method: Co-deliver your RNP with a linearized double-stranded DNA (dsDNA) donor template featuring long homology arms (e.g., 1000 bp). One study demonstrated that this approach achieved 50% integration efficiency in CHO-K1 cells, significantly outperforming standard methods [3].
- Synergize with Small Molecules: Adding small molecules that modulate DNA repair pathways can boost specific editing outcomes. For instance, Repsox, an inhibitor of the TGF-β pathway, has been shown to enhance NHEJ efficiency [5].
Experimental Protocols for Enhancing Editing Efficiency
Protocol 1: Enhancing NHEJ Efficiency with Small Molecules
This protocol is adapted from a study that successfully increased CRISPR/Cas9-mediated NHEJ gene editing efficiency in porcine PK15 cells [5].
- Cell Preparation: Culture PK15 cells in DMEM supplemented with 15% FBS. Trypsinize and count the cells.
- Electroporation: For RNP delivery, pre-incubate 10 µg of Cas9 protein with 100 pmol of sgRNA at room temperature for 10 minutes to form the RNP complex. Electroporate 1 x 10^6 cells with the RNP complex using a square-wave electroporator (e.g., CUY21EDIT II) with parameters set to 150 V, 10 ms, and 3 pulses [5].
- Small Molecule Treatment: Immediately after electroporation, add the optimal concentration of a small molecule to the culture medium. Based on the study:
- Repsox: Showed the greatest effect, increasing NHEJ efficiency by 3.16-fold.
- Zidovudine, GSK-J4, IOX1: Also showed significant improvements (1.17-fold, 1.16-fold, and 1.12-fold, respectively) [5].
- Analysis: Harvest cells 48-72 hours post-electroporation. Analyze editing efficiency via T7E1 assay, TIDE analysis, or next-generation sequencing.
Protocol 2: In Vivo Transfection via Electroporation
This protocol outlines a method for in vivo gene editing in mouse seminiferous tubules, which can be adapted for other tissues [6].
- Animal Preparation: Anesthetize the mouse (e.g., 3-5 weeks old) via intraperitoneal injection of lidocaine. Ensure no response to external stimuli and stable vital signs.
- Surgical Exposure: Make a 1 cm incision to access the abdominal cavity. Gently extract the testes and place them on sterile, saline-moistened filter paper.
- Microinjection: Clamp the efferent ductules. Using a glass needle, inject your cargo (e.g., EGFP-N1 plasmid at 1 µg/µL) directly into the seminiferous tubules [6].
- Electroporation: Apply electrode forceps on both sides of the testis. Deliver electrical stimulation using a square-wave electroporation device (e.g., ECM 830) with parameters of 8 pulses at 50 ms per pulse [6].
- Post-operative Care: Return the testes to the abdominal cavity and suture the incision. Monitor the animal until it recovers from anesthesia.
Research Reagent Solutions
The table below lists key reagents and their functions as cited in the research.
| Reagent / Material | Function in Experiment | Citation |
|---|---|---|
| Cationic Hyper-Branched Cyclodextrin-based Polymer (Ppoly) | A nanocarrier for encapsulating and delivering RNP complexes, demonstrating high efficiency and low cytotoxicity. | [3] |
| Repsox | A small molecule TGF-β signaling pathway inhibitor used to enhance the efficiency of NHEJ-mediated gene editing. | [5] |
| PAMAM Dendrimer (G6-OH) | A hyper-branched polymeric nanoparticle used for covalent conjugation and delivery of Cas9 RNP. | [4] |
| Lipid Nanoparticles (LNPs) | Synthetic nanoparticles used for the in vivo encapsulation and delivery of CRISPR cargo (pDNA, mRNA, or RNP). | [2] [7] |
| CRISPRMAX | A commercial lipid-based transfection reagent used as a benchmark for comparing the performance of novel delivery systems. | [3] |
Cargo Selection and Cellular Processing Pathways
The following diagram illustrates the fundamental differences in how each cargo form is processed within the cell to achieve genome editing, which directly impacts editing kinetics and specificity.
Experimental Workflow for Cargo Delivery and Analysis
This workflow provides a generalized overview of the key steps involved in a CRISPR-Cas9 experiment, from cargo preparation to analysis.
The decision to use plasmid DNA, mRNA, or RNP complexes hinges on the specific requirements of your experiment regarding efficiency, specificity, timing, and safety. For the highest editing precision and lowest off-target effects, particularly in therapeutic contexts, the evidence strongly supports the use of RNP complexes. By leveraging the troubleshooting guides, protocols, and reagent information provided, researchers can systematically optimize their CRISPR-Cas9 workflows to achieve robust and reliable genomic editing.
For researchers and drug development professionals focused on optimizing CRISPR-Cas9 delivery, the ribonucleoprotein (RNP) complex method offers distinct advantages over DNA or mRNA-based approaches. RNP delivery involves the direct introduction of preassembled Cas9 protein and single-guide RNA (sgRNA) complexes into cells, enabling immediate genome editing activity. This method addresses critical challenges in therapeutic development, including off-target effects, immunogenicity, and timing control. This technical support center provides comprehensive guidance on leveraging RNP delivery's inherent strengths for your research, complete with troubleshooting advice and experimental protocols.
Core Advantages: The RNP Value Proposition
Rapid Editing Activity
Direct delivery of preassembled Cas9 RNP complexes enables immediate genome editing activity upon reaching the cell nucleus, eliminating the transcription and translation steps required for DNA or mRNA approaches [1] [8]. This rapid activity is particularly valuable for working with cells having low transcription and translation activity, including embryonic stem cells, induced pluripotent stem cells, and tissue stem cells [8].
Enhanced Specificity & Reduced Off-Target Effects
The transient nature of RNP activity—typically remaining in cells for only 6-24 hours—significantly reduces off-target effects by limiting the time window during which unintended genomic edits can occur [1] [8] [9]. Studies demonstrate that RNP delivery decreases off-target mutations relative to plasmid transfection methods [9]. The minimal duration of RNP activity also lowers risks of insertional mutagenesis and immune responses [8].
Table 1: Quantitative Comparison of RNP Delivery Advantages
| Performance Metric | RNP Delivery | DNA Plasmid Delivery | mRNA Delivery |
|---|---|---|---|
| Time to Activity | Immediate (hours) [8] | Delayed (24-72 hours) [8] | Moderate (12-48 hours) [8] |
| Duration of Activity | Short (transient, 6-24 hours) [8] | Prolonged (days) [8] | Moderate (hours to days) [8] |
| Off-Target Editing | Significantly reduced [8] [9] | Higher risk [1] [8] | Moderate risk [8] |
| Immune Response | Lower immunogenicity [8] | Higher immunogenicity [8] | Moderate immunogenicity [10] |
| Editing Efficiency in Hard-to-Transfect Cells | High [8] [9] | Variable [1] | Variable [1] |
Experimental Workflow for RNP Delivery
RNP Delivery Methods: Technical Comparison
Physical Delivery Approaches
Physical methods directly introduce RNPs into cells by temporarily disrupting cell membranes:
- Electroporation: Application of electrical pulses creates temporary pores in cell membranes. This method shows high efficiency in hematopoietic stem cells and immune cells [11] [8].
- Microinjection: Direct mechanical injection using glass micropipettes allows quantitative control of delivered RNP complexes, though it requires specialized equipment and expertise [8].
Synthetic Carrier Systems
Chemical and biological carriers facilitate RNP delivery through cellular uptake mechanisms:
- Lipid Nanoparticles (LNPs): Synthetic nanoparticles that encapsulate RNPs and facilitate cellular delivery through endocytosis. Recent advancements enable tissue-specific targeting through surface modifications [12] [1].
- Cell-Derived Nanovesicles (Gesicles): Naturally derived membrane vesicles that can package RNP complexes and achieve genome editing in broad cell types with reduced off-target effects [11].
Table 2: RNP Delivery Method Comparison for Experimental Planning
| Delivery Method | Mechanism | Best Applications | Efficiency | Technical Complexity |
|---|---|---|---|---|
| Electroporation [11] [8] | Electrical field-induced membrane pores | Suspension cells (HSCs, lymphocytes) [11] | High | Moderate |
| Microinjection [8] | Direct mechanical injection | Embryos, oocytes, single cells [8] | Very High | High |
| Lipid Nanoparticles [12] [1] | Endocytosis of encapsulated RNPs | In vivo delivery, therapeutic applications [12] | Moderate-High | Moderate |
| Gesicles/Nanovesicles [11] | Membrane fusion-mediated delivery | Broad cell types, reduced off-target editing [11] | Moderate | Low-Moderate |
Technical Support Center
Frequently Asked Questions (FAQs)
Q: What is the recommended molar ratio for forming Cas9 RNP complexes? A: Research indicates optimal gene editing occurs at Cas9:sgRNA molar ratios of 1:3 to 1:5. Studies using HeLa reporter cells demonstrated higher editing efficiency at these ratios compared to 1:1 ratio [12]. The increased sgRNA concentration promotes complete complex formation and enhances target recognition.
Q: How does RNP delivery reduce immune responses compared to other methods? A: RNP delivery minimizes immune activation because the Cas9 protein and modified sgRNAs are less immunogenic than viral vectors or bacterial DNA sequences present in plasmids [8]. Additionally, the transient presence of RNPs (versus sustained expression from DNA vectors) further reduces immune recognition [8].
Q: What storage conditions maintain RNP complex stability? A: RNP-loaded nanoparticles maintain size uniformity (PDI < 0.2) and constant gene editing activity when stored at 4°C for up to 60 days [12]. For long-term storage, consider freezing individual RNP components at -80°C and forming complexes immediately before use.
Q: Can RNP delivery be used for in vivo therapeutic applications? A: Yes, recent advances in LNP technology enable systemic RNP delivery to multiple tissues. Intravenous injection of RNP-loaded LNPs has achieved tissue-specific, multiplexed editing in mouse lungs, liver, and brain [12]. This approach has been used to restore dystrophin expression in DMD mouse models and significantly decrease serum PCSK9 levels [12].
Troubleshooting Guide
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low editing efficiency | Suboptimal RNP complex formation [9] | Verify guide RNA concentration and maintain recommended Cas9:sgRNA ratios (1:3-1:5) [12] [9] |
| Inadequate nuclear localization | Include nuclear localization signals on Cas9 protein [12] | |
| High cell toxicity | Excessive RNP concentration [9] | Titrate RNP dose; use modified sgRNAs to reduce required concentration [9] |
| Delivery method toxicity | Optimize electroporation parameters or try alternative delivery methods [8] | |
| Inconsistent editing between replicates | Variable RNP delivery | Standardize delivery protocol; use internal controls; consider gesicles for more consistent delivery [11] |
| Poor delivery in hard-to-transfect cells | Inefficient cellular uptake | Utilize selective organ targeting (SORT) nanoparticles or cell-penetrating peptide conjugates [1] |
Essential Research Reagent Solutions
Table 3: Key Reagents for RNP Experiments
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Cas9 Proteins | Electroporation-grade Cas9 [11] | Optimized for stem cells and primary cells; higher purity reduces toxicity |
| Guide RNAs | Chemically synthesized sgRNAs with 2'-O-methyl modifications [9] | Enhanced stability against nucleases; improved editing efficiency; reduced immune stimulation |
| Delivery Materials | Cationic lipids (DOTAP) [12], Gesicles [11] | DOTAP enables RNP encapsulation in LNPs; Gesicles provide natural delivery mechanism |
| Validation Tools | T7 Endonuclease I assay [12], NGS sequencing [9] | T7EI for quick efficiency estimate; NGS for comprehensive sequence analysis |
Advanced RNP Delivery Workflow
The inherent strengths of Cas9 RNP delivery—particularly its high specificity and rapid activity—make it an indispensable approach for researchers optimizing CRISPR-based therapies. By implementing the protocols, troubleshooting guides, and experimental strategies outlined in this technical support center, research teams can effectively leverage RNP delivery to advance their therapeutic development pipelines while minimizing off-target effects and immune responses. As delivery technologies continue evolving, particularly in nanoparticle design and modification strategies, RNP delivery is poised to remain at the forefront of precision genome editing for both research and clinical applications.
Technical Support & Troubleshooting Guide
Frequently Asked Questions (FAQs)
1. What are the primary safety advantages of using Cas9 mRNA over DNA-based delivery systems?
The primary safety advantages stem from the transient nature of mRNA and its cytoplasmic activity. Unlike plasmid DNA, mRNA does not need to enter the nucleus to be active and lacks the genetic elements required for integration into the host genome. This fundamentally eliminates the risk of unintended insertional mutagenesis. Furthermore, its short half-life limits the window of Cas9 protein expression, thereby reducing the probability of off-target editing events, which are more common with persistent Cas9 expression from DNA vectors [13].
2. Why is Lipid Nanoparticle (LNP) delivery often paired with Cas9 mRNA for in vivo applications?
LNPs are the leading non-viral delivery vehicle for Cas9 mRNA due to their proven clinical success, ease of assembly, and ability to protect the fragile mRNA cargo from degradation by nucleases in the blood [13] [1]. They form stable complexes with nucleic acids, exhibit low immunogenicity compared to viral vectors, and can be engineered for specific tissue targeting (e.g., to the liver or lungs) [13] [14]. Their use was validated in mRNA COVID-19 vaccines and is now being applied to CRISPR therapies [1] [7].
3. What are the common causes of low editing efficiency when using Cas9 mRNA and how can they be addressed?
Low editing efficiency can arise from several factors:
- mRNA Instability: Use engineered mRNA with optimized codons and modified nucleotides (e.g., pseudouridine) to enhance stability and translation efficiency while reducing immunogenicity [13].
- Inefficient Delivery: Ensure your LNP formulation or transfection reagent is optimized for your target cell type. Consider using selective organ targeting (SORT) LNPs for specific tissues [1].
- Suboptimal sgRNA Design: Utilize bioinformatics tools to design highly specific sgRNAs with high on-target activity. It is often necessary to test multiple sgRNAs for each target to identify the most effective one [15].
4. Can Cas9 mRNA therapies be re-dosed, and what are the considerations?
Yes, a significant advantage of LNP-delivered mRNA over viral vector delivery is the potential for re-dosing. Viral vectors often elicit strong immune responses against the vector itself, making repeated administration ineffective or dangerous. In contrast, LNPs do not trigger the same level of immune memory against the delivery vehicle. There are already clinical precedents: an infant with CPS1 deficiency safely received three LNP doses, and participants in an Intellia Therapeutics trial for hATTR received a second, higher dose, with each subsequent dose increasing therapeutic efficacy [7].
Troubleshooting Common Experimental Issues
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Low Cell Viability | Cytotoxicity from transfection reagents or immune response to exogenous mRNA. | Titrate LNP/nanoparticle doses to find optimal balance; use modified nucleotides in mRNA synthesis to dampen immune activation [13] [1]. |
| High Immunogenicity | Recognition of in vitro transcribed mRNA by cellular pattern recognition receptors (e.g., TLRs, RIG-I). | Incorporate purified, base-modified mRNAs (e.g., N1-methylpseudouridine) to avoid dsRNA contaminants and reduce TLR activation [13]. |
| Inconsistent Editing | Batch-to-batch variability in mRNA quality or LNP formulation; variable transfection efficiency. | Use high-quality, HPLC-purified mRNA; standardize LNP formulation protocols; include a reporter to monitor transfection efficiency [2] [15]. |
| Poor In Vivo Performance | Rapid degradation of mRNA in serum; accumulation in non-target organs. | Optimize LNP lipid composition for enhanced stability and organ selectivity (e.g., SORT molecules); utilize organ-specific promoters [1] [14]. |
Experimental Protocols & Workflows
Protocol 1: Assessing Cas9 mRNA Delivery and Editing Efficiency
This protocol outlines a standard workflow for evaluating the performance of a Cas9 mRNA-based editing system in vitro.
1. mRNA Preparation:
- Template Design: Use a plasmid DNA template containing Cas9 codon-optimized for your target organism, flanked by 5' and 3' UTRs known to enhance stability (e.g., β-globin UTRs). Include a poly(A) tail.
- In Vitro Transcription (IVT): Synthesize the mRNA using an IVT kit. To reduce immunogenicity, include modified nucleotides such as pseudouridine in the reaction [13].
- Purification: Purify the transcribed mRNA using HPLC or affinity chromatography to remove aberrant double-stranded RNA (dsRNA) contaminants, which are potent inducers of interferon responses [13].
2. Delivery:
- Complexation: Formulate the purified Cas9 mRNA and synthetic sgRNA at an optimal mass ratio with your chosen delivery vehicle (e.g., LNPs, commercial transfection reagents).
- Transfection: Deliver the complexes to your target cells. For hard-to-transfect cells like primary T-cells or stem cells, electroporation may yield higher efficiency than lipid-based methods [2] [16].
3. Validation:
- Efficiency Analysis: 72 hours post-transfection, harvest genomic DNA. Assess editing efficiency at the target locus using T7 Endonuclease I assay, TIDE analysis, or next-generation sequencing.
- Off-Target Assessment: Use GUIDE-seq or in silico prediction tools followed by deep sequencing of top potential off-target sites to profile specificity [17] [2].
- Functional Confirmation: Confirm successful gene knockout via western blot (for protein loss) or a functional assay relevant to the target gene.
Protocol 2: In Vivo Gene Editing via LNP-mRNA
This protocol summarizes the key steps for achieving gene editing in living animal models, as demonstrated in recent high-impact studies [14] [7].
1. LNP Formulation Optimization:
- Component Selection: Prepare an LNP mixture containing ionizable cationic lipids, phospholipids, cholesterol, and PEG-lipids. The exact composition can be tuned for organ tropism (e.g., adding SORT molecules for lung targeting).
- Encapsulation: Use microfluidics to mix the lipid solution with an aqueous buffer containing the Cas9 mRNA and sgRNA, forming stable, monodisperse LNPs encapsulating the cargo.
- Quality Control: Characterize LNPs for size (e.g., 80-100 nm via DLS), polydispersity index, and mRNA encapsulation efficiency [1] [14].
2. Administration and Analysis:
- Dosing: Administer the LNP-mRNA formulation intravenously via tail-vein injection in mice. Dose is typically measured as mg of mRNA per kg of animal weight.
- Tissue Harvesting: After an appropriate period (e.g., 7-14 days), harvest the target organs (e.g., liver, lungs).
- Editing Quantification: Extract genomic DNA from the tissue and quantify editing efficiency at the target locus using next-generation sequencing. Editing percentages are calculated as the proportion of indel-containing reads.
The table below summarizes quantitative data from a key study using this approach:
Table: In Vivo Editing Efficiency of LNP-Delivered CRISPR Systems [14]
| Cas9 Editor Formulation | Target Organ | Target Gene | Average Editing Efficiency | Key Finding |
|---|---|---|---|---|
| iGeoCas9 RNP-LNPs (Biodegradable) | Liver | Reporter (Ai9 mice) | 37% | Efficient editing in a majority of liver tissue. |
| iGeoCas9 RNP-LNPs (Biodegradable) | Liver | PCSK9 | 31% | Therapeutically relevant level of gene disruption. |
| iGeoCas9 RNP-LNPs (Cationic) | Lung | Reporter (Ai9 mice) | 16% | Significant editing across entire lung tissue. |
| iGeoCas9 RNP-LNPs (Cationic) | Lung | Disease-causing SFTPC | 19% | Major improvement over previous viral/nonviral methods. |
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for Cas9 mRNA Delivery Experiments
| Reagent / Material | Function | Key Considerations |
|---|---|---|
| Codon-Optimized Cas9 mRNA | Template for in vivo translation of the Cas9 nuclease. | Select mRNA with base modifications (e.g., pseudouridine) for enhanced stability and reduced immunogenicity [13]. |
| Synthetic sgRNA | Guides the Cas9 protein to the specific genomic target sequence. | High-purity, chemically modified sgRNA can improve stability and reduce off-target effects [13] [15]. |
| Ionizable Lipids | Key component of LNPs; enables self-assembly, encapsulation, and endosomal escape. | Critical for in vivo efficacy. New biodegradable ionizable lipids are improving safety profiles [1] [14]. |
| Selective Organ Targeting (SORT) Molecules | Lipids engineered to direct LNPs to specific tissues beyond the liver (e.g., lungs, spleen). | Essential for expanding therapeutic applications to other organs [1]. |
| Electroporation Systems | Physical delivery method using electrical pulses to create pores in cell membranes. | Preferred method for hard-to-transfect cells like primary immune cells and stem cells (e.g., in CASGEVY) [2] [16]. |
| CD73-IN-13 | CD73-IN-13, MF:C13H11F3N4O2, MW:312.25 g/mol | Chemical Reagent |
| Sonnerphenolic B | Sonnerphenolic B, MF:C18H18O3, MW:282.3 g/mol | Chemical Reagent |
Frequently Asked Questions (FAQs)
Q1: What are the key differences between delivering Cas9 as mRNA versus as a pre-assembled protein (RNP)?
The choice of cargo significantly impacts editing efficiency, specificity, and immunogenicity [2] [1].
- Cas9 mRNA: Once delivered into the cell, mRNA must be translated by ribosomes to produce the Cas9 protein, which then complexes with the gRNA to form the active RNP [2]. This process leads to a slower onset of editing activity and more prolonged Cas9 expression, which can increase the risk of off-target effects [1]. Furthermore, exogenous mRNA can be highly immunogenic, potentially triggering strong innate immune responses [16].
- Cas9 RNP (Ribonucleoprotein): This method involves delivering the pre-assembled complex of the Cas9 protein and guide RNA directly into the cell [2] [1]. RNPs are immediately active and have a shorter intracellular lifetime, leading to faster editing kinetics and a significant reduction in off-target effects due to transient activity [1] [17]. RNP delivery generally results in lower cytotoxicity and reduced immunogenicity compared to mRNA or DNA delivery methods [2].
Q2: Why is nuclear delivery a major barrier for CRISPR-Cas9 editing, especially in non-dividing cells?
The CRISPR-Cas9 complex must ultimately localize to the nucleus to access the genomic DNA. The nuclear envelope, which is intact in non-dividing cells, presents a formidable physical barrier [16]. The Cas9 protein and its RNP complex have a large molecular weight and lack the intrinsic ability to efficiently traverse the nuclear pore complex [16]. While many strategies fuse the Cas9 protein with Nuclear Localization Signals (NLSs) to facilitate nuclear import, this is not always fully efficient, and a significant portion of the editing machinery may fail to reach its target [18] [19]. Overcoming this barrier is a primary focus of delivery system optimization.
Q3: How can I mitigate immunogenic responses to CRISPR components?
Immunogenicity can be directed against both the delivery vehicle and the CRISPR cargo itself.
- Pre-existing Immunity: Pre-existing immunity to Cas9 proteins from common bacterial exposures has been documented in human serum, which could potentially neutralize the therapy [2].
- Delivery Vehicle Choice: Viral vectors, particularly adenoviruses, can provoke strong immune reactions [1] [16]. Non-viral methods, such as Lipid Nanoparticles (LNPs), are generally associated with milder immune responses [2] [1].
- Cargo Selection: Using RNP complexes can reduce immunogenicity compared to prolonged expression from DNA or mRNA [1]. Researchers are also exploring the use of engineered, humanized, or high-fidelity Cas9 variants to minimize immune recognition [2] [16].
Q4: What strategies can improve the stability and delivery efficiency of Cas9 RNPs?
Stability is crucial for ensuring that a sufficient amount of intact RNP reaches the nucleus.
- Advanced Nanocarriers: Lipid Nanoparticles (LNPs) and other nanocages (e.g., apoferritin) are highly effective at protecting RNPs from degradation during delivery [2] [19].
- Protein Engineering: Addressing Cas9 protein aggregation is critical, as aggregates can compromise encapsulation efficiency and cellular uptake [2]. Optimizing buffer conditions and protein formulations can help maintain Cas9 in a monodisperse state.
- Enhanced Nuclear Import: As mentioned, NLS tags are standard. Novel approaches are also being developed, such as using small molecule "nuclear triggers" (e.g., doxorubicin) to actively facilitate the nuclear translocation of RNP complexes, even in the absence of a traditional NLS [19].
Troubleshooting Guides
Problem 1: Low Gene Editing Efficiency
A lack of efficient editing can stem from failures at multiple points in the delivery and action pathway.
| Possible Cause | Verification Method | Proposed Solution |
|---|---|---|
| Inefficient cellular uptake | Measure transfection/transduction efficiency using a fluorescent reporter. | Optimize delivery vehicle parameters (e.g., N:P ratio for LNPs) [2]; try alternative delivery methods (e.g., electroporation for hard-to-transfect cells) [16]. |
| Poor endosomal escape | Use confocal microscopy with endosomal/lysosomal markers. | Switch to or formulate delivery vehicles known for enhanced endosomal escape, such as ionizable LNPs or polymers [1]. |
| Inefficient nuclear import | Use fluorescently labeled Cas9 to visualize localization. | Ensure Cas9 is fused with a strong Nuclear Localization Signal (NLS) [18]; consider novel nuclear delivery strategies [19]. |
| Low cargo activity/ stability | Perform gel electrophoresis or other assays to check cargo integrity pre-delivery. | Use fresh, high-quality RNPs; optimize RNP formation conditions; use codon-optimized mRNA if using mRNA cargo [17]. |
| Low expression in target cell type | Check Cas9 and gRNA expression levels via qPCR/Western blot. | Use a cell-type-specific promoter to drive expression if using nucleic acid cargo [17]. |
Experimental Protocol: Assessing Nuclear Localization of Cas9 RNP
- Labeling: Fluorescently label the Cas9 protein (e.g., with a green dye like FITC) and the gRNA (e.g., with a red dye like Cy5).
- Transfection: Deliver the labeled RNP complex into your target cells using your standard method.
- Staining: At a predetermined timepoint (e.g., 6-24 hours post-transfection), fix the cells and stain the nucleus with a blue fluorescent DNA dye (e.g., DAPI).
- Imaging: Analyze the cells using confocal microscopy.
- Analysis: Colocalization of the green (Cas9) and red (gRNA) signals with the blue (nucleus) signal indicates successful nuclear delivery. The absence of signal in the nucleus suggests a failure at the uptake or nuclear import stage.
This workflow helps diagnose whether the bottleneck is nuclear delivery or an earlier step:
Problem 2: High Off-Target Effects or Cell Toxicity
Unwanted editing and cell death are often linked to the persistence and quantity of active Cas9.
| Symptom | Possible Cause | Proposed Solution |
|---|---|---|
| High off-target editing | Prolonged Cas9 activity from DNA/mRNA expression; low-specificity gRNA. | Switch to RNP delivery for transient activity [1]; use high-fidelity Cas9 variants (e.g., SpCas9-HF1) [2] [17]; improve gRNA design using prediction tools [17]. |
| High cell toxicity/death | Excessive cargo load; immunogenic response; delivery method toxicity. | Titrate down the concentration of CRISPR components [17]; use RNP instead of plasmid DNA to reduce cytotoxicity [2]; optimize delivery vehicle (e.g., use LNPs instead of some viral vectors) [1]. |
| Inflammatory response | Recognition of CRISPR cargo (e.g., bacterial Cas9) or vehicle by the immune system. | Use purified RNP complexes; select non-viral delivery systems like LNPs [2] [7]; consider immunosuppressive agents if applicable to the model. |
Experimental Protocol: Titering RNP Complexes to Minimize Toxicity
- Preparation: Prepare a dilution series of your Cas9 RNP complex (e.g., 0.1 µM, 0.5 µM, 1 µM, 2 µM).
- Delivery: Transfect cells with each concentration using a consistent protocol.
- Assessment: 24-48 hours post-transfection, assess cell viability using a standard assay (e.g., MTT, CellTiter-Glo).
- Analysis: Calculate the percentage of viable cells for each concentration. Choose the lowest RNP concentration that yields acceptable editing efficiency (as measured by a separate assay) while maintaining high cell viability (>80%).
The table below summarizes quantitative data on editing efficiencies from various delivery methods and cargos as reported in the literature. This provides a benchmark for expected outcomes.
| Delivery Method / Cargo Type | Target / Model | Editing Efficiency | Key Findings | Citation |
|---|---|---|---|---|
| Electroporation of RNP (Ex vivo, CASGEVY) | BCL11A in HSPCs | Up to 90% indels | First FDA-approved ex vivo CRISPR therapy; demonstrates high efficiency in human hematopoietic stem cells. | [2] |
| LNP delivering mRNA | Various in vivo targets | Highly efficient (data varies) | Platform demonstrated during COVID-19; high translational potential for liver/lung targets. | [2] [7] |
| Apoferritin delivering RNP (no NLS) | Lcn2 in MDA-MB-231 cells (in vitro) | ~33% | Novel delivery system achieving significant editing without traditional NLS tags. | [19] |
| Apoferritin delivering RNP (no NLS) | copGFP in HeLa model (in vivo) | 16% | Proof-of-concept for in vivo therapeutic application of NLS-independent nuclear delivery. | [19] |
| Microinjection | Various embryos | ~40% (e.g., GFP in HepG2) | Direct physical method allowing precise control over delivered dose. | [2] |
The Scientist's Toolkit: Essential Research Reagents
This table lists key reagents and their functions for troubleshooting core challenges in Cas9 delivery.
| Reagent / Material | Function in Experiment | Key Consideration |
|---|---|---|
| High-Fidelity Cas9 Variant | Engineered nuclease with reduced off-target effects. | Essential for therapeutic applications where specificity is critical [2] [17]. |
| Ionizable Lipid Nanoparticles (LNPs) | Synthetic nanoparticles for encapsulating and delivering nucleic acids or RNPs. | Excellent for in vivo delivery; naturally target liver; can be engineered for other tissues (SORT-LNPs) [2] [1]. |
| Nuclear Localization Signal (NLS) Peptides | Short amino acid sequences fused to Cas9 to promote its import into the nucleus. | A critical component for efficient editing, especially in non-dividing cells [18] [16]. |
| Apoferritin Nanocages | A biological nanocage used as an alternative delivery vector for RNP complexes. | Can facilitate nuclear delivery via alternative pathways, potentially bypassing NLS requirements [19]. |
| Electroporation System | Physical method using electrical pulses to create transient pores in cell membranes for cargo delivery. | Highly effective for hard-to-transfect cells like primary cells and stem cells (ex vivo) [2] [16]. |
| Aristolan-1(10)-en-9-ol | Aristolan-1(10)-en-9-ol|Natural Sesquiterpene|RUO | Aristolan-1(10)-en-9-ol is a sesquiterpene with researched sedative effects via the GABAergic system. For Research Use Only. Not for human or veterinary use. |
| Ara-F-NAD+ sodium | Ara-F-NAD+ sodium, MF:C21H25FN7NaO13P2, MW:687.4 g/mol | Chemical Reagent |
Cellular Uptake and Intracellular Trafficking Pathways for Different Cargoes
Fundamental Concepts: Cargo Types and Their Intracellular Journeys
FAQ: What are the primary forms of CRISPR-Cas9 cargo, and how do their intracellular trafficking pathways differ?
Answer: CRISPR-Cas9 systems can be delivered into cells in three primary forms, each with distinct pathways for cellular uptake, intracellular trafficking, and nuclear entry. The choice of cargo significantly impacts editing efficiency, kinetics, and off-target effects.
The table below summarizes the key characteristics of each cargo type:
Table 1: Comparison of CRISPR-Cas9 Cargo Types and Their Properties
| Cargo Type | Composition | Primary Uptake Mechanism | Intracellular Trafficking & Processing | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| DNA (Plasmid) | DNA plasmid encoding Cas9 and sgRNA [1] | Endocytosis (varies by delivery vector) [20] | Endosomal escape → Nuclear import → Transcription → Translation → Nuclear import of Cas9 protein [20] | Sustained, long-term expression [13] | Risk of host genome integration; prolonged expression increases off-target effects [13] |
| mRNA | mRNA encoding Cas9 + separate sgRNA [1] | Endocytosis (varies by delivery vector) [20] | Endosomal escape → Cytoplasmic translation → Nuclear import of Cas9 protein [13] | No genomic integration risk; shorter half-life reduces off-target effects [13] | mRNA instability and susceptibility to nucleases; can trigger immune responses [13] |
| Ribonucleoprotein (RNP) | Pre-complexed Cas9 protein and sgRNA [1] | Endocytosis [21] or membrane fusion (specialized systems) [21] | Endosomal escape → Direct nuclear import (no translation needed) [21] | Immediate activity; highest precision and lowest off-target effects; transient existence [13] [21] [14] | Difficulties in large-scale production and in vivo delivery [13] |
The following diagram illustrates the general intracellular trafficking pathways for these cargo types, from cellular uptake to nuclear entry and editing.
Diagram 1: Intracellular trafficking pathways for DNA, mRNA, and RNP cargo. A critical bottleneck for all cargo types is endosomal escape; failure results in degradation.
Troubleshooting Common Experimental Challenges
FAQ: Our CRISPR editing efficiency is low. Is this a cargo problem or a delivery problem?
Answer: Low editing efficiency can stem from issues with either the cargo or the delivery vector, and often both. A systematic approach is needed to diagnose the problem. The following troubleshooting guide outlines common issues and solutions.
Table 2: Troubleshooting Guide for Low CRISPR Editing Efficiency
| Observed Problem | Potential Causes | Recommended Solutions & Experiments |
|---|---|---|
| Low Protein Expression (mRNA cargo) | mRNA instability or poor translation efficiency [13] [22]. | 1. Optimize mRNA sequence: Use codon optimization tools (e.g., RiboDecode) to enhance stability and translation [22]. 2. Incorporate modified nucleotides (e.g., m1Ψ) to reduce immunogenicity [22]. 3. Engineer UTRs: Introduce AU-rich elements (e.g., "AUUUA" repeats) in the 3' UTR to enhance stability and translation [23]. |
| High Cytotoxicity or Immune Response | Delivery vector toxicity; innate immune recognition of foreign nucleic acids [13]. | 1. Switch cargo type: Use RNP complexes to minimize TLR activation compared to mRNA [14]. 2. Purify RNP preparations to remove contaminants. 3. Use LNP vectors with low immunogenicity profiles [13]. |
| Inefficient Nuclear Delivery | Cargo fails to be imported into the nucleus [20]. | 1. Fuse Nuclear Localization Signals (NLS): Attach NLS to the C-terminus, N-terminus, or both termini of the Cas9 protein. Research shows different NLS configurations can yield similar high efficiency [24]. 2. Use peptide-based transduction domains to facilitate nuclear entry. |
| Poor Endosomal Escape | Cargo is trapped and degraded in endo-lysosomal compartments [20] [1]. This is a major bottleneck. | 1. Select vectors that promote endosomal escape: Use ionizable LNPs (iLNPs) that become positively charged in the acidic endosomal environment, disrupting the endosomal membrane [20] [14]. 2. Codelivery with endosomolytic agents (e.g., chloroquine) in vitro. |
| Rapid Clearance & Low Biodistribution (in vivo) | The delivery system is recognized and cleared by the host before reaching target cells [25]. | 1. PEGylate nanoparticles to prolong circulation time [20] [25]. 2. Use tissue-selective LNP formulations: Engineer LNPs with selective organ targeting (SORT) molecules to direct them to specific tissues like lung, spleen, or liver [1] [14]. |
FAQ: We are considering switching from Cas9 mRNA to RNP delivery. What are the critical experimental parameters for successful RNP delivery?
Answer: Transitioning to RNP delivery requires optimization of RNP complex formation, delivery vector selection, and experimental timing. Key parameters to consider are listed below.
Table 3: Critical Parameters for RNP Delivery
| Parameter | Considerations & Recommendations |
|---|---|
| RNP Complex Formation | Pre-complex Cas9 protein and sgRNA at an optimal molar ratio (e.g., 1:1.2 to 1:3) in a suitable buffer. Incubate for 10-30 minutes at room temperature before use to ensure proper complex formation [21]. |
| Delivery Vector | Lipid Nanoparticles (LNPs): Optimal for in vivo use. Ensure the LNP formulation is compatible with protein cargo to avoid denaturation. Thermostable Cas9 variants (e.e., iGeoCas9) are more resistant to formulation stress [14].Electroporation: Highly efficient for ex vivo applications. |
| Cellular Uptake Validation | Use flow cytometry to quantify the uptake of fluorescently labeled RNPs. Employ correlative light and electron microscopy (CLEM) to visualize intracellular trafficking and confirm endosomal escape [26]. |
| Timeline for Analysis | RNP action is rapid. Analyze editing efficiency 24-72 hours post-delivery, as the effects are transient due to protein turnover [21] [14]. |
Detailed Experimental Protocols
Protocol: Quantifying mRNA Cargo Internalization and Uptake Pathways
This protocol is adapted from methodologies used to assess mRNA internalization and trafficking in human primary cells [26].
Objective: To quantify the cellular uptake of mRNA complexes and identify the primary endocytic pathways involved.
Reagents Needed:
- Fluorescently labeled mRNA (e.g., Cy5-labeled)
- Transfection reagent (e.g., Lipofectamine 3000, 3DFect)
- Appropriate cell culture medium and plates
- Endocytic inhibitors: Chlorpromazine (clathrin-mediated inhibitor), Wortmannin (macropinocytosis inhibitor), Genistein (caveolae-mediated inhibitor)
- Flow cytometry buffer (e.g., PBS with 1% BSA)
- Fixative (e.g., 4% PFA) - if required for analysis
Procedure:
- Cell Seeding: Seed cells at an appropriate density (e.g., 5 x 10^4 cells per well in a 24-well plate) and culture until 60-80% confluent.
- Inhibitor Pre-treatment (Optional - for pathway identification): Pre-treat cells with endocytic inhibitors for 1 hour.
- Chlorpromazine: 10 μg/mL
- Wortmannin: 100 nM
- Genistein: 200 μM
- Include a vehicle control (e.g., DMSO).
- Transfection Complex Formation:
- Dilute fluorescently labeled mRNA in a serum-free medium.
- Mix the transfection reagent gently and add it to the diluted mRNA. Use the manufacturer's recommended ratio.
- Incubate the mixture for 15-20 minutes at room temperature to form complexes.
- Transfection:
- Wash cells once with PBS.
- Add the mRNA-transfection reagent complexes to the cells.
- Incubate for the desired time (e.g., 1-4 hours) at 37°C.
- Analysis:
- Flow Cytometry: After incubation, wash cells thoroughly with PBS to remove non-internalized complexes. Trypsinize cells, resuspend in flow cytometry buffer, and analyze immediately using a flow cytometer. Measure the fluorescence intensity of the labeled mRNA in at least 10,000 cells per sample. A significant reduction in fluorescence in inhibitor-treated samples indicates the involvement of that pathway.
- Microscopy (CLEM): For spatial resolution, cells can be fixed after uptake and processed for Correlative Light and Electron Microscopy (CLEM) to visualize the mRNA in endosomes, lysosomes, or the cytosol [26].
Protocol: Assessing RNP Delivery and Editing in Vitro using LNPs
This protocol is based on successful RNP delivery using the MITO-Porter system and other LNP formulations [21] [14].
Objective: To deliver functional Cas9 RNP complexes into cells using lipid nanoparticles and assess genome editing efficiency.
Reagents Needed:
- Purified Cas9 protein (with NLS, e.g., C-NLS-Cas9)
- Target-specific sgRNA
- LNP formulation components (e.g., ionizable lipid, phospholipid, cholesterol, PEG-lipid) or a commercial RNP transfection reagent
- Microfluidic device (e.g., iLiNP device) or standard LNP preparation equipment
- HEPES buffer
- Target cells
Procedure:
- RNP Complex Formation:
- Combine Cas9 protein and sgRNA at a molar ratio of 1:1.2 to 1:3 in a buffer containing HEPES.
- Incubate for 10-30 minutes at room temperature to form the RNP complex [21].
- LNP Encapsulation (using a microfluidic device):
- Prepare the organic phase: Dissolve lipid components (e.g., DOPE, Sphingomyelin, cationic lipid, PEG-lipid) in ethanol.
- Prepare the aqueous phase: The RNP complex in HEPES buffer.
- Use a microfluidic device to mix the organic and aqueous phases at a defined total flow rate and flow rate ratio (e.g., 3:1 aqueous-to-organic ratio) to form LNPs [21].
- Dialyze the resulting LNP formulation against a buffer (e.g., PBS or HEPES) to remove residual ethanol.
- Characterization of RNP-LNPs:
- Measure particle size, polydispersity index (PdI), and zeta potential using dynamic light scattering.
- Determine encapsulation efficiency using a Ribogreen assay or similar.
- Cell Treatment and Analysis:
- Apply the RNP-LNPs to target cells. Optimize the dose based on particle number or RNP concentration.
- Incubate for 24-72 hours.
- Assess Editing Efficiency:
- Genomic DNA Extraction: Harvest cells and extract genomic DNA.
- T7 Endonuclease I (T7E1) Assay: PCR-amplify the target region, denature and reanneal the amplicons, and treat with T7E1 enzyme, which cleaves mismatched heteroduplex DNA. Analyze cleavage products by gel electrophoresis to calculate indel percentage [24].
- Next-Generation Sequencing (NGS): For the most accurate quantification of indels and mutation spectra.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for Studying Cargo Uptake and Trafficking
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Ionizable Lipid Nanoparticles (iLNPs) | Non-viral delivery vector that enables endosomal escape via protonation in acidic endosomes [20] [14]. | In vivo delivery of mRNA or RNP to liver and lung tissue [14]. |
| MITO-Porter System | A specialized LNP designed for mitochondrial targeting, delivering cargo via membrane fusion [21]. | Direct delivery of RNPs to mitochondria for mtDNA editing [21]. |
| Endocytic Inhibitors (Chlorpromazine, Wortmannin, Genistein) | Pharmacological tools to block specific endocytic pathways (clathrin-mediated, macropinocytosis, caveolae-mediated) [26]. | Identifying the primary route of cellular entry for a delivery vector [26]. |
| RiboDecode | A deep learning framework for mRNA codon optimization, enhancing translation efficiency and protein expression [22]. | Designing highly expressive mRNA cargo for therapeutic applications. |
| Nuclear Localization Signals (NLS) | Short amino acid sequences that facilitate active nuclear import when fused to proteins [24]. | Enhancing nuclear delivery of Cas9 protein in DNA, mRNA, and RNP formats. Research shows C-terminal, N-terminal, or dual NLS fusion can be similarly effective [24]. |
| Thermostable Cas9 Variants (e.g., iGeoCas9) | Engineered Cas9 proteins with high stability, making them more resistant to stress during LNP formulation and extending functional half-life [14]. | Achieving high-efficiency editing in vivo with RNP-LNPs, particularly in non-liver tissues like the lung [14]. |
| MTR-106 | MTR-106, MF:C28H27N7O2S, MW:525.6 g/mol | Chemical Reagent |
| Ajugamarin F4 | Ajugamarin F4, MF:C29H42O9, MW:534.6 g/mol | Chemical Reagent |
Advanced Delivery Systems and Workflows for Protein and mRNA Cargoes
Viral Vector Selection Guide
The choice between Adeno-Associated Viruses (AAV) and Lentiviruses (LV) is fundamental and depends on your experimental goals, cargo, and target cells. The table below summarizes the core characteristics of each vector to guide your selection.
Table 1: Key Characteristics of AAV vs. Lentiviral Vectors
| Characteristic | Adeno-Associated Virus (AAV) | Lentivirus (LV) |
|---|---|---|
| Primary Cargo | DNA (ssDNA) [27] | RNA (which is reverse-transcribed to DNA) [27] |
| Packaging Capacity | ~4.7 kb [28] [29] | ~8-12 kb [29] |
| Genomic Integration | Largely non-integrating (episomal) [29] [27] | Integrates into host genome [29] [27] |
| Ideal Application | In vivo delivery; transient expression; gene editing with small Cas orthologs [27] | Ex vivo delivery; stable, long-term expression; delivery of large genetic payloads [27] |
| Typical Expression Kinetics | Onset within days to weeks; can be long-term in non-dividing cells [29] | Onset within days; stable, long-term expression in dividing cells [29] |
| Immune Response | Generally low immunogenicity [28] [27] | Can elicit immune responses; higher safety profile than early retroviruses [27] |
| Tropism (Targeting) | Determined by capsid serotype (e.g., AAV8 for liver, AAV9 for CNS) [28] [29] | Determined by envelope pseudotype (e.g., VSV-G for broad tropism) [27] |
Viral Vector FAQs and Troubleshooting
AAV (Adeno-Associated Virus)
Q: My gene of interest, including Cas9 and gRNA, exceeds the 4.7 kb packaging limit of AAV. What are my options?
A: The AAV size limitation is a common challenge. You can consider these strategies:
- Use Smaller Cas9 Orthologs: Package your entire system in a single AAV by using compact Cas proteins. Staphylococcus aureus Cas9 (SaCas9, 1053 aa) and Campylobacter jejuni Cas9 (CjCas9, 984 aa) have been successfully used in vivo [28].
- Utilize a Dual AAV System: Split the Cas9 and gRNA expression cassettes across two separate AAVs. This requires co-infection and relies on intracellular reassembly, which can reduce overall efficiency [28] [1].
- Explore Split Intein Systems: Engineer SpCas9 to be split and packaged into two AAVs, with full-length protein reconstitution post-transduction via protein trans-splicing [28].
Q: I am not seeing the expected editing efficiency in my in vivo model. What could be wrong?
A: Low in vivo efficiency can stem from several factors:
- Serotype Selection: Ensure you are using an AAV serotype that efficiently transduces your target tissue (e.g., AAV8 for liver, AAV9 for heart and CNS) [28] [29].
- Promoter Compatibility: The promoter must be functional in your target cell type. Use tissue-specific promoters to ensure expression is restricted to the desired cells.
- Pre-existing Immunity: Pre-existing antibodies against the AAV serotype in your animal model can neutralize the virus. Screening for antibodies or using less common serotypes can mitigate this [29].
- Titer and Potency: Verify the functional titer of your viral prep and ensure the delivered dose is sufficient for your target organ.
Lentivirus (LV)
Q: My lentiviral production yields are low. How can I improve the titer?
A: Low titer is often related to upstream production factors [30] [31]:
- Plasmid DNA Quality: Do not use mini-prep DNA for transfection. Use high-quality, endotoxin-free midi- or maxi-prep DNA purified using a kit designed for lentiviral work [30] [31].
- Cell Health: Use healthy Lenti-X 293T cells at low passage number (< passage 16). Ensure cells are >90% confluent at the time of transfection [30] [31].
- Toxic Gene Products: If your gene of interest is large or toxic, it can inhibit viral production. Consider using an inducible system or testing a control vector to isolate the issue [30].
- Vector Integrity: Lentiviral vectors can recombine in standard E. coli. Use low-recombination bacterial strains like Stbl3 for plasmid propagation and validate plasmid integrity by restriction digest before use [30] [31].
Q: The transduction efficiency in my target cells is poor, even with a high-titer stock. What can I do?
A: Transduction efficiency depends on the target cell type. Consider these enhancements [30] [31]:
- Use a Transduction Enhancer: Add Polybrene (typically 4-8 µg/mL) to the transduction medium to neutralize charge repulsion between the viral particle and cell membrane. Note that some cell types are sensitive to Polybrene [30].
- Employ Spinoculation (Spinfection): Centrifuging the plate after adding the virus (e.g., 2000 x g for 30-60 minutes at 32°C) can significantly increase infection efficiency by forcing virus-cell contact [31].
- Concentrate Your Virus: Use a concentration reagent like Lenti-X Concentrator or ultracentrifugation to increase the effective viral titer [31].
- Use a Binding Matrix: For sensitive or hard-to-transduce cells like primary T cells, use RetroNectin to pre-coat plates. This binds the virus and co-localizes it with cells, improving transduction while removing inhibitors [31].
Research Reagent Solutions
The following table lists key reagents and their functions for successful viral vector production and transduction.
Table 2: Essential Reagents for Viral Vector Research
| Reagent / Kit Name | Primary Function | Brief Explanation |
|---|---|---|
| Stbl3 E. coli [30] | Cloning of Lentiviral Constructs | Bacterial strain with a recA13 mutation that minimizes unwanted recombination between the LTRs of lentiviral plasmids. |
| Lenti-X 293T Cell Line [31] | Lentivirus Packaging | A specially optimized HEK-293T-based cell line for high-titer lentivirus production when used with compatible packaging systems. |
| Lenti-X Concentrator [31] | Virus Concentration | A chemical precipitation method to concentrate lentiviral supernators, increasing viral titer up to 100-fold without ultracentrifugation. |
| RetroNectin Reagent [31] | Transduction Enhancement | A recombinant fibronectin fragment used to pre-coat plates, enhancing transduction of hard-to-transduce cells (e.g., primary T cells) by co-localizing virus and cells. |
| Polybrene [30] | Transduction Enhancement | A cationic polymer that reduces electrostatic repulsion between viral particles and the cell membrane, thereby increasing infection efficiency for many cell types. |
| Lenti-X GoStix [31] | Rapid Titer Estimation | A dipstick test for p24 capsid protein, providing a quick (10-minute) qualitative assessment of lentivirus production in supernatant. |
| Lenti-X qRT-PCR Titration Kit [31] | Viral Titer Determination | Quantifies the number of viral RNA genome copies per mL via qRT-PCR. Note: this measures physical particles, not all of which are infectious. |
| NucleoBond Xtra Maxi Kit [31] | Plasmid DNA Preparation | A gravity-flow column for purifying high-quality, "transfection-grade" plasmid DNA suitable for lentiviral packaging transfections. |
Experimental Workflow and Protocols
Workflow for AAV-Mediated In Vivo CRISPR Delivery
The diagram below outlines a standard workflow for producing and using AAV to deliver CRISPR components in vivo.
Detailed Protocol: AAV Production via Triple Transfection This protocol describes the production of recombinant AAV using the widely adopted three-plasmid transfection method in HEK293 cells [29].
Plasmid Co-transfection:
- Culture HEK293 cells (adherent or suspension) to an appropriate density.
- Co-transfect the cells with three plasmids:
- Transfer Plasmid (GOI): Contains your gene of interest (e.g., SaCas9 and gRNA) flanked by AAV Inverted Terminal Repeats (ITRs).
- Packaging Plasmid (Rep/Cap): Provides the AAV replication (Rep) and capsid (Cap) proteins. The Cap plasmid determines the serotype (e.g., AAV8, AAV9).
- Adenoviral Helper Plasmid: Supplies essential adenoviral genes (E4, E2a, VA) required for AAV replication.
- Use a transfection reagent optimized for high efficiency, such as polyethylenimine (PEI).
Harvest and Clarification:
- 48-72 hours post-transfection, harvest the cells and supernatant.
- Perform freeze-thaw cycles to lyse the cells and release the virus.
- Clarify the lysate by centrifugation to remove cell debris.
Purification and Concentration:
- Purify the AAV from the crude lysate. Common methods include iodixanol gradient ultracentrifugation or affinity chromatography columns.
- Concentrate the virus if necessary. The final product should be dialyzed into a suitable buffer like PBS.
Titration:
- Determine the viral genome titer (vg/mL) using quantitative PCR (qPCR). This is critical for dosing in animal experiments.
Workflow for Lentiviral-Mediated Ex Vivo CRISPR Delivery
The diagram below outlines a standard workflow for using lentivirus for ex vivo gene editing, such as in primary T cells for CAR-T therapy.
Detailed Protocol: Lentiviral Transduction of Adherent Cells This protocol is for transducing standard adherent cell lines. For primary or hard-to-transduce cells, additional optimization with reagents like RetroNectin is recommended [30] [31].
Day 0: Plate Cells:
- Plate your target cells so they will be 50-80% confluent at the time of transduction (typically the next day). Ensure the culture medium does not contain antibiotics.
Day 1: Transduction:
- Prepare the viral transduction mixture. Replace the cell culture medium with fresh medium containing the lentivirus at the desired Multiplicity of Infection (MOI). For initial experiments, a range of MOIs should be tested.
- Add a transduction enhancer, such as Polybrene, to a final concentration of 4-8 µg/mL.
- Optional but Recommended: Perform spinoculation by centrifuging the culture plate at 2000 x g for 30-90 minutes at 32°C.
- Incubate the cells with the virus-polybrane mixture for 4-24 hours.
Day 2: Refresh Medium:
- Remove the medium containing the virus and replace it with fresh, complete growth medium.
Day 3 Onwards: Analysis and Selection:
- If your vector contains a fluorescent marker, analyze transduction efficiency by flow cytometry or fluorescence microscopy 48-72 hours post-transduction.
- If your vector contains an antibiotic resistance gene, begin selection with the appropriate antibiotic 48-72 hours post-transduction. Maintain selection pressure for several days to kill non-transduced cells.
Lipid Nanoparticles (LNPs) as a Versatile Platform for mRNA and RNP Delivery
Lipid Nanoparticles (LNPs) have emerged as a leading non-viral delivery platform for genetic medicines, playing a pivotal role in the advancement of CRISPR-Cas9-based genome editing. For researchers optimizing the delivery of CRISPR-Cas9 components, a critical decision lies in choosing the appropriate cargo form: Cas9 mRNA or Cas9/sgRNA ribonucleoprotein (RNP) complexes [13]. While DNA forms of CRISPR-Cas9 offer sustained expression, they carry risks of host genome integration and higher off-target effects [13]. In contrast, mRNA and RNP forms provide transient activity, which minimizes off-target risks and eliminates genome integration concerns [13] [1]. This technical support article provides a comprehensive guide to troubleshooting LNP-based delivery for both Cas9 mRNA and RNPs, addressing key challenges in encapsulation, cellular uptake, and editing efficiency to support your research in developing precise and efficient gene therapies.
Core Concepts: Cargo Selection for CRISPR-LNP Systems
The choice between mRNA and RNP cargo significantly influences experimental design, editing kinetics, and safety profiles. The table below compares these two primary approaches for delivering CRISPR-Cas9 via LNPs.
Table 1: Comparison of Cas9 mRNA vs. RNP Delivery via LNPs
| Characteristic | Cas9 mRNA LNPs | Cas9 RNP LNPs |
|---|---|---|
| Cargo Form | mRNA encoding Cas9 protein + sgRNA | Pre-complexed Cas9 protein and sgRNA |
| Onset of Action | Requires translation (hours) | Immediate (minutes to hours) |
| Editing Duration | Moderate (days) | Short (hours to days) |
| Off-Target Risk | Moderate (prolonged expression) | Lower (transient activity) |
| Manufacturing Complexity | Moderate (stable mRNA) | Higher (protein stability) |
| Immune Recognition | Higher (can activate TLRs, RIG-I) [13] | Lower |
| Packaging Capacity | Suitable for larger cargo [13] | Limited by protein size |
| Ideal Applications | In vivo editing requiring sustained Cas9 expression | High-precision editing with minimized off-target effects |
Troubleshooting Guide: Common LNP Delivery Challenges
Poor Encapsulation Efficiency
Problem: Low encapsulation of mRNA or RNP cargo into LNPs results in reduced delivery efficiency and therapeutic payload.
Solutions:
- Optimize Lipid Ratios: Systematically adjust the ionizable lipid to phospholipid ratio. A typical starting point is a molar ratio of 50:10:38.5:1.5 for ionizable lipid:DSPC:cholesterol:DMG-PEG2000 [32].
- Modify N/P Ratio: The nitrogen (from cationic lipids) to phosphate (from RNA) ratio critically influences encapsulation. Test ratios between 6:1 and 8:1 for mRNA encapsulation [32].
- Refine Manufacturing Parameters: In microfluidic mixing, ensure proper total flow rates and a 3:1 aqueous-to-solvent flow rate ratio [32]. Adjust mixing parameters to achieve a turbulent flow regime without compromising mRNA integrity.
Inefficient Cellular Uptake and Endosomal Escape
Problem: LNPs are internalized but fail to release their cargo into the cytoplasm, leading to lysosomal degradation.
Solutions:
- Ionizable Lipid Selection: The ionizable lipid is critical for endosomal escape. Screen different ionizable lipids (e.g., SM-102, ALC-0315, MC3, C12-200) for your specific cell type [32]. Note that in vitro performance does not always predict in vivo efficacy [32].
- Modulate Cholesterol Content: Recent studies show that reducing cholesterol density in LNPs can significantly enhance mRNA uptake and endosomal escape in dendritic cells [33]. Test cholesterol percentages from 20% to 40% molar ratio.
- Surface Functionalization: Conjugate cell-penetrating peptides (CPPs) onto the LNP surface to improve cellular uptake [34]. For targeted delivery, incorporate SORT (Selective Organ Targeting) molecules into the formulation to direct LNPs to specific tissues beyond the liver [1] [35].
Limited In Vitro-In Vivo Correlation (IVIVC)
Problem: LNP formulations that perform well in cell culture models show poor efficacy in animal models.
Solutions:
- Use Physiologically Relevant Models: Immortalized cell lines may not accurately predict in vivo performance. Incorporate primary cells or immune cells (e.g., dendritic cells, macrophages) in your screening pipeline [32].
- Characterize Critical Quality Attributes (CQAs): Rigorously analyze particle size, PDI, zeta potential, and encapsulation efficiency. All LNP formulations should have comparable physicochemical properties (size 70-100 nm, low PDI, high mRNA encapsulation) before in vivo testing [32].
- Employ Computational Modeling: Utilize molecular dynamics (MD) simulations to predict LNP behavior and stability in biological environments. Coarse-grained MD can model LNP-membrane interactions and endosomal escape mechanisms [36].
Low Gene Editing Efficiency
Problem: Despite successful delivery, the resulting gene editing rates are insufficient for therapeutic applications.
Solutions:
- Optimize sgRNA Co-encapsulation: For RNP delivery, ensure proper stoichiometry between Cas9 protein and sgRNA during pre-complexing. Purify RNPs before encapsulation to remove unbound components.
- Validate mRNA Integrity and Purity: For mRNA delivery, use HPLC-purified mRNA with modified nucleosides (e.g., N1-methylpseudouridine) to reduce immunogenicity and enhance translation [13] [37]. Ensure the mRNA has a optimized 5' cap and 3' poly(A) tail.
- Cell-Type Specific Formulations: Tailor LNP composition to target cells. For example, BLANs with low cholesterol density showed exceptional mRNA uptake and PD-L1 knockout efficiency in dendritic cells [33].
Experimental Protocols
Protocol 1: Formulating CRISPR mRNA-LNPs via Microfluidics
This protocol describes the preparation of LNPs encapsulating Cas9 mRNA and sgRNA for genome editing applications.
Materials:
- Lipids: Ionizable lipid (e.g., SM-102, ALC-0315), DSPC, cholesterol, DMG-PEG2000 [32]
- Aqueous Phase: Cas9 mRNA and sgRNA in citrate buffer (pH 4) [32]
- Equipment: Microfluidic mixer (e.g., NanoAssemblr Ignite), Amicon spin filters
Procedure:
- Prepare Lipid Stock: Dissolve lipids in ethanol at a molar ratio of 50:10:38.5:1.5 (ionizable lipid:DSPC:cholesterol:DMG-PEG2000) to a total lipid concentration of 10-12.5 mg/mL.
- Prepare Aqueous Phase: Dissolve Cas9 mRNA and sgRNA in 50 mM citrate buffer (pH 4) to a final mRNA concentration of 70 μg/mL [32].
- Mixing: Use a microfluidic device with a 3:1 aqueous-to-organic flow rate ratio and a total flow rate of 12 mL/min [32].
- Buffer Exchange: Dialyze or use tangential flow filtration against PBS (pH 7.4) to remove ethanol and adjust pH.
- Concentration: Concentrate LNPs using Amicon Ultra centrifugal filters (100 kDa MWCO).
- Characterization: Measure particle size (target 70-100 nm), PDI (<0.2), zeta potential, and encapsulation efficiency (>90%) [32].
Protocol 2: Assessing Gene Editing Efficiency In Vitro
Materials:
- Target cell line (e.g., HEK293, HeLa, THP-1) [32]
- mRNA-LNPs or RNP-LPs
- Lysis buffer and genomic DNA extraction kit
- T7 Endonuclease I or TIDE analysis reagents
Procedure:
- Cell Seeding: Seed cells in 24-well plates at 70-80% confluence.
- Transfection: Treat cells with LNPs at various concentrations (e.g., 50-200 ng/mL mRNA). Include untreated controls.
- Incubation: Incubate for 48-72 hours to allow for editing and protein turnover.
- Genomic DNA Extraction: Harvest cells and extract genomic DNA.
- PCR Amplification: Amplify the target genomic region using specific primers.
- Editing Analysis:
- T7E1 Assay: Denature and reanneal PCR products, digest with T7 Endonuclease I, and analyze fragments by gel electrophoresis.
- TIDE Analysis: Sequence PCR products and use decomposition software to quantify insertion/deletion mutations.
- Cell Viability: Perform MTT or CellTiter-Glo assays in parallel to assess cytotoxicity.
Research Reagent Solutions
Table 2: Essential Reagents for LNP Development and Characterization
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | SM-102, ALC-0315, DLin-MC3-DMA (MC3), C12-200 [32] | Form core structure; enable endosomal escape via protonation | pKa should be ~6.5 for optimal endosomal escape; significantly affects efficacy [32] |
| Structural Lipids | DSPC, DOPE [32] [37] | Enhance structural integrity and stability | DSPC provides bilayer stability; DOPE promotes hexagonal phase for endosomal escape |
| Stabilizing Agents | Cholesterol, DMG-PEG2000, ALC-0159 [32] [37] | Modulate membrane fluidity, reduce particle aggregation, prolong circulation | Cholesterol density affects cellular uptake and endosomal escape [33] |
| mRNA Modifications | N1-methylpseudouridine, 5-methoxyuridine [13] [37] | Reduce immunogenicity, enhance stability and translation efficiency | Critical for minimizing TLR7/8 activation and RIG-I signaling [13] |
| Characterization Tools | Dynamic Light Scattering, RiboGreen assay, TEM [38] | Measure size, PDI, encapsulation efficiency, and morphology | Essential Critical Quality Attributes (CQAs) for reproducible manufacturing [38] |
Frequently Asked Questions (FAQs)
Q1: Why do my LNP formulations show good in vitro transfection but poor in vivo editing efficiency? This is a common challenge due to the complex in vivo environment. The formation of a "protein corona" on LNPs in blood can alter their cellular tropism, typically directing them to the liver via ApoE-mediated uptake [35]. Furthermore, in vitro cell lines may not accurately mimic target cells in vivo [32]. To address this, incorporate immune cells or primary cells in your screening and consider implementing SORT molecules to achieve extra-hepatic targeting [1].
Q2: What is the best method to improve endosomal escape of LNPs? The selection of the ionizable lipid is the most critical factor. Screen different ionizable lipids (e.g., SM-102, ALC-0315) as their chemical structure determines protonation behavior and membrane fusion capacity [32]. Additionally, recent research shows that modulating cholesterol density in LNPs can significantly enhance endosomal escape in specific cell types like dendritic cells [33].
Q3: How can I scale up LNP production without compromising quality? Maintaining consistent particle size and low polydispersity during scale-up requires careful control of mixing parameters. Implement quality-by-design (QbD) principles and use scalable technologies like controlled microfluidics or tangential flow filtration [38]. Rigorous in-process controls for critical quality attributes (size, PDI, encapsulation) are essential throughout scale-up.
Q4: Can I use the same LNP formulation for both Cas9 mRNA and RNPs? While possible, optimization is often required. RNP encapsulation may benefit from adjusted lipid ratios and different stabilization strategies due to the larger size and different charge characteristics of the protein-RNA complex. Some studies suggest incorporating additional helper lipids or peptides to stabilize protein cargo during encapsulation and release.
Q5: How can I reduce the immunogenicity of CRISPR-LNPs? For mRNA LNPs, use nucleoside-modified mRNA (e.g., N1-methylpseudouridine) and HPLC purification to remove double-stranded RNA impurities [13]. For both mRNA and RNP delivery, ensure high encapsulation efficiency to minimize exposure of nucleic acids to extracellular immune sensors. PEGylated lipids can also reduce immune recognition, though anti-PEG immunity is a potential concern with repeated dosing.
FAQs & Troubleshooting Guide
Q1: What are the primary advantages of using VLPs for RNP delivery in neuronal cells compared to AAVs or LNPs?
VLPs offer a unique combination of benefits for neuronal RNP delivery:
- Transient Activity & Reduced Immunogenicity: As VLPs deliver pre-assembled Cas9 RNP, the editing machinery is active for a short duration, minimizing off-target effects and reducing the risk of an immune response against persistently expressed Cas9. This is a significant advantage over AAVs, which lead to long-term Cas9 expression [39] [13].
- High Editing Efficiency: The RIDE (Ribonucleoprotein delivery) VLP platform demonstrated editing efficiency comparable to lentiviral vectors and higher than lipid nanoparticles (LNPs) in various cell types [39].
- Programmable Tropism: The surface of VLPs can be engineered to display specific ligands or antibodies, enabling targeted delivery to neurons and other hard-to-transfect cells, thereby improving specificity and reducing off-target effects [39] [40].
Q2: My VLP preps show low gene editing efficiency in primary neuronal cultures. What could be going wrong?
Low efficiency can stem from multiple points in the workflow. Consider the following troubleshooting steps:
- Verify RNP Packaging: Confirm that the Cas9 protein and gRNA are efficiently encapsulated within the VLPs. This can be checked using techniques like Western blot for Cas9 and RT-qPCR for gRNA on purified VLP lysates. The packaging is often dependent on specific molecular interactions, such as between the MS2 coat protein and MS2 stem loops incorporated into the gRNA [39].
- Check Viral Titer and Purity: Low particle titer will directly lead to low transduction rates. Use methods like p24 ELISA for lentiviral-based VLPs or other appropriate assays to quantify physical and functional titers. Ensure your preps are free of excessive cellular debris.
- Optimize Transduction Parameters: For neuronal cultures, factors like the age of the culture, the presence of neurotrophic factors during transduction, and the multiplicity of infection (MOI) need optimization. Pilot experiments with a GFP-reporting VLP can help establish optimal conditions.
- Assess Gating Strategy: The use of a constitutively expressed fluorescent marker (e.g., GFP) packaged alongside the RNP cargo is crucial for accurately identifying and sorting successfully transduced neurons for downstream analysis [39].
Q3: I am concerned about off-target effects. How do VLP-delivered RNPs compare to other delivery methods in this regard?
VLP-delivered RNPs are among the best strategies for minimizing off-target effects. Because the RNP complex is active immediately upon delivery but has a short half-life, the window for editing is limited. This transient activity significantly reduces the chance of Cas9 making cuts at unintended, off-target sites with similar sequences. Studies have shown that the RIDE VLP system induced fewer off-target effects than lentiviral vectors, which lead to long-term nuclease expression [39]. Furthermore, the RNP form of CRISPR-Cas9 itself has been reported to have the lowest rate of off-target effects compared to DNA and mRNA forms [13].
Q4: Can VLPs deliver other CRISPR-based editors besides standard Cas9 nuclease?
Yes, the VLP technology is adaptable. Research has demonstrated that the RIDE system can also deliver base editor proteins, achieving editing efficiencies of up to 69% in some cases [39]. Furthermore, streamlined SFV-based VLP platforms have been engineered to deliver not only RNPs but also mRNA and protein cargos, showcasing their versatility as a delivery platform [40].
Key Experimental Protocols
Protocol: Assessing VLP-mediated Gene Editing in Neuronal Cultures
This protocol outlines the steps to transduce primary neurons with CRISPR-Cas9 RNPs delivered via VLPs and to quantify the editing outcome.
Materials:
- Primary neuronal cultures (e.g., cortical or hippocampal neurons from rodent E18 brains or human iPSC-derived neurons).
- Purified VLP prep (e.g., RIDE system pseudotyped with VSV-G or neuron-targeting envelopes).
- Neurobasal medium with B27 supplement and GlutaMAX.
- Poly-D-lysine coated culture plates.
- Phosphate Buffered Saline (PBS).
- Fixative (e.g., 4% PFA) and Permeabilization buffer (e.g., 0.1% Triton X-100).
- Antibodies for immunostaining (e.g., against target protein or Cas9).
- Lysis buffer for genomic DNA extraction.
- PCR purification kit.
- T7 Endonuclease I or next-generation sequencing (NGS) library prep reagents.
Procedure:
- Culture Preparation: Plate primary neurons on poly-D-lysine coated plates in complete Neurobasal medium. Allow neurons to mature in vitro for at least 7-10 days before transduction.
- VLP Transduction:
- Thaw VLP stock on ice.
- Replace the neuronal culture medium with a minimal volume of fresh, pre-warmed medium.
- Add the calculated volume of VLPs to achieve the desired MOI. Include a control with non-targeting gRNA VLPs.
- Return the cells to the incubator (37°C, 5% CO2) for 6-24 hours.
- After the incubation, carefully remove the medium containing VLPs and replace it with fresh, complete Neurobasal medium.
- Incubation: Allow the gene editing to proceed for 3-7 days, with half-medium changes every 3-4 days.
- Efficiency Analysis (72 hours post-transduction):
- Microscopy: If using a fluorescent reporter, image live cells to assess the transduction efficiency.
- Immunostaining: Fix cells and stain for Cas9 protein and neuronal markers (e.g., MAP2) to confirm delivery and cell-type specificity.
- Genomic Editing Analysis (5-7 days post-transduction):
- DNA Extraction: Wash cells with PBS, lyse, and extract genomic DNA.
- PCR Amplification: Design primers flanking the target site and amplify a 400-800 bp region.
- Editing Quantification:
- T7EI Assay: Purify the PCR product, denature and reanneal, then treat with T7 Endonuclease I. Run the products on an agarose gel. The percentage of indels can be calculated from the band intensities [39].
- NGS (Gold Standard): Purify the PCR product and prepare libraries for deep sequencing. This provides the most accurate measurement of indel frequency and can be used for comprehensive off-target analysis.
Protocol: In Vivo Assessment of VLP-mediated Editing in Mouse Brain
This methodology is based on studies demonstrating therapeutic efficacy in Huntington's disease models [39].
Materials:
- Adult mice (e.g., Huntington's disease model mice).
- Stereotaxic injection apparatus.
- Purified, concentrated VLP preparation.
- Anesthetic (e.g., ketamine/xylazine).
- Artificial cerebrospinal fluid (aCSF).
Procedure:
- VLP Preparation: Concentrate and purify VLPs via ultracentrifugation. Resuspend the final pellet in sterile aCSF. Keep on ice until injection.
- Stereotaxic Injection:
- Anesthetize the mouse and secure it in the stereotaxic frame.
- Make a small craniotomy at the coordinates targeting the brain region of interest (e.g., striatum).
- Load the VLP solution into a Hamilton syringe with a fine-gauge needle.
- Slowly lower the needle to the target depth and infuse the VLP solution (e.g., 2-3 µL) at a slow, constant rate (e.g., 0.2 µL/min).
- Leave the needle in place for an additional 5-10 minutes post-injection to prevent backflow.
- Slowly retract the needle and suture the scalp.
- Post-operative Care: Monitor animals until they recover from anesthesia and provide analgesic care as per institutional guidelines.
- Analysis:
- Tissue Collection: At the experimental endpoint (e.g., 2-4 weeks post-injection), perfuse and harvest the brain.
- Assessment:
- Process brain sections for immunohistochemistry to detect edited protein (e.g., mutant huntingtin) and neuronal health markers.
- Microdissect the injected region for genomic DNA extraction. Quantify editing efficiency via T7EI assay or NGS as described in the in vitro protocol.
- For behavioral models, conduct relevant motor or cognitive tests to assess functional recovery.
Table 1: Editing Efficiency of VLP Platforms in Various Models
| VLP Platform | Target Cell/Tissue | Target Gene | Editing Efficiency | Key Finding | Citation |
|---|---|---|---|---|---|
| RIDE (Lentiviral-based) | Patient iPSC-derived neurons | Huntingtin (HTT) | Efficient editing (data tolerated in NHP) | Significantly ameliorated disease symptoms in Huntington's disease model | [39] |
| RIDE (Lentiviral-based) | Mouse Retinal Pigment Epithelium (RPE) | Vegfa | 38% indel frequency | ~60% decrease in VEGF-A levels; 43% reduction in choroidal neovascularization area | [39] |
| SFV-based VLP | Various cell lines (in vitro) | DsRed (reporter) | Successful knockout | Platform successfully delivered functional Cas9 RNP; demonstrated broad packaging capacity (up to 10 kb) | [40] |
| Customizable VLPs | Neurons (in vivo) | Not Specified | Effective delivery achieved | Streamlined SFV-based system engineered for enhanced blood-brain barrier (BBB) penetration | [40] |
Table 2: Comparison of CRISPR-Cas9 Delivery Formats
| Delivery Format | Advantages | Disadvantages / Challenges |
|---|---|---|
| VLP-delivered RNP | - Transient activity, lowest off-target effects [13]- High efficiency comparable to LVs [39]- Programmable cell tropism [39]- Reduced immunogenicity vs. DNA-based delivery [39] | - Manufacturing complexity [13]- Engineering challenges for targeted delivery [39] |
| Viral Vector (AAV/LV) | - Long-term expression (AAV)- High transduction efficiency for many cells | - Persistent Cas9 expression increases off-target/immune risks [13]- Limited packaging capacity (AAV) [13]- Risk of genomic integration (LV) [13] |
| LNP-delivered mRNA | - No genome integration risk [13]- Transient expression | - shorter half-life, susceptibility to nucleases [13]- Can induce immune responses [13]- Less established for neuron-targeting |
Research Reagent Solutions
Table 3: Essential Research Reagents for VLP-based RNP Delivery
| Reagent | Function in the Workflow | Example/Note |
|---|---|---|
| MS2-modified gRNA | Enables specific packaging of the gRNA (and pre-bound Cas9) into VLPs via interaction with the MS2 coat protein. | A critical component of the RIDE system; two copies of the MS2 stem loop are inserted into the gRNA backbone [39]. |
| MS2-fused GagPol | Forms the structural core of the lentiviral-based VLP and provides the MS2 coat protein for gRNA/RNP recruitment. | Often used with an integrase-deficient (D64V) mutation to prevent genomic integration [39]. |
| Envelope Proteins (e.g., VSV-G) | Pseudotypes the VLP, determining its tropism and enabling entry into target cells. | VSV-G offers broad tropism. For specific neuronal targeting, envelopes from other viruses (e.g., Rabies-G) or engineered versions can be used [39] [40]. |
| Capsid Protein (e.g., SFV C protein) | In non-lentiviral systems like the SFV-based VLP, the capsid protein binds and packages the cargo (mRNA, RNP). | Can be fused to Cas9 via a cleavable linker for direct RNP packaging [40]. |
| Fluorescent Reporter (e.g., GFP) | Packaged as a separate mRNA or protein cargo to visually identify successfully transduced cells for sorting or analysis. | Crucial for quantifying transduction efficiency and gating in flow cytometry [39]. |
Workflow and Pathway Diagrams
VLP for RNP Delivery Workflow
Troubleshooting Low VLP Editing
Electroporation is a highly efficient physical transfection method that uses controlled electrical pulses to create temporary pores in cell membranes, allowing biological materials like nucleic acids or proteins to enter cells [41] [42]. For researchers optimizing Cas9 protein versus mRNA delivery in ex vivo applications, electroporation offers a versatile platform to compare these cargo types directly in primary cells and cell lines. This technique has become a research standard due to its reproducibility, high efficiency, applicability to various cell types, and relatively low toxicity compared to some chemical methods [42] [43].
In the context of CRISPR-Cas9 genome editing, electroporation enables the delivery of all three primary cargo forms: DNA plasmids encoding Cas9 and guide RNA, Cas9 mRNA combined with guide RNA, and preassembled Cas9 ribonucleoprotein (RNP) complexes [1]. The choice between these cargo types significantly impacts key experimental outcomes, including editing efficiency, off-target effects, and cellular toxicity. This technical resource provides comprehensive guidance for implementing ex vivo electroporation specifically for CRISPR delivery optimization, with detailed troubleshooting protocols to address common experimental challenges.
Key Concepts and Fundamental Principles
How Electroporation Works
The fundamental principle of electroporation involves applying brief, high-voltage electrical pulses to cells in suspension between two electrodes. This electric field induces a transmembrane potential which causes the reversible breakdown of the cellular membrane, forming temporary pores that allow molecules like CRISPR components to enter the cell [41] [42]. The process involves two critical variables: field strength (measured as voltage per distance, typically V/cm) and pulse length (duration of the electrical pulse) [42].
After electroporation, the cell membrane recovers, and the delivered cargo can function within the cell. For CRISPR applications, each cargo type has distinct advantages: DNA plasmids enable sustained expression but carry integration risks; mRNA offers transient expression without genomic integration; while RNP complexes provide immediate activity with potentially reduced off-target effects due to shorter persistence [13] [1].
Electroporation Workflow for CRISPR Delivery
The diagram below illustrates the core workflow for ex vivo electroporation when delivering different CRISPR cargo types:
Research Reagent Solutions
The table below outlines essential materials and reagents required for ex vivo electroporation experiments in CRISPR research:
| Reagent/Equipment | Function & Importance in CRISPR Workflows |
|---|---|
| Electroporation Instrument | Generates controlled electrical pulses. Square wave generators often preferred for mammalian cells [42]. |
| Electroporation Cuvettes/Plates | Contain cell-DNA mixture during pulse. Gap width critical for field strength calculation [43]. |
| Electroporation Buffer | Maintains optimal conductivity and pH. Composition varies by cell type; isotonic and buffered [43]. |
| Cas9 Cargo | DNA, mRNA, or RNP format. Purity and quality dramatically impact efficiency and cell viability [1]. |
| Cell Culture Reagents | For pre- and post-electroporation cell handling. Critical for maintaining viability of edited cells [44] [43]. |
Experimental Protocols and Methodologies
Standard Protocol for Mammalian Cell Electroporation
The following detailed protocol adapts established methodologies for CRISPR delivery optimization [43]:
Cell Preparation
- Grow cells to late-log phase. For stable transformation, prepare 5 × 10^6 cells per transfection; for transient expression, prepare 1–4 × 10^7 cells.
- Harvest cells by centrifugation at 640 × g for 5 minutes at 4°C.
- For adherent cells: first trypsinize and inactivate trypsin with serum.
Buffer Exchange
- Resuspend cell pellet in half its original volume of ice-cold electroporation buffer.
- Harvest cells again by centrifugation as above.
- Resuspend cells at optimal density: 1 × 10^7/ml for stable transformation; higher concentrations (up to 8 × 10^7/ml) for transient expression.
- Transfer 0.5-ml aliquots into electroporation cuvettes placed on ice.
Cargo Addition
- Add purified CRISPR cargo to cell suspension in cuvettes:
- For DNA: 1-10 µg for stable transformation; 10-40 µg for transient expression.
- For RNP complexes: Pre-complex Cas9 protein with sgRNA at optimal molar ratio (typically 1:2 to 1:3) for 10-15 minutes at room temperature before electroporation.
- Mix DNA/cell suspension by flicking the cuvette and incubate 5 minutes on ice.
- Add purified CRISPR cargo to cell suspension in cuvettes:
Electroporation
- Place cuvette in electroporation apparatus at room temperature.
- Apply electrical pulse(s) at optimized voltage and capacitance settings.
- Specific parameters vary significantly by cell type (see Optimization Guide in Troubleshooting section).
- Return cuvette to ice for 10 minutes after electroporation.
Post-Electroporation Recovery and Analysis
- Dilute transfected cells 20-fold in nonselective complete medium.
- Rinse cuvette with this same medium to recover all transfected cells.
- For stable transformation: Grow cells 48 hours in nonselective medium before transferring to selective medium.
- For transient expression: Incubate cells 50-60 hours before harvesting for analysis.
Cargo-Specific Considerations
For Cas9 mRNA Delivery:
- Combine purified Cas9 mRNA with sgRNA at optimal concentration.
- mRNA should include 5' cap and 3' poly-A tail for enhanced stability and translation.
- Consider nucleotide modifications (e.g., pseudouridine) to reduce immunogenicity and enhance stability [13].
For RNP Complex Delivery:
- Pre-assemble Cas9 protein with sgRNA in a molar ratio of 1:2 to 1:3.
- Incubate 10-20 minutes at room temperature to allow complex formation before electroporation.
- RNP complexes typically require lower voltage parameters than nucleic acid cargoes.
Optimization and Troubleshooting Guide
Frequently Asked Questions
Q1: We're experiencing low transfection efficiency with our primary T-cells using Cas9 RNP. What parameters should we adjust first?
Low efficiency in hard-to-transfect cells like primary T-cells often results from suboptimal electrical parameters. Focus on these adjustments:
- Field Strength: Gradually increase voltage (typically 1200-1600V for 2mm gap cuvettes) while monitoring viability.
- Pulse Length: For square wave instruments, try increasing pulse duration (0.5-10ms range).
- Cell Health: Ensure cells are in log-phase growth and highly viable before electroporation.
- RNP Concentration: Increase RNP complex concentration while maintaining appropriate Cas9:sgRNA ratio.
- Buffer Composition: Test specialized buffers formulated for sensitive primary cells.
Q2: Our electroporated cells show poor viability post-transfection, particularly with Cas9 mRNA. How can we improve cell survival?
High cell death can be addressed through multiple approaches:
- Pulse Optimization: Reduce voltage or pulse duration - high efficiency and viability require careful balance.
- Recovery Conditions: Use pre-warmed complete medium with enhanced serum (20-30%) immediately after pulsing.
- Cargo Amount: Reduce mRNA concentration - too much nucleic acid can be toxic.
- Recovery Temperature: For some cell types, immediate placement at 37°C improves recovery; for others, gradual warming from 4°C to 37°C works better.
- Additive Incorporation: Test viability-enhancing additives like Rho kinase (ROCK) inhibitor in recovery medium.
Q3: We get inconsistent editing results between replicates. What are the key factors affecting reproducibility?
Improve reproducibility by standardizing these elements:
- Cell Preparation: Maintain consistent cell culture conditions and harvest at same confluence.
- Cargo Quality: Use highly purified, endotoxin-free preparations for all cargo types.
- Temperature Control: Keep cells on ice before electroporation and return to ice immediately after.
- Instrument Calibration: Regularly verify electroporator output parameters.
- Timing: Minimize time between cell harvesting and electroporation.
Advanced Optimization Data
The table below summarizes optimized electroporation parameters from published studies for different cell types relevant to CRISPR research:
| Cell Type | Cargo Type | Voltage (V) | Pulse Length (ms) | Number of Pulses | Efficiency (%) | Key Findings |
|---|---|---|---|---|---|---|
| Chick Retinal Cells [44] | DNA Plasmid | Not specified | Not specified | Multiple | 22-25% | Five-fold improvement over other methods; no effect on cell survival/differentiation |
| Mammalian Cells (General) [43] | DNA | 200-350V (for 0.4cm gap) | 10-20ms (exponential decay) | 1 | Varies | Protocol optimized for both stable and transient transfection |
| Hard-to-Transfect Cells [41] | Various | Cell-specific | Cell-specific | 1- multiple | >90% possible | Modern systems distribute pulse equally, maintaining stable pH for enhanced viability |
Technical Specifications and Equipment Guide
Electroporation System Configuration
The diagram below illustrates the relationship between electroporation system components and their role in the CRISPR delivery process:
Cargo-Specific Efficiency Considerations
When comparing Cas9 protein versus mRNA delivery via electroporation, several critical factors influence experimental outcomes:
Cas9 mRNA Advantages:
- No risk of host genome integration [13]
- Reduced off-target effects compared to DNA due to shorter persistence [1]
- Cytoplasmic expression without nuclear entry requirement [13]
Cas9 RNP Advantages:
- Immediate activity with fastest editing onset [1]
- Lowest off-target effects due to shortest persistence [13] [1]
- No transcriptional or translational delays
Key Efficiency Metrics:
- Delivery Efficiency: Percentage of cells receiving cargo
- Editing Efficiency: Percentage of cells with desired genetic modification
- Cell Viability: Percentage of cells surviving the process
- Functional Titer: Number of successfully edited cells per reaction
For all cargo types, careful optimization of electroporation parameters is essential to maximize the ratio of editing efficiency to cellular toxicity. The optimal balance varies significantly based on target cell type and specific application requirements.
Lipid Nanoparticles (LNPs) represent the most clinically advanced delivery system for nucleic acids, including messenger RNA (mRNA). A predominant characteristic of conventional LNPs is their natural tendency to accumulate in the liver following systemic administration. This liver-specific tropism is largely due to the interaction between apolipoprotein E (ApoE) adsorbed onto the LNP surface and the low-density lipoprotein (LDL) receptors abundantly expressed on hepatocytes [45]. The liver's anatomy, particularly the fenestrated endothelium of liver sinusoidal endothelial cells (LSECs), further facilitates the uptake of nanoparticles from the circulation [45].
This case study explores the formulation of LNPs for effective mRNA delivery to the liver, a critical requirement for emerging gene therapies, including those utilizing CRISPR-Cas9. We will examine the core components of LNPs, troubleshooting common challenges, and provide detailed protocols for researchers aiming to optimize delivery efficiency and specificity for hepatic gene editing applications.
LNP Formulation Components and Functions
A typical LNP formulation is composed of four key lipid components, each playing a distinct role in the structure, stability, and function of the nanoparticle.
Table 1: Core Components of Liver-Targeted LNPs and Their Functions
| Component | Key Function | Examples | Impact on Liver Delivery |
|---|---|---|---|
| Ionizable Lipid | - mRNA complexation & encapsulation- Endosomal escape | DLin-MC3-DMA, ALC-0315, SM-102 [46] | Core driver; pKa dictates ApoE binding & hepatocyte uptake via LDL receptor [45]. |
| Helper Phospholipid | - Structural stability- Promotes membrane fusion | DSPC, DOPE [46] | Influences membrane integrity & endosomal escape; DOPE may enhance efficacy [46]. |
| Cholesterol | - Modulates membrane fluidity & rigidity- Enhances stability | - | Contributes to liver tropism; its removal can reduce hepatic accumulation [47]. |
| PEG-lipid | - Reduces aggregation & opsonization- Controls particle size | DMG-PEG2000, ALC-0159 [48] | Prevents RES uptake, prolonging circulation; rapidly dissociates in vivo to allow cellular uptake [48]. |
Troubleshooting Common LNP Delivery Challenges
Problem 1: Low Transfection Efficiency in Target Liver Cells Question: "My LNPs are reaching the liver, but I'm observing poor mRNA translation and low levels of the desired protein. What could be the issue?" Answer: Low transfection efficiency often stems from inadequate endosomal escape, where the mRNA remains trapped and degraded inside the endosome.
- Potential Cause 1: Suboptimal Ionizable Lipid Structure. The ionizable lipid's ability to become protonated in the acidic endosomal environment is critical for membrane disruption.
- Solution: Design or select ionizable lipids with a pKa between 6.0 and 6.5 [45]. Consider lipids with biodegradable ester cores (e.g., nAcx-Cm lipids) that enhance mRNA release and safety profiles [47]. A combinatorial library approach can help identify optimal structures [47].
- Potential Cause 2: Incorrect Helper Lipid Selection. The helper lipid can influence the lamellar to hexagonal phase transition necessary for membrane fusion.
- Solution: Test different helper lipids. DOPE is often preferred over DSPC for mRNA delivery, as it more readily adopts the hexagonal phase that promotes endosomal escape [46].
Problem 2: Persistent Off-Target Accumulation and Toxicity Question: "My LNP formulation shows significant accumulation in off-target tissues like the spleen, and I observe signs of hepatotoxicity. How can I improve specificity and safety?" Answer: Off-target accumulation can be influenced by LNP composition and is closely linked to toxicity concerns, such as those observed in some LNP-based COVID-19 vaccines [49].
- Potential Cause 1: Cholesterol-Mediated Hepatic Uptake. Cholesterol in LNPs facilitates lipoprotein coating, enhancing interaction with hepatic cells [47].
- Solution: Reformulate LNPs with reduced cholesterol or explore cholesterol-free LNP systems. Studies show that removing cholesterol can specifically address the challenge of unwanted hepatic accumulation [47].
- Potential Cause 2: Activation of the Reticuloendothelial System (RES). Kupffer cells, the liver's resident macrophages, can rapidly sequester LNPs, reducing delivery to hepatocytes and potentially causing immune reactions [45].
- Solution: Optimize the molar ratio and structure of the PEG-lipid. Using PEG-lipids with shorter lipid tails (e.g., C14 DMG-PEG2000) allows for faster dissociation post-administration, improving cellular uptake and reducing RES recognition [48].
Problem 3: Short Duration of Transgene Expression Question: "I need sustained protein expression for my therapeutic application, but my current mRNA-LNPs only provide transient expression. What are my options?" Answer: mRNA is inherently transient. For prolonged expression, consider switching the cargo from mRNA to plasmid DNA (pDNA).
- Solution: Utilize pDNA-LNPs. Research has demonstrated that optimized pDNA LNPs can mediate prolonged transgene expression in the liver compared to mRNA-based technologies [50]. A multi-step screening platform can identify pDNA LNP formulations effective for liver-targeted, extended expression [50].
Advanced Strategies for Enhanced Liver Targeting
Strategy 1: Modulating LNP Physicochemical Properties
- Size: LNPs smaller than 100 nm in diameter can more easily traverse the fenestrations in LSECs to reach hepatocytes [45].
- Surface Charge: At physiological pH, LNPs should have a neutral or slightly negative surface charge to reduce non-specific interactions and improve serum resistance, while the ionizable lipid becomes positively charged in the acidic endosome to facilitate escape [47] [46].
Strategy 2: Selective Organ Targeting (SORT) The SORT methodology involves adding a fifth, supplemental lipid to conventional four-component LNPs. For enhanced liver targeting, the addition of SORT molecules based on ionizable lipids has been shown to increase specificity and functional delivery to hepatocytes [45].
Essential Experimental Protocols
Protocol 1: Formulating LNPs via Microfluidic Mixing
This is the standard method for producing monodisperse, stable LNPs.
- Prepare Lipid Stock: Dissolve the ionizable lipid, helper phospholipid, cholesterol, and PEG-lipid in ethanol at the desired molar ratio (e.g., 50:10:38.5:1.5) [51].
- Prepare Aqueous Phase: Dilute the mRNA in a sodium acetate buffer (e.g., 25 mM, pH 5.0) [51].
- Mixing: Use a microfluidic device to rapidly mix the ethanolic lipid phase with the aqueous mRNA phase at a standard flow rate (e.g., 1:3 volumetric ratio) [51] [48].
- Buffer Exchange & Purification: Dialyze the formed LNPs against a neutral pH buffer (e.g., Tris-HCl, pH 7.4) or use Tangential Flow Filtration (TFF) to remove ethanol and adjust the buffer [51].
- Characterization: Measure particle size, polydispersity index (PDI), and zeta potential using Dynamic Light Scattering (DLS). Determine encapsulation efficiency using a Ribogreen assay [51].
Protocol 2: In Vivo Screening for Liver-Targeted Expression
- Animal Model: Use immunocompetent mice (e.g., C57BL/6 or BALB/c).
- Administration: Inject LNPs intravenously via the tail vein. A common dose for screening is 0.5-1 mg mRNA per kg body weight.
- Analysis:
- Bioluminescence Imaging: If using luciferase-encoding mRNA, inject D-luciferin substrate and image at multiple time points (e.g., 6 h, 24 h, 48 h) to track the location and duration of protein expression [51].
- Biodistribution: Post-imaging, euthanize the animals, collect organs (liver, spleen, lungs, heart, kidneys), and perform ex vivo imaging to quantify mRNA accumulation and expression in each tissue [51].
- Functional Delivery: For gene editing, harvest liver tissue after 3-7 days and analyze genomic DNA for indel efficiency using next-generation sequencing (NGS) or T7E1 assays.
Diagram 1: LNP Formulation and Characterization Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for LNP Development and Testing
| Reagent / Material | Function / Application | Key Details |
|---|---|---|
| Ionizable Lipids (e.g., DLin-MC3-DMA) | Core functional lipid for mRNA complexation and endosomal escape. | pKa ~6.4; critical for ApoE binding and LDL receptor-mediated hepatocyte uptake [45] [46]. |
| DMG-PEG2000 | PEG-lipid for stability, size control, and reducing RES clearance. | C14 tail allows for rapid dissociation in vivo; typically used at 1.5-2 mol% [46] [48]. |
| Microfluidic Device | Enables reproducible, scalable production of monodisperse LNPs. | Standardized mixing of ethanolic lipid and aqueous mRNA phases [51]. |
| Ribogreen Assay Kit | Quantifies mRNA encapsulation efficiency (EE%). | Measures fluorescence before/after LNP disruption; target EE% >90% [51]. |
| Luciferase-Encoding mRNA | Reporter mRNA for in vivo screening of delivery efficiency. | Allows non-invasive bioluminescence imaging to track location and duration of protein expression [51] [50]. |
| (R)-MPH-220 | (R)-MPH-220, MF:C20H21N3O3S, MW:383.5 g/mol | Chemical Reagent |
| D-Allose-13C | D-Allose-13C, MF:C6H12O6, MW:181.15 g/mol | Chemical Reagent |
Diagram 2: Mechanism of LNP Uptake and mRNA Delivery in Hepatocytes
What are the core differences between Cas9 protein (RNP) and mRNA cargo for CRISPR-Cas9 delivery?
The choice between Cas9 protein complexed as a Ribonucleoprotein (RNP) and Cas9-encoding mRNA is a fundamental decision in CRISPR experimental design, impacting editing kinetics, specificity, and technical workflow. The table below summarizes their core characteristics.
Table 1: Core Characteristics of Cas9 RNP versus mRNA Cargo
| Feature | Cas9 RNP (Protein + sgRNA) | Cas9 mRNA (+ sgRNA) |
|---|---|---|
| Cargo Composition | Preassembled complex of Cas9 protein and single-guide RNA (sgRNA) [1] | mRNA molecule encoding the Cas9 protein, plus a separate sgRNA [1] |
| Onset of Activity | Immediate upon delivery; no translation required [1] | Delayed; requires cellular machinery to translate mRNA into functional Cas9 protein [13] |
| Duration of Activity | Short, transient (hours to a few days) due to rapid protein degradation [13] [1] | Moderate; depends on mRNA stability and translation kinetics, but longer than RNP [13] |
| Typical Editing Efficiency | High; fast activity can lead to efficient editing [52] [53] | Can be high, but depends on translation efficiency [54] |
| Off-Target Risk | Generally lower; transient activity reduces chance of unintended cuts [13] [1] | Potentially higher; longer expression window increases opportunity for off-target activity [13] |
| Risk of Genomic Integration | None; no DNA template involved [52] | Very low; mRNA acts in the cytoplasm and does not enter the nucleus [13] |
| Immunogenicity | Typically low [1] | Can be higher; exogenous mRNA can trigger innate immune responses [13] [55] |
| Production Complexity | High; requires expression and purification of active Cas9 protein, raising cost and complexity [13] | Lower; in vitro transcription (IVT) of mRNA is a well-established and scalable process [55] |
| Stability | Protein is sensitive to denaturation; requires careful handling and storage [13] | Inherently unstable; requires encapsulation in delivery vectors (e.g., LNPs) for protection [56] [13] |
How do I troubleshoot low knockout efficiency in my CRISPR experiments?
Low knockout efficiency can stem from multiple factors. The following checklist and table guide you through systematic troubleshooting.
Table 2: Troubleshooting Guide for Low Knockout Efficiency
| Problem Area | Potential Cause | Solution & Optimization Strategy |
|---|---|---|
| sgRNA Design | Suboptimal sgRNA sequence with low activity or specificity [15] | - Use bioinformatics tools (e.g., Benchling, CRISPR Design Tool) to predict high-efficiency sgRNAs [15].- Design and test 3-5 different sgRNAs per target gene to identify the most effective one [15] [53]. |
| Cargo Delivery | Low transfection efficiency; cargo not reaching enough cells [15] | - For RNP/mRNA: Optimize transfection parameters. Use lipid-based reagents (e.g., Lipofectamine, DharmaFECT) or lipid nanoparticles (LNPs). For hard-to-transfect cells, use electroporation [15].- For mRNA: Ensure it is properly modified and encapsulated to enhance stability and translation [13] [55]. |
| Cargo Integrity & Dosage | Degraded cargo or incorrect cell-to-cargo ratio [53] | - For RNP: Verify protein activity and complex formation. Use chemically modified sgRNAs to enhance stability (e.g., 2'-O-methyl-3'-thiophosphonoacetate modifications) [53].- For mRNA: Ensure high-quality, capped, and polyadenylated mRNA. Optimize the amount of mRNA delivered [55]. |
| Cellular & Biological Factors | High activity of DNA repair pathways or cell-type specific resistance [15] | - Use stable cell lines expressing Cas9 (e.g., inducible Cas9 systems) for more consistent and reproducible editing [15] [53].- Consider the cell cycle; dividing cells often show higher HDR efficiency. |
| Validation Methods | Ineffective sgRNA that induces mutations but does not disrupt protein function [53] | - Employ functional validation (e.g., Western blotting) to confirm protein loss, not just genetic INDEL detection. This identifies "ineffective sgRNAs" [15] [53]. |
What are the key delivery vehicles for in vivo and ex vivo administration?
The delivery vehicle is critical for transporting CRISPR cargo to target cells. The choice depends heavily on the application (in vivo vs. ex vivo) and the cargo type.
Table 3: Comparison of Key CRISPR-Cas9 Delivery Vehicles
| Delivery Vehicle | Description | Best For | Advantages | Disadvantages & Challenges |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | Synthetic, spherical vesicles that encapsulate nucleic acids or proteins [56] [1]. | In vivo delivery of mRNA and RNP [13] [1]. Ex vivo transfection. | - Low immunogenicity [1].- Scalable production [1].- Can be engineered for organ targeting (e.g., SORT-LNPs) [1].- Protects cargo from degradation [56]. | - Can trigger inflammatory responses [13].- Requires endosomal escape for functional delivery [1].- Potential cytotoxicity at high doses. |
| Adeno-Associated Viruses (AAVs) | Small, non-pathogenic viral vectors with tissue-specific tropism [13] [1]. | In vivo delivery requiring long-term expression. | - High transduction efficiency [13].- Long-lasting expression [13].- Mild immune response compared to other viruses [1]. | - Very limited packaging capacity (~4.7 kb), insufficient for SpCas9 and sgRNA from a single vector [13] [1].- Risk of prolonged off-target effects and genomic integration events [13].- Pre-existing immunity in populations. |
| Virus-Like Particles (VLPs) | Engineered particles containing viral capsid proteins but no viral genetic material [13] [1]. | Ex vivo and in vivo delivery of RNP complexes. | - Transient, RNP-like activity reduces off-target risks [1].- No risk of genomic integration [1].- Potential for cell-specific targeting [1]. | - Complex and challenging manufacturing [13] [1].- Issues with stability and scalability [1].- Cargo size limitations [1]. |
| Electroporation | Physical method using an electric field to create temporary pores in the cell membrane [15]. | Ex vivo delivery of all cargo types (RNP, mRNA, DNA), especially in hard-to-transfect cells like primary T cells or stem cells. | - High efficiency for many cell types [15].- Direct delivery into cytoplasm. | - Can cause significant cell death if not optimized [15].- Not suitable for in vivo applications.- Requires specialized equipment. |
Can you provide a sample experimental protocol for comparing RNP and mRNA delivery ex vivo?
The following workflow provides a standardized protocol for a head-to-head comparison of RNP and mRNA delivery in a cell culture model, ensuring a fair and interpretable assessment of editing efficiency.
Detailed Protocol Steps:
Step 1: Cargo Preparation
- For RNP: Complex purified S. pyogenes Cas9 protein with synthetic, chemically modified sgRNA at a molar ratio of 1:1.2 to 1:1.5 (Cas9:sgRNA). Incubate for 10-20 minutes at room temperature to form the functional RNP complex [53].
- For mRNA: Use a codon-optimized Cas9 mRNA sequence containing nucleotide modifications (e.g., pseudouridine) to enhance stability and reduce immunogenicity [13] [55]. For transfection, complex this mRNA with your chosen delivery vehicle (e.g., lipid nanoparticles or a commercial transfection reagent) according to the manufacturer's instructions.
Step 2: Cell Transfection
- Culture and harvest your target cells (e.g., HEK293, iPSCs). Count and resuspend the cells in an appropriate buffer.
- Split the cell suspension into two equal aliquots for RNP and mRNA delivery.
- For a fair comparison, use the same delivery method for both cargos. Electroporation is often preferred for its high efficiency, especially for RNP delivery [15]. Alternatively, use an optimized lipid-based transfection reagent.
- Deliver the precomplexed RNP and the mRNA/delivery-reagent complex into their respective cell aliquots using identical physical parameters (e.g., voltage, pulse time for electroporation).
Step 3: Post-Transfection Analysis
- Allow the cells to recover and express the edited genome for 48-72 hours.
- Harvest cells and analyze:
- INDEL Efficiency: Extract genomic DNA. Amplify the target region by PCR and analyze using the T7 Endonuclease I (T7EI) assay or, for higher accuracy, Next-Generation Sequencing (NGS). Tools like ICE (Inference of CRISPR Edits) can calculate the percentage of INDELs from sequencing data [53].
- Protein Knockout Confirmation: Perform Western blotting to confirm the loss of target protein expression. This is crucial to identify "ineffective sgRNAs" that create mutations but do not disrupt the protein [53].
- Cytotoxicity: Perform a cell viability assay (e.g., MTT, flow cytometry with viability dye) to assess any differences in toxicity between the two delivery methods.
What are essential research reagent solutions for CRISPR-Cas9 workflows?
The following table lists key reagents and their functions crucial for successful CRISPR experiments.
Table 4: Research Reagent Solutions for CRISPR Workflows
| Reagent / Tool | Function & Application | Key Considerations |
|---|---|---|
| Chemically Modified sgRNA | Synthetic sgRNA with modifications (e.g., 2'-O-methyl-3'-thiophosphonoacetate) to enhance nuclease resistance and stability within cells [53]. | Critical for improving RNP half-life and editing efficiency, especially in primary cells [53]. |
| Codon-Optimized Cas9 mRNA | mRNA engineered for enhanced translation efficiency in the target organism (e.g., human cells), often with nucleotide modifications (pseudouridine) to reduce immunogenicity [13] [55]. | Increases Cas9 protein yield per mRNA molecule, improving overall editing efficiency. |
| Lipid Nanoparticles (LNPs) | A leading non-viral delivery vector for in vivo applications, encapsulating and protecting mRNA or RNP cargo [56] [13]. | Look for organ-targeted LNP formulations (e.g., SORT-LNPs). Optimization of lipid composition is key for efficacy and reducing toxicity [1]. |
| Stable Inducible Cas9 Cell Lines | Cell lines (e.g., hPSCs-iCas9) engineered to express Cas9 protein only upon induction (e.g., with doxycycline) [53]. | Provides a homogeneous, reproducible system that eliminates delivery variability, leading to highly consistent and efficient editing [15] [53]. |
| Bioinformatics sgRNA Design Tools | Online platforms (e.g., Benchling, CRISPR Design Tool, CCTop) for predicting sgRNA on-target efficiency and potential off-target sites [15] [53]. | Benchling has been shown in some studies to provide highly accurate predictions [53]. Always design multiple sgRNAs per target. |
| INDEL Analysis Software | Algorithms like ICE (Synthego) and TIDE that deconvolute Sanger sequencing data to quantify editing efficiency [53]. | ICE has been validated to show high sensitivity and accuracy compared to T7EI assay and other tools [53]. |
Solving Efficiency Hurdles: Off-Target Effects, Immunogenicity, and Editing Kinetics
For researchers optimizing Cas9 protein versus mRNA delivery, controlling off-target effects is a critical challenge that can confound experimental results and diminish therapeutic potential [57]. Off-target activity refers to unintended DNA cleavage at sites other than the intended on-target site, which can lead to erroneous phenotypes, genotoxic effects, and confusion in data interpretation [58]. This guide provides targeted strategies and troubleshooting advice to enhance the specificity of your genome editing experiments, with particular focus on high-fidelity Cas variants and emerging Anti-CRISPR technologies.
FAQs: Addressing Common Experimental Challenges
Q1: What are the primary causes of off-target effects in CRISPR-Cas9 experiments?
Off-target effects primarily occur when the Cas9-sgRNA complex binds and cleaves DNA at sites with sequence similarity to the intended target. Key factors include:
- Mismatch tolerance: Cas9 can tolerate up to 3-5 base pair mismatches, particularly in the PAM-distal region of the sgRNA guiding sequence [57]
- Non-canonical PAM sequences: Cas9 can sometimes recognize and cleave DNA at sites with non-NGG PAM sequences [57]
- Excess energy hypothesis: Wild-type SpCas9 possesses more binding energy than needed for optimal on-target recognition, enabling cleavage at mismatched off-target sites [59]
- Prolonged Cas9 expression: Sustained nuclease expression from plasmid or viral vectors increases the window for off-target cleavage [13]
Q2: How do high-fidelity Cas9 variants reduce off-target effects while maintaining on-target activity?
High-fidelity variants are engineered through rational protein design to reduce non-specific DNA contacts while preserving on-target cleavage efficiency:
- SpCas9-HF1: Contains four alanine substitutions (N497A, R661A, Q695A, and Q926A) that disrupt hydrogen bonds to the target DNA phosphate backbone. This variant exhibits undetectable off-target activity with >85% of sgRNAs tested while retaining comparable on-target activity to wild-type SpCas9 [59]
- eSpCas9: Engineered to reduce non-specific binding to the non-target DNA strand [57]
- SuperFi-Cas9: A recently developed variant with extreme-low mismatch rates and near wild-type cleavage efficiency, designed based on structural insights into mismatch surveillance [60]
Table 1: Comparison of High-Fidelity Cas9 Variants
| Variant | Key Mutations | On-Target Efficiency | Off-Target Reduction | PAM Requirement |
|---|---|---|---|---|
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | >70% of wild-type for 86% of sgRNAs [59] | Undetectable with most sgRNAs [59] | NGG |
| eSpCas9 | Not specified | Comparable to wild-type [57] | Significant reduction [57] | NGG |
| SpCas9-HF1 | Mismatch surveillance optimization | Near wild-type [60] | Extreme-low mismatch rates [60] | NGG |
Q3: What delivery strategies minimize off-target effects for Cas9 protein versus mRNA approaches?
The form of CRISPR-Cas9 delivery significantly impacts off-target profiles:
- RNP (Ribonucleoprotein) complexes: Pre-assembled Cas9 protein-gRNA complexes enable rapid editing with minimal persistence, significantly reducing off-target effects compared to plasmid-based delivery [9] [2]
- mRNA delivery: Provides transient Cas9 expression without genomic integration risk, offering a balance between editing efficiency and reduced off-target potential [13]
- Plasmid/viral vectors: Lead to prolonged Cas9 expression, increasing off-target risks, though still widely used for their high transduction efficiency [13]
Table 2: Off-Target Profiles by Delivery Method
| Delivery Method | Editing Persistence | Relative Off-Target Risk | Key Advantages |
|---|---|---|---|
| RNP Complexes | Shortest (hours-days) | Lowest [9] [2] | Rapid clearance, high specificity |
| mRNA | Short (days) | Low [13] | No genome integration, controlled expression |
| Plasmid DNA | Moderate (days-weeks) | Moderate [13] | Simple production, high expression |
| Viral Vectors | Longest (weeks-months) | Highest [13] | High transduction efficiency, stable for hard-to-transfect cells |
Q4: How can Anti-CRISPR proteins be utilized to control off-target effects?
Anti-CRISPR (Acr) proteins are natural inhibitors encoded by bacteriophages to evade bacterial CRISPR immune systems. They offer a powerful "off-switch" for CRISPR activity:
- Mechanisms of action: Acr proteins employ diverse inhibitory strategies including (a) interruption of CRISPR-Cas complex assembly, (b) interference with target DNA binding, (c) blocking of target DNA/RNA cleavage, and (d) enzymatic modification or degradation of signaling molecules [61]
- Experimental applications: Acr proteins enable temporal control over genome editing windows, potentially constraining off-target effects by limiting the active timeframe [61]
- Specificity: Different Acr proteins target distinct CRISPR-Cas systems, allowing selective inhibition (e.g., AcrIIA4 inhibits SpyCas9) [61]
Q5: What sgRNA design strategies enhance targeting specificity?
Careful sgRNA design is crucial for minimizing off-target effects:
- Truncated sgRNAs: Using shorter guide sequences (17-18 nt instead of 20 nt) can reduce off-target cleavage without compromising on-target activity [62] [57]
- GG20 strategy: Replacing GX19 sgRNAs with two guanines at the 5' end (ggX20 sgRNAs) significantly reduces off-target effects [57]
- GC content optimization: Maintaining 40-60% GC content in the sgRNA seed region stabilizes the DNA:RNA duplex and improves specificity [57]
- Chemical modifications: Incorporating 2'-O-methyl-3'-phosphonoacetate in the ribose-phosphate backbone reduces off-target cleavage while maintaining on-target performance [57]
Experimental Protocols for Off-Target Assessment
Protocol 1: GUIDE-seq for Genome-Wide Off-Target Detection
GUIDE-seq (genome-wide unbiased identification of DSBs enabled by sequencing) provides comprehensive off-target profiling [59]:
- Transfection: Co-transfect cells with Cas9-sgRNA expression plasmids and a double-stranded oligodeoxynucleotide (dsODN) tag
- Tag integration: The dsODN tag integrates into double-strand break sites during repair
- Library preparation: Extract genomic DNA and prepare sequencing libraries using tag-specific primers
- Sequencing and analysis: Perform high-throughput sequencing and computational analysis to identify tag integration sites
Protocol 2: RNP Delivery for Reduced Off-Target Editing
For minimal off-target effects using ribonucleoprotein complexes [9]:
- Complex assembly: Incubate purified Cas9 protein (10-50 µM) with sgRNA at 1:1.2-1.5 molar ratio in optimized buffer for 10-20 minutes at 25°C
- Delivery: Introduce RNP complexes into cells via electroporation (for hard-to-transfect cells) or lipid nanoparticles
- Validation: Assess editing efficiency 48-72 hours post-delivery via T7EI assay or sequencing
Protocol 3: High-Fidelity Variant Validation
When implementing high-fidelity Cas9 variants [59]:
- Plasmid selection: Choose appropriate expression vectors for high-fidelity variants (e.g., SpCas9-HF1)
- Control experiments: Always include wild-type Cas9 controls to benchmark on-target efficiency
- Multi-sgRNA testing: Test 2-3 sgRNAs per target to identify optimal guides
- Off-target assessment: Employ GUIDE-seq or targeted sequencing of predicted off-target sites
Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Minimization
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| High-Fidelity Cas9 Variants | SpCas9-HF1, eSpCas9, SuperFi-Cas9 | Reduce non-specific DNA cleavage while maintaining on-target activity [59] [60] |
| Modified sgRNAs | 2'-O-methyl-3'-phosphonoacetate modified guides | Enhance stability and specificity of DNA recognition [57] |
| Anti-CRISPR Proteins | AcrIIA4, AcrIIC1, AcrVA1 | Provide temporal control as CRISPR "off-switches" [61] |
| Delivery Systems | Lipid nanoparticles, Electroporation systems | Enable efficient RNP or mRNA delivery [13] [2] |
| Off-Target Detection | GUIDE-seq reagents, T7 Endonuclease I | Identify and quantify off-target editing events [59] |
Decision Framework for Experimental Design
Optimizing Cas9 delivery efficiency while minimizing off-target effects requires a multifaceted approach combining high-fidelity variants, strategic delivery methods, and careful experimental design. For therapeutic applications where specificity is paramount, the combination of mRNA or RNP delivery with engineered high-fidelity Cas9 variants represents the current gold standard. As CRISPR technologies evolve, emerging tools including Anti-CRISPR proteins and improved computational prediction algorithms will provide researchers with increasingly sophisticated control over genome editing outcomes.
Troubleshooting Guide: Common mRNA Instability Issues in CRISPR-Cas9 Research
This guide addresses frequent challenges faced when working with mRNA-based CRISPR-Cas9 systems, helping researchers identify and resolve issues that compromise editing efficiency.
1. Problem: Low Cas9 Protein Expression Despite High-Quality mRNA
- Potential Causes: Rapid mRNA degradation, inefficient translation initiation, or strong immune response in target cells.
- Solutions:
2. Problem: High Off-Target Editing with mRNA-CRISPR Systems
- Potential Causes: Prolonged Cas9 expression beyond therapeutic window.
- Solutions:
3. Problem: Inconsistent Editing Efficiency Across Cell Types
- Potential Causes: Cell-type specific variations in translation machinery or mRNA degradation pathways.
- Solutions:
4. Problem: Poor mRNA Delivery and Cellular Uptake
- Potential Causes: Inefficient encapsulation or nanoparticle instability.
- Solutions:
Frequently Asked Questions (FAQs)
Q1: What are the key advantages of using mRNA over DNA for delivering CRISPR-Cas9 systems?
mRNA offers several advantages for CRISPR-Cas9 delivery: it eliminates the risk of host genome integration, provides transient expression that reduces off-target effects, and acts directly in the cytoplasm without needing nuclear entry [13]. Unlike DNA-based systems, mRNA does not require nuclear localization and enables instantaneous translation, making it particularly suitable for therapeutic applications where controlled, temporary expression is desirable.
Q2: How do nucleoside modifications improve mRNA stability and reduce immunogenicity?
Nucleoside modifications, such as replacing uridine with pseudouridine or N1-methylpseudouridine, mask mRNA from the innate immune system by preventing recognition by Toll-like receptors (TLR7 and TLR8) [63]. This reduces interferon responses and prevents translational shutdown while maintaining—and in some cases enhancing—protein expression capabilities. These modifications also increase resistance to enzymatic degradation by cellular nucleases.
Q3: What is the difference between traditional codon optimization and newer deep learning approaches?
Traditional codon optimization methods rely on predefined rules like Codon Adaptation Index (CAI) to match codon usage bias of highly expressed genes [66]. In contrast, deep learning frameworks like RiboDecode directly learn from large-scale ribosome profiling data (Ribo-seq) to explore a broader sequence space and generate context-aware optimized sequences [22]. These AI-driven approaches can account for cellular environment and demonstrate superior performance across different mRNA formats, including modified and circular mRNAs.
Q4: How important are UTRs in optimizing mRNA-based CRISPR systems?
UTRs are critical determinants of mRNA stability, localization, and translational efficiency. The 5'UTR significantly influences translation initiation by affecting ribosome scanning and loading, while the 3'UTR contains elements that regulate mRNA stability and degradation [64] [63]. Research shows that engineered UTRs can increase protein expression by 2-5 fold compared to standard globin UTRs commonly used in therapeutic mRNA [64]. Deep learning models can now design de novo UTRs specifically optimized for gene editing applications.
Q5: What are the main delivery challenges for mRNA-based CRISPR therapies?
The primary delivery challenges include: (1) protecting mRNA from serum nucleases during transit to target cells, (2) achieving efficient cellular uptake and endosomal escape, (3) minimizing immune recognition, and (4) ensuring tissue-specific delivery where needed [13] [67]. Lipid nanoparticles (LNPs) currently represent the gold standard for in vivo delivery, offering protection, efficient cellular uptake, and endosomal escape capabilities [13] [65].
Table 1: Comparison of mRNA Stabilization Approaches
| Technique | Key Improvement | Experimental Validation | Impact on Expression |
|---|---|---|---|
| Nucleoside Modification (N1-methylpseudouridine) | Reduced immunogenicity; Enhanced translation | In vivo mouse models; Multiple vaccine trials | Up to 2-3x increase in protein production compared to unmodified mRNA [63] |
| Codon Optimization (RiboDecode AI) | Improved translation efficiency; Context-aware design | In vitro translation; In vivo mouse neutralization assays | 10x stronger antibody responses; Equivalent efficacy at 1/5 dose [22] |
| 5'UTR Engineering (Optimus 5-Prime) | Enhanced ribosome loading; Reduced secondary structure | Massively parallel reporter assays (MPRAs); Gene editing efficiency tests | 2-5x increase in translation efficiency compared to standard UTRs [64] |
| Position-specific 2'-F Modification (1st nucleoside in codon) | Increased nuclease resistance without compromising translation | Cell-free translation systems; LC-MS validation | Significant stability improvement while maintaining >85% translational activity [68] |
| Poly(A) Tail Modifications (2'-F every 2nt) | Enhanced stability and translational activity | Sandwich ELISA evaluation of peptide production | Further enhanced protein production beyond non-modified poly(A) [68] |
Table 2: Performance Comparison of CRISPR-Cas9 Delivery Formats
| Delivery Format | Editing Efficiency | Off-Target Effects | Duration of Expression | Key Limitations |
|---|---|---|---|---|
| DNA (AAV vectors) | High, sustained | Higher due to persistent expression | Long-term (weeks to months) | Limited packaging capacity; Host genome integration risk [13] |
| mRNA (LNP delivery) | High, transient | Lower due to short half-life | Short-term (days) | Requires cold chain; Potential immunogenicity [13] |
| RNP (VLP delivery) | Moderate to high | Lowest among formats | Very short (hours) | Difficult manufacturing; Lack of efficient in vivo delivery vectors [13] |
Experimental Protocols for Key mRNA Optimization Techniques
Protocol 1: Assessing mRNA Stability and Translation Efficiency
Purpose: Systematically evaluate modified mRNA constructs for Cas9 expression.
Materials:
- N1-methylpseudouridine triphosphates for IVT [63]
- CleanCap AG co-transcriptional capping analog [65]
- In vitro transcription kit with T7 RNA polymerase
- HeLa cell-free translation system [68]
- Lipid nanoparticles for delivery optimization [65]
Method:
- Synthesize mRNA constructs using modified nucleosides via in vitro transcription.
- Purify mRNA using magnetic bead-based purification systems [65].
- Encapsulate in LNPs using microfluidic mixing technology.
- Transfert target cells (HEK293T, HepG2, or primary T-cells based on application).
- Assess editing efficiency via T7E1 assay or next-generation sequencing.
- Measure mRNA stability using qPCR over time course (0, 2, 4, 8, 24 hours).
- Evaluate immune activation via interferon-alpha ELISA.
Protocol 2: High-Throughput UTR Screening Using MPRA
Purpose: Identify optimal UTR sequences for enhanced Cas9 expression.
Materials:
- Randomized UTR library templates
- In vitro transcription reagents
- Polysome profiling equipment
- High-throughput sequencing platform
Method:
- Clone 5'UTR library with random sequences (25-50nt) upstream of Cas9 CDS.
- Perform in vitro transcription to generate mRNA library.
- Transfert cells and incubate for 8 hours with cycloheximide treatment.
- Perform polysome profiling to separate transcripts by ribosome number.
- Sequence fractions and calculate Mean Ribosome Load (MRL) for each UTR.
- Train deep learning models (e.g., Optimus 5-Prime) on MRL data [64].
- Design optimized UTRs using gradient descent (Fast SeqProp) or generative networks (DENs).
Essential Research Reagent Solutions
Table 3: Key Reagents for mRNA-Based CRISPR Research
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Modified Nucleosides | N1-methylpseudouridine, Pseudouridine, 5-methylcytidine | Reduce immunogenicity; Enhance stability and translation | N1-methylpseudouridine generally outperforms pseudouridine [63] |
| Capping Analogs | CleanCap, ARCA | Enhance translation initiation; Protect from 5' exonuclease | CleanCap provides superior cap1 structure compared to earlier technologies [65] |
| Codon Optimization Tools | RiboDecode, LinearDesign | Enhance translation efficiency; Optimize mRNA stability | RiboDecode shows superior performance across different mRNA formats [22] |
| Delivery Systems | LNPs, VLPs, Electroporation | Protect mRNA; Facilitate cellular uptake | LNP formulation must be optimized for specific cell/tissue targets [13] [65] |
| Stability-Enhancing Additives | Trehalose, Sucrose | Cryoprotection during storage | Critical for maintaining LNP integrity during freeze-thaw cycles [65] |
Visualization of mRNA Optimization Strategies
Optimization Strategy Relationships
mRNA Therapeutic Development Workflow
Addressing Cas9 Protein Aggregation for Improved Encapsulation and Efficiency
Frequently Asked Questions (FAQs)
Q1: What is Cas9 protein aggregation and why is it a problem for gene editing efficiency? Cas9 protein aggregation refers to the abnormal association of Cas9 proteins, forming insoluble assemblies that can range from small dimers to large particles. This is a significant problem because these aggregated particles can exceed the optimal size range for efficient cellular delivery, compromising the ability to deliver functional Cas9 into cells and thereby reducing genome editing efficiency [69]. Furthermore, aggregation can be triggered by environmental stresses common during experimental handling, such as temperature fluctuations and pH adjustments [69].
Q2: How does Cas9 aggregation specifically impact different delivery cargo types (plasmid, mRNA, RNP)? Aggregation impacts pre-assembled Ribonucleoprotein (RNP) complexes most directly, as they involve the delivery of the functional Cas9 protein itself. Aggregated Cas9 in RNP preparations leads to decreased functional uptake and editing. For plasmid and mRNA cargo types, which rely on in vivo translation to produce Cas9, aggregation is less of a direct delivery issue but can still affect the stability and functionality of the translated protein if it misfolds post-synthesis [69].
Q3: What are the primary causes of Cas9 aggregation during experimental preparation? The primary causes include:
- Environmental Stress: Fluctuations in temperature and pH during storage, handling, or formulation can induce protein denaturation and subsequent aggregation [69].
- Electrostatic Interactions: The net positive charge of Cas9 can lead to undesirable interactions with other molecules or surfaces during the preparation of delivery complexes, potentially promoting aggregation [69].
- Formulation Conditions: Suboptimal buffer composition, ionic strength, or concentration can destabilize the native protein structure [69].
Q4: What strategies can be used to prevent or minimize Cas9 aggregation? Key strategies include:
- Optimized Buffer Formulation: Using buffers with appropriate pH, ionic strength, and additives (e.g., stabilizers, surfactants) to maintain Cas9 solubility [69].
- Proper Handling and Storage: Maintaining consistent, low temperatures and avoiding repeated freeze-thaw cycles [69].
- AI-Guided Protein Engineering: Developing engineered Cas9 variants with mutations that enhance stability and solubility, thereby reducing inherent aggregation propensity [70].
- Advanced Delivery Vehicles: Utilizing lipid nanoparticles (LNPs) or other nano-carriers designed to encapsulate and protect Cas9 from the aggregation-inducing extracellular environment [69].
Troubleshooting Guides
Problem: Low Gene Editing Efficiency Suspected from Cas9 Aggregation
Step 1: Visual Inspection and Simple Assays
- Action: Visually inspect the Cas9 protein or RNP solution for cloudiness or particulate matter. Perform a quick centrifugation test; significant pellet formation after a low-speed spin may indicate aggregation.
- Rationale: Gross aggregation is often detectable without sophisticated equipment, providing an initial diagnostic [69].
Step 2: Characterize Particle Size and Distribution
- Action: Use Dynamic Light Scattering (DLS) to measure the hydrodynamic diameter and polydispersity of your Cas9 sample. Compare the size profile to a freshly prepared or known good batch.
- Rationale: Aggregation causes a measurable increase in particle size and heterogeneity. DLS provides quantitative data to confirm aggregation and its extent [69].
Step 3: Identify the Root Cause Refer to the following table to diagnose and address the specific cause of aggregation.
Table: Troubleshooting Cas9 Aggregation
| Observed Symptom | Potential Root Cause | Corrective Action |
|---|---|---|
| Cloudiness after thawing | Repeated freeze-thaw cycles or improper freezing | Aliquot Cas9 into single-use volumes. Flash-freeze in liquid nitrogen and store at -80°C. Avoid frost-free freezers. |
| Precipitation upon dilution | Shift to non-optimal pH or buffer conditions | Dialyze or dilute into a validated storage buffer. Ensure compatibility between your Cas9 buffer and all downstream reagents. |
| Low editing efficiency with high protein concentration | Concentration-induced aggregation | Dilute the stock to the working concentration in a stabilizing buffer. Avoid storing Cas9 at high concentrations for long periods. |
| Aggregation in final LNP formulation | Unfavorable electrostatic interactions during encapsulation | Re-optimize the charge ratio between cationic lipids and the anionic RNP complex. Incorporate stabilizing polymers [69]. |
Step 4: Implement Preventive Measures
- For RNP Delivery: Switch to freshly prepared RNP complexes for critical experiments. Ensure gRNA is pure and free of contaminants.
- For All Cargo Types: Adhere strictly to recommended storage conditions. Consider using engineered, high-stability Cas9 variants (e.g., AI-designed mutants) that are less prone to aggregation [70].
Problem: Optimizing Lipid Nanoparticles (LNPs) to Prevent Cas9 RNP Aggregation
Background: LNPs are a leading non-viral delivery platform. Their formulation is critical to prevent aggregation and ensure efficient encapsulation of CRISPR cargo [1] [69].
Protocol: Formulating and Testing LNPs for Cas9 RNP Delivery
LNP Preparation via Microfluidic Mixing:
- Prepare an ethanol phase containing ionizable lipid, helper phospholipid, cholesterol, and PEG-lipid.
- Prepare an aqueous phase containing your purified Cas9 RNP complex in a suitable buffer.
- Mix the two phases rapidly using a microfluidic device at a controlled flow rate and ratio (typically 3:1 aqueous-to-ethanol) to form LNPs through spontaneous nanoprecipitation [69].
LNP Purification and Characterization:
- Dialyze or use tangential flow filtration against a suitable buffer (e.g., PBS) to remove ethanol and non-encapsulated material.
- Use DLS to measure LNP size and polydispersity index (PDI). Aim for a size of 80-150 nm and PDI < 0.2.
- Measure zeta potential to assess surface charge.
- Use a Ribogreen assay to quantify RNP encapsulation efficiency [69].
Functional Validation in Cell Culture:
- Treat target cells (e.g., HEK293T) with the formulated LNPs.
- Measure editing efficiency 48-72 hours post-transfection via next-generation sequencing (NGS) of the target locus [70].
Table: LNP Formulation Parameters and Their Impact on Aggregation & Efficiency
| Parameter | Impact on Aggregation | Impact on Editing Efficiency | Optimization Goal |
|---|---|---|---|
| Ionizable Lipid : RNP Charge Ratio | Incorrect ratio leads to incomplete encapsulation and external aggregation. | Optimal charge balance maximizes encapsulation and endosomal escape. | Find the ratio that yields >90% encapsulation and high cell viability. |
| PEG-Lipid Content (%) | High PEG content improves colloidal stability and prevents LNP fusion. | Excess PEG can inhibit cellular uptake and endosomal escape. | Typically 1.5-5 mol%; balance stability with functional delivery. |
| Final LNP Size (nm) | Larger particles (>200 nm) may indicate aggregation and are less efficiently internalized. | Smaller particles (~80-100 nm) often show superior tissue penetration and cellular uptake. | Target 80-150 nm with low PDI. |
| Buffer Composition (aqueous phase) | Non-physiological pH or osmolality can denature Cas9 during formulation. | Maintains Cas9 protein integrity and activity post-encapsulation. | Use a validated, Cas9-stable buffer at neutral pH. |
Experimental Protocols & Workflows
Protocol 1: Assessing Cas9 Aggregation and Its Impact on Editing Efficiency
This protocol allows researchers to quantitatively link the physical state of their Cas9 protein to its functional performance.
Materials:
- Purified Cas9 protein (wild-type and/or engineered variants)
- Dynamic Light Scattering (DLS) instrument
- Cell line for testing (e.g., HEK293T)
- Transfection reagent (e.g., LNP formulation kit)
- Next-Generation Sequencing (NGS) platform
Method:
- Induce Controlled Stress: Subject aliquots of Cas9 protein to different stress conditions (e.g., 37°C for 1 hour, vortexing, pH shift).
- Measure Aggregation: Analyze the stressed samples and a control sample (stored at -80°C) using DLS to determine the hydrodynamic diameter and PDI.
- Formulate RNPs: Complex the stressed and control Cas9 samples with a target-specific gRNA to form RNPs.
- Transfect Cells: Deliver the RNPs into your target cell line using a consistent method (e.g., LNP transfection). Include an untreated control.
- Quantify Editing Efficiency: Harvest cells after 48-72 hours, extract genomic DNA, and amplify the target locus by PCR for NGS analysis. Calculate the percentage of indels or precise edits.
Workflow for assessing Cas9 aggregation impact.
Protocol 2: Evaluating Engineered Cas9 Variants for Enhanced Stability
This methodology is adapted from recent studies using AI to design high-performance Cas9 variants with improved properties, which can include reduced aggregation [70].
Materials:
- Plasmids encoding wild-type and AI-engineered Cas9 variants (e.g., AncBE4max-AI-8.3) [70]
- HEK293T cells or other relevant cell lines
- sgRNA plasmids targeting standard genomic loci
- Flow cytometer with cell sorting capability
- NGS platform
Method:
- Cell Transfection: Co-transfect HEK293T cells with plasmids encoding the Cas9 variant (wild-type or AI-engineered) and the corresponding sgRNA.
- Cell Selection: Approximately 48 hours post-transfection, enrich successfully transfected cells (e.g., based on a fluorescent marker like mCherry) using fluorescence-activated cell sorting (FACS).
- Genomic Analysis: Extract genomic DNA from the sorted cell population. Amplify the target genomic loci by PCR and subject the amplicons to NGS.
- Efficiency Calculation: Analyze the NGS data to calculate the percentage of edited sequences for each Cas9 variant at each target site.
- Stability Inference: Consistently higher editing efficiency from engineered variants, particularly at challenging sites, indicates improved functional stability and reduced aggregation, as demonstrated by the 2-3 fold increase in efficiency shown by AncBE4max-AI-8.3 [70].
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Reagents for Investigating Cas9 Aggregation
| Reagent / Tool | Function / Application | Key Consideration |
|---|---|---|
| Dynamic Light Scattering (DLS) | Measures hydrodynamic diameter and polydispersity to quantify protein aggregation. | Critical for pre-experiment quality control of Cas9 protein and formulated LNPs. |
| AI-Engineered Cas9 Variants | High-performance Cas9 with point mutations (e.g., G1218R, C80K) that improve stability and editing efficiency. | Reduces inherent aggregation propensity. Can provide a 2-3 fold efficiency increase [70]. |
| Ionizable Lipid Nanoparticles (LNPs) | A leading non-viral delivery vehicle for encapsulating and protecting RNP complexes. | Must be optimized for charge ratio and PEG content to prevent aggregation during formulation [69]. |
| Cationic Polymers (e.g., PEI) | Can form polyplexes with CRISPR cargo but requires careful optimization to avoid inducing aggregation. | Simpler than LNPs but may have higher cytotoxicity; less ideal for sensitive applications [1]. |
| Next-Generation Sequencing (NGS) | The gold standard for quantitatively measuring on-target genome editing efficiency. | Essential for functionally validating that aggregation-mitigation strategies successfully restore high editing rates. |
| KR-39038 | KR-39038, MF:C24H32ClFN6O, MW:475.0 g/mol | Chemical Reagent |
AI and Machine Learning for Optimizing LNP Formulations and mRNA Codon Sequences
Frequently Asked Questions (FAQs)
FAQ 1: How can AI models improve mRNA codon sequences beyond traditional optimization methods? Traditional codon optimization methods, like the Codon Adaptation Index (CAI), rely on predefined rules and often fail to capture the complex factors governing mRNA translation and stability. AI models, such as RiboDecode, address this by using deep learning to directly learn from large-scale experimental data (e.g., ribosome profiling or Ribo-seq) [22] [71]. This data-driven approach allows AI to explore a vast sequence space and generate optimized mRNA sequences that significantly enhance protein expression. For instance, RiboDecode has been shown to substantially improve protein expression in vitro and induce stronger immune responses in vivo compared to past methods [22].
FAQ 2: What specific properties of Lipid Nanoparticles (LNPs) can AI help optimize? AI and machine learning models can predict and optimize key LNP properties that influence mRNA delivery efficiency. Researchers have used models like COMET and LightGBM to optimize the following [72] [73]:
- Transfection Efficiency: The ability of LNPs to deliver mRNA into cells and produce the encoded protein.
- Cell-Type Specific Targeting: Designing LNPs that preferentially deliver their payload to specific cell types (e.g., liver cells or Caco-2 cells).
- Stability: Formulating LNPs that can withstand processes like lyophilization (freeze-drying) to extend shelf-life.
- Ionizable Lipid Structure: Identifying optimal chemical structures, such as the number of carbons in the lipid tail, which is a key factor for potency [73].
FAQ 3: My AI-optimized mRNA sequence shows high protein expression in vitro, but the therapeutic effect in vivo is lacking. What could be wrong? High protein expression does not always directly correlate with the desired therapeutic outcome, such as a robust immune response. This can be due to several factors:
- Antigen-Specific Immune Dynamics: The immune system's response to an antigen is complex. One study found that while a novel loop structure in the poly(A) tail significantly increased antigen translation, it did not lead to a statistically significant difference in T-cell immunity compared to other structures, though it did improve antibody responses [74].
- Cellular Context: The AI model may not have been trained on data relevant to your specific target cell type or in vivo environment. Ensure your optimization framework, like RiboDecode, is context-aware and can account for different cellular environments [22].
- Delivery Efficiency: The LNP may not be effectively reaching the target cells in vivo. Consider using AI to re-optimize the LNP formulation for improved in vivo targeting [72].
FAQ 4: Are AI-optimized components compatible with different mRNA formats used in CRISPR research? Yes, a key advantage of advanced AI optimization frameworks is their robustness across various mRNA formats. For example, the RiboDecode platform has demonstrated robust performance across different mRNA formats, including unmodified, m1Ψ-modified, and circular mRNAs [22] [71]. This is particularly important for CRISPR therapy research, which may utilize mRNA to deliver Cas9 nucleases, base editors, or prime editors.
Troubleshooting Guides
Issue 1: Poor Transfection Efficiency with New AI-Designed LNP Formulations
This guide addresses low protein output after implementing a new AI-predicted LNP formulation.
- Problem: Low mRNA translation efficiency observed in cell culture or in vivo models after switching to a new AI-designed LNP.
- Solution: Follow the diagnostic workflow below to identify and correct the issue.
Diagnostic Steps:
- Verify mRNA Integrity: Before encapsulation, check the quality of the mRNA via gel electrophoresis. Degraded mRNA will not translate efficiently, regardless of the LNP quality [74].
- Check LNP Physical Properties: Use Dynamic Light Scattering (DLS) to measure the particle size and polydispersity index (PDI). Abnormal sizes or a high PDI can indicate unstable formulations or improper assembly, leading to poor delivery [74].
- Confirm Component Compatibility: If the AI model introduced a new type of material (e.g., a branched polymer like PBAE), ensure it is compatible with the other LNP components (ionizable lipid, cholesterol, PEG-lipid, helper lipid). Incompatibilities can disrupt nanoparticle self-assembly [72].
- Re-calibrate the AI Model: AI models like COMET improve with more data. If the initial predictions are suboptimal, generate a small set of experimental results using the new components or target cells and feed this data back into the model to refine its predictions [72].
Issue 2: Suboptimal In Vivo Performance of AI-Optimized Cas9 mRNA
This guide addresses when an AI-optimized Cas9 mRNA sequence shows great performance in cells but fails in animal models.
- Problem: AI-optimized Cas9 mRNA does not yield the expected gene editing outcomes in vivo.
- Solution: Systematically check the following areas.
Diagnostic Steps:
- Confirm Cellular Context Awareness: Was the mRNA sequence optimized with the target tissue in mind? AI models like RiboDecode can incorporate cellular context (e.g., gene expression profiles from RNA-seq) to improve translation in specific environments. Verify that the optimization considered the context of your target organ [22].
- Re-evaluate the LNP Formulation: The default tendency of LNPs is to accumulate in the liver [7]. If your target is not the liver, your AI-optimized mRNA may not be reaching the correct cells. Explore using AI models to design LNPs with affinity for different organs [72].
- Consider Re-dosing: Unlike viral delivery, LNP-based mRNA delivery may allow for safe re-dosing. Early clinical trials for in vivo CRISPR therapies have successfully administered multiple doses to increase the percentage of edited cells [7]. A single dose of your optimized mRNA might be insufficient.
- Analyze the Optimization Parameters: Understand what the AI model optimized for. If it solely maximized protein expression (translation), it might have overlooked other factors crucial for Cas9 activity, such as the timing and duration of expression. Consider using a joint optimization strategy that balances translation and mRNA stability (MFE) [22].
Key Experimental Data and Protocols
Performance Comparison of mRNA Optimization Techniques
The table below summarizes quantitative data from key studies, providing a benchmark for comparing AI-driven and traditional methods.
Table 1: Comparative Performance of mRNA Sequence Optimization Methods
| Method | Type | Key Metric | Performance Result | Experimental Context |
|---|---|---|---|---|
| RiboDecode [22] [71] | Deep Learning | Protein Expression | Substantial improvements in vitro | Various cell lines |
| Neutralizing Antibody Response | ~10x stronger vs. unoptimized | In vivo (Mice, Influenza HA mRNA) | ||
| Therapeutic Dose Efficiency | Equivalent effect at 1/5th the dose | In vivo (Mice, Optic Nerve Crush Model) | ||
| LinearDesign [73] | Computational Linguistics | Anti-spike IgG Antibody Titer | 57- to 128-fold increase vs. benchmark | In vivo (Mice, SARS-CoV-2 Spike mRNA) |
| Neutralizing Antibody Titer | 9- to 20-fold increase vs. benchmark | In vivo (Mice, SARS-CoV-2 Spike mRNA) | ||
| Poly(A) Tail Loop (A50L50LO) [74] | Structural Engineering | Bioluminescence Signal | Highest sustained expression vs. linear tails | In vitro & In vivo (Mice, Luciferase mRNA) |
| Protein Expression (hEPO) | Highest expression level vs. other structures | In vivo (Mice, hEPO mRNA) |
Performance Comparison of AI-Optimized LNP Formulations
Table 2: Efficacy of AI-Designed Lipid Nanoparticles (LNPs)
| AI Model / Approach | Prediction Accuracy | Key Finding / Optimized Property | Experimental Validation |
|---|---|---|---|
| COMET [72] | N/A | Predicted novel LNP formulations with higher efficiency than commercial benchmarks. | Increased mRNA delivery and protein production in mouse skin cells and Caco-2 cells. |
| LightGBM [73] | R² = 0.94 (Model Fit) | Identified carbon tail length of ionizable lipid as most critical for transfection. | High correlation (R² = 0.83) between predicted and actual nanoluciferase activity for novel lipids. |
| High-Throughput Screening + ML [73] | N/A | Accelerated discovery of ionizable lipids for mRNA delivery and gene editing. | Effective delivery of EGFP reporter and Cre recombinase mRNA. |
Detailed Experimental Protocol: Validating AI-Optimized mRNA In Vivo
The following workflow details a standard method for testing the efficacy of an AI-optimized mRNA, such as one encoding Cas9 or an antigen, in a mouse model [22] [74].
Protocol Steps:
- LNP Formulation: Encapsulate the AI-optimized mRNA and a control mRNA in LNPs using standard microfluidic mixing techniques.
- LNP Characterization: Characterize the formulated mRNA-LNPs using Dynamic Light Scattering (DLS) to ensure a consistent particle size (e.g., 70-100 nm) and low polydispersity index (PDI < 0.3). Measure encapsulation efficiency to confirm most mRNA is protected within the particles [74].
- Animal Dosing: Administer the mRNA-LNP complex to mice (e.g., C57BL/6) via an appropriate route. Intramuscular (IM) injection is common for vaccines, while intravenous (IV) is used for systemic delivery. Include groups for the optimized sequence, the original sequence, and a negative control [22] [74].
- Expression Analysis (Early Time Points: 6-48 hours):
- Bioluminescence Imaging: If the mRNA encodes a reporter like firefly luciferase (F/L), monitor protein expression over time using an In Vivo Imaging System (IVIS) [74].
- ELISA / Blood Tests: If the mRNA encodes a secreted protein (e.g., human Erythropoietin, hEPO), collect serum at various time points and measure protein concentration using ELISA [22] [74].
- Functional Assessment (Later Time Points: Days to Weeks):
- Immune Profiling: For vaccines, collect serum to measure antigen-specific antibody titers via ELISA and isolate splenocytes to analyze T-cell responses (e.g., via flow cytometry for activation markers like CD69/CD25 or cytokine production) [74].
- Gene Editing Analysis: For Cas9 mRNA, harvest target tissues after a suitable period. Extract genomic DNA and analyze the target locus using next-generation sequencing (NGS) to quantify indel percentages and assess editing efficiency [7].
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for AI-Guided LNP and mRNA Research
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Ionizable Lipids | Critical LNP component for entraping mRNA and enabling endosomal escape. | SM-102, ALC-0315. AI models can design novel variants by predicting the effect of tail length [72] [73]. |
| Helper Lipids | Stabilize the LNP structure and fluidity. | Cholesterol, DSPC. |
| PEG-Lipids | Shield LNPs, reduce clearance, and modulate circulation time. | DMG-PEG, ALC-0159. |
| Branched Polymers (PBAEs) | Can be added to LNPs as a fifth component to potentially enhance performance. | COMET AI model has been used to optimize LNP formulations containing PBAEs [72]. |
| CleanCap Cap Analog | Enables co-transcriptional capping of in vitro transcribed (IVT) mRNA, enhancing translation efficiency. | Used in high-throughput screening of LNPs [73]. |
| N1-methylpseudouridine | Modified nucleoside that replaces uridine in IVT mRNA to reduce immunogenicity and increase translation. | Key modification used in clinical mRNA vaccines [74]. |
| AI-Optimized Poly(A) Tail | mRNA structural element that significantly enhances stability and translation efficiency. | A50L50LO structure (a loop-forming poly(A) tail) showed superior performance in vivo [74]. |
This technical support guide provides detailed protocols and troubleshooting for researchers utilizing Lipid Nanoparticle Spherical Nucleic Acids (LNP-SNAs) to enhance the delivery and efficiency of CRISPR-Cas9 systems, a critical focus in optimizing Cas9 protein versus mRNA delivery strategies.
Performance Data and Key Advantages of LNP-SNAs
LNP-SNAs represent a significant structural advance over conventional LNPs. Their unique architecture, which features a dense shell of DNA surrounding a lipid core, enhances cellular targeting and uptake [75]. The table below summarizes key performance metrics established through foundational experiments.
Table 1: Quantitative Performance Comparison of LNP-SNAs vs. Standard LNPs
| Performance Metric | Standard LNPs | LNP-SNAs | Experimental Context |
|---|---|---|---|
| Cellular Uptake Efficiency | Baseline | 2-3 times higher [76] [75] | Various human and animal cell types [76] |
| Gene Knockout (INDEL) Efficiency | Baseline | 2-3 times higher frequency [75] | Human pluripotent stem cells (hPSCs) [77] |
| Precise Editing (HDR) Efficiency | ~8% | ~21% (2.6x increase) [75] | Delivery of Cas9, sgRNA, and DNA repair template [76] |
| Cytotoxicity | Baseline | Significantly reduced [76] [75] | In vitro cell cultures [76] |
Core Experimental Protocol: LNP-SNA Synthesis and Testing
This section details the foundational methodology for synthesizing LNP-SNAs and testing their efficacy in gene-editing experiments [76].
Synthesis of LNP-SNAs
- Step 1: Core Formation. Prepare the standard LNP core using a microfluidic device. The core encapsulates the full CRISPR toolkit: Cas9 protein (or mRNA), guide RNA (sgRNA), and, for precise editing, a single-stranded DNA repair template (ssODN) [76].
- Step 2: SNA Shell Assembly. Incubate the pre-formed LNPs with short, synthetic DNA strands. These strands spontaneously assemble into a dense, spherical nucleic acid shell around the LNP core, forming the complete LNP-SNA [76].
- Step 3: Purification. Purify the resulting LNP-SNAs via dialysis or tangential flow filtration to remove unencapsulated cargo and excess DNA strands. Final products should be characterized for size, charge (zeta potential), and concentration [76].
In Vitro Gene-Editing Workflow
- Step 1: Cell Seeding. Plate relevant cell lines (e.g., HEK293, human bone marrow stem cells, primary lymphocytes) at an appropriate density 24 hours before treatment [76].
- Step 2: Transfection. Apply the LNP-SNA formulation to cells. The optimal dose will require titration; a starting point of 100 nM total RNA is recommended [76].
- Step 3: Incubation and Analysis. Incubate cells for 48-72 hours to allow for gene editing and protein expression.
- Efficiency Analysis: Harvest cells and extract genomic DNA. Use targeted next-generation sequencing (NGS) or T7E1 assays to quantify INDEL frequencies at the target locus [77].
- HDR Analysis: For edits involving a repair template, use droplet digital PCR (ddPCR) or NGS to detect the precise incorporation of the desired sequence [75].
- Cell Viability: Assess cytotoxicity using assays like MTT or CellTiter-Glo alongside an untreated control [76].
Frequently Asked Questions (FAQ) and Troubleshooting
Table 2: Troubleshooting Guide for Common LNP-SNA Experimental Challenges
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Low Editing Efficiency | Ineffective sgRNA; Poor LNP-SNA uptake. | Pre-validate sgRNAs using algorithms like Benchling [77]. Confirm cellular uptake using flow cytometry with a fluorescently labeled LNP-SNA. |
| Low HDR Efficiency | Dominant NHEJ repair pathway; Poor repair template delivery. | Use the HDR Enhancer Protein to bias repair toward HDR [78]. Ensure repair template is co-encapsulated and of high quality. |
| High Cell Toxicity | Excessive LNP-SNA dose; Contaminants. | Titrate the LNP-SNA dose to find the optimal balance. Ensure proper purification and use fresh, sterile formulations. |
| Inconsistent Results | Variability in LNP-SNA synthesis; Cell passage number. | Standardize synthesis protocol (time, temperature, pressure). Use low-passage-number cells and consistent culture conditions. |
Q: Can LNP-SNAs be used for in vivo applications? A: Yes, the modular nature of the LNP-SNA platform allows the surface DNA to be engineered for specific cell and tissue targeting, making it promising for in vivo therapeutic delivery. Commercial development for clinical trials is underway [76] [75].
Q: How does the performance of AI-designed editors like OpenCRISPR-1 relate to delivery? A: The development of highly active and specific editors like OpenCRISPR-1, which is over 400 mutations away from SpCas9, underscores the critical need for efficient delivery systems [79]. Advanced delivery platforms like LNP-SNAs are essential to fully leverage the potential of these novel, AI-generated proteins.
Essential Research Reagent Solutions
The table below lists key reagents and their critical functions for successful LNP-SNA-based CRISPR experiments.
Table 3: Key Reagents for LNP-SNA CRISPR Experiments
| Reagent / Material | Function / Role | Example / Note |
|---|---|---|
| Ionizable Lipid | Forms LNP core; enables endosomal escape. | SM-102 is used in clinical LNP formulations [80]. |
| Alt-R HDR Enhancer Protein | Boosts precise gene editing rates. | Increases HDR efficiency by up to 2-fold in challenging cells [78]. |
| Validated sgRNA | Directs Cas9 to specific genomic target. | Use algorithms (e.g., Benchling) for prediction and validate experimentally [77]. |
| High-Fidelity Cas9 | Catalyzes DNA cleavage with minimal off-target effects. | Available as protein or mRNA for encapsulation. |
| DNA Repair Template | Provides donor DNA for precise edits (HDR). | Single-stranded ODNs (ssODNs) are commonly used [76]. |
FAQs: Navigating Cell Type and Delivery Method Selection
Q1: What is the fundamental difference in how viral vectors transduce dividing versus non-dividing cells?
The key difference lies in their ability to access the host cell nucleus. Lentiviral vectors (LVs) are proficient at infecting both dividing and non-dividing cells because their pre-integration complex can actively import into the nucleus through the nuclear pore complex [1] [81]. In contrast, adeno-associated viruses (AAVs) efficiently transduce non-dividing cells, as their single-stranded DNA genome does not require nuclear envelope breakdown for access [1] [82]. However, dividing cells can lead to a dilution of the AAV genome in daughter cells if the DNA does not integrate.
Q2: Why is mRNA delivery often considered to have a lower risk of off-target effects than plasmid DNA delivery?
mRNA delivery offers transient expression of the Cas9 protein. Once delivered into the cytoplasm, mRNA is translated into protein but is then rapidly degraded by cellular machinery. This short half-life limits the window of time during which Cas9 is active, thereby reducing the probability of it cleaving at unintended, off-target sites [13] [81]. Plasmid DNA, however, must enter the nucleus where it can persist for much longer, leading to sustained Cas9 expression and a higher potential for off-target activity [81].
Q3: How can I tailor my delivery system to target a specific tissue, like the liver or lungs?
Tissue targeting, or tropism, is achieved by selecting delivery vehicles with inherent or engineered specificity.
- AAV Serotypes: Different naturally occurring AAV serotypes have affinities for different tissues. For example, AAV9 exhibits strong tropism for the liver, heart, and skeletal muscle, while AAVrh10 is often used for lung and central nervous system targeting [81].
- Engineered Lipid Nanoparticles (LNPs): The surface of LNPs can be modified with specific molecules, such as antibodies or peptides, that bind to receptors on the target tissue. The novel SORT (Selective Organ Targeting) LNP technology allows for precise targeting of tissues like the lung, spleen, and liver by incorporating specific lipid molecules [1].
- Viral Pseudotyping: Lentiviruses can be "pseudotyped" with envelope proteins from other viruses (e.g., the VSV-G protein) to alter and broaden their cellular tropism [1].
Q4: My target cell type is difficult to transfect (e.g., primary T cells or neurons). What is the recommended delivery method?
For hard-to-transfect cells, electroporation of mRNA or Ribonucleoprotein (RNP) complexes is often the most effective method due to its high efficiency across a broad range of cell types [82] [81]. While it can be stressful to cells, optimizing pulse parameters can improve viability. For in vivo applications targeting neurons, AAV vectors (e.g., AAV9 or AAVrh10) or Integrase-Deficient Lentivirus (IDLV) are preferred due to their ability to efficiently transduce non-dividing neurons with a favorable safety profile [81].
Troubleshooting Guides
Problem: Low Editing Efficiency in Non-Dividing Cells
Potential Causes and Solutions:
- Cause 1: Use of a vector incapable of nuclear entry.
- Cause 2: Rapid degradation of CRISPR cargo before action.
- Cause 3: The delivery vehicle lacks tropism for your target cell.
Problem: High Cytotoxicity or Immune Response
Potential Causes and Solutions:
- Cause 1: The delivery method itself is physically damaging.
- Cause 2: Prolonged expression of CRISPR components triggering an immune response.
- Cause 3: Contamination with immunogenic components.
- Solution: Use high-purity, endotoxin-free plasmids for DNA delivery. For protein delivery, ensure the RNP preparation is free from toxic contaminants [81].
Problem: Specificity - High Off-Target Editing
Potential Causes and Solutions:
- Cause 1: Persistent expression of Cas9 nuclease.
- Solution: Utilize self-inactivating delivery systems. RNP delivery is ideal as it is active for only a short period. Alternatively, deliver Cas9 as mRNA instead of plasmid DNA. For viral vectors, consider doxycycline-inducible systems to control the timing and duration of Cas9 expression [81].
- Cause 2: High concentration of CRISPR components.
- Solution: Titrate the amounts of Cas9 and gRNA (whether as DNA, mRNA, or RNP) to find the minimum dose that achieves efficient on-target editing. Using excessive amounts increases the risk of off-target cleavage [83].
- Cause 3: The guide RNA sequence has high similarity to off-target sites.
- Solution: Meticulously design gRNAs using specialized software to minimize homology with other genomic regions. Avoid gRNAs with fewer than 2 mismatches in the PAM-proximal "seed" region to potential off-target sites [83].
Data Presentation: Delivery Method Comparison
Table 1: Quantitative Comparison of CRISPR Delivery Vehicle Efficiency by Cell Type
| Delivery Vehicle | Cargo Format | Dividing Cells (e.g., HEK293, iPSCs) | Non-Dividing Cells (e.g., Neurons, Cardiomyocytes) | Primary T Cells | Key Considerations |
|---|---|---|---|---|---|
| Lentivirus (LV) | DNA | High | High [1] [81] | High [81] | - Risk of insertional mutagenesis.- Suitable for long-term expression. |
| Adeno-Associated Virus (AAV) | DNA | Moderate (genome dilution) [81] | High [1] [81] | Moderate [81] | - Limited cargo capacity (~4.7 kb).- Low immunogenicity. |
| Adenovirus (AdV) | DNA | High | High [1] | High | - Can trigger strong immune responses.- Large cargo capacity. |
| Lipid Nanoparticles (LNPs) | mRNA / RNP | Moderate to High [82] | Low to Moderate [13] | Moderate to High (with optimization) [82] | - Low immunogenicity.- Efficiency is cell-type dependent. |
| Electroporation | mRNA / RNP | High [82] [81] | Moderate (e.g., neurons) [81] | High [82] [81] | - Can cause significant cell stress.- Very high efficiency for RNP delivery. |
Table 2: Pros and Cons of Cas9 Cargo Formats
| Cargo Format | Editing Onset | Expression Duration | Risk of Genomic Integration | Relative Cost | Ideal Use Case |
|---|---|---|---|---|---|
| Plasmid DNA | Slow (24-48h) | Prolonged | Moderate [81] | Low | Basic research where cost is a primary factor and off-targets can be tolerated. |
| mRNA | Fast (4-24h) | Transient (days) | None [13] [81] | Moderate | In vivo therapies requiring safety and transient activity. |
| Ribonucleoprotein (RNP) | Immediate (0-8h) | Very Short (hours) | None [81] | High | Clinical applications (ex vivo), and hard-to-transfect cells where precision and high efficiency are critical. |
Experimental Protocols
Protocol 1: Electroporation of CRISPR RNP into Primary T Cells
This protocol is optimized for high efficiency and cell viability in hard-to-transfect, non-dividing, or slowly dividing primary immune cells [81].
- Isolate and Activate T Cells: Isolate primary human T cells from whole blood or PBMCs. Activate the cells using CD3/CD28 activation beads for 24-48 hours.
- Prepare RNP Complex: For a single reaction, complex 10-40 µg of purified Cas9 protein with a 1.2x molar ratio of synthetic sgRNA. Incubate at room temperature for 10-20 minutes to form the RNP.
- Prepare Cells for Electroporation: Wash the activated T cells to remove serum and resuspend them in an electroporation-compatible buffer (e.g., P3 buffer for Lonza 4D-Nucleofector) at a concentration of 5-10 million cells per 100 µL.
- Electroporation: Mix 100 µL of cell suspension with the pre-formed RNP complex. Transfer the entire mixture to a certified electroporation cuvette. Electroporate using a pre-optimized program for primary T cells (e.g., "EO-115" on a Lonza 4D-Nucleofector X Unit).
- Recovery and Culture: Immediately after pulsing, add 500 µL of pre-warmed, serum-free culture medium to the cuvette. Gently transfer the cells to a culture plate containing complete medium supplemented with IL-2 (50-100 U/mL). Culture at 37°C and 5% CO₂.
- Analysis: Assess editing efficiency 48-72 hours post-electroporation via T7E1 assay or next-generation sequencing (NGS).
Protocol 2: LNP Formulation for mRNA Delivery In Vivo
This protocol outlines the preparation of LNPs for the in vivo delivery of Cas9 mRNA, enabling targeted tissue delivery [1] [13].
- Prepare Lipid Mixture: Create an ethanol-phase lipid mixture containing ionizable cationic lipid, phospholipid, cholesterol, and PEG-lipid at a defined molar ratio (e.g., 50:10:38.5:1.5 mol%). The ionizable lipid is critical for endosomal escape.
- Prepare Aqueous Phase: Dissolve the Cas9 mRNA in an acidic aqueous buffer (e.g., 10 mM citrate, pH 3.0) to stabilize the nucleic acid.
- Rapid Mixing (Nanoprecipitation): Use a microfluidic mixer or turbulent jet mixing to rapidly combine the ethanol-phase lipid mixture with the aqueous-phase mRNA solution at a defined flow rate ratio (typically 3:1 aqueous-to-ethanol). This spontaneous process forms mRNA-encapsulating LNPs.
- Buffer Exchange and Dialysis: Dialyze the formed LNP suspension against a large volume of PBS (pH 7.4) for several hours at 4°C to remove residual ethanol and adjust the pH to physiological conditions.
- Characterization and Storage: Determine the particle size and polydispersity (PDI) via dynamic light scattering (e.g., Z-average ~80 nm, PDI < 0.2). Measure mRNA encapsulation efficiency using a Ribogreen assay. Filter-sterilize the LNPs and store at 4°C for short-term use.
Signaling Pathways and Workflows
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Delivery Optimization
| Reagent / Kit Name | Function / Description | Key Application |
|---|---|---|
| Lipofectamine CRISPRMAX | A lipid-based transfection reagent specifically optimized for the delivery of CRISPR RNP complexes into a wide range of cell lines. | Simplifies RNP delivery for in vitro studies in standard cell lines [13]. |
| Lonza 4D-Nucleofector System | An electroporation platform with pre-optimized programs for hundreds of cell types, including primary and hard-to-transfect cells. | Gold-standard method for high-efficiency RNP or mRNA delivery into primary T cells, HSCs, and neurons [82] [81]. |
| AAVpro Helper Free System (Takara Bio) | A system for producing high-titer, pure AAV vectors of various serotypes without the need for helper virus co-infection. | Generating AAVs with specific tissue tropism (e.g., AAV9 for liver/CNS) for in vivo delivery [81]. |
| Precision gRNA Synthesis Kit (Synthego) | A kit for high-yield, chemically modified synthetic gRNA production. Chemical modifications enhance gRNA stability and editing efficiency. | Producing research-grade gRNAs for use with RNP complexes or mRNA co-delivery [1]. |
| GenCrispr Cas9 SmartNuclease (VectorBuilder) | A ready-to-transfect, endotoxin-free plasmid encoding Cas9, often with a fluorescent marker for easy tracking of transfected cells. | A reliable DNA-based delivery option for initial proof-of-concept experiments in easily transfected cell lines [81]. |
Benchmarking Success: Efficacy, Safety, and Clinical Trial Outcomes
FAQs and Troubleshooting Guides
This section addresses common challenges in CRISPR-Cas9 experiments, focusing on the critical choice between delivering the Cas9 enzyme as protein or mRNA.
FAQ: Cas9 Protein vs. mRNA – Key Considerations
What are the fundamental trade-offs between using Cas9 protein (as RNP) versus mRNA for delivery?
The choice between Cas9 protein (often as a Ribonucleoprotein complex, or RNP) and mRNA significantly impacts the kinetics, persistence, and safety profile of your gene editing. The table below summarizes the core quantitative and qualitative differences.
| Metric | Cas9 RNP (Protein + gRNA) | Cas9 mRNA + gRNA |
|---|---|---|
| Time to Initial Activity | Minutes to hours [1]. Pre-formed complex is immediately active in the cell. | Hours [1]. Requires delivery into the cytoplasm, translation into protein, and nuclear localization. |
| Editing Efficiency | Often higher and more consistent, especially in hard-to-transfect cells [1]. | Can be variable; depends on the efficiency of mRNA translation in the target cell type [15]. |
| Duration of Activity | Short (hours to days) [1]. Rapid degradation by proteases limits activity window. | Moderate. Longer than RNP due to sustained translation from mRNA, but shorter than DNA plasmid delivery [1]. |
| Precision & Off-Target Effects | Higher precision, lower off-targets [1]. Short activity window reduces time for off-target cleavage. | Potentially higher off-target risk [1]. Longer persistence of Cas9 activity increases the chance of cutting at unintended sites. |
| Toxicity & Immune Response | Lower cytotoxicity and immunogenicity [1]. As a protein, it avoids triggering nucleic acid sensing pathways. | Higher risk of immune activation. Exogenous mRNA can be recognized by cellular pathogen-recognition receptors [1]. |
| Cargo Stability | Protein complex is relatively stable but requires specific storage conditions. | mRNA is inherently unstable and requires careful handling and encapsulation to prevent degradation. |
Troubleshooting Guide: Low Editing Efficiency
Issue: My experiment is showing low knockout efficiency. How can I address this?
Low editing efficiency can stem from multiple factors. Follow this diagnostic guide to identify and resolve the problem.
Potential Cause 1: Suboptimal sgRNA Design
- Problem: The single-guide RNA (sgRNA) may have low binding affinity to the target DNA due to its sequence, GC content, or secondary structures [15].
- Solution:
Potential Cause 2: Inefficient Delivery
- Problem: The Cas9 cargo (whether mRNA or protein) is not effectively entering your target cells.
- Solution:
- For RNP Complexes: Optimize transfection protocols. For hard-to-transfect cells (e.g., primary cells, stem cells), use electroporation. For standard cell lines, high-efficiency lipid-based transfection reagents (e.g., Lipofectamine CRISPRMAX) can be effective [15] [1].
- For mRNA: Ensure the mRNA is properly capped, polyadenylated, and encapsulated in lipid nanoparticles (LNPs) to protect it from degradation and enhance cellular uptake [1].
Potential Cause 3: High Off-Target Activity
- Problem: Cas9 is cutting at unintended genomic sites, which can confound efficiency measurements and experimental outcomes [17].
- Solution:
- Use high-fidelity Cas9 variants (e.g., SpCas9-HF1, eSpCas9) that are engineered to reduce off-target cleavage while maintaining on-target activity [17].
- Utilize RNP complexes, as their transient activity inherently lowers off-target rates [1].
- Perform next-generation sequencing (NGS)-based methods to comprehensively assess off-target edits.
Potential Cause 4: Cell Line-Specific Variability
- Problem: Different cell lines have varying intrinsic DNA repair capacities and susceptibility to CRISPR editing [15].
- Solution:
- Use stably expressing Cas9 cell lines to ensure consistent and reproducible editor presence, which can improve knockout efficiency and reliability [15].
- Research the specific characteristics of your cell line and adjust protocols accordingly (e.g., HeLa cells have robust DNA repair mechanisms that can lower knockout success) [15].
Experimental Protocol: Comparing Cas9 Delivery Methods
This protocol provides a detailed methodology for directly comparing the editing efficiency, precision, and toxicity of Cas9 delivered as mRNA versus RNP in a cell culture model.
1. Objective To quantitatively compare the performance of Cas9 mRNA and Cas9 RNP in terms of on-target editing efficiency, off-target effects, and impact on cell viability.
2. Materials
- Research Reagent Solutions:
Item Function Cas9 mRNA Template for in vivo translation of the Cas9 nuclease. Synthetic sgRNA Guides the Cas9 protein to the specific genomic target sequence. Purified Cas9 Protein The core nuclease enzyme for DNA cleavage. Lipid Nanoparticles (LNPs) or Transfection Reagent Vehicle for delivering mRNA and sgRNA into cells. Electroporation System Physical method for delivering RNP complexes into cells. Cell Viability Assay (e.g., MTT) Measures potential toxicity of the delivery method and CRISPR components. Next-Generation Sequencing (NGS) Kit For high-sensitivity quantification of on-target and off-target edits.
3. Workflow
The following diagram illustrates the parallel experimental workflows for testing Cas9 mRNA and RNP.
4. Key Steps and Quantification
- Complex Formation & Transfection:
- mRNA Group: Complex a fixed amount (e.g., 1 µg) of Cas9 mRNA and sgRNA with a lipid-based transfection reagent. Transfert the complex into your target cells according to the manufacturer's protocol [1].
- RNP Group: Pre-complex a fixed amount (e.g., 2 µg) of purified Cas9 protein with sgRNA (molar ratio ~1:2) for 10-20 minutes at room temperature. Deliver the RNP complex into cells via electroporation using an optimized protocol [1].
- Incubation: Incubate the cells for 48-72 hours to allow for editing and expression analysis.
- Quantitative Analysis:
- Efficiency: Harvest genomic DNA and amplify the target locus by PCR. Use NGS to precisely quantify the percentage of insertions and deletions (indels) at the target site. Expected Outcome: RNP delivery often results in higher editing efficiency within a shorter time frame [1].
- Toxicity: At 48 hours post-transfection, perform a cell viability assay (e.g., MTT). Compare the results to an untreated control to calculate the relative toxicity of each method. Expected Outcome: RNP delivery typically shows lower cytotoxicity compared to mRNA lipofection [1].
- Precision: Use NGS-based off-target discovery methods (e.g., GUIDE-seq if using DNA delivery, or DISCOVER-Seq) to screen the top predicted off-target sites. Compare the frequency of indels at these sites between the two delivery groups. Expected Outcome: RNP delivery should demonstrate a significantly lower off-target profile due to its transient activity [1].
Decision Framework for Delivery Method
The following flowchart provides a logical pathway for choosing between Cas9 mRNA and RNP based on your primary experimental goal.
Frequently Asked Questions (FAQs) on mRNA-LNP Delivery & Experimental Design
Q1: What are the key advantages of using mRNA-LNP over viral vectors for in vivo CRISPR delivery? mRNA-LNP delivery offers several distinct advantages for in vivo CRISPR therapies. Unlike viral vectors such as AAV, mRNA-LNPs do not integrate into the host genome, eliminating the risk of insertional mutagenesis [13]. Their activity is transient, which reduces the duration of Cas9 exposure and limits the potential for off-target editing [13] [16]. Furthermore, the LNP platform allows for redosing, a significant clinical advantage as immune responses often preclude repeated administration of viral vectors [7].
Q2: Why is the liver a primary target for current mRNA-LNP CRISPR therapies, and how can other tissues be targeted? LNPs administered systemically (via IV injection) have a natural tropism for the liver due to their physicochemical properties and the physiological function of the liver in filtering particulates from the blood [7]. This makes liver-expressed proteins like TTR (for hATTR) and kallikrein (for HAE) ideal initial targets. To expand to other tissues, researchers are actively developing novel LNP formulations. For instance, Selective Organ Targeting (SORT) nanoparticles are engineered by adding specific lipid molecules to redirect LNPs to organs like the lungs and spleen [1].
Q3: What are the primary stability challenges with CRISPR mRNA, and how can they be mitigated in experimental design? CRISPR mRNA faces challenges of instability and degradation by nucleases in the blood, leading to a short half-life [13] [84]. It can also trigger unwanted immune responses through Toll-like receptors (TLR3, TLR7) and RIG-I [13]. Key mitigation strategies in your mRNA design should include:
- 5' and 3' UTR Optimization: Using optimized untranslated regions to enhance stability and translation efficiency [13].
- Nucleoside Modification: Incorporating modified nucleosides (e.g., pseudouridine) to reduce innate immune recognition [13].
- Codon Optimization: Engineering the mRNA sequence with human-preferred codons to improve protein yield [13].
Q4: How does the choice between Cas9 mRNA and Cas9 RNP (Ribonucleoprotein) impact an LNP experiment? The choice between mRNA and RNP cargo is fundamental and affects editing kinetics, safety, and formulation.
- Cas9 mRNA: Requires in vivo translation after delivery, leading to a slight delay in editing. The primary concerns are mRNA instability and potential immunogenicity [13] [84].
- Cas9 RNP: The pre-formed protein-RNA complex is immediately active upon delivery, leading to faster editing and a very short intracellular half-life that minimizes off-target effects [85]. A major technical hurdle has been encapsulating the large RNP complex in LNPs without denaturing the protein, though the use of thermostable Cas9 variants (e.g., iGeoCas9) is overcoming this challenge [85].
Troubleshooting Common Experimental Challenges
| Challenge | Possible Root Cause | Potential Solutions & Optimization Strategies |
|---|---|---|
| Low Editing Efficiency | - Inefficient cellular uptake or endosomal escape of LNPs.- Poor mRNA stability/translation.- Suboptimal LNP formulation for target cell. | - Optimize LNP composition with ionizable/cationic lipids to enhance endosomal escape [1] [85].- Implement mRNA engineering strategies (codon optimization, nucleoside modifications) [13].- Screen different LNP formulations with tissue-specific targeting molecules (e.g., SORT molecules) [1]. |
| High Immunogenicity | - Recognition of exogenous mRNA by host immune sensors (TLRs, RIG-I).- Immune reaction to LNP components. | - Use highly purified, nucleoside-modified mRNA to evade immune detection [13].- Include HPLC purification to remove double-stranded RNA impurities [13]. |
| Off-Target Effects | - Prolonged presence of active Cas9 nuclease.- Low specificity of the guide RNA (gRNA). | - Utilize RNP delivery for transient, short-lived activity [85].- Employ high-fidelity Cas9 variants and use computational tools to design and screen gRNAs for high specificity [16]. |
| Inefficient Delivery to Non-Liver Tissues | - Natural accumulation of standard LNPs in the liver after systemic administration. | - Develop novel LNP formulations with selective organ targeting (SORT) technology [1].- Explore local administration routes where feasible (e.g., inhalation for lung targets) [85]. |
Experimental Protocol: Analyzing mRNA-LNP Editing Efficiency for a Liver Target
This protocol outlines key steps for evaluating the efficacy of a CRISPR mRNA-LNP therapy targeting a liver gene in a preclinical model, based on methodologies from recent clinical trials [7].
Objective: To assess the in vivo genome editing efficiency and functional protein reduction following systemic administration of CRISPR mRNA-LNP.
Materials:
- Test Article: CRISPR mRNA-LNP formulation (e.g., containing Cas9 mRNA and sgRNA targeting TTR or KLKB1).
- Control: Control LNP (e.g., with non-targeting sgRNA).
- Animal Model: Wild-type or disease-model mice.
- Reagents: ELISA kits for target protein (TTR or kallikrein), PCR reagents for NGS, tissue collection supplies.
Method:
- LNP Administration: Administer a single intravenous injection of the CRISPR mRNA-LNP formulation to the animal model. Include a control group receiving control LNPs.
- Blood Collection: Collect peripheral blood samples at baseline and at regular intervals post-injection (e.g., weekly for 4-12 weeks).
- Functional Efficacy Analysis:
- Centrifuge blood samples to isolate plasma.
- Use a commercial ELISA kit to quantify the concentration of the target protein (e.g., TTR for hATTR models, kallikrein for HAE models) in the plasma. This functional readout directly correlates with therapeutic efficacy [7].
- Molecular Efficacy Analysis (Terminal Procedure):
- At the study endpoint, harvest target tissues (e.g., liver).
- Isolate genomic DNA from the tissue.
- Amplify the target genomic region by PCR and subject the product to next-generation sequencing (NGS).
- Analyze the NGS data with computational tools (e.g., CRISPResso2) to quantify the percentage of insertion/deletion (indel) mutations at the target site.
The workflow for this protocol is summarized in the following diagram:
Key Signaling Pathways in mRNA-LNP Delivery and Immune Recognition
Understanding the intracellular journey of mRNA-LNPs is crucial for troubleshooting. The following diagram illustrates the key pathways involved in delivery and the potential immune responses that can be activated.
Research Reagent Solutions for mRNA-LNP CRISPR Therapy Development
The table below lists essential tools and reagents critical for developing and optimizing in vivo mRNA-LNP CRISPR therapies.
| Research Reagent / Tool | Function & Application in Therapy Development |
|---|---|
| Ionizable/Cationic Lipids | A core component of LNPs that enables encapsulation of nucleic acids and facilitates endosomal escape upon ionization in the acidic endosome, critical for releasing cargo into the cytoplasm [1] [85]. |
| Nucleoside-Modified mRNA | mRNA engineered with modified nucleosides (e.g., pseudouridine) to reduce its immunogenicity by evading detection by pattern recognition receptors like TLRs, thereby enhancing protein expression [13]. |
| Compact Cas9 Orthologs | Smaller Cas9 proteins (e.g., SaCas9, CjCas9) that can be more easily packaged alongside sgRNAs into delivery vectors with limited capacity. They are also valuable for dual-AAV strategies, though less critical for the spacious LNPs [86]. |
| Thermostable Cas9 RNPs | Ribonucleoprotein complexes using engineered Cas9 proteins (e.g., iGeoCas9) with high thermal stability. These are ideal for LNP-RNP delivery as they withstand formulation conditions and enable highly efficient, transient editing with minimal off-target effects [85]. |
| Selective Organ Targeting (SORT) Molecules | Engineered lipids or molecules incorporated into LNPs to redirect them from the liver to specific extrahepatic tissues such as the lungs and spleen, greatly expanding the therapeutic applicability of the platform [1]. |
| High-Fidelity Cas9 Variants | Engineered Cas9 proteins with mutations that increase specificity and reduce off-target editing, a critical safety consideration for therapeutic genome editing [16]. |
CASGEVY (exagamglogene autotemcel) represents the first FDA-approved therapy developed with CRISPR technology for treating sickle cell disease (SCD) in patients aged 12 years and older with recurrent vaso-occlusive crises (VOCs) [87]. This one-time autologous gene therapy utilizes a non-viral delivery approach centered on ribonucleoprotein (RNP) electroporation to genetically modify a patient's own hematopoietic stem cells (HSCs) [88]. The therapeutic strategy involves precise CRISPR/Cas9 genome editing of the BCL11A gene to reactivate fetal hemoglobin (HbF) production, effectively mimicking the benign hereditary persistence of fetal hemoglobin (HPFH) phenotype observed in individuals who experience milder SCD symptoms [89] [88].
The RNP electroporation approach delivers preassembled complexes of Cas9 protein and guide RNA directly into target cells, contrasting with alternative methods that rely on plasmid DNA or mRNA delivery. This technique offers significant advantages for clinical applications, including reduced off-target effects, elimination of DNA vector integration concerns, and rapid clearance of editing components from treated cells [90]. The entire CASGEVY manufacturing process occurs ex vivo, where CD34+ HSCs are collected from the patient, genetically modified via RNP electroporation, expanded, and then reinfused after the patient receives conditioning chemotherapy [91] [92].
Experimental Protocols and Workflows
CASGEVY Manufacturing Process
The production of CASGEVY involves a multi-step protocol requiring specialized equipment and controlled manufacturing environments:
Stem Cell Collection: CD34+ hematopoietic stem cells are collected from the patient via apheresis after mobilization with granulocyte colony-stimulating factor (G-CSF) or other mobilizing agents. The process may require multiple cycles over up to one week to obtain sufficient cells [91] [92].
RNP Complex Formation: Recombinant Cas9 protein is complexed with synthetic guide RNA targeting the erythroid-specific enhancer region of the BCL11A gene. The complex is assembled in vitro at specific molar ratios and incubated to allow proper formation before electroporation [90] [88].
Electroporation Conditions: CD34+ cells are resuspended in electroporation buffer and subjected to electrical parameters optimized for human HSCs. The preassembled RNP complexes are delivered simultaneously via electroporation using cell-type specific settings [90] [88].
Quality Control and Expansion: Successfully edited cells undergo rigorous quality assessment, including measurement of editing efficiency, viability testing, and sterility checks. Cells are then expanded in culture media containing cytokines that support HSC maintenance [91].
Cryopreservation and Storage: The final product is cryopreserved in media containing DMSO and dextran 40, then stored at ultra-low temperatures until patient infusion [91].
Patient Conditioning and Infusion: Patients receive myeloablative conditioning with busulfan before infusion of the final CASGEVY product via intravenous administration [91] [92].
RNP Complex Preparation and Delivery
The core editing component preparation follows this detailed methodology:
Cas9 Protein Purification: His-tagged Cas9 protein is expressed in E. coli and purified using nickel-affinity chromatography, followed by buffer exchange into electroporation-compatible storage buffer [90].
Guide RNA Synthesis: Target-specific single-guide RNA (sgRNA) is synthesized in vitro using T7 RNA polymerase or purchased from commercial vendors. The sgRNA is designed to target the erythroid-specific enhancer region of BCL11A (chr2:60,466,389-60,466,411 in GRCh38) [90] [88].
RNP Complex Assembly: Purified Cas9 protein and sgRNA are mixed at a 1:1.2 molar ratio in electroporation buffer and incubated at room temperature for 10-15 minutes to form ribonucleoprotein complexes [90].
Electroporation Optimization: CD34+ cells are washed and resuspended at a concentration of 1-2×10^6 cells/mL in appropriate electroporation buffer. The Lonza 4D-Nucleofector system with cell-type specific settings (program DZ-100 for human HSCs) is typically employed [90].
Post-Electroporation Recovery: Immediately after electroporation, cells are transferred to pre-warmed culture medium containing cytokines (SCF, TPO, FLT3-L) and maintained at 37°C with 5% CO2 for recovery before quality assessment and expansion [90].
Troubleshooting Common Experimental Challenges
Frequently Encountered Issues and Solutions
Problem: Low Editing Efficiency in Hematopoietic Stem Cells
- Potential Causes: Suboptimal RNP concentration, inadequate electroporation parameters, or poor cell viability.
- Solutions: Titrate RNP concentrations (typically 2-4 μM Cas9), optimize cell density during electroporation (1-2×10^6 cells/100μL), and verify buffer composition. Test different electroporation programs specifically validated for HSCs [90].
Problem: Reduced Cell Viability Post-Electroporation
- Potential Causes: Excessive electrical parameters, improper post-electroporation handling, or suboptimal recovery conditions.
- Solutions: Reduce pulse parameters, implement immediate transfer to recovery medium with Rho kinase inhibitor (Y-27632), and ensure proper cytokine supplementation during recovery phase [90].
Problem: Inconsistent Engraftment of Edited Cells
- Potential Causes: Damage to stem cell potential during editing process, insufficient conditioning, or product quality issues.
- Solutions: Limit ex vivo culture time to <48 hours, maintain stemness-promoting cytokines (SCF, TPO, FLT3-L), and ensure high viability (>80%) pre-infusion [91] [87].
Problem: Variable HbF Induction Despite High Editing Rates
- Potential Causes: Heterogeneous editing outcomes, positional effects, or analysis timing issues.
- Solutions: Use digital PCR for precise quantification of editing outcomes, analyze multiple time points post-engraftment (3-6 months), and ensure measurement in erythroid progeny rather than stem cells [89] [93].
Research Reagent Solutions and Essential Materials
Table 1: Key Research Reagents for RNP Electroporation Experiments
| Reagent/Material | Function/Purpose | Examples/Specifications |
|---|---|---|
| Cas9 Protein | CRISPR endonuclease for targeted DNA cleavage | Recombinant His-tagged SpCas9, >90% purity, endotoxin-free [90] |
| Guide RNA | Targets Cas9 to specific genomic loci | Synthetic sgRNA targeting BCL11A enhancer region, HPLC-purified [88] |
| Electroporation System | Delivery of RNP complexes into cells | Lonza 4D-Nucleofector with X Unit, program DZ-100 [90] |
| CD34+ Selection Kit | Isolation of hematopoietic stem cells | immunomagnetic bead-based separation (e.g., Miltenyi CD34 MicroBead Kit) [89] |
| Cytokine Cocktail | Maintains stemness during culture | SCF (100ng/mL), TPO (100ng/mL), FLT3-L (100ng/mL) [89] |
| Editing Assessment Tools | Measures editing efficiency and specificity | NGS-based assays, CHANGE-seq for off-target profiling [93] |
Clinical Trial Outcomes and Efficacy Data
The phase 3 clinical trial (CLIMB SCD-121) demonstrated compelling efficacy for CASGEVY in treating severe sickle cell disease. This single-arm, open-label study enrolled patients aged 12-35 with SCD and at least two severe vaso-occlusive crises (VOCs) annually [87].
Table 2: Key Efficacy Endpoints from CASGEVY Clinical Trial
| Efficacy Parameter | Results | Patient Population | Follow-up Duration |
|---|---|---|---|
| Freedom from Severe VOCs | 29/31 patients (93.5%) [91] | Severe SCD with recurrent VOCs | ≥12 consecutive months |
| Freedom from VOC Hospitalizations | 30/30 patients (100%) [91] | Severe SCD with recurrent VOCs | ≥12 consecutive months |
| Duration of VOC Freedom | Median 22.2 months [91] | Responders (n=29) | Ongoing follow-up |
| Hemoglobin F Levels | Significant increase post-treatment [91] | All treated patients (n=44) | 6-24 months |
| Neutrophil Engraftment | 100% of patients [87] | All treated patients (n=44) | Median time: 29 days |
The trial demonstrated that a single treatment with CASGEVY resulted in sustained increases in fetal hemoglobin levels, with 96.7% of evaluable patients (29/30) free from severe VOCs for at least 12 consecutive months in the final study analysis [87]. The safety profile was consistent with that of autologous stem cell transplantation following myeloablative conditioning, with no malignancies reported due to treatment [87].
FAQs: Addressing Common Research Questions
What advantages does RNP electroporation offer over other CRISPR delivery methods? RNP electroporation provides rapid editing with minimal off-target effects due to transient Cas9 exposure, eliminates risks of viral vector integration, avoids promoter compatibility issues, and enables editing in hard-to-transfect primary cells like HSCs [90]. The rapid clearance of RNPs from cells (within 24-48 hours) reduces potential immune responses and minimizes off-target activity [90].
How is the risk of off-target editing assessed and mitigated in CASGEVY manufacturing? Comprehensive off-target assessment includes CHANGE-seq for unbiased genome-wide identification of potential off-target sites, followed by targeted sequencing of edited HSCs [93]. Additionally, careful guide RNA design avoids sequences with high similarity elsewhere in the genome, and the use of RNP delivery rather than sustained expression limits editing duration [93] [88].
What are the critical quality control checkpoints for the edited cell product? Key quality metrics include: viability (>80%), editing efficiency at target locus (typically >60%), CD34+ cell count and viability, sterility (bacterial/fungal culture), mycoplasma testing, endotoxin levels, and vector copy number (to confirm absence of plasmid integration) [91] [93].
How long does the complete CASGEVY manufacturing process take? From cell collection to final product release, the process requires approximately 6 months [91]. This includes cell collection, manufacturing, quality control testing, and shipment back to the treatment center.
What functional assays confirm biological activity of the edited cells? In vitro erythroid differentiation assays measure HbF production via HPLC or FACS, sickling assays under hypoxic conditions assess functional improvement, and colony-forming unit (CFU) assays demonstrate maintained stem cell potential post-editing [89] [93].
Troubleshooting Common Experimental Challenges
FAQ: Why is my gene editing efficiency low in human induced pluripotent stem cells (iPSCs)?
iPSCs are notoriously difficult to transfect and are classified as "hard-to-transfect" cells. Low editing efficiency typically stems from challenges in delivering CRISPR components across the cell membrane. [94]
- Problem: The Cas9-sgRNA ribonucleoprotein (RNP) complex or mRNA cannot efficiently cross the plasma membrane due to its large size and negative charge. [94] [95]
- Solution: Implement physical delivery methods like electroporation or nucleofection. These techniques use controlled electrical pulses to create transient pores in the cell membrane, allowing CRISPR components to enter the cytoplasm directly. [94] For iPSCs, the Neon Transfection System has been specifically developed as a flexible, user-friendly electroporation setup that improves delivery efficiency in these sensitive cells. [94]
FAQ: I am observing high variability in editing outcomes between differentiations of my iPSC line. Is this normal?
Yes, this is a recognized challenge in the field. iPSC derivation and differentiation are multistep processes where small variations at each stage can accumulate and generate significantly different outcomes. [96]
- Problem: Heterogeneity can arise from the genetic background of the donor, somatic mutations acquired during reprogramming or culture, and technical variations in differentiation protocols. [96]
- Solution:
- Standardize Protocols: Rigorously document and adhere to standardized culture and differentiation protocols to minimize technical variation. [96]
- Use Isogenic Controls: Generate genetically matched control lines. These are iPSC lines derived from the same individual but engineered to differ only at the specific locus being studied, making them the gold standard for controlling for genetic background effects. [96]
- Implement Quality Control: Perform regular genetic and functional quality control checks on your iPSC lines to monitor for genetic stability and pluripotency. [96]
FAQ: How can I reduce high off-target editing activity in my neuronal cultures?
Prolonged Cas9 activity increases the risk of off-target effects and genotoxicity. [97]
- Problem: Cas9 protein or mRNA persists in cells for too long, leading to unintended cleavage at genomic sites with similar sequences to your target.
- Solution:
- Use RNP Complexes: Deliver the CRISPR/Cas9 system as a pre-assembled Ribonucleoprotein (RNP) complex. This approach is the most rapid and offers minimal off-target effects because the complex degrades quickly after editing. [94]
- Employ Inducible Degradation Systems: A novel solution is the Cas9-degron (Cas9-d) system. This molecular glue system rapidly degrades Cas9 in the presence of the FDA-approved drug pomalidomide (POM), reducing on-target editing by 3- to 5-fold within hours. This system is reversible and has been shown to function in hiPSC-derived GABAergic neurons, providing precise temporal control over editing duration. [97]
FAQ: My mRNA-based CRISPR delivery is triggering an innate immune response in neurons. What can I do?
Exogenous mRNA can be recognized by pattern-recognition receptors in cells, triggering an innate immune response that shuts down translation and can lead to cytotoxicity. [55]
- Problem: The in vitro transcribed (IVT) mRNA is being recognized as a foreign pathogen.
- Solution: Use modified nucleotides in your IVT mRNA synthesis. Replacing uridine with naturally modified nucleotides like 5-methyluridine (m5U) or pseudouridine (Ψ) can cloak the mRNA from immune detection. This suppresses the innate immune response and significantly enhances the stability and translation efficiency of the mRNA, leading to higher protein yields. [55]
Experimental Protocols for Direct Cargo Comparison
Protocol: Side-by-Side Editing Efficiency and Kinetics in iPSCs vs. iPSC-Derived Neurons
Objective: To quantitatively compare the editing kinetics and outcome distribution of Cas9 protein (RNP) versus Cas9 mRNA delivery in a matched isogenic system.
Materials:
- Cell Models: Human iPSCs and their isogenic, differentiated counterpart (e.g., cortical neurons or GABAergic neurons). [98] [97]
- CRISPR Components:
- Cas9 Protein: Purified, active Cas9 nuclease.
- sgRNA: Target-specific single-guide RNA.
- Cas9 mRNA: Modified mRNA encoding Cas9 (e.g., with Ψ or m5U).
- Delivery Method: Electroporation/Nucleofection system (e.g., Neon System). [94]
- Analysis Tools: NGS library preparation kit, flow cytometer, cell culture reagents.
Methodology:
- Cell Preparation:
- Culture and maintain your human iPSC line under standard conditions.
- Differentiate a portion of the iPSCs into the desired neuronal cell type (e.g., GABAergic neurons) using a validated, standardized protocol. [97]
- On the day of the experiment, harvest and prepare single-cell suspensions of both iPSCs and neurons in an electroconductive buffer.
CRISPR Cargo Preparation:
- Condition A (RNP): Pre-complex purified Cas9 protein with sgRNA at a molar ratio of 1:2 to form RNP complexes. Incubate for 10-20 minutes at room temperature.
- Condition B (mRNA): Prepare a mixture of Cas9 mRNA and the same sgRNA.
Delivery via Electroporation:
- Combine cells with either the RNP complex or the mRNA/sgRNA mixture.
- Electroporate using parameters optimized for each cell type. For example:
- iPSCs: Voltage: 1100 V; Width: 20 ms; 2 pulses.
- Neurons: Voltage: 1350 V; Width: 10 ms; 3 pulses.
- Note: Optimal parameters must be determined empirically for your specific cell lines and equipment. [94]
Time-Course Sampling & Analysis:
- After electroporation, plate the cells and collect samples at multiple time points (e.g., 6, 12, 24, 48, 72 hours).
- Efficiency & Kinetics: Isolate genomic DNA from each sample and use next-generation sequencing (NGS) of the target locus to quantify the percentage of indels over time. This will reveal how quickly editing initiates and peaks for each cargo.
- Outcome Distribution: Analyze the NGS data to characterize the spectrum of insertions and deletions (indels) produced by each delivery method.
- Cell Health: Perform flow cytometry for apoptosis/viability markers (e.g., Annexin V, PI) at 24 and 72 hours to assess cytotoxicity.
Expected Outcomes:
- RNP Delivery: Faster onset of editing, peaking within 24-48 hours. Generally shows a lower spectrum of indel diversity and reduced off-target effects due to rapid degradation of the complex. [94]
- mRNA Delivery: Slower onset, as it requires translation of the mRNA into functional protein. Editing peaks later (e.g., 48-72 hours) and the Cas9 protein may persist longer, potentially increasing the risk of off-target effects. [94]
Table 1: Characteristic Comparison of CRISPR/Cas9 Delivery Cargos in Stem Cells and Neurons
| Parameter | Cas9 RNP Complex | Cas9 mRNA + sgRNA |
|---|---|---|
| Time to Onset of Editing | Rapid (hours) [94] | Delayed (requires translation) [94] |
| Editing Duration | Short (complex degrades quickly) [94] | Prolonged (protein persists) [94] [97] |
| Typical Editing Efficiency | High [94] | Variable, can be high [94] |
| Off-Target Risk | Lower [94] [97] | Higher (due to prolonged activity) [94] [97] |
| Immunogenicity | Minimal [94] | Moderate to High (can be mitigated with base modifications) [55] |
| Key Advantage | Fast, precise, minimal off-targets [94] | No need for protein purification [94] |
| Key Challenge | High production cost [94] | Risk of immune activation; stability issues [94] [55] |
Table 2: Cargo-Specific Considerations for Different Cell Models
| Consideration | iPSCs | Neurons (iPSC-Derived) |
|---|---|---|
| Optimal Cargo for High Efficiency | RNP via nucleofection [94] | RNP via nucleofection |
| Primary Delivery Challenge | Hard-to-transfect; sensitive to electroporation stress [94] | Delicate post-mitotic cells; sensitive to electroporation stress |
| Key Toxicity Concern | Maintaining pluripotency and genomic integrity post-editing [96] | General cell health and neuronal function (e.g., electrophysiology) |
| Prolonged Cas9 Activity Concern | Increased risk of genotoxicity and aberrant differentiation [97] | Increased off-target editing and potential for genotoxicity [97] |
Signaling Pathways and Experimental Workflows
Experimental Workflow for Head-to-Head Cargo Comparison
Intracellular Fate of Cas9 mRNA vs. RNP Cargos
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Cargo Optimization
| Reagent / Tool | Function/Purpose | Key Considerations |
|---|---|---|
| Purified Cas9 Protein | Forms the core of the RNP complex for direct delivery. [94] | Ensure high purity and nuclease-free status for optimal performance and low toxicity. |
| Modified Cas9 mRNA | Template for in-cell production of Cas9 protein; modified bases (Ψ, m5U) reduce immunogenicity. [55] | Critical for minimizing immune activation in sensitive cells like neurons. |
| sgRNA (synthesized) | Guides the Cas9 nuclease to the specific genomic target. [94] | Can be chemically modified to enhance stability. Must be HPLC-purified. |
| Electroporation/Nucleofection System | Enables physical delivery of cargo into hard-to-transfect cells. [94] | Parameters (voltage, pulse width) must be rigorously optimized for each cell type. |
| Cas9-degron (Cas9-d) System | Provides temporal control over Cas9 levels using Pomalidomide to rapidly degrade Cas9. [97] | A cutting-edge tool to minimize off-target effects; proven in hiPSC-derived neurons. |
| Isogenic iPSC Line Pairs | Genetically matched control and mutant lines. [96] | The gold standard for controlling genetic background in phenotype analysis. |
For researchers in gene editing and drug development, selecting the optimal delivery system for CRISPR-Cas9 components is a critical decision that profoundly influences experimental outcomes and therapeutic potential. A key differentiator among delivery platforms is their redosing potential—the capacity for repeated administration to achieve or maintain therapeutic efficacy. This technical resource center examines the distinct advantage of Lipid Nanoparticle (LNP)-based mRNA therapies over viral vectors in this crucial aspect, providing troubleshooting guidance and experimental protocols to support your research.
FAQs: Core Concepts for Researchers
Q1: What is the fundamental difference in redosing capability between LNP-mRNA and viral vectors?
The fundamental difference lies in their mechanisms of action and how the immune system recognizes them. LNP-delivered mRNA is a transient carrier of genetic instructions that does not integrate into the host genome and is degraded after its therapeutic protein is expressed. This transient nature means it can typically be redosed. In contrast, viral vectors often provoke persistent and robust immune responses against the viral capsid proteins. After initial exposure, the body develops neutralizing antibodies that effectively clear subsequent doses of the same vector, severely limiting or eliminating redosing potential [1] [99].
Q2: Why do Adenovirus-Associated Viruses (AAVs), a common viral vector, pose such a significant redosing challenge?
AAVs are highly prevalent in the human population, with many individuals having pre-existing immunity from natural infections. One study noted that pre-existing immunity to AAVs is present in a substantial proportion of the population [1]. This means that a first dose of an AAV-based therapeutic might be ineffective in some patients and that any initial administration will likely induce a strong, memory-based immune response that prevents effective re-administration [99].
Q3: For CRISPR delivery, how does the choice of cargo (Cas9 protein vs. Cas9 mRNA) impact the experimental outcome?
The choice of cargo affects editing efficiency, specificity, and duration of activity, which are critical for both safety and efficacy.
- Cas9 mRNA: Requires translation into protein within the cell, leading to a slight delay in activity and a longer window of Cas9 expression. This can increase the chance of off-target effects [2] [1].
- Cas9 Ribonucleoprotein (RNP): The pre-assembled complex of Cas9 protein and guide RNA is immediately active upon delivery. This results in a faster onset of editing and a shorter half-life, which significantly minimizes off-target effects and reduces the risk of immune activation against Cas9 [2] [1]. The transient nature of RNP activity is highly compatible with redosing regimens.
Q4: What are the primary analytical methods for characterizing LNP-mRNA batches to ensure consistent redosing efficacy?
Batch-to-batch consistency is paramount for reliable redosing in experiments. Key quality attributes and their analytical methods include [100]:
- mRNA Integrity/Purity: Capillary Gel Electrophoresis (CGE) to assess full-length mRNA and detect truncated species.
- Identity/Sequence Verification: Reverse Transcription PCR (RT-PCR) coupled with Sanger sequencing or direct RNA sequencing.
- Capping Efficiency: High-Performance Liquid Chromatography (HPLC) with UV or Mass Spectrometry detection to ensure proper 5' capping for efficient translation.
- Impurity Profile: Enzymatic-Linked Immunosorbent Assay (ELISA) or gel electrophoresis to detect double-stranded RNA (dsRNA) impurities that can trigger unwanted immune responses.
Troubleshooting Guides
Issue: Reduced Protein Expression Efficacy Upon Redosing
Potential Cause 1: Immune Activation Against LNP Components or mRNA While LNPs are less immunogenic than viruses, they can still stimulate the immune system. The ionizable lipid component or the mRNA itself (if unmodified) can trigger innate immune responses (e.g., via TLR signaling), potentially leading to accelerated clearance upon redosing [101] [55].
- Solution: Utilize mRNA with nucleoside modifications (e.g., pseudouridine). These modifications dampen innate immune recognition by pattern recognition receptors (PRRs), thereby enhancing protein expression and reducing immune-driven clearance [102] [55].
- Experimental Protocol:
- Transfert two cell lines (e.g., HEK-293 and a macrophage line like THP-1) with either unmodified or nucleoside-modified mRNA-LNPs.
- 24 hours post-transfection, measure secretion of pro-inflammatory cytokines (e.g., IFN-α, IL-6) using ELISA.
- In parallel, quantify target protein expression via Western blot or flow cytometry.
- Compare cytokine levels and protein expression between the two mRNA groups to validate the immune-dampening effect of nucleoside modifications.
Potential Cause 2: Anti-PEG Immunity Polyethylene glycol (PEG) is a common component of LNPs that provides a "stealth" layer. However, pre-existing anti-PEG antibodies are common and can accelerate blood clearance of the second dose, a phenomenon known as the ABC phenomenon.
- Solution: Explore PEG-free LNP formulations. Alternative polymers or lipids can be used to stabilize LNPs without invoking anti-PEG immunity.
- Experimental Protocol:
- Administer a primary dose of PEGylated LNP to a mouse model.
- After 14 days, administer a second dose of either the same PEGylated LNP or a non-PEGylated alternative.
- Collect plasma at various time points post-injection and quantify mRNA biodistribution and clearance using qRT-PCR.
- Compare the pharmacokinetic profiles of the two groups to assess the impact of anti-PEG immunity.
Issue: Inefficient In Vivo Delivery and Endosomal Escape
Problem: A major bottleneck for LNP efficacy is the failure to escape the endosomal compartment after cellular uptake, leading to lysosomal degradation of the mRNA payload.
- Solution: Optimize the ionizable lipid structure. The pKa of the ionizable lipid is critical for triggering the "proton sponge" effect and facilitating endosomal escape [103] [25].
- Experimental Protocol:
- Formulate LNPs with a library of ionizable lipids with varying pKa values (e.g., 5.8 - 6.8).
- Transfert a reporter cell line (e.g., HepG2) with LNP formulations encapsulating GFP mRNA.
- 24 hours post-transfection, analyze GFP expression via flow cytometry to determine transfection efficiency.
- Use a confocal microscopy assay with endosomal/lysosomal markers (e.g., LysoTracker) to visually confirm the co-localization and subsequent escape of LNPs from endosomes.
Table 1: Key Parameter Comparison: LNP-mRNA vs. Viral Vectors
| Parameter | LNP-mRNA | Viral Vectors (e.g., AAV) |
|---|---|---|
| Redosing Potential | High (transient expression, lower immunogenicity) | Very Low (potent, lasting anti-vector immunity) [1] [99] |
| Cargo Size Limit | High (~20 kb for saRNA) [55] | Limited (AAV: ~4.7 kb) [1] |
| Onset of Action | Hours to days (requires translation) | Days to weeks (requires transcription/translation) |
| Risk of Insertional Mutagenesis | None (cytoplasmic function) | Low but present (random integration possible) [1] |
| Primary Immune Concern | Innate immune activation (manageable with modifications) [55] | Adaptive humoral immunity (neutralizing antibodies) [99] |
Table 2: CRISPR Cargo Format Comparison for LNP Delivery
| Cargo Format | Editing Efficiency | Off-Target Risk | Duration of Activity | Immunogenicity |
|---|---|---|---|---|
| DNA Plasmid | Variable | High (prolonged Cas9 expression) | Long (days-weeks) | High (TLR9 activation) |
| Cas9 mRNA | High | Moderate | Moderate (days) | Moderate (manageable with modifications) [2] |
| Cas9 RNP | Highest | Lowest | Short (hours) | Lowest (immediate activity, no transcription/translation) [2] [1] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for LNP and Cargo Analysis
| Reagent / Assay | Function/Benefit | Key Consideration for Redosing |
|---|---|---|
| Nucleoside-Modified NTPs (e.g., N1-Methylpseudouridine) | Reduces mRNA immunogenicity, enhances translation [102] [55] | Critical for minimizing innate immune activation upon repeated doses. |
| Ionizable Lipids (e.g., DLin-MC3-DMA, SM-102) | Enables efficient encapsulation and endosomal escape. | pKa optimization is crucial for consistent performance across doses. |
| Anti-PEG Antibody ELISA Kit | Detects pre-existing and therapy-induced anti-PEG antibodies. | Essential for diagnosing and troubleshooting reduced efficacy of later doses. |
| Cytokine Profiling Array (e.g., for IFN-I, IL-6) | Measures innate immune activation post-dosing. | A key pharmacodynamic marker for assessing the "stealth" of your formulation. |
| dsRNA ELISA Kit | Quantifies dsRNA impurities in IVT mRNA preps [100]. | High dsRNA levels can trigger potent immune responses, compromising redosing. |
Visualizing Workflows and Mechanisms
Diagram 1: LNP-mRNA vs. Viral Vector Redosing Mechanisms
Diagram 2: Optimizing LNP-mRNA Formulation Workflow
Analyzing Immunogenic Profiles and Long-Term Safety Data Across Cargo Types
The immunogenic profile and long-term safety of CRISPR-Cas9 therapies are significantly influenced by the form of Cas9 cargo delivered. The three primary cargo types—DNA, mRNA, and Ribonucleoprotein (RNP) complexes—each interact uniquely with the host immune system, presenting distinct advantages and challenges for therapeutic development [13]. Understanding these differences is crucial for researchers and drug development professionals aiming to optimize editing efficiency while minimizing immune-related risks.
DNA-based cargo typically uses plasmids or viral vectors to encode Cas9, resulting in prolonged Cas9 expression that increases both editing opportunities and the risk of sustained immune activation and off-target effects [13]. mRNA cargo offers transient Cas9 expression, reducing the duration of immune exposure but potentially triggering potent innate immune responses through pattern recognition receptors [13]. RNP complexes, consisting of preassembled Cas9 protein and guide RNA, provide the most transient activity of all cargo types, limiting off-target effects but still potentially eliciting adaptive immune responses against the bacterial-derived Cas9 protein [13] [104].
Pre-existing immunity to Cas9 presents a particular concern, as many individuals have been exposed to the Streptococcus pyogenes bacterium from which the common SpCas9 is derived [105]. This pre-existing immunity can potentially lead to rapid clearance of CRISPR-containing cells, reduced editing efficiency, and adverse inflammatory responses. The following sections provide detailed troubleshooting guidance and comparative data to help researchers navigate these complex immunogenicity and safety considerations.
Comparative Immunogenicity Profiles of Cas9 Cargo Types
Table 1: Comparative Immunogenic Profiles and Safety Characteristics of Cas9 Cargo Types
| Cargo Type | Immune Activation Pathways | Key Immunogenic Components | Duration of Cas9 Expression | Risk of Pre-existing Immunity Impact |
|---|---|---|---|---|
| DNA (Plasmid/Viral) | Cytosolic DNA sensors (cGAS-STING), TLR9 | Bacterial DNA sequences, viral capsid proteins (if using viral delivery) | Days to weeks (prolonged) | High (for viral vectors); Moderate (for non-viral) |
| mRNA | TLR7/8, RIG-I-like receptors | 5' triphosphate, uridine-rich sequences, dsRNA impurities | Hours to days (transient) | Moderate |
| RNP (Ribonucleoprotein) | MHC class I and II presentation | Bacterial Cas9 protein epitopes | Hours (most transient) | High (for adaptive T-cell responses) |
Table 2: Editing Efficiency and Practical Implementation Considerations
| Cargo Type | Typical Editing Efficiency | Risk of Off-Target Effects | Key Advantages | Key Limitations |
|---|---|---|---|---|
| DNA (Plasmid/Viral) | Variable; can be high with viral vectors | High (due to prolonged expression) | Sustained expression, stable for in vivo delivery | High immunogenicity, insertional mutagenesis risk |
| mRNA | Moderate to High | Moderate | No genomic integration risk, transient expression | Innate immune activation, stability challenges |
| RNP (Ribonucleoprotein) | High in many systems | Lowest (due to transient activity) | Immediate activity, high precision, no genomic integration | Delivery challenges, potential adaptive immune responses |
FAQs: Addressing Common Immunogenicity and Safety Concerns
Q1: What are the primary immune recognition pathways for each Cas9 cargo type, and how can they be mitigated?
- DNA cargo is primarily recognized by endosomal Toll-like receptor 9 (TLR9) and cytosolic DNA sensors (e.g., cGAS-STING), leading to type I interferon responses [13] [99]. Mitigation strategies include: (1) using minimal bacterial backbone plasmids; (2) employing chromatin-insulating elements; and (3) selecting non-viral delivery vectors when possible to avoid viral protein immunity.
- mRNA cargo activates endosomal TLR7/TLR8 and cytosolic RIG-I/MDA5 pathways, triggering interferon-α and inflammatory cytokine production [13]. Effective mitigation approaches include: (1) incorporating modified nucleosides (e.g., pseudouridine, N1-methylpseudouridine); (2) implementing HPLC purification to remove dsRNA impurities; (3) using codon optimization; and (4) employing cap modifications (CleanCap technology).
- RNP cargo primarily elicits adaptive immune responses through MHC class I and II presentation of bacterial Cas9 epitopes, potentially leading to T-cell mediated clearance [104] [105]. Mitigation strategies include: (1) using Cas9 epitope prediction and engineering to remove immunodominant T-cell epitopes; (2) employing humanized or engineered Cas9 variants with reduced immunogenicity; and (3) utilizing transient immunosuppression regimens during treatment.
Q2: How does cargo choice impact long-term safety and persistence of editing?
The persistence of Cas9 expression directly correlates with both editing opportunities and safety risks. DNA cargo, particularly when delivered via viral vectors like AAV, can maintain Cas9 expression for months to years, potentially leading to sustained immune activation and increased off-target effects [13]. While this enables more editing opportunities, it raises significant long-term safety concerns. mRNA cargo typically persists for several days, offering a balance between editing efficiency and safety, with reduced off-target risks compared to DNA. RNP complexes have the shortest activity window (hours), minimizing off-target effects but potentially requiring higher initial doses, which could intensify acute immune responses [1] [104]. For long-term therapeutic applications where repeat dosing may be necessary, mRNA and RNP formats are generally preferred due to their transient nature and reduced risk of genomic integration.
Q3: What strategies exist to overcome pre-existing immunity to Cas9?
Pre-existing immunity to Cas9, particularly SpCas9, is present in a significant portion of the human population due to previous exposure to Streptococcus pyogenes [105]. This can lead to rapid clearance of Cas9-expressing cells and reduced therapeutic efficacy. Solutions include: (1) Serological screening of patients for pre-existing anti-Cas9 antibodies before treatment; (2) Cas9 ortholog switching to less common variants (e.g., SaCas9, NmCas9) with lower seroprevalence; (3) Epitope engineering to modify immunodominant regions while maintaining catalytic activity; (4) Transient immunosuppression using corticosteroids or other immunomodulators during the initial treatment phase; and (5) Delivery system optimization to shield Cas9 from immune recognition, such as using lipid nanoparticles with PEGylation or cell-specific targeting motifs.
Q4: What are the best practices for assessing immunogenicity in pre-clinical models?
Comprehensive immunogenicity assessment should include both in vitro and in vivo evaluations. Recommended practices include: (1) In vitro human PBMC assays to assess T-cell activation using CFSE dilution or ELISpot; (2) Human dendritic cell maturation assays measuring CD80/CD86 expression; (3) Seroprevalence studies using human serum samples to detect pre-existing antibodies; (4) Humanized mouse models engrafted with human immune systems; (5) Comprehensive cytokine profiling post-treatment; and (6) Immunohistochemical analysis of treated tissues for immune cell infiltration. These approaches should be complemented by standard editing efficiency assessments to balance efficacy and safety.
Experimental Protocols for Immunogenicity Assessment
Protocol 1: In Vitro Immune Activation Assay for Cas9 Cargo
Purpose: To evaluate the innate immune response potential of different Cas9 cargo formats in human peripheral blood mononuclear cells (PBMCs).
Materials:
- Freshly isolated human PBMCs from healthy donors
- Cas9 cargo preparations: plasmid DNA, modified mRNA, RNP complexes
- Positive controls: LPS (1μg/mL) for TLR4, R848 (1μM) for TLR7/8
- Cell culture media (RPMI-1640 with 10% FBS)
- ELISA kits for IFN-α, IFN-β, IL-6, TNF-α
- Flow cytometry antibodies for CD14, CD80, CD86, HLA-DR
Procedure:
- Isolate PBMCs from whole blood using Ficoll density gradient centrifugation.
- Seed 1×10^6 PBMCs per well in 24-well plates and allow to rest for 2 hours.
- Treat cells with Cas9 cargoes at concentrations reflecting planned in vivo doses (typically 1-5μg/mL for DNA and mRNA, 0.5-2μM for RNP).
- Include appropriate positive controls (LPS, R848) and negative controls (vehicle alone).
- Collect supernatant at 6 hours (for early cytokines) and 24 hours (for late cytokines).
- Analyze cytokine secretion using ELISA according to manufacturer protocols.
- At 24 hours, harvest cells for flow cytometry analysis of dendritic cell maturation markers.
- Perform statistical analysis using one-way ANOVA with post-hoc testing.
Troubleshooting: If high background activation is observed, ensure nucleic acid preparations are free of endotoxin using LAL testing. For RNP complexes, verify the absence of contaminating bacterial components from protein purification.
Protocol 2: In Vivo Persistence and Adaptive Immune Response Assessment
Purpose: To evaluate the persistence and adaptive immune response to different Cas9 cargo formats in a murine model.
Materials:
- C57BL/6 mice (6-8 weeks old)
- Cas9 cargo preparations with appropriate delivery vehicles (LNP for mRNA/RNP, electroporation for DNA)
- ELISA kits for anti-Cas9 IgG, IgM
- IFN-γ ELISpot kit
- T-cell media and peptides covering Cas9 protein sequence
Procedure:
- Divide mice into experimental groups (n=5-8) receiving DNA, mRNA, or RNP formulations.
- Administer Cas9 cargo via relevant route (IV, IM, or local administration).
- Collect serum samples weekly for 4 weeks to measure anti-Cas9 antibody titers via ELISA.
- At day 28, sacrifice animals and harvest spleens for T-cell analysis.
- Isolate splenocytes and perform IFN-γ ELISpot using pools of Cas9-derived peptides.
- For DNA cargo groups, analyze tissues for Cas9 persistence via qPCR at endpoint.
- Perform immunohistochemistry on target tissues to assess immune cell infiltration.
Troubleshooting: If unexpected clearance of Cas9 is observed, assess pre-existing immunity in animal colonies. Consider using humanized mouse models for more translational relevance to human immune responses.
Research Reagent Solutions for Immunogenicity Studies
Table 3: Essential Research Reagents for Cas9 Immunogenicity Profiling
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Immune Assay Kits | IFN-α/β ELISA, IFN-γ ELISpot, Multiplex Cytokine Panels | Quantifying innate and adaptive immune responses | Verify species reactivity; choose high-sensitivity kits for low-abundance cytokines |
| Cell Culture Models | Primary human PBMCs, Monocyte-derived Dendritic Cells, DC2.4 cell line | In vitro immunogenicity screening | Use multiple donors to account for human variability; include relevant positive controls |
| Detection Antibodies | Anti-CD80/86, HLA-DR, CD3/CD4/CD8, Cas9-specific antibodies | Flow cytometry, immunohistochemistry | Validate specificity; optimize titers to reduce background |
| Cas9 Antigens | Full-length recombinant Cas9 protein, Cas9 peptide pools (15-mers) | T-cell activation assays, antibody detection | Ensure proper folding of recombinant protein; include positive control peptides |
| Delivery Vehicles | LNPs, Electroporation systems, AAV vectors, Polyethylenimine (PEI) | Cargo-specific immunogenicity assessment | Compare multiple vehicles; optimize charge ratios for nucleic acid complexes |
Signaling Pathways in Cas9 Immunogenicity
Cas9 Cargo Immune Activation Pathways
Experimental Workflow for Comparative Immunogenicity Analysis
Immunogenicity Assessment Workflow
Conclusion
The choice between Cas9 protein and mRNA delivery is not a matter of one being universally superior, but rather hinges on the specific therapeutic context. Protein RNP delivery offers unparalleled speed and precision, making it ideal for ex vivo applications and situations where minimizing off-target effects is paramount. mRNA delivery, facilitated by advanced LNPs, provides a versatile and redosable platform for in vivo therapies, with a superior safety profile regarding host genome integration. The future of CRISPR therapeutics lies in the continued convergence of these approaches with cutting-edge technologies—from AI-driven design of both mRNA sequences and delivery vehicles to novel nanostructures that enhance cellular uptake. This synergy will pave the way for more potent, precise, and personalized genetic medicines, expanding the treatable disease landscape.
References
- 1. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 2. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.mdpi.com/1422-0067/26/9/4420]
- 3. Efficient Delivery of CRISPR-Cas9 RNP Complexes with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12608112/]
- 4. Efficient intracellular delivery of CRISPR-Cas9 ... [https://www.sciencedirect.com/science/article/abs/pii/S174801322500026X]
- 5. Improving the Efficiency of CRISPR/Cas9-Mediated Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384319/]
- 6. Optimization of in vivo delivery methods and their applications ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-025-01021-0]
- 7. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 8. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7738854/]
- 9. 5 tips when starting a CRISPR-Cas9 experiment | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/quick-tips-when-starting-a-crispr-experiment/]
- 10. Unlocking mRNA-driven CRISPR-Cas9 gene therapy via ... [https://pubmed.ncbi.nlm.nih.gov/41210587/]
- 11. CRISPR/Cas9 delivery methods [https://www.takarabio.com/learning-centers/gene-function/gene-editing/crisprcas9-delivery-methods?srsltid=AfmBOorz5l957WJLNB_xBQSkhQd-CGiwYVO9QP-oEfg9MGgPZYRVzLyx]
- 12. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing | Nature Communications [https://www.nature.com/articles/s41467-020-17029-3]
- 13. Unlocking mRNA-driven CRISPR-Cas9 gene therapy via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594903/]
- 14. Lung and liver editing by lipid nanoparticle delivery of a stable... [https://www.nature.com/articles/s41587-024-02437-3?error=cookies_not_supported&code=6c7d99a5-2a3d-48d9-9619-ddcd880f4d48]
- 15. Low Knockout Efficiency in Troubleshooting ... - CD Biosynsis CRISPR [https://www.biosynsis.com/blog/troubleshooting-low-knockout-efficiency-in-crispr-experiments/]
- 16. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 17. for Troubleshooting - Guide Editing Problems Common CRISPR Cas 9 [https://synapse.patsnap.com/article/troubleshooting-guide-for-common-crispr-cas9-editing-problems]
- 18. Nuclear Localization Signals Enable the Cellular Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12636420/]
- 19. Non-nuclear localization signal-guided CRISPR/Cas9 ... [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d4nr04762a]
- 20. Nanotechnology-Based Delivery of CRISPR/Cas9 for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346122/]
- 21. Lipid nanoparticle delivery of the CRISPR/Cas9 system ... [https://www.nature.com/articles/s41598-025-03671-8]
- 22. Deep generative optimization of mRNA codon sequences ... [https://www.nature.com/articles/s41467-025-64894-x]
- 23. Enhancing mRNA translation efficiency by introducing ... [https://www.sciencedirect.com/science/article/pii/S2162253125000393]
- 24. of Various Nuclear Localization Signal-Fused... Comparison [https://pmc.ncbi.nlm.nih.gov/articles/PMC5844304/]
- 25. Deciphering the biological fate of mRNA-LNP-based ... [https://www.sciencedirect.com/science/article/pii/S2211383525007737]
- 26. Inside the cell: Approaches to evaluating mRNA ... [https://pubmed.ncbi.nlm.nih.gov/40513751/]
- 27. AAV Vs. Lentiviral Vectors - Life in the Lab [https://www.thermofisher.com/blog/life-in-the-lab/aav-vs-lv/]
- 28. Overcoming the AAV Size Limitation for CRISPR Delivery [https://blog.addgene.org/a-match-made-in-heaven-crispr/cas9-and-aav]
- 29. AAV vs Lentivirus Resources - BioInnovatise [https://bioinnovatise.com/articles/aav-vs-lentivirus-which-viral-vector-is-right-for-my-research/]
- 30. Lentiviral Gene Delivery for Mammalian Expression— ... [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-expression-support-center/lentiviral-gene-delivery-support/lentiviral-gene-delivery-support-troubleshooting.html]
- 31. Tips for successful lentiviral transduction [https://www.takarabio.com/learning-centers/gene-function/viral-transduction/lentivirus/lentiviral-tips?srsltid=AfmBOopZzuDlZgrbhIKBRumuPUiQmz7CzPJz0FFVhz9PVSCkOEcsHzc9]
- 32. Exploring the Challenges of Lipid Nanoparticle Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC12031360/]
- 33. Optimized lipid nanoparticles enable effective CRISPR ... [https://www.sciencedirect.com/science/article/pii/S2211383524003526]
- 34. Enhanced CRISPR-Cas9 RNA system delivery using cell ... [https://www.sciencedirect.com/science/article/pii/S0168365924007545]
- 35. Lipid nanoparticles (LNPs) for in vivo RNA delivery and ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X23003058]
- 36. Challenges and opportunities in computational studies for ... [https://www.nature.com/articles/s44386-025-00024-3]
- 37. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 38. Lipid nanoparticle (LNP) manufacturing: Challenges & ... [https://www.susupport.com/blogs/knowledge/lipid-nanoparticle-lnp-manufacturing-challenges-solutions]
- 39. Customizable virus-like particles deliver CRISPR–Cas9 ... [https://www.nature.com/articles/s41565-024-01851-7]
- 40. Engineering a streamlined virus-like particle for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528395/]
- 41. Electroporation | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/methods/electroporation.html]
- 42. Electroporation Education [https://www.btxonline.com/technical-resources/electroporation-education.html]
- 43. Transfection by Electroporation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2975437/]
- 44. Ex vivo electroporation of retinal cells: a novel, high efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3615050/]
- 45. Why do lipid nanoparticles target the liver? Understanding of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11919328/]
- 46. Recent Advances in the Lipid Nanoparticle-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10059764/]
- 47. Reformulating lipid nanoparticles for organ-targeted mRNA ... [https://www.nature.com/articles/s41467-024-50093-7]
- 48. Role of PEGylated lipid in lipid nanoparticle formulation for ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925000811]
- 49. Liver injury after SARSâ€CoVâ€2 vaccination - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9348326/]
- 50. Multi-step screening of DNA/lipid nanoparticles and co- ... [https://www.nature.com/articles/s41467-022-31993-y]
- 51. Low-liver-accumulation lipid nanoparticles enhance the ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06924-2]
- 52. A comparison of three different delivery methods for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2023.1111110/full]
- 53. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 54. Delivery Strategies of the CRISPR-Cas9 Gene-Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5723556/]
- 55. Progress and prospects of mRNA-based drugs in pre- ... [https://www.nature.com/articles/s41392-024-02002-z]
- 56. Enhancing Vaccine Efficacy and Stability: A Review of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11353004/]
- 57. Recent Advancements in Reducing the Off-Target Effect of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10802171/]
- 58. Off-target Effects in CRISPR/Cas9-mediated Genome ... [https://www.sciencedirect.com/science/article/pii/S216225311630049X]
- 59. High-fidelity CRISPR-Cas9 variants with undetectable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4851738/]
- 60. promising for development of high-fidelity Cas9 variants [https://www.nature.com/articles/s41392-022-01139-z]
- 61. an advanced genome editor to amend the CRISPR gene editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC10328345/]
- 62. Latest Developed Strategies to Minimize the Off-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7407193/]
- 63. Nucleoside-modified messenger RNA [https://en.wikipedia.org/wiki/Nucleoside-modified_messenger_RNA]
- 64. Optimizing 5'UTRs for mRNA-delivered gene editing using ... [https://www.nature.com/articles/s41467-024-49508-2]
- 65. Overcoming RNA Bottlenecks - Part 1: How to Ensure ... [https://exothera.world/overcoming-rna-bottlenecks-part-1-how-to-ensure-consistent-formulation-stability/]
- 66. Codon-optimization in gene therapy: promises, prospects ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1371596/full]
- 67. Overcoming the Delivery Challenges in CRISPR/Cas9 ... [https://www.medsci.org/v22p3625.htm]
- 68. Position-specific ORF nucleoside-ribose modifications ... [https://www.nature.com/articles/s41467-025-65788-8]
- 69. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.preprints.org/manuscript/202503.2038]
- 70. AI-guided Cas9 engineering provides an effective strategy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583830/]
- 71. Deep generative optimization of mRNA codon sequences ... [https://pubmed.ncbi.nlm.nih.gov/41224770/]
- 72. How AI could speed the development of RNA vaccines and ... [https://news.mit.edu/2025/how-ai-could-speed-development-rna-vaccines-and-other-rna-therapies-0815]
- 73. TL Artificial intelligence applied to mRNA vaccines [https://www.trilinkbiotech.com/blog/artificial-intelligence-applied-to-mrna-vaccines/]
- 74. Loop structure in poly(A) tail of mRNA vaccine enhances ... [https://www.nature.com/articles/s41541-025-01287-7]
- 75. CMN Weekly (5 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-5-september-2025-your-weekly-crispr-medicine-news/]
- 76. CRISPR's Efficiency Triples with Spherical Nucleic Acid ... [https://www.mccormick.northwestern.edu/news/articles/2025/09/crisprs-efficiency-triples-with-spherical-nucleic-acid-delivery-system/]
- 77. Optimization of gene knockout approaches and sgRNA ... [https://pubmed.ncbi.nlm.nih.gov/41253931/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=125m97__nL7-trC8hegpj0e5Ib2U6PBxHcdjn9mBoioOuOOQkh&fc=None&ff=20251119115323&v=2.18.0.post22+67771e2]
- 78. Boost HDR Efficiency with new HDR Enhancer Protein | IDT [https://www.idtdna.com/pages/about/news/2025/09/25/integrated-dna-technologies-launches-new-enhancer-protein-to-accelerate-crispr-based-therapies-from-translational-research-to-clinical-breakthrough]
- 79. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 80. Structural characterization of mRNA lipid nanoparticles ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925007047]
- 81. CRISPR This Way: Choosing Between Delivery System [https://en.vectorbuilder.com/resources/vector-academy/lessons/crispr-delivery-systems.html]
- 82. Optimizing CRISPR delivery for drug discovery [https://www.drugdiscoverynews.com/optimizing-crispr-delivery-for-drug-discovery-16487]
- 83. CRISPR-Cas9 Troubleshooting | Blog [https://go.zageno.com/blog/crispr-cas9-troubleshooting]
- 84. Unlocking mRNA-driven CRISPR/Cas9 gene therapy via ... [https://www.sciencedirect.com/science/article/pii/S2162253125002914]
- 85. Lung and liver editing by lipid nanoparticle delivery of a ... [https://www.nature.com/articles/s41587-024-02437-3]
- 86. innovations and challenges in rAAV vector-based CRISPR ... [https://www.nature.com/articles/s41434-025-00573-2]
- 87. Researchers Publish Final Results of Key Clinical Trial for ... [https://www.chop.edu/news/researchers-publish-final-results-key-clinical-trial-gene-therapy-sickle-cell-disease]
- 88. How Does CASGEVY® Work? [https://www.casgevyhcp.com/mechanism-of-action]
- 89. CRISPR/Cas9 gene editing for curing sickle cell disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC8049447/]
- 90. CRISPR 101: Ribonucleoprotein (RNP) Delivery [https://blog.addgene.org/crispr-101-ribonucleoprotein-rnp-delivery]
- 91. CASGEVY® Clinical Trial and Results [https://www.casgevy.com/sickle-cell-disease/study-information]
- 92. How Does CASGEVY® Work? [https://www.casgevy.com/sickle-cell-disease/how-casgevy-works]
- 93. Development and IND-enabling studies of a novel Cas9 ... [https://www.sciencedirect.com/science/article/pii/S1525001624004775]
- 94. Recent Advances in CRISPR/Cas9 Delivery Approaches for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10661828/]
- 95. Delivery strategies for CRISPR/Cas genome editing tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9614410/]
- 96. Addressing variability in iPSC-derived models of human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6994963/]
- 97. Controlling CRISPR-Cas9 genome editing in human cells ... [https://www.sciencedirect.com/science/article/pii/S2162253125001945]
- 98. Induced pluripotent stem cells (iPSCs) [https://www.nature.com/articles/s41392-024-01809-0]
- 99. Overcoming the Delivery Challenges in CRISPR/Cas9 Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12434829/]
- 100. Current Analytical Strategies for mRNA -Based Therapeutics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11990077/]
- 101. Frontiers | Technological breakthroughs and advancements in the... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1524317/full]
- 102. Clinical development of therapeutic mRNA applications [https://www.sciencedirect.com/science/article/pii/S1525001625002084]
- 103. Pharmacometric modeling of lipid nanoparticle-encapsulated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418826/]
- 104. Efficient in vivo labeling of endogenous proteins with ... [https://www.nature.com/articles/s41467-025-58945-6]
- 105. Peeling back the layers of immunogenicity in Cas9-based ... [https://www.sciencedirect.com/science/article/pii/S1525001625005350]