Homology-Independent Knock-In in Zebrafish: A Robust Strategy for Precision Genome Editing
This article provides a comprehensive overview of homology-independent knock-in strategies for precise genome editing in zebrafish, a pivotal model in biomedical and drug discovery research.
Homology-Independent Knock-In in Zebrafish: A Robust Strategy for Precision Genome Editing
Abstract
This article provides a comprehensive overview of homology-independent knock-in strategies for precise genome editing in zebrafish, a pivotal model in biomedical and drug discovery research. We explore the foundational principles that distinguish homology-independent repair from homology-directed repair (HDR), detailing the core mechanisms like non-homologous end joining (NHEJ) that enable efficient integration of large DNA cassettes. The article offers a practical guide on methodology, from vector design to germline transmission, and presents crucial troubleshooting and optimization protocols, including the use of chemical modulators to enhance efficiency. Finally, we validate the approach through comparative analyses with other editing techniques and showcase its successful application in creating specific reporter lines and disease models, underscoring its transformative potential for functional genomics and the study of human diseases.
Beyond HDR: Understanding the Foundations of Homology-Independent Knock-In
Defining Homology-Independent vs. Homology-Directed Repair (HDR) Pathways
The development of CRISPR-Cas9 technology has revolutionized genetic research, enabling precise modifications in the genomes of model organisms like zebrafish. When CRISPR-Cas9 introduces a double-strand break (DSB) in DNA, the cell activates endogenous repair mechanisms to resolve the break. The two primary competing pathways for this repair are homology-directed repair (HDR) and homology-independent repair, which includes non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). Understanding the distinct mechanisms, applications, and limitations of these pathways is essential for designing effective genome editing experiments, particularly for knock-in strategies in zebrafish research.
Homology-directed repair is a precise DNA repair mechanism that utilizes homologous sequences—such as a sister chromatid or an exogenously supplied donor template—to accurately repair DSBs. In contrast, homology-independent pathways like NHEJ directly rejoin broken DNA ends without requiring a homologous template, often resulting in small insertions or deletions (indels). The choice between these pathways significantly impacts the outcome of genome editing experiments, making pathway selection a critical consideration in experimental design.
Molecular Mechanisms of DNA Repair Pathways
Homology-Directed Repair (HDR) Mechanism
HDR is a high-fidelity repair pathway that uses a homologous DNA template to accurately repair double-strand breaks. This pathway is active primarily in the S and G2 phases of the cell cycle when sister chromatids are available. The process begins with resection of the 5' ends at the break site, creating 3' single-stranded DNA overhangs. The recombinase RAD51 then coats these overhangs and facilitates strand invasion into a homologous template sequence. DNA polymerase extends the invading strand using the template, and the repair is completed through synthesis-dependent strand annealing (SDSA) or double-strand break repair (DSBR) pathways [1].
In CRISPR-mediated HDR applications, researchers supply an exogenous donor template containing the desired modification flanked by homology arms that match sequences surrounding the target site. The cell's repair machinery uses this template to incorporate precise genetic changes, including point mutations, insertions, or gene replacements. The efficiency of HDR is influenced by multiple factors, including template design, cell cycle stage, and the relative activity of competing repair pathways [2].
Homology-Independent Repair Mechanisms
Non-Homologous End Joining (NHEJ)
NHEJ is the dominant DSB repair pathway in most cells, functioning throughout the cell cycle. This pathway begins with the recognition of broken DNA ends by the Ku heterodimer (Ku70/Ku80), which recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form an active complex. The Artemis nuclease processes the DNA ends, and the XRCC4-DNA ligase IV complex ligates them back together. Unlike HDR, NHEJ does not require a homologous template and is therefore error-prone, often resulting in small insertions or deletions (indels) that can disrupt gene function [2].
NHEJ is particularly useful for generating gene knockouts, as the introduced indels can create frameshift mutations that prematurely truncate the encoded protein. While traditionally considered random, NHEJ can also be harnessed for precise knock-in strategies using carefully designed templates that leverage the pathway's end-joining capabilities [3].
Microhomology-Mediated End Joining (MMEJ)
MMEJ represents an alternative homology-independent pathway that utilizes short homologous sequences (5-25 bp) flanking the break site for repair. The key regulator of MMEJ is polymerase theta (Polθ), which aligns microhomologous regions before initiating DNA synthesis. MMEJ typically results in deletions flanked by microhomology regions and can be a backup pathway when NHEJ is compromised [4]. While MMEJ can be harnessed for specific editing applications, it often competes with HDR and can reduce precise editing efficiency.
Comparative Analysis of Repair Pathways
The table below summarizes the key characteristics of HDR and homology-independent repair pathways:
Table 1: Comparison of DNA Repair Pathways in Genome Editing
| Feature | HDR | NHEJ | MMEJ |
|---|---|---|---|
| Template Requirement | Requires homologous template (endogenous or exogenous) | No template required | No template required; uses microhomology regions |
| Fidelity | High precision, error-free | Error-prone, creates indels | Error-prone, creates deletions |
| Cell Cycle Phase | S and G2 phases | Active throughout cell cycle | Active throughout cell cycle |
| Key Proteins | RAD51, BRCA2, PALB2 | Ku70/Ku80, DNA-PKcs, XRCC4-LigIV | Polθ, PARP1, DNA Ligase I/III |
| Primary Applications | Precise knock-ins, point mutations, gene corrections | Gene knockouts, random mutagenesis | Gene knockouts, deletion studies |
| Efficiency in Zebrafish | Low (typically <10% without enhancement) | High (often >50%) | Variable |
| Advantages | High precision, versatile for various edits | Highly efficient, works in non-dividing cells | Can create specific deletion patterns |
| Disadvantages | Low efficiency, competes with NHEJ/MMEJ, requires donor design | Introduces random mutations, less precise | Limited control over outcomes, less characterized |
Experimental Protocols for HDR in Zebrafish
Optimized HDR Workflow for Zebrafish Embryos
The following protocol has been optimized for precise genome editing in zebrafish using HDR, incorporating recent advancements to enhance efficiency:
Reagent Preparation:
- CRISPR Components: Prepare high-efficiency sgRNA (≥60% cutting efficiency) and Cas9 protein (200-800 pg optimal amount) as ribonucleoprotein (RNP) complexes [1] [5].
- Donor Template: Design single-stranded oligodeoxynucleotide (ssODN) with 30-50 nt homology arms for point mutations or short insertions (<50 nt). For larger insertions, use long single-stranded DNA with 350-700 nt homology arms [6]. Position the desired edit within 10 nt of the Cas9 cut site.
- Template Enhancement: Incorporate silent mutations in the PAM sequence or seed region to prevent re-cutting of successfully edited alleles [6]. Chemically modified Alt-R HDR templates can improve integration efficiency [5].
- Small Molecule Inhibitors: Prepare 50 μM NU7441 (NHEJ inhibitor) dissolved in DMSO for microinjection [7].
Microinjection Procedure:
- Set up zebrafish mating pairs and collect embryos within 15 minutes post-fertilization.
- Prepare injection mixture containing:
- Cas9 RNP complex (final amount 200-800 pg)
- HDR donor template (50-100 pg)
- 50 μM NU7441 (or DMSO vehicle for controls)
- Microinject 1-2 nL of the mixture directly into the cell cytoplasm or yolk of 1-2 cell stage embryos.
- Maintain injected embryos at 28-32°C and monitor development.
Validation and Screening:
- At 24-48 hours post-fertilization, screen for somatic editing using appropriate methods (e.g., fluorescence if using a reporter system, PCR-based assays).
- For germline transmission, raise injected embryos (F0) to adulthood and outcross to wild-type fish.
- Screen F1 progeny for the desired edit using PCR, restriction fragment length analysis, or sequencing.
Table 2: Troubleshooting HDR in Zebrafish
| Problem | Potential Cause | Solution |
|---|---|---|
| Low HDR efficiency | High NHEJ/MMEJ competition | Add NHEJ inhibitors (NU7441); optimize Cas9 amount |
| Mosaic editing in F0 | Late editing after cell division | Inject at earliest embryonic stage; optimize injection site |
| Random integration | dsDNA template toxicity | Switch to single-stranded DNA templates |
| Cell death/toxicity | Excessive Cas9, inhibitor toxicity | Titrate Cas9 concentration; reduce inhibitor amount |
| No germline transmission | Edit not incorporated in germ cells | Increase sample size; use HDR-enhancing chemicals |
HDR Enhancement Using Small Molecule Inhibitors
Chemical inhibition of competing pathways significantly enhances HDR efficiency. The most effective inhibitor identified for zebrafish is NU7441, a DNA-PKcs inhibitor that blocks NHEJ. In quantitative studies, 50 μM NU7441 enhanced HDR-mediated repair up to 13.4-fold compared to DMSO controls [7]. This treatment increased the average number of successfully edited cells per embryo from 4.0 ± 3.0 to 53.7 ± 22.1 in a fluorescent reporter assay.
The HDRobust approach, which combines inhibition of both NHEJ and MMEJ, has demonstrated remarkable efficiency in human cells, achieving HDR rates of up to 93% (median 60%) [4]. While optimized for cell culture, this dual inhibition strategy presents a promising avenue for further optimization in zebrafish embryos.
Pathway Visualization and Experimental Design
DNA Repair Pathway Decision and HDR Enhancement Strategies
Emerging Alternatives and Complementary Technologies
Prime Editing
Prime editing represents a significant advancement beyond traditional HDR, enabling precise edits without requiring double-strand breaks or donor templates. This system uses a catalytically impaired Cas9 (nickase) fused to a reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that contains both the targeting sequence and the desired edit. In zebrafish, prime editing has demonstrated superior performance for certain applications, with one study reporting up to a fourfold increase in editing efficiency compared to HDR for base substitutions [5].
Two primary prime editing systems have been optimized for zebrafish:
- PE2: A nickase-based system most effective for nucleotide substitutions, achieving precision scores of 40.8% compared to 11.4% for nuclease-based systems [8].
- PEn: A nuclease-based system more effective for inserting short DNA fragments (3-30 bp) with higher efficiency than PE2 for these applications [8].
Base Editing
Base editors enable direct conversion of one nucleotide to another without inducing DSBs, making them valuable for specific point mutations. These include:
- Cytosine Base Editors (CBEs): Convert C:G to T:A base pairs
- Adenine Base Editors (ABEs): Convert A:T to G:C base pairs
Recent developments like the "near PAM-less" cytidine base editor (CBE4max-SpRY) have expanded the targeting scope in zebrafish, achieving editing efficiencies up to 87% at some loci [9]. Base editors are particularly valuable for modeling human genetic diseases caused by point mutations.
Essential Research Reagents and Tools
Table 3: Research Reagent Solutions for DNA Repair Studies in Zebrafish
| Reagent/Tool | Function | Examples/Specifications |
|---|---|---|
| CRISPR-Cas9 Components | Induces targeted double-strand breaks | High-efficiency sgRNA (>60%), Cas9 protein (200-800 pg optimal) [1] [5] |
| HDR Donor Templates | Provides homologous template for precise repair | ssODN (<200 nt), long ssDNA (>500 nt), homology arms (30-50 nt for ssODN; 350-700 nt for long ssDNA) [6] |
| NHEJ Inhibitors | Enhances HDR efficiency by blocking competing pathway | NU7441 (50 μM optimal), DNA-PKcs inhibitors [7] |
| HDR Enhancers | Stimulates homology-directed repair | RS-1 (RAD51 agonist), 15-30 μM [7] |
| Prime Editing Systems | Enables precise edits without DSBs or donors | PE2 (nickase-based), PEn (nuclease-based) [8] |
| Base Editors | Creates point mutations without DSBs | CBEs (C:G to T:A), ABEs (A:T to G:C) [9] |
| Validation Tools | Confirms editing efficiency and specificity | T7E1 assay, amplicon sequencing, fluorescence reporters [7] |
The strategic selection between homology-directed and homology-independent repair pathways is fundamental to successful genome engineering in zebrafish. While HDR enables precise modifications, its efficiency remains limited by competition with endogenous repair pathways. Recent advancements, including small molecule inhibition of NHEJ, optimized donor designs, and the development of novel editors like prime editors and base editors, have significantly improved the toolkit for precise genome modification.
For researchers designing knock-in experiments in zebrafish, we recommend a stratified approach: using HDR with NHEJ inhibition for medium-to-large insertions, prime editing for point mutations and small insertions, and base editing for specific nucleotide conversions. As these technologies continue to evolve, they will further enhance our ability to model human diseases and perform functional genomic studies in zebrafish.
In the context of zebrafish research, the pursuit of precise genomic integration is fundamental to creating accurate models for studying gene function and human diseases. The error-prone non-homologous end joining (NHEJ) pathway, once considered merely a source of stochastic indel mutations for gene knockouts, has been strategically repurposed as a powerful tool for targeted DNA integration. This application note details how homology-independent knock-in strategies exploit this competing DNA repair mechanism to achieve efficient transgene integration in zebrafish, bypassing the efficiency limitations of homology-directed repair (HDR) that have traditionally constrained precise genome editing in this model organism [10] [11].
The competitive balance between NHEJ and HDR pathways presents both a challenge and an opportunity for genome editors. While HDR is restricted to specific cell cycle phases (primarily S and G2), NHEJ operates throughout the cell cycle, making it the dominant repair pathway in most contexts [12] [13]. In normal human fibroblasts, NHEJ demonstrates higher activity than HR at all cell cycle stages, with its efficiency increasing as cells progress from G1 to G2/M phases [12]. This fundamental biological principle underpins the development of NHEJ-mediated knock-in approaches, which leverage the constant availability of this repair pathway in early zebrafish embryos to achieve high integration rates unattainable through HDR-based methods alone.
Molecular Mechanisms: NHEJ Versus HDR Pathways
Competitive Pathway Dynamics
Double-strand breaks (DSBs) induced by CRISPR/Cas9 activate competing DNA repair pathways, with the balance between these pathways determining editing outcomes. The NHEJ pathway operates throughout the cell cycle by directly ligating broken DNA ends, while HDR is restricted primarily to S and G2 phases where sister chromatids are available as templates [12] [13]. This temporal restriction significantly limits HDR efficiency in many experimental contexts.
In zebrafish embryos, the rapid cell cycles and developmental timing further constrain HDR efficacy, making NHEJ the predominant repair mechanism during early development [14]. Studies in normal human fibroblasts demonstrate that NHEJ activity increases progressively from G1 through S to G2/M phases, whereas HDR peaks during S phase and declines in G2/M [12]. This cell cycle dependency creates a narrow window for HDR efficiency while NHEJ remains constitutively active.
Strategic Exploitation of NHEJ for Integration
Homology-independent knock-in strategies deliberately leverage the NHEJ pathway's error-prone nature by designing donor constructs that are cleaved simultaneously with the genomic target. When both the chromosomal locus and donor plasmid experience DSBs, the cellular repair machinery frequently joins these fragments through NHEJ-mediated ligation [14]. This approach capitalizes on the natural efficiency of NHEJ while bypassing the complex machinery and cell cycle limitations of HDR.
The strategic innovation lies in designing donor vectors with CRISPR target sequences ("bait" sequences) that ensure co-cleavage of the donor and genomic target. This simultaneous cleavage creates compatible ends that NHEJ factors efficiently ligate, resulting in targeted integration without requiring homologous templates [14] [15]. By incorporating short homology arms (10-40 bp) flanking the genomic target in the donor vector, researchers can further enhance precise integration through microhomology-mediated mechanisms [15].
Quantitative Assessment of Editing Efficiencies
Comparative Performance of Knock-in Methods
Table 1: Efficiency Comparison of Genome Editing Methods in Zebrafish
| Method | Mechanism | Typical Efficiency | Key Advantages | Reported Applications |
|---|---|---|---|---|
| NHEJ-mediated Knock-in | Homology-independent ligation of co-cleaved donor | 22-85% (somatic) [14] [15] | Works throughout cell cycle; suitable for large inserts | eGFP to Gal4 line conversion [14] |
| HDR with ssODN | Homology-directed repair with single-stranded oligos | Variable (typically low) [10] | Precise edits; suitable for small changes | SNP introductions; small tag insertions [10] |
| HDR with Plasmid Donor | Homology-directed repair with double-stranded donor | Often <1.5% (germline) [10] | Can incorporate large inserts with high precision | Endogenous gene tagging [1] |
| NHEJ with Short Homology Arms | Enhanced microhomology-mediated integration | 77% with 40bp arms [15] | Improved precision over standard NHEJ | krtt1c19e-eGFP tagging [15] |
Critical Success Factors
Table 2: Parameters Influencing NHEJ-mediated Knock-in Efficiency
| Parameter | Optimal Condition | Impact on Efficiency | Experimental Evidence |
|---|---|---|---|
| sgRNA Efficiency | >60% indel rate [1] | Foundational for successful integration | High-efficiency sgRNAs resulted in 75% of injected embryos showing targeted integration [14] |
| Homology Arm Length | 10-40 bp [15] | 77% precise integration with 40bp arms vs 60% with 10bp | Precise integration rates increased with longer homology arms [15] |
| Donor Design | Bait sequences for co-cleavage [14] | Essential for NHEJ-mediated integration | No integration observed without donor cleavage [15] |
| Injection Timing | 1-2 cell stage [1] | Maximizes access to genome before rapid divisions | Standard practice for zebrafish genome editing [1] [15] |
| NHEJ Inhibition | Scr7 (DNA Ligase IV inhibitor) [13] | Up to 19-fold HDR increase in mammalian cells | Demonstrated in cell lines; potential application in zebrafish [13] |
Experimental Protocols for Zebrafish Knock-in
NHEJ-Mediated Knock-in Workflow
Detailed Step-by-Step Methodology
Step 1: Component Design and Preparation
sgRNA Selection: Identify genomic target sites using specialized tools (CHOP-CHOP or CRISPRscan) [10]. Select sgRNAs with demonstrated high efficiency (>60% indel rates) based on prior validation or prediction algorithms. For the bait sequence in the donor plasmid, choose an sgRNA with proven high cleavage efficiency (e.g., eGFP-gRNA with 66% efficiency) [14] [15].
Donor Vector Construction: Clone the desired insert (e.g., fluorescent protein, Gal4) into a suitable backbone. Incorporate the "bait" target sequence for co-cleavage on both sides of the insert. For enhanced precision, include short homology arms (10-40 bp) corresponding to sequences flanking the genomic cut site. Introduce silent mutations in the donor to prevent re-cleavage after integration [15].
Example: Auer et al. designed a donor plasmid containing eGFP bait sequences followed by E2A-KalTA4. This design enabled conversion of eGFP lines to Gal4 drivers with integration rates sufficient to observe RFP-positive cells in >75% of injected embryos when crossed with UAS:RFP reporters [14].
Step 2: Injection Mix Preparation
Formulate the injection mixture containing:
- Cas9 mRNA (100-300 ng/μL) or Cas9 protein (300-600 ng/μL)
- Genomic-targeting sgRNA (25-50 ng/μL)
- Bait-targeting sgRNA (25-50 ng/μL)
- Donor plasmid (25-100 ng/μL)
Optional: Include NHEJ inhibitors such as Scr7 (DNA Ligase IV inhibitor) to potentially shift balance toward HDR, though optimal concentrations for zebrafish require empirical determination [13].
Step 3: Microinjection Procedure
Inject 1-2 nL of the prepared mixture into the cytoplasm or cell body of 1-2 cell stage zebrafish embryos. The rapid cell divisions at this developmental stage necessitate early introduction of editing components to maximize distribution throughout the embryo [1] [15].
Step 4: Screening and Validation
Somatic Screening: For reporter integrations, screen injected embryos (F0) for expression patterns around 24-48 hours post-fertilization. The mosaic nature of F0 animals means expression will likely be restricted to a subset of cells.
Molecular Validation: For precise integration assessment, randomly select injected embryos for PCR analysis using primers flanking the target site and internal to the inserted sequence. Sequence PCR products to verify precise junction formation. Hisano et al. achieved 77% precise integration with 40bp homology arms, with sequence verification confirming accurate junctions [15].
Step 5: Germline Transmission
Raise injected embryos (F0 founders) to adulthood. Outcross to wild-type fish and screen F1 progeny for the integrated sequence. The germline transmission rate typically correlates with the somatic integration efficiency observed in F0 animals. Hisano et al. found that founders exhibiting broad eGFP expression as larvae were more likely to produce positive F1 progeny [15].
Research Reagent Solutions
Table 3: Essential Reagents for NHEJ-Mediated Knock-in in Zebrafish
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Nucleases | Cas9 mRNA, Cas9 protein | CRISPR/Cas9 system component; protein form may reduce off-target effects [10] |
| Targeting RNAs | sgRNAs (genomic & bait targets) | Guide Cas9 to specific genomic loci and donor bait sequences [14] |
| Donor Templates | Plasmid donors with bait sequences | Template for integration; include bait sites and optional homology arms [14] [15] |
| NHEJ Modulators | Scr7 (DNA Ligase IV inhibitor) | Shifts repair balance toward HDR; use requires concentration optimization [13] |
| Validation Tools | Junction PCR primers, sequencing primers | Essential for confirming precise integration events and germline transmission [15] |
| Reporter Systems | Fluorescent proteins (eGFP, mCherry), Gal4 | Enable visual screening of successful integration events [14] [16] |
Troubleshooting and Optimization Guidelines
Addressing Common Challenges
Low Integration Efficiency: When encountering insufficient integration rates, first verify sgRNA cutting efficiency using T7E1 assay or sequencing of the target locus in injected embryos. Ensure donor plasmid concentration is optimized (typically 25-100 ng/μL) and consider incorporating short homology arms (20-40 bp) to enhance precise integration through microhomology-mediated mechanisms [15].
Vector Backbone Integration: A common issue with NHEJ-mediated approaches is random integration of entire plasmid backbone. To prevent this, design donors with Cas9 cleavage sites flanking only the insert of interest, enabling precise excision from the backbone. Hisano et al. implemented this strategy by placing eGFP-gRNA target sequences on both sides of the eGFP and polyA signal sequence, successfully generating backbone-free integrations [15].
Mosaicism in Founders: The mosaic nature of F0 founders necessitates screening multiple offspring from each founder to identify germline transmission events. Focus breeding efforts on founders that showed widespread somatic integration as larvae, as these demonstrate higher likelihood of germline transmission [15].
Advanced Optimization Strategies
For difficult-to-edit loci, consider employing dual sgRNA approaches to create defined deletions followed by NHEJ-mediated integration into the deletion site. Additionally, testing Cas9 protein versus mRNA delivery may improve efficiency for some targets, as protein delivery accelerates nuclear activity in early embryos [10]. When targeting essential genes, validate that integration events do not disrupt critical gene functions through functional assays where possible.
Precise genome editing in zebrafish has been fundamentally limited by the inefficiency of Homology-Directed Repair (HDR) and the high prevalence of somatic mosaicism in F0 embryos. This application note details robust experimental protocols that leverage homology-independent knock-in strategies to overcome these challenges. By utilizing non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) pathways, researchers can achieve high-efficiency integration of large DNA cassettes, significantly accelerating the generation of knock-in zebrafish models for drug discovery and functional genomics.
In zebrafish, precise genome editing using conventional HDR-based approaches faces two significant hurdles. First, HDR competes inefficiently with the dominant non-homologous end joining (NHEJ) pathway, which introduces random indels at the target site [7]. Second, the extremely rapid early cell divisions in zebrafish embryos create a narrow window for DNA repair before the first cell division, resulting in somatic mosaicism where F0 embryos contain multiple, different editing events [17]. This mosaicism complicates phenotypic analysis and requires extensive outcrossing to obtain stable germline transmissions.
Homology-independent knock-in strategies bypass these limitations by utilizing alternative DNA repair pathways—NHEJ and MMEJ—that are more active during early embryonic stages. These methods facilitate direct ligation of double-strand breaks in donor vectors with breaks at the genomic target site, enabling highly efficient integration without requiring homologous templates [14] [18].
Quantitative Analysis of Editing Efficiency
The tables below summarize key performance metrics for various knock-in strategies, providing researchers with comparative data for experimental planning.
Table 1: Efficiency Comparison of Knock-in Strategies in Zebrafish
| Strategy | Repair Mechanism | Typical Efficiency Range | Key Advantages | Reported Cassette Size |
|---|---|---|---|---|
| Conventional HDR | Homology-Directed Repair | 1-5% | Precise integration; seamless junctions | Limited by homology arm design |
| Chemical-Enhanced HDR | HDR with NHEJ inhibition | Up to 13.4-fold improvement over HDR [7] | Enhanced precision; uses standard donor design | Similar to conventional HDR |
| NHEJ-Mediated Knock-in | Non-Homologous End Joining | >75% of embryos show integration [14] | Very high efficiency; simple vector design | Up to 5.7 kb demonstrated [14] |
| MMEJ-Mediated Knock-in | Microhomology-Mediated End Joining | High efficiency with precise deletion | Predictable deletions; reduced collateral damage | Varies with microhomology arms |
Table 2: Chemical and Physical Enhancement of Genome Editing Efficiency
| Treatment | Concentration/ Condition | Effect on Efficiency | Key Findings | Potential Drawbacks |
|---|---|---|---|---|
| NU7441 (NHEJ inhibitor) | 50 µM | 13.4-fold HDR increase [7] | Shifts repair equilibrium toward HDR | Requires optimization of delivery |
| RS-1 (RAD51 agonist) | 15-30 µM | Modest HDR increase (1.5-fold) [7] | Stimulates HDR pathway | Limited effect as standalone treatment |
| Temperature Reduction | 12°C for 30-60 min | Increased mutagenesis rate [17] | Extends single-cell stage by 30-60 min | Prolonged development time |
Homology-Independent Knock-in Protocols
NHEJ-Mediated Knock-in Workflow
This protocol enables highly efficient integration of DNA cassettes through direct ligation of cleaved ends, achieving reporter integration in >75% of injected embryos [14].
Experimental Workflow:
Donor Vector Design:
- Incorporate sgRNA target sequences ("bait" sites) flanking the insert cassette
- Include desired promoter and reporter gene (e.g., KalTA4, eGFP)
- Ensure reading frame maintenance with E2A peptide linkers if needed [14]
Zebrafish Embryo Preparation:
- Collect freshly fertilized zebrafish eggs (within 15 minutes post-fertilization)
- Prepare injection setup with standard microinjection equipment
Injection Mix Preparation:
- 150 ng/µL donor plasmid DNA
- 150 ng/µL Cas9 mRNA or protein
- 50 ng/µL sgRNA targeting genomic locus
- 1 µL phenol red indicator (optional)
- Nuclease-free water to 10 µL total volume
Microinjection:
- Inject 1-2 nL into the cell cytoplasm or yolk of 1-cell stage embryos
- Incubate injected embryos at 28°C in E3 embryo medium
Post-injection Processing:
- For temperature-modulated editing: Transfer embryos to 12°C for 30-60 minutes post-injection, then return to 28°C [17]
- Screen for successful integration via fluorescence (48-72 hpf)
- Raise positive founders to adulthood for germline transmission analysis
MMEJ-Mediated Knock-in Protocol
MMEJ utilizes short microhomology sequences (20-40 bp) flanking the insert to direct integration, resulting in predictable deletions at the target site [18].
Key Protocol Modifications:
Donor Vector Design for MMEJ:
- Incorporate 20-40 bp microhomology arms homologous to sequences flanking the target site
- Position microhomology arms to generate precise deletions upon integration
- Include reporter cassette with appropriate regulatory elements
Injection Mix:
- 100-200 ng/μL MMEJ donor vector
- 50 ng/μL Cas9 protein
- 30 ng/μL sgRNA targeting genomic region between microhomology arms
- Optional: 50 μM NU7441 for NHEJ inhibition [7]
Validation:
- Confirm precise junction sequences by PCR and sequencing
- Assess phenotypic consequences of predictable deletions
Signaling Pathways and Molecular Mechanisms
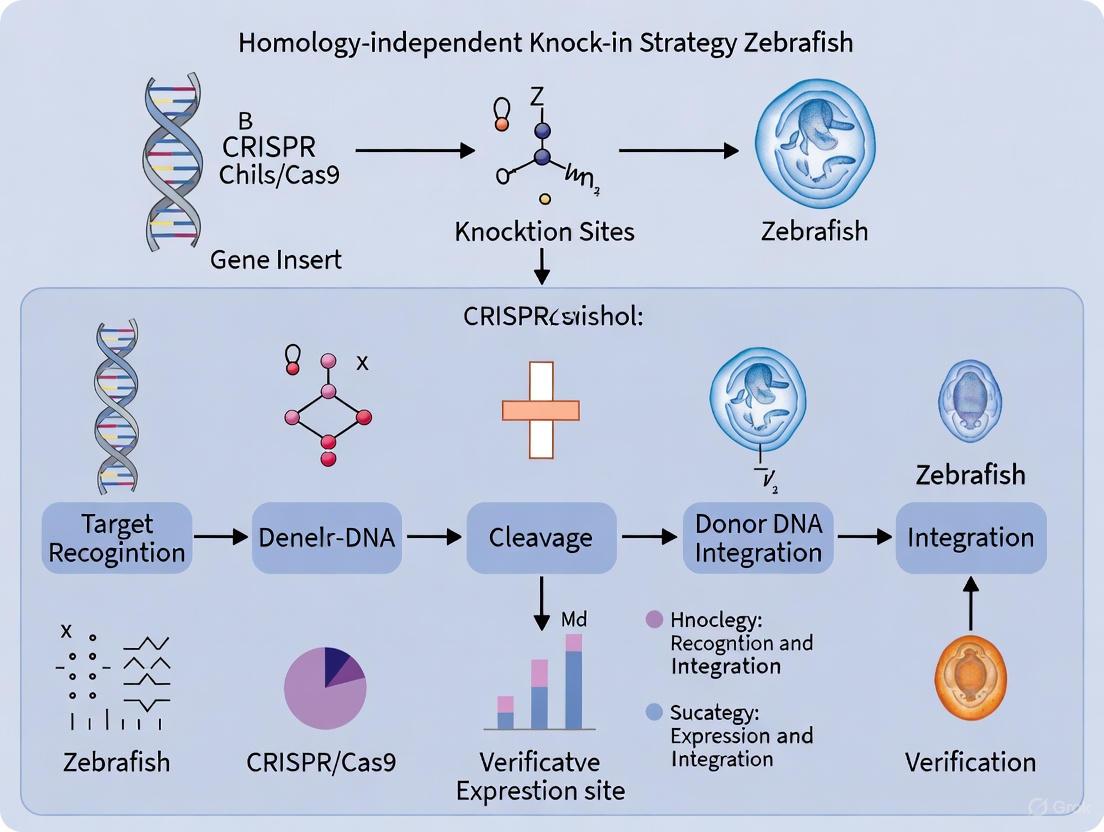
The diagram below illustrates the DNA repair pathways exploited for homology-independent knock-in in zebrafish embryos, highlighting how targeted double-strand breaks lead to successful gene integration.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Homology-Independent Knock-in in Zebrafish
| Reagent/Category | Specific Examples | Function & Application | Optimization Notes |
|---|---|---|---|
| Nuclease Systems | CRISPR/Cas9 (sgRNA + Cas9 protein/mRNA) | Induces targeted double-strand breaks at genomic locus and donor vector | Cas9 protein provides immediate activity; mRNA allows sustained expression |
| Donor Vectors | NHEJ: Bait-containing plasmids; MMEJ: Microhomology-flanked cassettes | Template for integration; design determines pathway utilization | For NHEJ: Include sgRNA target sites flanking insert; for MMEJ: 20-40 bp homology arms |
| Chemical Enhancers | NU7441 (50 µM), RS-1 (15-30 µM) | Modulate DNA repair pathways; inhibit NHEJ or stimulate HDR | Co-injection with editing components; optimal concentrations vary [7] |
| Physical Modulators | Temperature reduction (12°C) | Extends single-cell stage window for editing | Apply for 30-60 minutes post-injection [17] |
| Visual Reporters | eGFP, tdTomato, KalTA4-UAS systems | Enable rapid screening of successful integration | Tissue-specific promoters allow domain-restricted expression validation |
| (R)-ZG197 | (R)-ZG197, MF:C28H35F3N4O3, MW:532.6 g/mol | Chemical Reagent | Bench Chemicals |
| Phidianidine B | Phidianidine B|1301638-42-5|CAS Number | Phidianidine B is a marine alkaloid for neuroscience and pharmacology research. It is a potent DAT inhibitor and μ-opioid receptor ligand. For Research Use Only. | Bench Chemicals |
Troubleshooting and Optimization Guide
- Low Integration Efficiency: Verify sgRNA activity using T7E1 assay or sequencing; increase donor plasmid concentration; optimize Cas9:sgRNA ratio; implement temperature reduction to 12°C post-injection [17].
- High Embryo Mortality: Reduce injection volume to 1 nL; titrate Cas9 concentration; include phenol red for visualization; use smaller needle apertures.
- Mosaicism Persistence: Inject at earliest possible developmental stage (within 15 minutes post-fertilization); use Cas9 protein instead of mRNA for immediate activity; consider oocyte injection with rainbow trout ovarian fluid preservation [17].
- Off-target Integration: Include multiple sgRNAs for donor linearization; validate integration site with junction PCR and sequencing; use bioinformatics tools to predict and avoid off-target sites.
Homology-independent knock-in methods represent a paradigm shift in zebrafish genome engineering, effectively addressing the long-standing challenges of low HDR efficiency and somatic mosaicism. By leveraging the innate efficiency of NHEJ and MMEJ repair pathways, researchers can achieve integration rates exceeding 75% in F0 embryos, dramatically accelerating the generation of precise genetic models. These protocols provide robust frameworks for implementing these advanced techniques, empowering drug development professionals and researchers to more effectively link genetic modifications to phenotypic outcomes in zebrafish systems.
The emergence of homology-independent knock-in strategies represents a pivotal advancement in zebrafish genome engineering, fundamentally shifting the paradigm from traditional homologous recombination-based methods. Prior to these developments, targeted insertion of foreign DNA cassettes into the zebrafish genome remained challenging due to the characteristically low efficiency of homology-directed repair (HDR) in this model organism [19] [14]. The zebrafish model itself offers unique advantages for developmental studies, including external fertilization, optical transparency during embryogenesis, and high fecundity—with single mating pairs producing 70-300 embryos—making it particularly suitable for large-scale genetic studies [20]. The breakthrough came with the adaptation of CRISPR/Cas9 technology, which leveraged the more active non-homologous end joining (NHEJ) pathway in early zebrafish embryos to enable efficient integration of large DNA fragments without requiring extensive homologous arms [14] [21]. This historical progression from HDR-dependent to homology-independent mechanisms has dramatically expanded the zebrafish genetic toolbox, permitting researchers to create sophisticated reporter lines, lineage tracing tools, and disease models with unprecedented efficiency and precision.
The Evolution of Knock-In Strategies: From Concept to Mainstream Application
Foundational Studies and Key Technological Breakthroughs
The historical development of homology-independent knock-in strategies in zebrafish unfolded through a series of methodological innovations that progressively addressed the limitations of previous approaches. The initial proof-of-concept study in 2014 demonstrated that concurrent cleavage of both the genome and a donor plasmid by CRISPR/Cas9 could facilitate targeted integration via NHEJ repair pathways, achieving integration of DNA cassettes up to 5.7 kb [14]. This foundational work established the core principle that would underpin subsequent refinements: the use of "bait" sequences in donor plasmids that could be cleaved by sgRNAs complementary to the genomic target site, thus creating compatible ends for ligation [14] [22].
Subsequent innovations focused on optimizing multiple aspects of the methodology. Researchers explored various template designs, including the use of chemically modified double-stranded DNA donors with 5' AmC6 modifications that significantly enhanced integration efficiency by reducing degradation and multimerization [23]. The field also witnessed the development of versatile targeting strategies, including 5' knock-in upstream of the start codon, 3' knock-in preceding the stop codon, and intronic insertions, each offering distinct advantages for preserving endogenous gene function or enabling specific genetic manipulations [16] [23]. As the methodology matured, applications expanded from simple reporter lines to more sophisticated genetic tools, including inducible Cre systems for lineage tracing and conditional mutagenesis [23]. The progression of these techniques is summarized in Table 1, which highlights key milestones in the evolution of homology-independent knock-in methods.
Table 1: Historical Timeline of Key Methodological Developments
| Year | Development | Key Innovation | Significance | Citation |
|---|---|---|---|---|
| 2014 | Initial homology-independent knock-in | Concurrent genome & plasmid cleavage | Demonstrated NHEJ-mediated integration of large cassettes (>5.7 kb) | [14] |
| 2014 | Expanded application to endogenous loci | Modified donor with hsp70 promoter | Achieved germline transmission at multiple endogenous loci (>25% efficiency) | [22] |
| 2016 | Systematic comparison in human cells | Direct HDR vs NHEJ efficiency comparison | Quantitatively demonstrated superiority of NHEJ for large insertions | [21] |
| 2017 | Endogenous promoter-driven reporters | Knock-in upstream of ATG without gene disruption | Faithful recapitulation of endogenous expression patterns | [16] |
| 2023 | Chemical modification of dsDNA donors | 5' AmC6 modified PCR fragments | Enhanced integration efficiency; cloning-free approach | [23] |
| 2025 | Quantitative parameter optimization | Long-read sequencing analysis | Identified optimal conditions for precise insertion | [19] |
Quantitative Advancements in Efficiency and Precision
The progression of homology-independent knock-in methods has been characterized by significant improvements in both efficiency and precision, with recent studies achieving remarkable success rates. Early efforts demonstrated the feasibility of the approach but with variable efficiency—the seminal 2014 study reported successful conversion of eGFP to Gal4 in approximately 22% of injected embryos exhibiting recapitulated expression patterns [14]. Subsequent optimization studies achieved germline transmission rates exceeding 20% for precise insertions across multiple loci, representing a substantial improvement over traditional HDR-based approaches that typically yielded efficiencies below 5% [19] [23].
Parameter optimization has been instrumental in these efficiency gains. Comparative studies identified that chemically modified templates significantly outperformed those released in vivo from plasmids, while both Cas9 and Cas12a nucleases demonstrated similar efficacy for targeted insertion [19]. The distance between the double-strand break and the insertion site emerged as a critical factor, with closer proximities favoring precise editing rates [19]. Furthermore, the elimination of non-homologous base pairs in homology templates consistently improved outcomes, highlighting the importance of molecular precision in template design [19]. The quantitative progression of these efficiency improvements across key studies is detailed in Table 2, illustrating the collective impact of methodological refinements.
Table 2: Evolution of Knock-In Efficiency Across Methodological Generations
| Study Focus | Template Type | Nuclease | Maximum Efficiency Achieved | Key Determinants of Efficiency |
|---|---|---|---|---|
| Initial proof-of-concept [14] | Plasmid with bait sequence | Cas9 | 22% (transient) | sgRNA efficiency; concurrent cleavage |
| Endogenous locus targeting [22] | Plasmid with bait sequence & hsp70 promoter | Cas9 | 25% (germline) | sgRNA activity; promoter selection |
| 3' knock-in lineage tracing [23] | AmC6-modified dsDNA PCR fragments | Cas9 RNP | 20% (germline) | Chemical modifications; RNP delivery |
| Multi-locus optimization [19] | Chemically modified ssODNs | Cas9/Cas12a | >20% (germline across 4 loci) | Template design; break-to-insert distance |
Experimental Protocols and Methodological Guidelines
Core Protocol: Homology-Independent Knock-In via NHEJ
The following protocol represents a synthesis of the most effective methodologies developed across the historical progression of homology-independent knock-in techniques, incorporating key refinements that maximize efficiency and reproducibility.
Reagent Preparation
sgRNA Design and Synthesis:
- Design sgRNAs targeting the genomic region of interest using tools like CHOP-CHOP [16].
- Select target sites approximately 200-600 bp upstream of the gene start codon for 5' integrations or immediately upstream of the stop codon for 3' integrations [23] [22].
- Critical: Verify target specificity and minimize off-target effects by screening against the zebrafish genome.
- Synthesize sgRNAs using in vitro transcription with T7 or U6 promoters [16].
Donor Template Construction:
- For plasmid-based donors: Clone a "bait" sequence (e.g., Gbait, Tbait, Mbait) upstream of the insertion cassette, followed by your promoter-reporter/payload and polyA signal [14] [22].
- For dsDNA donors: Amplify insertion cassettes using PCR primers with 5' AmC6 modifications and 35-50 bp homology arms [23].
- Optional: Incorporate the hsp70 minimal promoter to enhance expression levels and enable enhancer trapping [22].
Cas9 Preparation:
- Prepare Cas9 as mRNA or recombinant protein. Recent evidence supports superior performance of preassembled Cas9/gRNA ribonucleoprotein complexes (RNPs) for early integration [23].
Microinjection Procedure
Injection Mixture Preparation:
- Combine the following components in nuclease-free water:
- 100-200 ng/μL donor template (plasmid or dsDNA)
- 25-50 ng/μL sgRNA (genomic target)
- 25-50 ng/μL sgRNA (donor bait target, if using plasmid donor)
- 300-500 ng/μL Cas9 mRNA or 100-200 ng/μL Cas9 protein
- Phenol red tracer (0.1%)
- Combine the following components in nuclease-free water:
Zebrafish Embryo Injection:
- Inject 1-2 nL of the mixture into the cell cytoplasm or yolk of one-cell stage zebrafish embryos.
- Maintain injected embryos at 28.5°C in E3 embryo medium [16].
Post-Injection Screening:
- For fluorescent reporters: Screen live embryos at 24-48 hpf for mosaic expression patterns.
- Raise approximately 50-100 injected embryos to adulthood to establish founder lines.
Founder Identification and Line Establishment
- Outcrossing and Germline Transmission:
- Outcross potential founder (F0) fish to wild-type partners.
- Screen F1 embryos for transgene expression or use PCR genotyping to identify germline transmission.
- Establish stable lines from positive founders.
Diagram Title: Homology-Independent Knock-In Workflow
Advanced Applications: Lineage Tracing and Conditional Systems
The historical development of knock-in methodologies has enabled increasingly sophisticated genetic applications, particularly in the realm of lineage tracing and conditional systems. The 3' knock-in approach has proven exceptionally valuable for these applications, as it permits the insertion of genetic cassettes immediately upstream of the stop codon, thereby preserving endogenous gene function while adding reporter or recombinase capabilities [23].
Protocol: 3' Knock-In for Lineage Tracing
Donor Design for Lineage Tracing:
- Design a donor cassette containing (in frame): self-cleavable peptide (P2A), fluorescent protein, second self-cleavable peptide (T2A), and Cre recombinase (iCre or CreERT2) [23].
- Flank the cassette with 35-50 bp homology arms targeting the region immediately upstream of the stop codon.
- Incorporate synonymous mutations in the homology arm to prevent re-cleavage of the knock-in allele [23].
Template Generation:
- Amplify the donor cassette using PCR with 5' AmC6-modified primers to enhance stability and integration efficiency.
- Purify PCR products using gel extraction or column purification.
Embryo Injection and Screening:
- Co-inject 100-200 ng/μL of the AmC6-modified dsDNA donor with preassembled Cas9 RNP complexes.
- Screen F0 embryos for fluorescence patterns matching expected endogenous expression.
- Raise mosaic founders with high integration efficiency (>30% mosaic fluorescence) for germline transmission [23].
Lineage Tracing Experiments:
- For CreERT2 lines: Treat adult fish or embryos with 4-hydroxytamoxifen to induce recombination.
- Analyze labeled lineages using fluorescence microscopy at desired timepoints.
The historical optimization of homology-independent knock-in techniques has identified critical reagents that consistently contribute to experimental success. Table 3 summarizes these essential research solutions, their specific functions, and optimization notes derived from the methodological evolution detailed in the literature.
Table 3: Essential Research Reagent Solutions for Homology-Independent Knock-In
| Reagent Category | Specific Examples | Function & Mechanism | Optimization Notes | Citations |
|---|---|---|---|---|
| Nuclease Systems | Cas9 mRNA, Cas9 RNP, Cas12a | Creates targeted DSBs for donor integration | RNP complexes enable earlier integration; Cas9 & Cas12a show similar efficacy | [19] [23] |
| Template Types | Plasmids with bait sequences, AmC6-modified dsDNA, ssODNs | Provides donor DNA for integration | Chemically modified templates outperform unmodified; AmC6 modifications reduce degradation | [19] [23] |
| Bait Sequences | Gbait (eGFP-derived), Tbait (Tet1-derived), Mbait (Mc4r-derived) | Enables concurrent donor cleavage for NHEJ | Efficiency varies by sequence; test multiple baits if initial failure | [14] [22] |
| Promoter Systems | hsp70 minimal promoter, endogenous promoters | Drives transgene expression; hsp70 enables enhancer trapping | hsp70 increases expression level; endogenous promoters ensure faithful expression | [16] [22] |
| Genetic Elements | 2A self-cleaving peptides, loxP sites, fluorescent reporters | Enables multicistronic expression, conditional systems, and visualization | 2A peptides maintain endogenous gene function while adding reporters | [23] [22] |
The zebrafish research community has developed extensive curated databases that are invaluable for knock-in experimental design:
- ZFIN (Zebrafish Information Network): Provides comprehensive information on genetic sequences, mutations, and validated reagents [20].
- ZIRC (Zebrafish International Resource Center): Maintains and distributes zebrafish lines, including wild-type strains and genetic mutants [20].
- CHOP-CHOP: Web tool for sgRNA design and off-target prediction [16].
Current State and Future Perspectives
The historical trajectory of homology-independent knock-in strategies in zebrafish research has transformed this model organism into a premier system for precise genetic manipulation. The current state of the field is characterized by remarkably high efficiencies, with germline transmission rates consistently exceeding 20% across multiple loci when optimized parameters are employed [19] [23]. The methodology has evolved from a novel approach to a standardized technique capable of generating a diverse array of genetic tools, including fluorescent reporters, Cre drivers, and conditional alleles with high fidelity to endogenous expression patterns [16] [23].
Recent advances in long-read sequencing technologies have further accelerated methodological refinements by enabling comprehensive quantification of editing outcomes, revealing previously unappreciated factors influencing knock-in efficiency [19]. The integration of chemical modifications in donor templates represents another significant advancement, addressing long-standing challenges related to template stability and concatemerization in vivo [19] [23]. As these methodologies continue to mature, their application is expanding to include increasingly sophisticated genetic manipulations, such as dual-recombinase systems for intersectional labeling and complex disease modeling.
The historical progression from initial discovery to widespread application demonstrates how homology-independent knock-in strategies have effectively addressed the unique challenges of zebrafish genome engineering. By leveraging the naturally active NHEJ pathway in early embryos and systematically optimizing critical parameters, researchers have established a robust and efficient platform for reverse genetic approaches that continues to drive innovation in developmental biology, disease modeling, and functional genomics.
A Step-by-Step Guide to Implementing Homology-Independent Knock-In
This application note provides a detailed protocol for the design and implementation of donor plasmids incorporating 'bait' sequences for CRISPR/Cas9-mediated homology-independent knock-in in zebrafish. The homology-independent approach leverages the non-homologous end joining (NHEJ) DNA repair pathway to enable efficient integration of large DNA cassettes (>5.7 kb) into the zebrafish genome [14]. By incorporating specific bait sequences into donor plasmids that are cleaved concurrently with the genomic target site, researchers can achieve knock-in efficiencies exceeding 25% for stable transgenic founder generation [22]. This guide outlines the molecular design principles, provides quantitative performance data, and details step-by-step protocols for implementing this powerful genome engineering strategy.
The concurrent cleavage strategy for knock-in in zebrafish represents a significant advancement over homology-directed repair (HDR) methods, which typically exhibit low efficiency in this model organism [10]. This approach utilizes the cell's endogenous NHEJ pathway to integrate linearized donor DNA fragments into targeted genomic double-strand breaks (DSBs) [14]. The core innovation involves designing donor plasmids with specific 'bait' sequences that are cleaved by CRISPR/Cas9 simultaneously with the chromosomal target, creating compatible ends that facilitate ligation via NHEJ repair mechanisms [24] [22].
This method offers several advantages for zebrafish research: it circumvents the need for extensive homology arms, simplifies donor construction, enables insertion of large DNA cassettes, and demonstrates high efficiency across multiple genomic loci [23] [22]. The technique has been successfully applied to generate reporter lines, convert existing transgenic lines, and create targeted mutations at endogenous loci [14] [16] [24].
Bait Sequence Design and Selection
Characteristics of Effective Bait Sequences
Effective bait sequences share several key characteristics that optimize CRISPR/Cas9 cleavage efficiency and minimize off-target effects:
- Length: Typically 20-23 nucleotides [24] [22]
- PAM Sequence: Must include an appropriate protospacer adjacent motif (NGG for S. pyogenes Cas9) [14]
- Uniqueness: Should not appear elsewhere in the donor plasmid or zebrafish genome to prevent unintended cleavage [24]
- Efficiency: Should be highly amenable to Cas9 cleavage based on established sgRNA design principles [10]
Validated Bait Sequences
The table below summarizes bait sequences successfully implemented in zebrafish studies:
Table 1: Validated Bait Sequences for Zebrafish Knock-In
| Bait Name | Sequence (5' to 3') | PAM | Reported Efficiency | Application | Citation |
|---|---|---|---|---|---|
| Tbait | GGCTGCTGTCAGGGAGCTCATGG | CGG | >50% founder generation | Medaka and zebrafish transgenesis | [24] |
| Gbait | eGFP-derived sequence | NGG | Successful line conversion | GFP to Gal4 conversion | [22] |
| Mbait | Rat Mc4r-derived sequence | NGG | High efficiency | Reporter integration | [22] |
| eGFP 1 | eGFP-targeting sequence | NGG | 66% indel mutation rate | KalTA4 integration | [14] |
Bait Sequence Incorporation into Donor Plasmids
Bait sequences should be positioned immediately upstream of the insertion cassette in the donor plasmid [24] [22]. The cassette typically includes a minimal promoter (e.g., hsp70) followed by the gene of interest (reporter, Cre recombinase, etc.) [22]. Strategic placement ensures clean cleavage and release of the linear insert while preventing damage to the functional elements of the cassette.
Quantitative Performance Data
Knock-in Efficiency Across Loci
The concurrent cleavage method has demonstrated robust performance across multiple genomic loci in zebrafish:
Table 2: Knock-in Efficiency Across Zebrafish Genomic Loci
| Target Locus | Insert Size | Knock-in Efficiency | Expression Pattern | Citation |
|---|---|---|---|---|
| neurod:eGFP | E2A-KalTA4 | >75% injected embryos showed targeted integration | Recapitulated endogenous neurod pattern | [14] |
| evx2 | Gal4 | 12% founder efficiency (2/17 fish) | Broad CNS expression matching Evx2 | [22] |
| eng1b | Gal4 | 3% founder efficiency (1/40 fish) | MHB and muscle pioneers | [22] |
| krt92 | p2A-EGFP-t2A-CreERT2 | 5.1% mosaic embryos; 50% of mosaics produced founders | Skin epithelium | [23] |
| otx2 | Venus | Successful reporter line generation | Midbrain-hindbrain boundary | [16] |
| pax2a | turboRFP | Successful reporter line generation | Midbrain-hindbrain boundary | [16] |
Factors Influencing Efficiency
Several critical factors significantly impact knock-in efficiency:
- sgRNA efficiency: Designs with higher indel formation rates (>70%) substantially improve knock-in success [10]
- Donor format: 5' modified double-stranded DNA donors with AmC6 modifications increase integration efficiency 5-fold compared to unmodified donors [23]
- Developmental timing: Early integration events produce higher mosaicism, improving germline transmission rates [23]
- Component delivery: RNP complex injection yields higher efficiency than DNA/mRNA formats [23] [25]
Step-by-Step Experimental Protocol
Donor Plasmid Construction
Materials:
- Base plasmid with minimal promoter (e.g., hsp70) and reporter/driver gene
- Oligonucleotides containing bait sequence
- Standard molecular cloning reagents
Procedure:
- Insert selected bait sequence immediately upstream of the promoter element using standard molecular cloning techniques
- Verify bait sequence incorporation and orientation by Sanger sequencing
- For 3' knock-in approaches, include 2A peptide sequences (p2A, t2A) to enable multicistronic expression [23]
- For epitope tagging or precise mutations, design donors with in-frame integrations
CRISPR Component Preparation
Materials:
- Cas9 expression vector (e.g., pCS2-hSpCas9) [24]
- sgRNA cloning vector (e.g., pDR274) [24]
- In vitro transcription kits
Procedure:
- Design sgRNAs targeting both the genomic locus and bait sequence
- Synthesize sgRNAs using T7 or SP6 in vitro transcription systems [24]
- Transcribe Cas9 mRNA from linearized template DNA
- Complex sgRNAs with Cas9 protein to form RNPs for improved efficiency [23]
Zebrafish Embryo Injection
Materials:
- One-cell stage zebrafish embryos
- Microinjection apparatus
- Phenol red injection marker
Injection Solution Formulation:
- 9 ng/μl sgRNA for bait sequence digestion [24]
- 18 ng/μl sgRNA for genomic target digestion [24]
- 200 ng/μl Cas9 mRNA or 300-500 ng/μl Cas9 protein for RNP formation [23]
- 9 ng/μl donor plasmid or 25-50 ng/μl PCR-amplified donor fragment [23] [24]
- Optional: 5' AmC6-modified primers for PCR-amplified donors to prevent degradation [23]
Procedure:
- Prepare injection solution and centrifuge briefly to remove particulates
- Load injection needles with prepared solution
- Inject approximately 1-2 nL into the cell cytoplasm or yolk of one-cell stage embryos
- Culture injected embryos at 28.5°C and monitor for development
Screening and Validation
Initial Screening:
- Assess mosaic expression in injected F0 embryos at 24-48 hpf
- Raise embryos showing correct expression patterns to adulthood
Founder Identification:
- Outcross potential F0 founders to wild-type fish
- Screen F1 progeny for transgene expression or using PCR genotyping
- For PCR screening, use one primer outside the targeted integration site and one within the transgene
Molecular Validation:
- Confirm precise integration junctions by Sanger sequencing
- Verify expression pattern recapitulates endogenous gene expression
- Assess potential off-target integrations through systematic PCR screening
Experimental Workflow for Bait Sequence-Mediated Knock-In
Troubleshooting Guide
Table 3: Troubleshooting Common Issues
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low mosaic rate in F0 | Inefficient sgRNAs | Test multiple sgRNAs; select those with >70% indel efficiency [10] |
| No germline transmission | Late integration events | Use RNP complexes and 5' modified donors for earlier integration [23] |
| Random integration | Off-target cleavage | Verify bait sequence uniqueness; use BLAST against zebrafish genome |
| Incomplete expression pattern | Epigenetic silencing | Include insulator elements; test multiple integration events |
| High embryo mortality | Injection toxicity | Titrate component concentrations; use phenol red as injection marker |
Research Reagent Solutions
Table 4: Essential Reagents for Concurrent Cleavage Knock-In
| Reagent Category | Specific Examples | Function | Source/Reference |
|---|---|---|---|
| Bait Sequences | Tbait, Gbait, Mbait | Donor plasmid linearization | [24] [22] |
| Cas9 Source | Cas9 mRNA, recombinant Cas9 protein | Targeted DNA cleavage | [24] [25] |
| Donor Vectors | Tbait-hs-lRl-GFP, Mbait-hs-lRl-GFPTx | Template for integration | [22] |
| sgRNA Templates | pDR274 vector, PCR templates | Guide RNA synthesis | [24] |
| Modification Reagents | AmC6-modified primers | Donor protection and efficiency enhancement | [23] |
| Reporter Cassettes | GFP, RFP, Gal4, CreERT2 | Visualizing and manipulating targeted cells | [14] [23] [22] |
Applications in Zebrafish Research
The concurrent cleavage knock-in strategy has enabled numerous advanced applications in zebrafish research:
- Lineage tracing: Knock-in of CreERT2 cassettes enables temporal control of recombination for fate mapping studies [23]
- Endogenous reporting: Precisely tagged genes report native expression patterns without disruptive random integration [16]
- Driver line generation: Tissue-specific Gal4 lines allow targeted manipulation of specific cell populations [14] [22]
- Disease modeling: Precise insertion of human disease-associated mutations creates accurate models [10]
- Multiplexed editing: Simultaneous targeting of multiple loci enables complex genetic engineering [24]
Molecular Mechanism of Bait Sequence-Mediated Knock-In
The incorporation of bait sequences into donor plasmids for concurrent cleavage with genomic targets represents a highly efficient and robust method for achieving homology-independent knock-in in zebrafish. This approach consistently yields high rates of targeted integration across diverse genomic loci, simplifies donor construction by eliminating the need for extensive homology arms, and supports the insertion of large genetic cassettes. As CRISPR/Cas9 technology continues to evolve, further refinements to bait sequence design and delivery methods promise to enhance the precision and efficiency of this already powerful genome engineering strategy, solidifying its position as a fundamental technique in zebrafish genetic research.
The homology-independent knock-in strategy has emerged as a powerful and efficient alternative to homology-directed repair (HDR) for generating targeted insertions in the zebrafish genome. Unlike HDR, which remains challenging due to its low efficiency, homology-independent insertion leverages the error-prone non-homologous end joining (NHEJ) pathway, enabling highly efficient integration of DNA cassettes. The success of this approach critically depends on the precise formulation of the co-injection mix, comprising sgRNA, Cas9 nuclease, and donor DNA. This protocol details the optimization of these components based on recent quantitative studies, providing researchers with a robust framework for achieving high rates of precise germline transmission.
Key Reagent Solutions for Homology-Independent Knock-In
Table 1: Essential Reagents for Homology-Independent Knock-In in Zebrafish
| Reagent Type | Specific Examples & Key Features | Primary Function in the Protocol |
|---|---|---|
| CRISPR Nuclease | SpCas9: Creates blunt-end DSBs. [19] | Generates a targeted double-strand break (DSB) in the genome to initiate repair. |
| LbCas12a: Creates 5-nt 5' overhangs; may improve HDR at some loci. [19] | ||
| Donor Template | Plasmid DNA with "bait" sequences: Linearized in vivo by co-injected nuclease (e.g., I-SceI or Cas9). [19] [14] | Serves as the template for integration into the genomic DSB via NHEJ. |
| Chemically modified double-stranded DNA templates: Features modifications that reduce degradation, outperforming plasmid templates. [19] | ||
| Chemical Enhancers | NU7441 (DNA-PK inhibitor): Shifts DNA repair equilibrium toward HDR; can enhance HDR-mediated repair up to 13.4-fold. [7] | Increases the frequency of precise, HDR-based editing events. |
| Screening Tools | Fluorescent PCR and Capillary Electrophoresis (CRISPR-STAT): Detects precise knock-in by analyzing PCR product size changes. [26] | Enables efficient, PCR-based screening for successful knock-in events without cloning. |
| Long-read sequencing (Pacific Biosciences): Accurately quantifies all repair events, including large inserts, overcoming short-read sequencing bias. [19] |
Optimized Co-Injection Ratios and Formulations
The composition of the co-injection mix is a primary determinant of knock-in efficiency. The table below summarizes optimal concentrations and ratios derived from empirical data.
Table 2: Quantitative Optimization of Co-Injection Components
| Component | Recommended Concentration/Ratio | Impact on Efficiency & Key Findings |
|---|---|---|
| sgRNA | Highly active sgRNA (e.g., >70% indel rate in pre-testing). [10] | Foundational prerequisite. Efficiency of the initial DSB is the most critical factor for successful knock-in. [10] |
| Cas9 Protein (RNP) | Pre-complexed with sgRNA as a ribonucleoprotein (RNP) complex. | Direct RNP delivery increases mutagenesis efficiency and can reduce off-target effects. [27] |
| Donor DNA Template | Chemically modified double-stranded DNA templates. | Quantitative side-by-side comparison showed chemically modified templates outperform those released from a plasmid in vivo. [19] |
| NHEJ Inhibitor (NU7441) | 50 µM. | In an HDR reporter assay, this concentration enhanced HDR-mediated repair up to 13.4-fold compared to DMSO control. [7] |
| Targeting Strategy | Donor plasmid contains the same sgRNA target ("bait") sequence as the genomic locus. | Enables concurrent cleavage of the genome and donor plasmid, facilitating efficient homology-independent integration via NHEJ. [14] |
Step-by-Step Experimental Protocol
Protocol Workflow
The following diagram outlines the complete experimental workflow for homology-independent knock-in in zebrafish, from sgRNA design to founder identification:
Detailed Protocol Instructions
Step 1: Design and Validation of sgRNA
- Design: Select a target site near the desired integration point using reliable design tools (e.g., CCTop, CRISPRscan). The spacer sequence should directly precede a 5'-NGG-3' PAM sequence. [10]
- Validation: Synthesize the sgRNA and validate its cutting efficiency before knock-in attempts. Inject the sgRNA and Cas9 mRNA/protein into wild-type embryos, extract genomic DNA from a pool of embryos at 24-48 hpf, and assess the indel mutation rate via T7 endonuclease I (T7EI) assay or deep sequencing. Proceed only with sgRNAs that show high activity (>70% indels in a pool of embryos). [10]
Step 2: Preparation of the Donor Template
- Vector Construction: For homology-independent knock-in, clone your cargo (e.g., reporter gene, epitope tag) into a plasmid backbone. Crucially, flank this cargo with the identical sgRNA target sequence ("bait" sequence) that is being used to target the genomic locus. [14]
- Template Format: While early studies used plasmids linearized by a co-injected nuclease, recent quantitative comparisons show that chemically modified double-stranded DNA templates significantly outperform unmodified plasmid-based templates. Use templates with stability-enhancing modifications (e.g., 5' and 3' end modifications) to reduce degradation in the embryo. [19]
Step 3: Formulation of the Co-Injection Mix
- RNP Complex Formation: Pre-complex the purified Cas9 protein with the validated sgRNA at a molar ratio of 1:2 to 1:3 (Cas9:sgRNA) by incubating at 37°C for 10-15 minutes. This forms the ribonucleoprotein (RNP) complex. [27]
- Final Mix Composition: Combine the following components in nuclease-free water to a final volume suitable for injection:
- RNP complex (final Cas9 concentration: 100-200 µg/mL)
- Donor DNA template (final concentration: 25-50 ng/µL)
- NHEJ inhibitor, NU7441 (final concentration: 50 µM) [7]
- Critical Note: The use of an NHEJ inhibitor like NU7441 is a key optimization. It chemically reprograms the embryo's DNA repair machinery, shifting the balance away from error-prone NHEJ and favoring precise HDR, even in a homology-independent knock-in context. [7]
Step 4: Microinjection and Embryo Handling
- Microinjection: Load the co-injection mix into a needle and inject 1-2 nL directly into the cytoplasm of one-cell stage zebrafish embryos.
- Embryo Care: After injection, incubate embryos in egg water at 28.5°C. Remove unfertilized or dead embryos after a few hours. Raise the injected embryos (F0 founders) to adulthood under standard laboratory conditions.
Step 5: Somatic and Germline Screening
- Somatic Screening (CRISPR-STAT): At 1-3 days post-fertilization (dpf), pool a subset of injected embryos and extract genomic DNA. Perform fluorescent PCR with primers flanking the target site. Analyze the PCR products using capillary electrophoresis. Successful precise integration will produce a distinct peak corresponding to the expected size increase, allowing for rapid assessment of knock-in efficiency before raising founders to adulthood. [26]
- Germline Transmission Screening: When F0 founders reach adulthood, perform a fin clip biopsy to screen for the presence of the knock-in allele in somatic tissue. For founders that test positive, outcross them to wild-type fish. Collect and pool embryos from these crosses, and screen the F1 progeny using the same PCR-based methods to confirm germline transmission and establish stable lines. [26]
Troubleshooting and Technical Notes
- Low Integration Efficiency: If efficiency is low, first re-confirm the activity of your sgRNA. Ensure the donor plasmid is being effectively linearized in vivo by verifying the "bait" target site is identical to the genomic target. Switching to a chemically modified donor template can also yield significant improvements. [19]
- High Embryo Mortality: High mortality can result from injecting too much volume or concentration of the RNP complex. Titrate the Cas9 concentration downward. Toxicity can also occasionally come from in vitro-transcribed gRNAs; using chemically synthesized gRNAs can mitigate this. [28]
- Imprecise Integration: The nature of the NHEJ pathway means that a fraction of integration events will be imprecise. This makes careful screening of the final integrated sequence in F1 animals essential. Long-read sequencing is the most reliable method for characterizing these complex editing outcomes. [19] [26]
Within the broader thesis on advancing homology-independent knock-in strategies in zebrafish, mastering the critical parameters of single guide RNA (sgRNA) efficiency and the precise timing of embryonic injection is paramount. Unlike knock-outs, which rely on the error-prone non-homologous end joining (NHEJ) pathway, homology-independent knock-in requires the precise integration of a DNA cassette into a targeted genomic double-strand break (DSB) [14]. This method leverages the cell's endogenous repair machinery to insert large DNA fragments, such as fluorescent reporters or transcriptional activators, without the need for a homologous template [14] [16]. The success of this sophisticated editing approach is exquisitely sensitive to the quality of the sgRNA and the developmental stage of the embryo at the moment of injection, which collectively determine the rate of mosaicism and the likelihood of germline transmission [29] [30]. This application note details the protocols and parameters essential for optimizing these factors, providing a reliable framework for researchers in drug development and genetic research.
The efficiency of CRISPR-mediated knock-in is influenced by a quantifiable set of factors. The data below summarize critical parameters from key studies.
Table 1: Key Parameters Affecting Knock-in Efficiency
| Parameter | Impact on Efficiency | Optimal Condition / Value | Key Findings |
|---|---|---|---|
| sgRNA Efficiency | Directly correlates with knock-in rate [14] [30] | High-efficiency sgRNA (e.g., >66% indel rate) [14] | An sgRNA with 66% indel frequency achieved knock-in in >75% of injected embryos [14]. |
| Cas9 Format | Affects speed and potency of DSB creation [30] | Cas9 protein [30] | Cas9 protein significantly outperformed mRNA, yielding ~5.1% vs. ~0.9% HDR efficiency at one locus [30]. |
| Donor DNA Conformation | Influences HDR pathway engagement [31] [30] | Circular plasmid with flanking CRISPR sites [31] or PAM-distal ssODN [30] | A circular "bait" plasmid increased phenotypic rescue to 46% of larvae [31]. PAM-distal ssODNs outperformed PAM-proximal conformations [30]. |
| Injection Timing | Reduces mosaicism by targeting the one-cell stage [29] | Injection into the zygote shortly after fertilization [29] | Injection into porcine zygotes after IVF resulted in the highest rate of mutant blastocysts, reducing mosaicism [29]. |
| Concentration of Components | High concentration increases biallelic editing but can be toxic [29] | Cas9 protein: 20-100 ng/µL; sgRNA: 20-100 ng/µL [29] | Increasing Cas9/gRNA from 20 ng/µL to 100 ng/µL significantly increased biallelic mutations in porcine blastocysts (0% to 16.7%) [29]. |
Table 2: Homology-Independent vs. Homology-Directed Knock-in Strategies
| Feature | Homology-Independent Knock-in [14] | Homology-Directed Repair (HDR) [31] |
|---|---|---|
| Core Mechanism | NHEJ-mediated ligation of DSBs in genome and donor plasmid. | Homology-directed repair using a template with homologous arms. |
| Donor Template | Circular plasmid flanked by sgRNA target sites ("bait" plasmid). | Long linear DNA fragments or ssODNs with silent mutations in the PAM site. |
| Typical Efficiency | Very high (>75% of embryos with targeted integration) [14]. | Low to moderate (initially ~1%, up to 46% with optimized circular donor) [31]. |
| Primary Advantage | High efficiency for integrating large cassettes; simple design. | High precision for introducing single nucleotide changes. |
| Key Application | Converting existing transgenes (e.g., eGFP to Gal4); creating reporter lines. | Precise single nucleotide polymorphism (SNP) exchanges; phenotypic rescue of mutations. |
Experimental Workflow and Protocol
The following diagram and detailed protocol outline the key steps for performing a homology-independent knock-in in zebrafish, highlighting the critical points of intervention for optimizing sgRNA efficiency and injection timing.
Diagram 1: Homology-Independent Knock-in Workflow. The process highlights critical steps for sgRNA validation and zygote injection.
Detailed Protocol for Homology-Independent Knock-in
sgRNA Design, Synthesis, and Validation
The first critical parameter is the generation of a highly efficient sgRNA.
- Design: Identify a 20-nucleotide target sequence immediately upstream of the NGG Protospacer Adjacent Motif (PAM) within the 5' genomic region upstream of the ATG start codon of your gene of interest [16]. Use web tools like CHOP-CHOP or ZiFiT Targeter for design and to check for potential off-target sites with ≤2 bp mismatches [31] [16].
- Synthesis: Clone the target-specific oligonucleotide into an sgRNA expression vector (e.g., pDR274). Perform in vitro transcription using a kit such as the Ambion MEGAshortscript T7 Kit, followed by purification [31] [16].
- Validation of Efficiency: This is a crucial and often overlooked step.
- Co-inject the sgRNA with Cas9 mRNA or protein into wild-type embryos at the one-cell stage.
- At 24-48 hours post-fertilization (hpf), pool 10 embryos and extract genomic DNA.
- Perform PCR amplification of the target genomic locus.
- Quantify the indel mutation efficiency using the Inference of CRISPR Edits (ICE) tool or next-generation sequencing (NGS). An effective sgRNA should induce indels in >66% of sequenced alleles [14] [30]. Do not proceed with knock-in experiments with a poorly performing sgRNA.
Donor Plasmid Construction for Homology-Independent Integration
- Clone an 853 bp - 1 kb genomic fragment (the "bait") from your target locus into a standard vector (e.g., pGEM-T, pCS2+) [31] [16].
- Insert your desired cargo (e.g., the coding sequence for the fast-maturing fluorescent protein Venus) upstream of the bait sequence's ATG start codon.
- Critical Step: Ensure the bait sequence is flanked by the same sgRNA target sites used for the genomic target. This allows the Cas9 complex to linearize the donor plasmid in vivo, providing the necessary ends for NHEJ-mediated integration [31] [14].
Microinjection into Zebrafish Zygotes
The timing of this step is the second critical parameter for minimizing mosaicism.
- Prepare the Injection Mix:
- Perform Microinjection:
- Collect zebrafish zygotes within 15-30 minutes post-fertilization.
- Using a microinjector and a fine needle, immobilize the zygotes and inject ~1 nL of the injection mix directly into the cell cytoplasm [29] [16].
- The injection must be performed at the one-cell stage to ensure the edited allele is present in as many cells as possible, including the germline, thereby reducing mosaicism [29].
Screening and Germline Transmission
- Somatic Screening: At 1-3 dpf, screen injected embryos (F0) for successful integration using fluorescence microscopy (e.g., for Venus expression) [16].
- Early Genotyping (Highly Recommended): Use the Zebrafish Embryo Genotyper (ZEG) device to extract a tiny amount of genomic DNA from 72 hpf embryos with minimal lethality. Use NGS to quantify the somatic editing efficiency in individual embryos [30].
- Raising Founders: Select and raise only the embryos with the highest somatic editing efficiency to adulthood (F0 founders).
- Germline Screening: Outcross F0 founder fish to wild-types. Screen the resulting F1 progeny at 3 dpf for the presence of the knock-in allele via fluorescence or PCR. A founder with germline transmission will produce a percentage of positive F1 offspring [31].
The Scientist's Toolkit: Essential Reagents
Successful execution of this protocol relies on key reagents and tools, summarized below.
Table 3: Essential Research Reagent Solutions
| Reagent / Tool | Function / Application | Example Products / Notes |
|---|---|---|
| sgRNA in vitro Transcription Kit | Synthesizes high-quality, functional sgRNA. | Ambion MEGAshortscript T7 Kit [31]. |
| Recombinant Cas9 Protein | Provides immediate nuclease activity, leading to higher editing efficiency and reduced mosaicism compared to mRNA [30]. | Guide-it Recombinant Cas9 (Takara Bio) [29]. |
| Homology-Independent Donor Plasmid | Serves as the template for knock-in. The "bait" sequence is flanked by sgRNA sites for in vivo linearization. | Can be cloned into pGEM-T Easy or pCS2+ [31] [16]. |
| Microinjection System | For precise delivery of CRISPR components into the cytoplasm of single-cell zygotes. | FemtoJet 4i microinjector and Femtotips II needles (Eppendorf) [29]. |
| Zebrafish Embryo Genotyper (ZEG) | Enables minimally invasive early genotyping to select embryos with high editing efficiency for raising [30]. | A microdevice for extracting genomic DNA from 72 hpf embryos. |
| ICE Analysis Software | A computational tool for robust quantification of indel frequencies from sequencing data to validate sgRNA efficiency [30]. | Inference of CRISPR Edits (Synthego). |
| ARS-2102 | ARS-2102, MF:C28H31ClF2N6O2, MW:557.0 g/mol | Chemical Reagent |
| ZG1077 | ZG1077, MF:C33H33F2N5O5S, MW:649.7 g/mol | Chemical Reagent |
The rigorous application of the protocols outlined herein—emphasizing sgRNA validation and precise zygotic injection—is fundamental for exploiting homology-independent knock-in strategies in zebrafish. By quantitatively assessing sgRNA efficiency and meticulously controlling the timing of the procedure, researchers can significantly increase the yield of non-mosaic, germline-transmitting founders. This methodological framework empowers scientists to reliably generate sophisticated genetic models, thereby accelerating functional genomics research and the development of novel therapeutic strategies.
The zebrafish has solidified its role as a premier vertebrate model for studying development, physiology, and disease, owing to external fertilization, optical clarity of embryos, and high fecundity [16]. A significant technological breakthrough for this model has been the development of CRISPR/Cas9-mediated genome editing, which enables precise genetic manipulations. While early genetic studies relied on random transgenesis or labor-intensive homologous recombination, recent advances have established homology-independent knock-in strategies as a powerful and efficient alternative [32] [10]. These methods primarily exploit cellular DNA repair pathways like non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) to integrate reporter cassettes, epitope tags, and recombinases directly into the genome without the need for long homology arms [33] [10].
This application note details practical protocols and quantitative data for generating key genetic tools—reporter alleles, Cre-driver lines, and conditional loss-of-function alleles—using these streamlined approaches. By leveraging methods such as Homology-Mediated End Joining (HMEJ), researchers can now achieve high-efficiency targeted integration, overcoming previous challenges associated with traditional homology-directed repair (HDR) in zebrafish [34] [33] [35].
Key Applications and Quantitative Performance
The following table summarizes the performance of homology-independent knock-in strategies across various genetic loci and desired applications, providing researchers with realistic expectations for project planning.
Table 1: Performance Metrics of Homology-Independent Knock-In Strategies in Zebrafish
| Target Gene | Application | Knock-In Strategy | Key Metric | Efficiency/Result |
|---|---|---|---|---|
| otx2, pax2a [16] | Fluorescent Reporter (Venus) | CRISPR/HDR (5' knock-in) | Endogenous expression | Faithful recapitulation, no disturbance of native expression |
| sox11a [34] [35] | Epitope Tag (MYC) | HDR with modified donors | Germline Transmission | Successful line establishment with functional characterization |
| ascl1b [36] | Cre Driver (2A-Cre) | Short HDR (48 bp arms) | Germline Transmission Rate | 100% (3/3 F0 founders) |
| olig2 [36] | Cre Driver (2A-Cre) | Short HDR (48 bp arms) | Germline Transmission Rate | 10% (2/20 F0 founders) |
| neurod1 [36] | Cre Driver (2A-Cre) | Short HDR (48 bp arms) | Germline Transmission Rate | 20% (1/5 F0 founders) |
| nkx6.1, id2a [23] | Lineage Tracing (2A-FP-2A-Cre) | 3' HDR with AmC6-modified dsDNA | Founder Rate | High germline transmission from mosaic F0 |
| noto [33] | Reporter (2A-TagRFP) | HMEJ (24 bp homology) | Precise Integration | 95% of sequenced junctions (19/20) |
| noto [33] | Reporter (2A-TagRFP) | HMEJ (48 bp homology) | Precise Integration | 79% of sequenced junctions (15/19) |
| rbbp4, rb1 [37] | Conditional Loss-of-Function (UFlip) | GeneWeld (48 bp arms) | Allele Recovery Frequency | 4% to 14% |
Experimental Protocols
Protocol 1: Generating Fluorescent Reporter Lines via 5' HMEJ
This protocol is adapted from studies targeting genes like otx2 and pax2a to create transcriptional reporter lines that faithfully recapitulate endogenous gene expression [16] [33].
Reagents and Equipment
- Cas9 protein (e.g., Alt-R S.p. Cas9 Nuclease V3) or Cas9 mRNA
- Target-specific sgRNA and Universal Guide RNA (UgRNA)
- Donor plasmid (e.g., pGTag series) containing:
- Fluorescent reporter (e.g., Venus, TagRFP)
- UgRNA recognition sites flanking the cassette
- Short homology arms (24-48 bp) to the genomic target
- Microinjection apparatus
- Zebrafish embryos at one-cell stage
Procedure
- Design and Preparation: Design a target-specific sgRNA to a site within the 5'UTR or upstream of the ATG start codon of your gene of interest. Clone 24-48 bp homology arms corresponding to the sequences directly flanking the genomic cut site into a donor vector containing your reporter and the UgRNA sites [33].
- Reagent Injection: Co-inject into the cytoplasm of one-cell stage zebrafish embryos [33]:
- Cas9 protein (e.g., 500 pg) or Cas9 mRNA
- Target-specific sgRNA (e.g., 250 pg)
- UgRNA (to linearize the donor in vivo)
- Donor plasmid (e.g., 25-50 pg)
- Screening and Raising: Screen injected F0 embryos for mosaic fluorescence expression at 1-3 days post-fertilization (dpf). Raise embryos with robust, pattern-specific fluorescence to adulthood.
- Founder Identification: Outcross adult F0 fish to wild-type partners and screen F1 progeny for the fluorescent reporter. Confirm precise genomic integration via PCR and sequencing of the junction fragments.
Protocol 2: Creating Endogenous Cre-Driver Lines Using Short Homology
This method uses short homology arms to knock-in a 2A-Cre cassette into a coding exon, ensuring Cre expression is under the control of the native promoter [36].
Reagents and Equipment
- Cas9 mRNA
- Two sgRNAs: one for the genomic target, one for the donor plasmid (UgRNA)
- Donor plasmid with 2A-Cre-2A-Fluorophore cassette flanked by 48 bp homology arms
- Tg(UAS:Reporter) zebrafish line for functional Cre testing
Procedure
- Target Selection: Design a genomic sgRNA to a coding exon of the target gene (e.g., ascl1b, olig2). Prepare a donor plasmid with 48 bp homology arms and a 2A-Cre-2A-Fluorophore (e.g., EGFP) cassette [36].
- Mosaic Analysis: Inject the donor plasmid, genomic sgRNA, UgRNA, and Cas9 mRNA into embryos from a Tg(UAS:Reporter) line. Widespread reporter expression in F0 embryos indicates successful somatic integration and functional Cre activity.
- Germline Transmission: Raise injected F0 embryos to adulthood and outcross to wild-type fish. Screen F1 embryos for the integrated cassette (via fluorophore in the lens or other secondary marker).
- Line Validation: Identify F1 offspring with precise integration by junction PCR and sequencing. Establish stable lines and confirm cell-type-specific Cre activity by crossing with reporter lines.
Protocol 3: Engineering Conditional Loss-of-Function Alleles with the UFlip System
The UFlip system allows for the creation of Cre-regulatable alleles for conditional gene inactivation or rescue, using a single targeting event [37].
Reagents and Equipment
- Cas9 protein or mRNA
- Genomic target sgRNA and UgRNA
- UFlip donor vector (contains floxed 2A-mRFP gene trap and BFP secondary marker)
- Cre mRNA or transgenic Cre driver lines
Procedure
- Vector Design: Design 48 bp homology arms to an intron of your target gene (e.g., rbbp4, rb1) and clone them into the UFlip vector. The vector contains a floxed gene trap cassette that, in the "off" orientation, splices into the endogenous mRNA, truncating the functional protein and expressing mRFP [37].
- Knock-In Generation: Co-inject the UFlip donor, genomic sgRNA, UgRNA, and Cas9 into one-cell stage embryos. Raise F0 embryos and outcross to identify founders transmitting the allele (screened via BFP in the heart or lens).
- Phenotypic Validation: Characterize stable UFlip "off" allele lines. The expression of mRFP should mirror the endogenous gene, and homozygous fish should exhibit the expected loss-of-function phenotype due to gene trapping.
- Conditional Manipulation: To invert the cassette and "turn on" the gene, introduce Cre recombinase (via mRNA injection or by crossing with a tissue-specific Cre driver). This results in loss of mRFP and phenotypic rescue. Conversely, starting with a phenotypically silent UFlip "on" allele, Cre introduction inverts the cassette to the "off" state, inducing mRFP expression and gene knockout in a spatially and temporally controlled manner [37].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Homology-Independent Knock-Ins in Zebrafish
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| pGTag Vector Series [33] | Modular donor plasmids with sites for easy cloning of short homology arms and cargo (e.g., fluorescent proteins, Cre). | Standardized vector backbone for HMEJ-based knock-ins. |
| Universal gRNA (UgRNA) [33] | A pre-validated, highly efficient sgRNA sequence that targets a site in the donor plasmid, but not the zebrafish genome, for in vivo linearization. | Liberating homology arms from the donor plasmid inside the embryo to dramatically boost integration efficiency. |
| Alt-R CRISPR-Cas9 System [34] [35] | Synthetic, chemically modified crRNAs and tracrRNAs complexed with Cas9 protein as a ribonucleoprotein (RNP). | Increases editing efficiency and can reduce off-target effects in knock-in experiments. |
| 5' Modified dsDNA Donors [23] | PCR-amplified double-stranded DNA donors synthesized with 5' end modifications (e.g., AmC6 linkers) on the primers. | Streamlined cloning-free 3' knock-in; AmC6 modification protects the donor and increases integration efficiency. |
| UFlip Vector [37] | A universal targeting vector containing a floxed, invertible gene trap cassette (2A-mRFP) and a secondary BFP marker. | A single vector system for generating Cre/lox-regulated conditional rescue and inactivation alleles. |
| CD73-IN-11 | CD73-IN-11, MF:C14H10F3N5O2, MW:337.26 g/mol | Chemical Reagent |
| HS-243 | 3-Nitro-N-(1-propyl-1H-benzo[d]imidazol-2-yl)benzamide | 3-Nitro-N-(1-propyl-1H-benzo[d]imidazol-2-yl)benzamide is a benzimidazole-based compound for research use only. Explore its potential in anticancer and antimicrobial studies. NOT FOR HUMAN OR VETERINARY USE. |
Visual Workflow for Knock-In Strategy and Experimental Logic
The following diagram illustrates the core workflow and molecular mechanism of the HMEJ-based knock-in strategy.
HMEJ Knock-In Workflow from Design to Validation
Molecular Mechanism of HMEJ Integration
The ability to precisely modify the genome of model organisms like the zebrafish (Danio rerio) has been revolutionized by the advent of CRISPR/Cas9 technology. Within the realm of genome engineering, homology-independent knock-in strategies have emerged as a powerful and efficient alternative to traditional homology-directed repair (HDR). These methods leverage the cell's endogenous non-homologous end joining (NHEJ) pathway to integrate exogenous DNA cassettes at targeted genomic loci, bypassing the need for homologous arms and the typically low efficiency of HDR in zebrafish [38] [14]. This case study focuses on a specific application of this approach: the conversion of existing enhanced Green Fluorescent Protein (eGFP) transgenic lines into Gal4 driver lines using the "Gbait" strategy, a technique that enables in-depth analysis and manipulation of specific cell populations [39].
The Gbait strategy is particularly valuable because it repurposes the well-defined expression patterns of numerous existing eGFP lines. Instead of undertaking the laborious process of generating new, specific Gal4 lines de novo, researchers can directly convert these eGFP lines, effectively harnessing their established regulatory elements to control the expression of the potent transcriptional activator Gal4. Once expressed, Gal4 can then be used to drive the expression of any gene placed downstream of an Upstream Activating Sequence (UAS), allowing for targeted expression of reporters, calcium indicators, optogenetic tools, or dominant-negative proteins in the cell type of interest [39] [38]. This protocol has been successfully implemented to generate stable converted transgenic lines within a timeframe of approximately four months, significantly accelerating functional genetic studies [39].
Experimental Principle and Workflow
The core principle of the Gbait conversion strategy involves the CRISPR/Cas9-mediated co-cleavage of a donor plasmid and a genomic eGFP locus, followed by the NHEJ-mediated integration of the donor cassette into the genome.
Conceptual Workflow
The diagram below illustrates the logical sequence of the eGFP-to-Gal4 conversion process.
Molecular Mechanism
The process begins with the microinjection of a mixture containing a single-guide RNA (sgRNA) designed to target the eGFP coding sequence, Cas9 nuclease mRNA, and a donor plasmid into single-cell embryos of a double-transgenic line that possesses both the target eGFP transgene and a UAS:RFP reporter [39] [14]. The donor plasmid is engineered to carry an optimized version of the Gal4 gene, KalTA4, which is linked via an E2A peptide linker to ensure multicistronic expression. Crucially, the plasmid also contains a "Gbait" sequence—a fragment of the eGFP gene that is targeted by the same sgRNA [39] [14].
Upon expression of the sgRNA and Cas9, double-strand breaks are introduced concurrently at two locations: the genomic eGFP locus and the Gbait sequence on the donor plasmid. The cellular NHEJ repair machinery then ligates these broken ends, resulting in the integration of the entire donor plasmid into the eGFP locus. When the integration occurs in the correct orientation and reading frame (theoretically in 1 out of 6 events), the expression of the eGFP gene is disrupted, and the KalTA4 protein is produced from the same promoter and regulatory elements that formerly controlled eGFP [14]. The successful conversion is visualized in the injected (F0) generation by the loss of eGFP fluorescence and the concomitant activation of RFP in the same cell population, indicating that the KalTA4 protein is being expressed and is functional [39] [14].
Key Research Reagent Solutions
The following table details the essential reagents and materials required for the successful implementation of the Gbait knock-in strategy.
Table 1: Essential Research Reagents for Gbait-Mediated Knock-In
| Reagent/Material | Function and Key Features | Reference |
|---|---|---|
| Donor Plasmid | Contains the Gbait sequence (for linearization), an E2A peptide sequence (for multicistronic expression), and the KalTA4 coding sequence (an optimized Gal4 variant). | [39] [14] |
| eGFP-targeting sgRNA | Single-guide RNA designed to complement a sequence within the eGFP coding region, guiding Cas9 to create a double-strand break in both the genomic locus and the donor plasmid. | [39] [14] |
| Cas9 mRNA | mRNA encoding the Cas9 nuclease, which complexes with the sgRNA to execute targeted DNA cleavage. | [39] [14] |
| UAS:RFP Reporter Line | A transgenic zebrafish line used as an injection host. RFP expression serves as a live, visual marker for successful Gal4 conversion in mosaic F0 embryos. | [39] [14] |
| Target eGFP Transgenic Line | The specific zebrafish line with known eGFP expression pattern that is to be converted into a Gal4 driver line. | [39] |
Detailed Experimental Protocol
Preparation of Molecular Reagents
- sgRNA Template and Synthesis: Design a sgRNA targeting a sequence within the eGFP open reading frame. The template can be generated by cloning annealed oligonucleotides into a sgRNA expression vector (e.g., pDR274) or via a PCR-based approach, followed by in vitro transcription [14].
- Cas9 mRNA Synthesis: Linearize a plasmid containing the Cas9 coding sequence (e.g., pT3TS-nCas9n). Perform in vitro transcription using an mRNA synthesis kit to generate capped and polyadenylated mRNA, and purify the product [14].
- Donor Plasmid Construction: Clone the KalTA4 coding sequence, preceded by an E2A peptide linker, into a suitable plasmid backbone. Immediately upstream of the E2A sequence, insert a ~200-500 bp fragment of the eGFP gene ("Gbait") that contains the target site for the sgRNA described above [39] [14].
Zebrafish Embryo Microinjection
- Setup: Set up mating pairs for the target eGFP transgenic line crossed with a UAS:RFP reporter line to generate double-transgenic embryos for injection.
- Injection Mix: Prepare a microinjection mixture containing:
- Microinjection: Inject approximately 1 nL of the mixture into the cell cytoplasm or yolk of one-cell stage embryos.
- Incubation: Incubate injected embryos at 28.5°C in egg water.
Screening and Establishment of Stable Lines
- F0 Mosaic Screening: Between 1 to 3 days post-fertilization (dpf), screen injected embryos under a fluorescence microscope. Identify mosaic individuals that show a clear conversion from eGFP to RFP fluorescence within the expected expression domain. A successful injection session should yield RFP-positive cells in >75% of embryos, with a significant portion (e.g., 22%) showing broad conversion [14].
- Raising Founders: Raise approximately 30-80 embryos showing mosaic RFP expression to adulthood. These are the potential F0 founder fish [39] [22].
- Germline Transmission Screening: Outcross each adult F0 founder to wild-type or UAS:GFP fish. Collect and screen the resulting F1 progeny for ubiquitous and non-mosaic GFP (or RFP) expression, which indicates stable germline transmission of the converted allele.
- Genotypic Confirmation: Perform genomic PCR on DNA from F1 embryos using primers flanking the integration site and internal to the KalTA4 sequence to confirm precise knock-in. Sequence the PCR product to verify the junction sequences [39] [40].
- Line Establishment: Raise PCR-positive F1 fish to establish stable, homozygous Gal4 knock-in lines.
Efficiency and Validation Data
The Gbait strategy has proven to be a highly efficient method for generating knock-in lines. The tables below summarize key quantitative outcomes from seminal studies.
Table 2: Knock-in Efficiency from Key Studies
| Study Target | Injected Embryos Showing Mosaic Conversion | Founders Screened | Stable Lines Obtained (Germline Transmission Rate) | Reference |
|---|---|---|---|---|
| neurod:eGFP to KalTA4 | >75% (293/388 embryos); 22% with broad conversion | N/A | 2 stable lines within 4 months | [39] [14] |
| evx2 Locus (Enhancer Trap) | N/A | 17 fish | 2 founders (11.8%) | [22] |
| eng1b Locus (Enhancer Trap) | N/A | 40 fish | 1 founder (2.5%) | [22] |
Table 3: Validation of Knock-in Line Functionality
| Validation Method | Key Finding | Implication | Reference |
|---|---|---|---|
| Expression Pattern Comparison | eGFP expression in heterozygous Tg[pax2a-hs:eGFP] embryos completely recapitulated endogenous pax2a expression domains. |
Confirms that the knocked-in reporter is under the control of native regulatory elements without disruption. | [40] |
| Homozygous Phenotype Analysis | Homozygous Tg[pax2a-hs:eGFP] embryos exhibited loss of the midbrain-hindbrain boundary, identical to the pax2a mutant (noi). |
Demonstrates that the knock-in allele is loss-of-function, validating precise integration into the native locus. | [40] |
| Gal4/UAS Effector Expression | Conversion of eGFP to Gal4 enabled stable expression of UAS-driven transgenes (e.g., RFP, GFP) in the original eGFP pattern. | Validates the functional utility of the converted line for driving effectors in specific cell populations. | [39] [22] |
Applications in Neurobiology and Beyond
The creation of specific Gal4 lines via the Gbait strategy has opened new avenues for sophisticated genetic manipulations, particularly in the field of neurobiology.
- Cell-Specific Gene Inactivation: The converted Gal4 lines can be combined with UAS-driven Cas9 to create a versatile platform for conditional gene knockout in specific cell types or at specific developmental times. This approach circumvents embryonic lethality and compensatory mechanisms often associated with constitutive mutants [38].
- Neuronal Circuit Analysis: The Gal4/UAS system can be used to express optogenetic actuators (e.g., Channelrhodopsin) or calcium indicators (e.g., GCaMP) in defined neuronal populations. This allows researchers to precisely manipulate or monitor neuronal activity in living, behaving zebrafish [38].
- Lineage Tracing and Clonal Analysis: When combined with Cre/lox and Brainbow technologies, this system enables permanent labeling of Cas9-expressing cells. This facilitates genetic lineage tracing of mutant cells and allows for the investigation of cell-autonomous gene function at single-cell resolution within a wild-type tissue environment [38].
The following diagram summarizes the core molecular components and their interactions in the Gbait knock-in system, providing an overview of how the strategy is integrated into a full research workflow.
Maximizing Efficiency: Troubleshooting and Protocol Optimization
In zebrafish research, the emergence of sophisticated knock-in strategies, particularly homology-independent methods, has revolutionized our ability to create precise genetic models. However, a significant challenge persists: accurately measuring the outcomes of these editing experiments. Traditional short-read sequencing methods often fail to provide a comprehensive picture of editing events, especially for larger insertions or complex rearrangements. This application note explores how long-read sequencing technologies are transforming the quantification of genome editing outcomes, enabling researchers to obtain robust, detailed analyses of their knock-in experiments.
The critical limitation of short-read sequencing (e.g., Illumina) for analyzing knock-in events lies in its read-length constraints. Confirming precise knock-in requires sequencing reads that encompass the entire insert and flanking homology regions, which often exceeds the capabilities of standard short-read platforms. Furthermore, size bias during PCR amplification and library preparation can lead to the under-representation of larger precisely edited fragments, while complex rearrangement events may be missed entirely if primer-binding sites are altered [19]. Long-read sequencing platforms, such as those from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), overcome these limitations by generating reads tens of kilobases long, allowing the entire edited locus to be captured in a single, continuous sequence [19] [41].
Advantages of Long-Read Sequencing for Editing Analysis
Long-read sequencing provides several distinct advantages for analyzing homology-independent knock-in outcomes in zebrafish:
- Comprehensive Variant Detection: It enables the identification of precise knock-in events, imprecise integrations, indels, and complex structural variations simultaneously from a single data set [19] [41].
- Phasing Capability: Long reads can determine whether multiple edits or mutations lie on the same DNA molecule (in cis) or on different alleles (in trans), which is crucial for accurately characterizing mosaic founders [41].
- Simplified Workflow: The process of validating edited alleles is streamlined, as it avoids the need for multiple overlapping Sanger sequencing reads or intricate in silico assembly of complex allelic variants [41].
The following diagram illustrates the core workflow for utilizing long-read sequencing in genome editing analysis:
Quantitative Data on Editing Outcomes
The application of long-read sequencing has provided quantitative evidence of its superiority for assessing editing experiments. The table below summarizes key performance metrics from recent studies:
Table 1: Quantitative Performance of Long-Read Sequencing in Genome Editing Analysis
| Metric | Reported Performance | Experimental Context |
|---|---|---|
| Sequence Recapitulation Accuracy | >99.9% (excluding homopolymers) [41] | ONT sequencing of wild-type zebrafish amplicons (0.9-2 kb) with HAC basecalling. |
| Precise Insertion Germline Transmission Rate | >20% at four tested loci [19] | Zebrafish knock-in using optimized, chemically modified dsDNA templates. |
| Founder Identification Rate | 5.1% (8/158 injected embryos) [23] | 3' knock-in at the krt92 locus using AmC6-modified dsDNA donors. |
| HDR Efficiency by Phenotypic Rescue | Up to 98.5% (pigmentation rescue) [42] | zLOST method for tyr gene correction in albino zebrafish. |
| Required Sequencing Depth | 100X-300X for high accuracy [41] | ONT sequencing with HAC basecalling to achieve >99.9% recall after filtration. |
Further analysis reveals how different experimental parameters influence the success of knock-in experiments. The following table compares the efficiency of various template types and methods:
Table 2: Comparison of Knock-in Template and Method Efficiencies in Zebrafish
| Template/Method Type | Key Characteristics | Reported Efficiency/Performance |
|---|---|---|
| Chemically Modified dsDNA (AmC6) [19] [23] | 5' end-protected PCR amplicons; reduces degradation and concatemerization. | High; >20% germline transmission; enables early integration in F0 mosaics. |
| Long ssDNA (lssDNA) [42] | zLOST method; long single-stranded DNA template. | Very High; up to 98.5% phenotypic rescue (somatic); up to 31.8% germline transmission. |
| Plasmid-based Template [19] | Template released in vivo by co-injected nuclease (e.g., I-SceI or Cas9). | Lower performance compared to chemically modified synthetic templates. |
| Prime Editing (PEn) [8] | Nuclease-based; inserts short DNA fragments without donor template. | High precision for inserts up to 30 bp; effective for stop codon and NLS insertion. |
| NHEJ-mediated Knock-in [14] | Homology-independent; concurrent cleavage of genome and donor plasmid. | High efficiency (>75% injected embryos showed integration); suitable for large cassettes. |
Detailed Experimental Protocol
Protocol: Long-Read Sequencing for Analyzing Knock-in Outcomes in Zebrafish
Objective: To accurately sequence and quantify the spectrum of genome editing outcomes (precise knock-in, indels, complex rearrangements) at a targeted locus in injected zebrafish embryos (F0) or their progeny (G1) using long-read sequencing [19] [41].
Materials:
- Biological Sample: Pooled genomic DNA from ~10 mosaic F0 zebrafish embryos or fin clips from individual G1 fish [41].
- PCR Reagents: High-fidelity DNA polymerase and associated buffers.
- Primers: Sequence-specific primers designed to amplify the entire edited locus, including flanking regions (generating a 0.9-2 kb amplicon, depending on the insert size) [41].
- Library Prep Kit: Compatible with the chosen long-read sequencing platform (e.g., ONT Ligation Sequencing Kit).
- Barcodes: Native Barcodes for multiplexing samples [41].
- Sequencing Equipment: Oxford Nanopore MinION or PromethION system, or PacBio Sequel system [19] [41].
Procedure:
DNA Extraction and Quality Control:
- Extract genomic DNA using a standard protocol (e.g., Phenol-Chloroform extraction or commercial kits). Ensure DNA is of high quality and integrity.
- Quantify DNA using a fluorometric method (e.g., Qubit). Verify the absence of significant degradation by gel electrophoresis or similar.
Target Amplification:
- Design primers to amplify the target locus, ensuring the amplicon spans the entire knock-in cassette and sufficient flanking sequence for unambiguous alignment.
- Perform PCR amplification using a high-fidelity polymerase to minimize PCR errors.
- Purify the PCR amplicons using magnetic beads or columns and re-quantify.
Library Preparation and Barcoding:
- Follow the manufacturer's instructions for your chosen long-read sequencing platform. For ONT:
- Dilute the amplicon to the recommended concentration.
- Proceed with end-prep and adapter ligation steps.
- Incorporate Native Barcodes during library preparation to enable multiplexing of different samples or loci in a single sequencing run [41].
- Pool the barcoded libraries if multiplexing.
- Follow the manufacturer's instructions for your chosen long-read sequencing platform. For ONT:
Sequencing:
- Load the prepared library onto the sequencer (e.g., a MinION R9.4 or newer flow cell).
- Initiate the sequencing run, typically for 24-72 hours, to achieve sufficient depth (>100X coverage per amplicon) [41].
Data Analysis:
- Basecalling and Demultiplexing: Use the platform's software (e.g., Guppy for ONT) for basecalling. The "High Accuracy Calling" (HAC) model is recommended for superior accuracy [41]. Subsequently, demultiplex the barcoded samples.
- Read Filtering: Apply a quality filter (e.g., using Filtlong) to retain high-quality reads. A quality threshold of q84 to q98 is effective without excessively reducing depth [41].
- Alignment: Align the filtered reads to the reference genome sequence (GRCz11) using an aligner like Minimap2 [41].
- Variant Calling and Consensus Building: Use the alignment to identify variants and generate consensus sequences. A consensus threshold of 60% is effective for accurate sequence recapitulation [41].
- Visualization and Quantification: Manually inspect a subset of aligned reads in a viewer like IGV to validate complex events. Quantify the frequency of precise knock-in, indels, and other repair outcomes from the sequencing data.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Long-Read Sequencing of Edited Loci
| Item | Function/Description | Example Use Case |
|---|---|---|
| High-Fidelity DNA Polymerase | Ensures accurate amplification of the target locus from genomic DNA for sequencing. | Generating amplicons for ONT library prep with minimal PCR-induced errors [41]. |
| 5' Chemically Modified Primers (e.g., AmC6) [23] | Primers with AmC6 modifications used to generate dsDNA donor templates; enhances knock-in efficiency by reducing degradation. | Producing highly efficient dsDNA donors for 3' knock-in to generate reporter lines. |
| Long ssDNA (lssDNA) Donor Template [42] | A long single-stranded DNA template for HDR; can be produced enzymatically. | zLOST method for highly efficient precise mutation introduction and gene correction. |
| Cas9 Nuclease (as protein or mRNA) | Creates a double-strand break at the target genomic locus to initiate repair. | Used in conjunction with a donor template (e.g., lssDNA or dsDNA) for knock-in [19] [23]. |
| Oxford Nanopore Ligation Sequencing Kit | Prepares the amplified DNA library for loading onto Nanopore flow cells. | Standardized library construction for ONT sequencing of targeted amplicons [41]. |
| Native Barcodes (ONT) | Allows for multiplexing of multiple samples (e.g., different loci or individuals) in a single sequencing run. | Cost-effective sequencing of several amplicons simultaneously by pooling them before loading [41]. |
| RMS-07 | RMS-07, MF:C35H40N8O2, MW:604.7 g/mol | Chemical Reagent |
| MK2-IN-4 | MK2-IN-4, CAS:1105658-32-9, MF:C25H24N4O2, MW:412.5 g/mol | Chemical Reagent |
The integration of long-read sequencing into the analysis of homology-independent knock-in experiments in zebrafish provides an unparalleled level of insight into editing outcomes. This approach moves beyond simple confirmation of presence/absence to offer a quantitative, detailed profile of all editing events within a sample. As the methods for both genome editing and sequencing continue to advance, the combination of efficient knock-in strategies with robust long-read analysis will undoubtedly accelerate the creation and validation of sophisticated zebrafish models for biomedical research and drug development.
In zebrafish genome editing, achieving precise homology-directed repair (HDR) remains a significant challenge due to the dominance of the error-prone non-homologous end joining (NHEJ) pathway. NHEJ is the primary DNA repair mechanism in most organisms, including zebrafish, and actively competes with the less efficient HDR pathway [7]. This competition drastically reduces the rate of precise knock-in events, making the generation of clean, seamlessly integrated lines labor-intensive and inefficient. Chemical reprogramming of the DNA repair landscape offers a powerful strategy to shift this balance. By using small-molecule inhibitors to suppress the NHEJ pathway, researchers can effectively enhance the efficiency of HDR, enabling more reliable and efficient genome engineering in zebrafish models [7] [43].
The Scientific Basis: Manipulating DNA Repair Pathways
The Competition Between NHEJ and HDR
When a CRISPR/Cas9-induced double-strand break (DSB) occurs, the cell activates multiple repair mechanisms. The key pathways involved are:
- Non-Homologous End Joining (NHEJ): An error-prone pathway that ligates DNA ends without a template, operating throughout the cell cycle. It is the dominant pathway in zebrafish embryos [7] [10].
- Homology-Directed Repair (HDR): A precise pathway that uses a homologous DNA template (such as a donor plasmid or single-stranded oligodeoxynucleotide) to repair the break, but is restricted to late S and G2 phases of the cell cycle [43].
- Microhomology-Mediated End Joining (MMEJ): An alternative, error-prone repair pathway that utilizes microhomology sequences flanking the break site [44].
The objective of chemical reprogramming is to tip the balance away from NHEJ and in favor of HDR.
Mechanism of Action of NHEJ Inhibitors
A critical regulator of the NHEJ pathway is the DNA-dependent protein kinase (DNA-PK) complex. The catalytic subunit, DNA-PKcs, is a prime target for pharmacological inhibition [43]. Small-molecule inhibitors such as NU7441 and KU-0060648 act by blocking DNA-PKcs activity. This disruption prevents the initiation of the classical NHEJ repair cascade, thereby reducing the frequency of mutagenic indel events and allowing more DSBs to be channeled into the HDR pathway [43]. Recent research also highlights the role of the MMEJ pathway. Inhibiting DNA-PKcs with a next-generation compound like AZD7648 can shift DSB repair towards MMEJ, and when combined with inhibition of the MMEJ factor Polθ, can further enhance HDR outcomes, although this advanced strategy is yet to be fully validated in zebrafish [44].
The following diagram illustrates how NHEJ inhibitors shift the DNA repair balance to favor precise Homology-Directed Repair.
Quantitative Data on NHEJ Inhibitors
The effectiveness of NHEJ inhibitors in enhancing HDR has been quantitatively demonstrated in zebrafish models. The following table summarizes key performance data for small-molecule inhibitors from published studies.
Table 1: Efficacy of Small-Molecule Inhibitors in Enhancing HDR in Zebrafish
| Inhibitor | Target | Optimal Concentration | Reported HDR Enhancement | Key Findings |
|---|---|---|---|---|
| NU7441 | DNA-PKcs | 50 µM (injected) | Up to 13.4-fold increase in somatic HDR events [7] | - Dramatic increase in HDR efficiency quantified by a visual reporter assay.- Increase in somatic HDR correlated directly with germline transmission [7]. |
| KU-0060648 | DNA-PKcs | 250 nM (cell culture) [43] | Increased HDR frequency [43] | - Compatible with Cas9-editing technology.- Reduces NHEJ frequency while increasing HDR [43]. |
| RS-1 | RAD51 activator | 15-30 µM (injected) | Modest but significant increase (DMSO: 4.8 ± 3.0 vs 30 µM: 7.3 ± 5.3 red fibers) [7] | - Directly stimulates the HDR pathway.- Effect is modest compared to NU7441 [7]. |
| SCR7 | DNA Ligase IV | Not effective in zebrafish [7] | No significant effect [7] | - Reported to have species-specific effects.- Shown to be ineffective in the zebrafish model tested [7]. |
| AZD7648 | DNA-PKcs | Information for zebrafish not specified in search results | Shifts DSB repair towards MMEJ; enhances HDR in mouse embryos and human cells [44] | - A next-generation, highly potent and selective DNA-PKcs inhibitor.- Effect in zebrafish is an area for future validation. |
Detailed Experimental Protocol for Zebrafish Knock-In
This protocol integrates the use of NU7441 to enhance HDR efficiency for knocking a reporter sequence into a zebrafish gene, based on optimized methods from recent literature [7] [45] [19].
Reagents and Equipment
- Zebrafish: Wild-type (AB strain) or appropriate transgenic line.
- CRISPR Components: Cas9 protein (e.g., recombinant S. pyogenes Cas9) and synthetic crRNA/tracrRNA or sgRNA.
- HDR Template: A PCR-generated, linear double-stranded DNA template with short homology arms (30-60 bp). Using chemically modified templates (e.g., with 5' phosphorylation) is recommended as they outperform unmodified templates [45] [19].
- NHEJ Inhibitor: NU7441, reconstituted in DMSO to create a stock solution (e.g., 50 mM).
- Injection Equipment: Standard zebrafish microinjection setup.
- Other Reagents: Phenol red injection buffer, E3 embryo medium.
Step-by-Step Procedure
The workflow for the knock-in experiment, from preparation to screening, is outlined below.
1. Preparation of the Knock-In Mixture - CRISPR RNP Complex Formation: Complex high-efficiency sgRNA (or crRNA:tracrRNA duplex) with Cas9 protein to form a ribonucleoprotein (RNP) complex. Incubate at room temperature for 10-15 minutes. - Final Injection Mixture: Combine the following components to create the final injection mix: - Pre-complexed Cas9 RNP - PCR-generated HDR template (50-100 ng/µL) - NU7441 stock solution to a final concentration of 50 µM [7] - Phenol red (0.1-0.5%) for visualization - Critical Note: A control injection with DMSO vehicle instead of NU7441 should always be included to accurately assess the enhancement effect.
2. Microinjection into Zebrafish Embryos - Inject 1-2 nL of the final mixture into the cytoplasm or yolk of 1-cell stage zebrafish embryos. - The use of Cas9 protein (RNP) is highly recommended over Cas9 mRNA as it leads to faster editing and reduces off-target effects [45].
3. Embryo Handling and Screening - After injection, maintain embryos in E3 medium at 28.5°C. - Screen injected (F0) embryos for successful knock-in between 3 and 4 days post-fertilization (dpf). The screening method depends on the knock-in: - For fluorescent reporters, screen directly for fluorescence under a microscope [45] [16]. - For non-visible knock-ins (e.g., epitope tags), use PCR-based genotyping of pooled embryos or, ideally, long-read sequencing (e.g., PacBio) for accurate quantification of precise repair events [19].
4. Raising Founders and Germline Transmission - Raise all embryos that show positive screening signals (mosaic F0 founders) to adulthood. - Outcross adult F0 fish to wild-type partners and screen the resulting F1 progeny for ubiquitous expression of the knock-in allele to identify germline-transmitting founders.
Table 2: Key Research Reagent Solutions for Enhanced Knock-In in Zebrafish
| Reagent / Solution | Function / Purpose | Examples / Notes |
|---|---|---|
| CRISPR RNP Complex | Induces a site-specific double-strand break (DSB) at the target genomic locus. | Using synthetic crRNA/tracrRNA and recombinant Cas9 protein is the gold standard for high efficiency and low toxicity [45] [35]. |
| Linear dsDNA HDR Template | Serves as the donor template for precise integration via HDR. | PCR-generated templates with short homology arms (30-100 bp) are highly effective and cloning-free [45]. Chemical modifications (e.g., 5' phosphorylation) enhance template stability and HDR efficiency [19]. |
| NHEJ Inhibitors (Small Molecules) | Shifts the DNA repair balance from error-prone NHEJ to precise HDR. | NU7441 is a well-validated DNA-PKcs inhibitor in zebrafish [7]. AZD7648 is a potent next-generation inhibitor to explore [44]. |
| HDR Enhancers (Small Molecules) | Directly stimulates the activity of the HDR repair machinery. | RS-1, a RAD51 stimulator, shows a modest but significant enhancement of HDR [7]. |
| Long-Range PCR and Sequencing | For accurate genotyping and quantification of precise knock-in events. | Pacific Biosciences (PacBio) long-read sequencing is ideal for quantifying heterogeneous knock-in outcomes, as it avoids the size bias of short-read sequencing [19]. |
Troubleshooting and Technical Notes
- Low HDR Efficiency: Ensure your sgRNA has a very high cutting efficiency (>60-70%). The distance between the Cas9 cut site and the intended insertion point should be minimal (ideally <20 bp) [46] [10]. Always use a high-quality, purified HDR template.
- Toxicity from Inhibitors: While NU7441 at 50 µM was not reported to affect embryo survival, dose optimization may be necessary for different chemical batches or specific zebrafish lines [7]. Monitor embryo survival and morphology closely.
- Mosaicism in F0: Injected F0 animals are invariably mosaic because editing occurs after the first cell division. Therefore, it is essential to raise multiple F0 founders and screen their F1 progeny to find those that transmit the knock-in allele through the germline [7] [45].
- Template Design is Critical: The HDR template must be designed to alter the PAM site or the sgRNA binding sequence to prevent re-cleavage of the successfully edited allele [45] [46].
Chemical reprogramming using NHEJ inhibitors like NU7441 represents a straightforward and highly effective strategy to overcome the major bottleneck of low HDR efficiency in zebrafish genome engineering. When combined with optimized protocols—such as the use of Cas9 RNP, PCR-generated templates with short homology arms, and long-read sequencing for validation—this approach enables researchers to achieve high rates of precise knock-in and robust germline transmission. This methodology significantly advances the generation of sophisticated zebrafish models for functional genomics and disease modeling.
Within zebrafish research, achieving precise genomic knock-ins via homology-independent strategies has been a significant challenge due to the characteristically low efficiency of homology-directed repair (HDR). The choice of donor template is a critical factor influencing the success of these genome-editing experiments. Recent quantitative side-by-side comparisons have definitively established that chemically modified templates significantly outperform traditional plasmid-based templates for precise targeted insertion. These synthetic, chemically protected templates resist degradation and concatemerization in vivo, leading to a marked increase in germline transmission rates of precise edits, consistently achieving rates greater than 20% across multiple loci [19]. This application note details the experimental evidence and provides protocols for implementing these superior template engineering strategies within the context of homology-independent knock-in research in zebrafish.
Quantitative Comparison of Template Performance
Key Findings from Side-by-Side Comparisons
A comprehensive 2025 study systematically compared editing outcomes using long-read sequencing to quantify precise insertion rates across various template types and CRISPR nucleases. The research evaluated double-stranded DNA (dsDNA) templates where the homology-directed repair (HDR) template was released in vivo from a plasmid through digestion with a co-injected nuclease (I-SceI meganuclease or Cas9), and compared them to synthetic, chemically modified templates [19].
The central finding was that chemically modified templates consistently demonstrated superior performance over those released from plasmids. This performance advantage is attributed to the enhanced stability of chemically modified templates within the cellular environment; they are less susceptible to degradation and avoid concatemerization, thereby increasing the effective concentration of intact, functional templates available for the DNA repair machinery [19].
Table 1: Summary of Template Performance in Zebrafish Knock-In
| Template Type | Key Characteristics | Reported Germline Transmission Rate | Primary Advantages |
|---|---|---|---|
| Chemically Modified dsDNA | Synthetic templates with chemical protections (e.g., 5' AmC6) on primers or donor blocks [19] [23]. | >20% (across four loci) [19] | Enhanced stability, reduced degradation/concatemerization, high efficiency, commercial availability. |
| Plasmid-Released Template | Linear template released in vivo by co-injected nuclease (I-SceI or Cas9) [19]. | Lower than chemically modified templates [19] | Familiar technology, suitable for very large inserts. |
| PCR-Amplified dsDNA (5' AmC6 modified) | Cloning-free PCR amplicons with 5' AmC6-modified primers, flanked by homology arms [23]. | Founder rates from 11.5% to 20% [23] | No complex cloning, high scalability, good efficiency. |
Impact of Template Purity and Design
Beyond chemical modification, template design and preparation are crucial. The use of nanoplasmid DNA—a minimized backbone optimized for gene therapy that prevents transgene silencing—has shown promise in mammalian T-cell engineering via HITI, yielding high cell numbers and efficient integration [47]. Furthermore, the presence of non-homologous base pairs in homology arms has been shown to significantly reduce precise editing rates, underscoring the need for meticulous template design [19].
Experimental Protocols for High-Efficiency Knock-In
Protocol 1: Knock-In Using Chemically Modified Donor Blocks
This protocol is adapted from studies achieving high-efficiency epitope tagging and is based on commercially available, synthetic CRISPR reagents [34].
- Target Site and Donor Design: Use a web-based HDR design tool (e.g., Integrated DNA Technologies' Alt-R CRISPR HDR Design Tool). Select a target site in the 5'UTR, approximately 16 base pairs upstream of the start codon. Design the donor template with the insert (e.g., MYC tag) placed immediately after the start codon, flanked by asymmetric homology arms (e.g., 40 bp left arm, 80 bp right arm) [34].
- Reagent Preparation: Order the donor as a synthesized, chemically modified "Alt-R HDR Donor Block." Complex the Alt-R CRISPR-Cas9 crRNA with tracrRNA to form a guide RNA (gRNA) duplex. Use a high-quality Cas9 protein (e.g., Alt-R S.p. Cas9 Nuclease V3) [34].
- Microinjection Mix Preparation: Combine the following components in nuclease-free water:
- gRNA: 250 pg
- Cas9 protein: 500 pg
- Chemically modified HDR donor template: 37.5 pg
- A tracking dye (e.g., Dextran red)
- Zebrafish Embryo Injection: Microinject 1-2 nL of the prepared mix into the cell of one-cell stage zebrafish embryos.
- Screening and Validation: Extract genomic DNA from injected embryos or adult fin clips. Screen for successful insertion using PCR with primers flanking the target site and confirm precise integration by Sanger sequencing [34].
Protocol 2: Cloning-Free 3' Knock-In with 5' AmC6-Modified dsDNA
This streamlined protocol from Mi & Andersson (2023) enables efficient 3' knock-in for lineage tracing without disruptive cloning steps [23].
- Donor Template PCR Amplification: Design a vector containing the insert cassette (e.g., fluorescent protein followed by a self-cleavable 2A peptide and Cre recombinase). Amplify the donor template via PCR using primers that include:
- Long ( ~900 bp) or short homology arms matching the genomic sequence immediately surrounding the stop codon of the target gene.
- 5' AmC6 modifications on both primers to protect the dsDNA product from degradation and multimerization.
- Ribonucleoprotein (RNP) Complex Assembly: In vitro, pre-assemble the Cas9/gRNA RNP complex by incubating purified Cas9 protein with the target-specific sgRNA.
- Microinjection: Co-inject the following into one-cell stage embryos:
- The pre-assembled Cas9/gRNA RNP complex.
- The PCR-amplified, AmC6-modified dsDNA donor.
- Founder Identification: Raise injected (F0) embryos. Screen for mosaic founders displaying strong fluorescence in the expected pattern. Outcross potential founders and screen the F1 generation for germline transmission, which can be expected in 11.5-20% of offspring from successful founders [23].
Signaling Pathways and Workflows
The following diagram illustrates the core mechanistic difference leading to the superior performance of chemically modified templates.
The Scientist's Toolkit: Essential Reagents for Template Engineering
Successful implementation of high-efficiency knock-in relies on a specific set of reagents designed for stability and precision.
Table 2: Key Research Reagent Solutions for Template Engineering
| Reagent / Solution | Function & Description | Application Note |
|---|---|---|
| Alt-R HDR Donor Blocks (IDT) | Chemically modified, double-stranded DNA fragments. Designed as HDR templates with user-defined homology arms and insert sequences. | The chemical modifications enhance stability in vivo. Superior to plasmid-released templates for insertions like epitope tags [19] [34]. |
| 5' AmC6-Modified Primers | PCR primers with a 5' C6 amino linker modification. Used to generate protected, cloning-free dsDNA donor templates. | Prevents degradation of PCR-amplified donors. Injection of donors made with these primers results in high germline transmission rates for 3' knock-in [23]. |
| Cas9 Ribonucleoprotein (RNP) | Pre-complexed complex of purified Cas9 protein and target-specific sgRNA. | Direct delivery of the editing machinery, leading to high efficiency and reduced off-target effects compared to mRNA injection. Used in both featured protocols [34] [23]. |
| Nanoplasmid DNA | A minimized plasmid backbone with a size of ~430 bp, containing an R6K origin and an antibiotic-free selection system. | Prevents transgene silencing post-integration. While evidenced in mammalian cells [47] [48], the backbone optimization principle is highly relevant for future template engineering in zebrafish. |
| NHEJ Inhibitors (e.g., NU7441) | Small-molecule inhibitor of DNA-PK, a key enzyme in the non-homologous end joining (NHEJ) pathway. | Shifts DNA repair equilibrium toward HDR. In zebrafish, injection of NU7441 enhanced HDR-mediated repair efficiency up to 13.4-fold in a somatic reporter assay [7]. |
| KTX-582 | KTX-582, MF:C45H51F3N8O7, MW:872.9 g/mol | Chemical Reagent |
| Nidurufin | Nidurufin, CAS:99528-66-2, MF:C20H16O8, MW:384.3 g/mol | Chemical Reagent |
Homology-independent knock-in strategies have emerged as powerful techniques in zebrafish research, enabling precise genomic modifications for modeling human diseases and studying gene function. Unlike homology-directed repair (HDR), these methods leverage alternative DNA repair pathways such as non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) to integrate exogenous DNA sequences into specific genomic loci. While offering substantial advantages, researchers face significant challenges including off-target effects, low efficiency, and embryo viability when implementing these approaches. This application note provides detailed protocols and strategic frameworks to overcome these hurdles, facilitating more robust and reliable experimental outcomes in zebrafish genome engineering.
Core Challenges and Quantitative Assessments
The successful implementation of homology-independent knock-in strategies requires a thorough understanding of common pitfalls and their quantitative impact on experimental outcomes. The table below summarizes key challenges and performance metrics based on recent studies.
Table 1: Performance Metrics of Homology-Independent Knock-In Strategies in Zebrafish
| Challenge | Performance Metric | Baseline Efficiency | Optimized Efficiency | Key Factors Influencing Outcome |
|---|---|---|---|---|
| Editing Efficiency | Germline transmission rate | Highly variable, often <5% [19] | 20-56% with optimized protocols [19] [49] | Cas9 amount (200-800 pg optimal) [5], template design [19], gRNA quality [49] |
| Off-Target Effects | Mutation rate at non-target sites | Variable depending on gRNA design | Significant reduction with prime editing [5] | gRNA specificity, nuclease type (Cas9 vs. Cas12a) [19], delivery method |
| Embryo Viability | Survival to adulthood | Influenced by injection technique and reagent toxicity | Improved with direct cytoplasmic injection [5] | Injection volume, reagent concentration, injection site (yolk vs. cell) [5] |
| Template Integration | Precise insertion rate | Low with unmodified templates [19] | Substantial improvement with chemically modified templates [19] [23] | Template modification (Alt-R, AmC6) [5] [23], homology arm design, distance from DSB [19] |
The Scientist's Toolkit: Essential Reagents and Solutions
Table 2: Research Reagent Solutions for Homology-Independent Knock-In in Zebrafish
| Reagent Category | Specific Solution | Function & Mechanism | Application Notes |
|---|---|---|---|
| Nucleases | S. pyogenes Cas9 | Generates blunt-end DSBs; most widely used nuclease | Optimal amount 200-800 pg per injection [5] |
| L. bacterium Cas12a | Generates 5-nt 5' overhang; different PAM (TTTN) | Similar performance to Cas9 for targeted insertion [19] | |
| Template Design | Chemically modified dsDNA (Alt-R) | Reduced degradation and concatemerization | Outperforms plasmid-based templates [5] [19] |
| 5' AmC6-modified PCR primers | Protection from exonucleases; prevents multimerization | Enables cloning-free 3' knock-in strategy [23] | |
| Delivery Enhancers | Preassembled Cas9/gRNA RNP complexes | Enables early integration; improves editing efficiency | Direct injection into cell cytoplasm recommended [23] |
| LiCl-purified gRNA | Removes impurities; increases knock-in efficiency | Critical for high-efficiency KI (>50% germline transmission) [49] |
Optimized Experimental Protocols
Protocol 1: HITI-based Knock-In Using Chemically Modified Templates
This protocol outlines the steps for implementing Homology-Independent Targeted Integration (HITI) for precise insertion of genetic elements, adapted from successful applications in zebrafish and mammalian systems [19] [50].
Reagent Preparation:
- gRNA Design: Select target site in intronic region whenever possible to avoid disrupting coding sequences. Verify specificity using Cas-OFFinder or similar tools to minimize off-target effects [50].
- Cas9 Protein: Use high-quality, commercially available Cas9 protein at concentration of 500 ng/μL.
- Donor Template: Generate double-stranded DNA template with 50-800 bp homology arms using PCR amplification with 5' AmC6-modified primers [23]. For insertions >1 kb, use chemically modified Alt-R templates [19].
Microinjection Setup:
- Prepare injection mixture containing:
- 100-200 ng/μL Cas9 protein
- 50-100 ng/μL gRNA
- 20-50 ng/μL chemically modified donor template
- 1× injection buffer
- Load injection needles using fine-tip micropipettes to avoid introducing air bubbles.
- Calibrate injection volume to 1-2 nL per embryo using a micrometer.
Embryo Injection and Screening:
- Inject directly into the cell cytoplasm of one-cell stage zebrafish embryos immediately following fertilization [5].
- Transfer injected embryos to embryo medium and maintain at 28.5°C.
- For fluorescent reporters, screen F0 embryos at 24-48 hpf for mosaic expression patterns.
- Raise high-quality mosaic F0 founders (showing >30% correct expression pattern) to adulthood [23].
- Outcross F0 adults to wild-type fish and screen F1 progeny for germline transmission using PCR and sequencing.
Protocol 2: Prime Editing for Precise Sequence Modifications
Prime editing represents a recent advancement that can surpass traditional HDR in efficiency while minimizing off-target effects [5].
Workflow Optimization:
- Prime Editing Guide RNA (pegRNA) Design: Design pegRNA with 10-15 nt homology arm and primer binding site complementary to the target strand.
- Ribonucleoprotein Complex Formation: Preassemble prime editor protein with pegRNA at molar ratio of 1:3 (protein:RNA) and incubate at 37°C for 10 minutes.
- Microinjection: Co-inject preassembled RNP complexes (100-200 ng/μL) with donor template (20-50 ng/μL) into the cell cytoplasm.
- Validation: Use targeted sequencing of injected embryos to quantify editing efficiency at 24 hpf. Prime editing can achieve up to 4-fold higher efficiency than HDR for specific targets [5].
Visualizing the Knock-In Workflow and Strategic Decision Process
The following diagram illustrates the complete experimental workflow for homology-independent knock-in in zebrafish, integrating both HITI and prime editing approaches:
Diagram 1: Comprehensive workflow for homology-independent knock-in strategies in zebrafish.
The selection of appropriate strategy and optimization parameters is critical for success. The following decision framework illustrates key considerations:
Diagram 2: Strategic decision framework for selecting and optimizing knock-in approaches.
Advanced Applications and Implementation
Dual-Function Allele Engineering
The dual-cassette donor strategy enables simultaneous conditional knockout and gene tagging in a single integration event [49]. This approach utilizes a "PoNe" (Positive-Negative) donor design containing:
- Positive Cassette (Po-cassette): Maintains target gene expression with in-frame fluorescent reporter fusion (separated by 2A peptide) flanked by loxP sites.
- Negative Cassette (Ne-cassette): Contains dual polyadenylation signals and mutated exon sequence to ensure complete gene disruption after Cre recombination.
Implementation of this strategy at the tbx5a locus achieved germline transmission rates up to 56% following preselection of F0 embryos with correct reporter expression [49].
Troubleshooting Common Implementation Issues
Table 3: Troubleshooting Guide for Homology-Independent Knock-In Experiments
| Problem | Potential Causes | Solutions | Validation Methods |
|---|---|---|---|
| Low editing efficiency | Suboptimal gRNA efficiency, template degradation, insufficient Cas9 | Use LiCl-purified gRNA [49], chemically modified templates [19], optimize Cas9 concentration (200-800 pg) [5] | T7E1 assay on pooled embryos, NGS of target locus |
| High off-target effects | Low gRNA specificity, excessive nuclease amount | Use CHOP-CHOP or CRISPOR for gRNA design [16], employ prime editing where possible [5] | GUIDE-seq [50], whole-genome sequencing of established lines |
| Poor embryo viability | Toxicity from injection components, mechanical damage | Optimize injection volume (1-2 nL), use purified proteins instead of mRNA [23] | Survival rate monitoring at 24 hpf |
| No germline transmission | Low mosaicism in F0, inadequate screening | Preselect F0 with >30% correct expression pattern [23], increase number of outcrossed F0 | Junction PCR, fluorescence screening in F1 |
Homology-independent knock-in strategies in zebrafish have evolved substantially, with modern techniques addressing previous limitations in efficiency and precision. The implementation of chemically modified templates, advanced nuclease systems, and optimized delivery methods has enabled germline transmission rates exceeding 20% across multiple loci. Prime editing emerges as a particularly promising approach, offering up to fourfold efficiency improvements over conventional HDR with reduced off-target effects. By adhering to the protocols and strategic frameworks outlined in this application note, researchers can effectively navigate the challenges of off-target effects, low efficiency, and embryo viability to successfully generate precise genetic models in zebrafish.
| Reagent Category | Specific Product/System | Function in Knock-in Experiment | Key Considerations for Zebrafish |
|---|---|---|---|
| CRISPR Nuclease | SpCas9 Nuclease, LbCas12a (Cpf1) Nuclease | Induces a precise double-strand break (DSB) at the target genomic locus. | Cas9 uses an NGG PAM; Cas12a uses a T-rich PAM (TTTV), expanding targetable sites [51]. |
| Guide RNA | Target-specific sgRNA (for Cas9), crRNA (for Cas12a) | Directs the Cas nuclease to the specific DNA sequence for cleavage. | For Cas12a, crRNAs can be arranged in arrays for multiplexed editing [51]. |
| Repair Template | Chemically modified single-stranded oligodeoxynucleotides (ssODNs) or dsDNA donors with long homology arms | Provides the exogenous DNA sequence for integration into the genome via Homology-Directed Repair (HDR). | Chemically modified templates (e.g., with AmC6) significantly outperform plasmid-based templates and reduce random integration [52] [23]. |
| Microhomology Template | dsDNA donor with short homology arms (e.g., ~50 bp) | Facilitates integration via Microhomology-Mediated End Joining (MMEJ), an alternative HDR pathway. | Effective when combined with 5' AmC6-modified primers on the PCR-amplified donor [23]. |
| Injection Material | Pre-assembled Cas9/gRNA Ribonucleoprotein (RNP) Complexes | Allows for rapid activity upon injection, reducing mosaicism and improving editing efficiency. | Co-injection of RNPs with the DNA donor is a standard method for zebrafish embryo injection [23]. |
The selection of the appropriate CRISPR nuclease is a critical first step in designing a successful homology-independent knock-in experiment in zebrafish. While both Cas9 and Cas12a are widely used, they possess distinct molecular characteristics that influence their application. Cas9 induces a blunt-ended double-strand break and requires a G-rich protospacer adjacent motif (PAM) sequence (NGG) downstream of the target site [51] [53]. In contrast, Cas12a creates a staggered cut with a 4-5 nucleotide 5' overhang and recognizes a T-rich PAM (TTTV) upstream of the target site [51] [54]. This difference in PAM requirement allows Cas12a to access genomic regions that may be inaccessible to Cas9, thereby expanding the potential target space [55].
Beyond PAM preferences, the nature of the DSB influences the repair outcome. The sticky ends generated by Cas12a are theorized to be more favorable for certain homology-independent repair pathways that rely on microhomology, potentially offering an advantage for specific knock-in strategies [54].
Comparative Performance Data for Cas9 and Cas12a
The following table summarizes key performance metrics for Cas9 and Cas12a, based on empirical data from zebrafish and other model systems.
| Performance Metric | Cas9 | Cas12a | Experimental Context & Notes |
|---|---|---|---|
| On-target Efficiency | Very High | Variable, can be high with optimization | In zebrafish, side-by-side comparisons show they can perform similarly for targeted insertion [52]. In plants, Cas9 often shows higher efficiency [51] [54]. |
| Precise Knock-in Rate (HDR) | Can achieve >20% germline transmission in zebrafish with optimized parameters [52] | Can achieve >20% germline transmission in zebrafish with optimized parameters [52] | Efficiency for both is highly dependent on the distance between the DSB and the insertion point, and template design [52]. |
| Indel Pattern | Predominantly small insertions and deletions (<10 bp); balanced insertions vs. deletions [56] [53] | Tends to produce larger deletions; predominantly deletions over insertions [56] [53] [54] | The staggered-end break of Cas12a and its interaction with exonucleases promotes larger deletions, which can be advantageous for gene disruptions [56]. |
| Specificity (Off-target) | High-fidelity variants available; can show more off-targets in some early studies [51] [53] | Generally high specificity with fewer off-targets reported in multiple systems [51] [53] [54] | Cas12a's longer PAM and other mechanistic differences may contribute to its high observed specificity [53]. |
| Multiplexing Capacity | Requires multiple individual sgRNA expression cassettes | Native ability to process a single CRISPR RNA array into multiple crRNAs [51] [54] | This inherent feature of Cas12a simplifies simultaneous targeting of multiple genomic loci. |
Experimental Protocol: HDR-mediated Knock-in in Zebrafish
This protocol details the steps for achieving precise knock-in of exogenous DNA using Cas9 or Cas12a and chemically modified repair templates, based on methodologies that have yielded high germline transmission rates in zebrafish [52] [23].
Stage 1: Target Selection and Reagent Design (4-5 Days)
- Target Locus Identification: Select a target site within the gene of interest. For 3' knock-in, target a site 20-30 base pairs upstream of the stop codon to ensure the knock-in cassette is in-frame while preserving endogenous gene function [23].
- gRNA/crRNA Design: Design sgRNAs (for Cas9) or crRNAs (for Cas12a) using standard tools. Ensure the target sequence is unique within the genome to minimize off-target effects.
- Repair Template Design:
- Template Type: Use a single-stranded DNA (ssODN) or a PCR-amplified double-stranded DNA (dsDNA) donor as the repair template. Chemically modified templates have been shown to outperform plasmid donors [52].
- Homology Arms: For dsDNA donors, incorporate homology arms. Long arms (~800-900 bp) can enhance HDR, while short arms (~50 bp) can be used with MMEJ [23].
- Synonymous Mutations: Introduce silent mutations in the gRNA/crRNA target sequence within the repair template to prevent re-cleavage of the successfully edited allele [23].
- Chemical Modification: Synthesize the dsDNA donor via PCR using primers with 5' AmC6 modifications. This modification protects the donor DNA and significantly boosts knock-in efficiency [23].
Stage 2: Preparation of Injection Materials (2-3 Days)
- Synthesize gRNA/crRNA: Produce the guide RNA using in vitro transcription from a DNA template or purchase synthetic guides.
- Produce Cas9/Cas12a Protein or mRNA: For highest efficiency and reduced mosaicism, use purified Cas9 or Cas12a protein. Alternatively, in vitro transcribed mRNA can be used.
- Assemble RNP Complexes: Pre-incubate the Cas protein with the gRNA/crRNA at a molar ratio of 1:2 to 1:5 (Cas:guide) for 10-20 minutes at room temperature to form Ribonucleoprotein (RNP) complexes.
- Prepare Injection Mixture: Combine the following in nuclease-free water:
- RNP complexes (final concentration: 200-400 ng/µL for Cas protein)
- Chemically modified repair template (final concentration: 50-100 ng/µL)
- Phenol red dye (0.1%) for visualization.
Stage 3: Zebrafish Embryo Microinjection (1 Day)
- Inject 1-2 nL of the prepared mixture directly into the cytoplasm or cell yolk of one-cell stage zebrafish embryos.
- Incubate injected embryos in egg water at 28.5°C.
Stage 4: Screening and Validation (3-4 Weeks)
- Mosaic Founder (F0) Screening: At 1-3 days post-fertilization (dpf), screen injected embryos for evidence of correct knock-in using fluorescence microscopy if a reporter is included.
- Raise Founders: Raise mosaic F0 embryos to adulthood.
- Outcross and Identify Founders: Outcross adult F0 fish to wild-type partners. Screen the resulting F1 offspring for the presence of the knock-in allele via PCR genotyping and DNA sequencing.
- Establish Stable Lines: Positive F1 fish can be raised to establish stable transgenic lines.
The workflow below visualizes this protocol.
Figure 1: Workflow for HDR-mediated knock-in in zebrafish.
Advanced Strategy: Enhancing Deletions with Exonuclease Fusions
For applications requiring larger deletions, such as removing non-coding regulatory elements or entire gene clusters, fusing exonucleases to Cas nucleases is a highly effective strategy. Research in plants and zebrafish has shown that fusing exonucleases like sbcB (a 3' to 5' exonuclease) to either Cas9 or Cas12a can significantly increase the size of the induced deletions [56].
This approach works by promoting more extensive resection of the DNA ends after the initial Cas-induced break, which favors repair pathways like Microhomology-Mediated End Joining (MMEJ) that result in larger deletions [56]. The diagram below illustrates how this fusion protein operates.
Figure 2: Mechanism of exonuclease-fused Cas for large deletions.
In the context of homology-independent knock-in in zebrafish, both Cas9 and Cas12a are powerful and capable of achieving high rates of precise integration when paired with optimized, chemically modified repair templates [52]. The choice between them should be guided by the specific needs of your experiment.
- Choose Cas9 if: Your target site is flanked by an NGG PAM, you require the highest possible on-target cutting efficiency, or your experimental system is already optimized for Cas9.
- Choose Cas12a if: Your target locus is in an A/T-rich region with a TTTV PAM, your experimental design requires multiplexing several guides, or your priority is to minimize potential off-target effects.
- Universal Critical Factor: Regardless of nuclease choice, the use of chemically modified repair templates (e.g., AmC6-modified dsDNA donors) and pre-assembled RNP complexes is key to maximizing knock-in efficiency and generating robust zebrafish models for drug development and biomedical research [52] [23].
Validating and Comparing Knock-In Strategies for Robust Research Outcomes
Within the expanding toolkit for zebrafish genome engineering, achieving high-efficiency germline transmission of precise knock-in alleles remains a significant challenge. While homology-directed repair (HDR) has been the traditional focus, homology-independent strategies present a promising alternative that can circumvent the cell cycle limitations and low efficiency often associated with HDR. Recent methodological refinements have made germline transmission rates exceeding 20% an attainable goal for precise insertions across multiple loci. This Application Note details the protocols and parameters essential for consistently achieving this efficiency benchmark, with a specific focus on homology-independent integration mechanisms.
Quantitative Analysis of Optimized Knock-In Parameters
Recent systematic comparisons have identified critical factors influencing knock-in efficiency. The following table summarizes key parameters and their impact on achieving high-rate germline transmission.
Table 1: Parameters for High-Efficiency Germline Transmission
| Parameter | Optimal Condition | Impact on Germline Transmission | Key Supporting Evidence |
|---|---|---|---|
| Template Type | Chemically modified double-stranded DNA (dsDNA) | Significantly outperforms plasmid-derived templates [19]. | Founder rates >20% across four distinct loci [19]. |
| CRISPR Nuclease | Cas9 or Cas12a | Both nucleases perform similarly for targeted insertion, offering target site flexibility [19]. | Comparable precise editing rates between Cas9 and Cas12a [19]. |
| Homology Arm Length | 25-35 bp for MMEJ-based strategies | Shorter arms are sufficient for microhomology-mediated end joining (MMEJ), simplifying donor construction [57]. | High KI efficiency with the S-25 donor (25 bp arms) [57]. |
| Donor Design | 5'-end chemical modifications (e.g., AmC6) | Prevents donor degradation and multimerization, boosting integration efficiency [23]. | >5-fold increase in knock-in efficiency observed in zebrafish models [23]. |
| Screening Workflow | Fluorescence enrichment + junction PCR | Streamlines identification of true founders with germline transmission [57]. | Enabled efficient germline transmission screening for 33 connexin genes [57]. |
Experimental Protocols for High-Efficiency Knock-In
Protocol: 3' Knock-In Using Chemically Modified dsDNA Donors
This protocol outlines a cloning-free, MMEJ-based strategy for C-terminal gene tagging, which preserves endogenous gene function and has proven effective for lineage tracing [23].
Reagents and Materials
- 5' AmC6-Modified Primers: For generating protected PCR amplicons.
- Template Plasmid: Containing the cargo (e.g., fluorescent protein - 2A peptide - Cre recombinase).
- High-Fidelity DNA Polymerase: For PCR amplification of the donor.
- Cas9 Protein: For pre-assembling Ribonucleoprotein (RNP) complexes.
- sgRNA: Targeting the desired locus just upstream of the stop codon.
Procedure
- Donor DNA Synthesis:
- Amplify the knock-in cassette (e.g., p2A-EGFP-t2A-CreERT2) using PCR primers with 5' AmC6 modifications.
- Design the forward and reverse primers to include 25-35 bp homology arms corresponding to the sequences immediately flanking the target DSB site.
- Incorporate synonymous mutations in the homology arm within the sgRNA target sequence to prevent re-cleavage of the integrated donor [23].
- Purify the PCR amplicon.
RNP Complex Assembly:
- Pre-assemble Cas9 protein and sgRNA at a molar ratio of 1:2 to form RNP complexes. Incubate at 37°C for 10 minutes.
Microinjection into Zebrafish Embryos:
- Prepare the injection mixture containing:
- Cas9 RNP complexes (e.g., 300 ng/µL Cas9, 150 ng/µL sgRNA)
- Chemically modified dsDNA donor (e.g., 50-100 ng/µL)
- Inject 1-2 nL of the mixture into the cell of one-cell stage zebrafish embryos.
- Prepare the injection mixture containing:
Founder (F0) Screening and Raising:
- Raise injected embryos. Screen for mosaic F0 individuals exhibiting strong fluorescence in expected patterns at 2-5 days post-fertilization, indicating early integration.
- Raise these high-mosaicism F0 fish to adulthood.
Germline Transmission Screening:
- Outcross adult F0 fish with wild-type partners.
- Screen the resulting F1 progeny (e.g., 200 embryos) for ubiquitous fluorescence.
- Perform junction PCR and sequencing on genomic DNA from fluorescent F1 embryos to confirm precise in-frame integration.
Protocol: The S-NGG-25 MMEJ-Mediated Knock-In Strategy
This protocol leverages a simplified donor design with short homology arms and a single gRNA cutting site to achieve highly efficient, seamless integration [57].
Reagents and Materials
- S-NGG-25 Donor Plasmid: Contains the insert (e.g., eGFP) flanked by 25 bp homology arms and a single
lamGoldensgRNA site. - Cas9 mRNA: For in vivo expression of Cas9.
- Target sgRNA: Specific to the genomic locus.
lamGoldensgRNA: To linearize the donor plasmid in vivo.
Procedure
- Donor Preparation:
- Use a donor plasmid where the insert is flanked by 25 bp homology arms and a single
lamGoldensgRNA site adjacent to one arm. - The
lamGoldensequence is: 5'-GTGGTTCACGTCACCGCGCGCGG-3' [57].
- Use a donor plasmid where the insert is flanked by 25 bp homology arms and a single
Microinjection Mixture:
- Co-inject into one-cell stage embryos:
- Cas9 mRNA
- Target-specific sgRNA
lamGoldensgRNA- S-NGG-25 donor plasmid
- Co-inject into one-cell stage embryos:
Identification and Validation of Founders:
- Raise injected F0 embryos and identify potential founders based on somatic fluorescence.
- Outcross fluorescent F0 fish.
- Screen F1 offspring using a combined approach of fluorescence observation and junction PCR on caudal fin clips for robust and reliable identification of germline-transmitting founders [57].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Homology-Independent Knock-In
| Reagent / Solution | Function / Explanation | Example Application |
|---|---|---|
| AmC6-Modified Primers | 5' end-group modification that protects linear dsDNA donors from degradation and concatemerization, dramatically improving HDR and MMEJ efficiency. | Generating stable, PCR-amplified donors for 3' knock-in [23]. |
| Cas9 RNP Complexes | Pre-complexed Cas9 protein and sgRNA. Offers rapid activity, reduced off-target effects, and higher efficiency in early embryos compared to mRNA injections. | Standard delivery method for CRISPR cutting in protocols achieving >20% transmission [23]. |
| S-NGG-25 Donor | A donor design featuring short (25 bp) homology arms and a single sgRNA site for in vivo linearization. Optimized for highly efficient MMEJ-mediated integration. | Targeted insertion of fluorescent tags into multiple connexin genes [57]. |
| Polq Knockdown + AZD7648 | A combination strategy that manipulates DNA repair pathways. AZD7648 inhibits NHEJ, shifting repair towards MMEJ/HDR, while Polq knockdown blocks MMEJ, further favoring HDR. | Universal strategy in mouse embryos to achieve up to 90% knock-in efficiency; potentially applicable to zebrafish [44]. |
Visualizing the High-Efficiency Knock-In Workflow
The following diagram illustrates the logical workflow and decision points for the optimized knock-in strategies described in this note.
The consistent achievement of germline founder rates greater than 20% for precise knock-ins in zebrafish is now a feasible reality. Success hinges on the adoption of optimized, homology-independent strategies that utilize chemically modified dsDNA donors or streamlined MMEJ donors like S-NGG-25, coupled with efficient RNP-based delivery and refined screening protocols. These advanced methods provide researchers and drug development professionals with a robust framework for rapidly generating high-quality zebrafish models, thereby accelerating functional genomics and translational research.
Within zebrafish research, the selection of an appropriate genome-editing strategy is pivotal for the success of knock-in experiments. Homology-Directed Repair (HDR) has traditionally been the method for achieving precise gene modifications. However, the emergence of homology-independent strategies, notably Homology-Independent Targeted Integration (HITI), presents a powerful alternative. This application note provides a head-to-head comparison of these two methodologies, framing them within the context of a broader thesis on advancing knock-in strategies in zebrafish. We present structured quantitative data, detailed protocols, and clear pathway diagrams to equip researchers with the necessary information to select and optimize the most suitable approach for their specific experimental goals, thereby enhancing the efficiency and scope of genetic engineering in this model organism.
Quantitative Comparison at a Glance
The following tables summarize the key performance metrics and general characteristics of HDR and homology-independent methods based on data from zebrafish studies.
Table 1: Comparative Performance Metrics in Zebrafish
| Performance Metric | Homology-Directed Repair (HDR) | Homology-Independent Integration (HITI) |
|---|---|---|
| Reported Knock-in Efficiency | Highly variable (e.g., ~1.5% in early studies) [14] | Up to 75% of injected embryos showing targeted integration [14] [58] |
| Germline Transmission Rate | Can be correlated with optimized somatic HDR rates (~5.6% transmission reported with NHEJ inhibition) [7] | High germline transmission expected from high somatic efficiency; specific rates depend on construct and locus [14] |
| Ideal Template Size | Best for smaller inserts (e.g., point mutations, small tags); efficiency decreases with larger payloads [1] | Highly effective for large cassettes (>5 kb) [14] [47] |
| Cell Cycle Dependence | Requires S/G2 phases; inefficient in non-dividing cells [2] | Cell cycle-independent; functions in both dividing and non-dividing cells [59] |
| Mosaicism in P0 | Often high [7] | Can be high, but high-efficiency injection reduces screening burden [14] |
Table 2: General Characteristics and Workflow
| Characteristic | Homology-Directed Repair (HDR) | Homology-Independent Integration (HITI) |
|---|---|---|
| Underlying Mechanism | Uses endogenous homologous recombination machinery with a donor template containing homology arms [2] | Leverages the error-prone NHEJ pathway to ligate broken ends from the genome and the donor [14] [59] |
| Template Design | Requires long homology arms (often >500 bp) flanking the insert [1] | Requires short "bait" sequences on the donor plasmid that match the genomic target site [14] [16] |
| Key Advantage | High precision; seamless integration | High efficiency; works in non-dividing cells; simpler template design |
| Primary Limitation | Low efficiency, especially in vivo | Potential for off-target integration; insertions can be bidirectional without optimized design [60] |
The Scientist's Toolkit: Essential Research Reagents
This section details the crucial reagents and their functions for performing HDR and HITI knock-ins in zebrafish.
Table 3: Key Reagent Solutions for Zebrafish Knock-in
| Research Reagent | Function/Description | Application Notes |
|---|---|---|
| CRISPR-Cas9 System | Engineered nuclease (e.g., Cas9 protein or mRNA) and sgRNA to create a site-specific double-strand break (DSB). | The core nuclease for both HDR and HITI. Using Cas9 protein can increase efficiency and reduce mosaicism [7]. |
| HDR Donor Template | Single-stranded oligonucleotide (ssODN) or double-stranded DNA (dsDNA) plasmid with long homology arms (>500 bp) surrounding the desired insertion. | Used for HDR. ssODNs are ideal for point mutations. For larger insertions, dsDNA with long homology arms is required [1]. |
| HITI Donor Template | Double-stranded DNA plasmid (e.g., nanoplasmid) containing the insert flanked by sgRNA target sequences ("bait" sequences). | The bait sequences are cut by Cas9, creating ends compatible with the genomic DSB for repair via NHEJ [14] [47]. |
| Chemical Inhibitors (NU7441) | A small-molecule inhibitor of DNA-PK, a key kinase in the NHEJ pathway. | Shifts repair balance towards HDR. Shown to enhance HDR efficiency in zebrafish embryos by up to 13.4-fold [7]. |
| Chemical Enhancers (RS-1) | A small-molecule agonist of the RAD51 protein, which stimulates the HDR pathway. | Can provide a modest increase in HDR efficiency (~1.5-fold in zebrafish) [7]. |
Experimental Protocols
Protocol A: Enhancing HDR with Chemical Reprogramming
This protocol is adapted from a study that used chemical inhibition to significantly boost HDR efficiency in zebrafish embryos [7].
1. Reagent Preparation: - sgRNA and Cas9: Synthesize sgRNA targeting the genomic locus of interest. Use high-purity Cas9 mRNA or recombinant Cas9 protein. - HDR Donor Template: For a fluorescent reporter knock-in (e.g., tdTomato), prepare a dsDNA donor plasmid with homology arms (e.g., 303 bp LHA, 1022 bp RHA). Include a Cas9 target site within the homology arm to linearize the donor in vivo and boost efficiency [7]. - Chemical Modulators: Prepare stock solutions of NU7441 (50 µM final concentration) and/or RS-1 (15-30 µM final concentration) in DMSO.
2. Microinjection Mix Preparation: - Combine the following in a nuclease-free tube: - Cas9 protein (e.g., 300 ng/µL) or mRNA - sgRNA (e.g., 50 ng/µL) - HDR donor DNA (e.g., 50 ng/µL) - NU7441 (50 µM final concentration) - Optional: Fluorescent tracer dye (e.g., phenol red) - Centrifuge the mix briefly and keep it on ice until injection.
3. Zebrafish Embryo Injection: - Inject 1-2 nL of the prepared mix directly into the cell cytoplasm of 1-cell stage zebrafish embryos. - Maintain injected embryos in standard E3 embryo medium at 28.5°C.
4. Screening and Validation: - Screen F0 embryos for successful editing. For a fluorescent reporter, visualize under a fluorescence microscope at 24-72 hours post-fertilization (hpf). - For germline transmission, raise injected (F0) embryos to adulthood and outcross to wild-type fish. Screen the F1 offspring for the presence of the knock-in.
Protocol B: Efficient HITI-Mediated Knock-in
This protocol is based on a highly efficient homology-independent knock-in method developed for zebrafish [14] [58].
1. Reagent Preparation: - sgRNA and Cas9: Synthesize a highly efficient sgRNA targeting the genomic locus. Test efficiency prior to knock-in experiments (target >60% indel rate) [1]. - HITI Donor Plasmid: Clone the desired cassette (e.g., KalTA4, Venus) into a plasmid backbone. On both ends of the insert, include the same sgRNA target sequence ("bait") used for the genomic locus. Disrupt the PAM site on the donor to prevent re-cleavage after integration [14].
2. Microinjection Mix Preparation: - Combine the following in a nuclease-free tube: - Cas9 mRNA (e.g., 150 ng/µL) - sgRNA (e.g., 50 ng/µL) - HITI donor plasmid (e.g., 25 ng/µL) - Optional: Fluorescent tracer dye - Centrifuge the mix briefly and keep it on ice.
3. Zebrafish Embryo Injection: - Inject 1-2 nL of the mix into the cell cytoplasm of 1-cell stage zebrafish embryos. - Maintain injected embryos in standard E3 embryo medium at 28.5°C.
4. Screening and Validation: - For cassette exchange (e.g., eGFP to Gal4), screen for loss of the original fluorescence (eGFP) and gain of the new reporter (e.g., RFP under UAS control) in F0 embryos [14]. - For insertion into an endogenous locus without a visible marker, a PCR-based genotyping assay (e.g., junction PCR) on pooled embryos or fin clips from raised founders is required.
Signaling Pathways and Workflows
The following diagrams illustrate the core DNA repair mechanisms and experimental workflows for HDR and HITI.
DNA Repair Pathway Mechanics
HITI Experimental Workflow
Within the context of homology-independent knock-in strategies in zebrafish, functional validation is a critical step to confirm that the inserted transgene is under the precise control of the endogenous promoter and that the resulting fusion protein exhibits correct expression fidelity. This ensures that the observed expression patterns, dynamics, and levels accurately reflect those of the native gene, which is paramount for meaningful biological conclusions. This Application Note details a suite of protocols and quantitative benchmarks for rigorous functional validation, enabling researchers to verify that their knock-in lines faithfully recapitulate endogenous gene behavior.
Quantitative Benchmarks for Knock-In Validation
The table below summarizes key quantitative metrics from recent studies for assessing knock-in efficiency and validation, providing benchmarks for experimental planning and evaluation.
Table 1: Key Performance Metrics for Zebrafish Knock-In and Functional Validation
| Metric | Reported Value / Range | Application / Significance | Source |
|---|---|---|---|
| Somatic Knock-In Efficiency (F0) | 10% - 40% of injected embryos [45] | Initial screening for successful integration; varies by locus and method. | PCR tagging with short homology arms [45] |
| Germline Transmission Rate (Optimized) | >20% founder rate for precise insertions [19] | Efficiency of generating stable, heritable lines. | Chemically modified dsDNA templates [19] |
| Germline Founder Mosaicism | 5.8% - 35.4% [57] | Proportion of F1 progeny inheriting the knock-in allele from a founder. | S-25 MMEJ strategy for connexin genes [57] |
| Gene Knockdown Efficiency (Ribozyme) | Up to 20-fold reduction in mRNA [61] | Validation of functional loss-of-effect (e.g., for ribozyme-based strategies). | T3H48 self-cleaving ribozyme [61] |
| Base Editing Efficiency (F0) | Up to 87% at specific loci [9] | High-efficiency introduction of point mutations for functional testing. | "Near PAM-less" CBE4max-SpRY system [9] |
Experimental Workflow for Functional Validation
The following diagram illustrates the integrated workflow for generating a knock-in zebrafish line and performing subsequent functional validation to ensure expression fidelity.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below catalogs essential reagents and their functions for executing the described knock-in and validation protocols.
Table 2: Essential Reagents for Knock-In Generation and Functional Validation
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| CRISPR-Cas9 RNP Complex | Preassembled Cas9 protein and sgRNA for high-efficiency DSB induction. | Co-injection with HDR template for precise integration [45] [23]. |
| Chemically Modified dsDNA Donor | PCR-amplified dsDNA with 5' AmC6 modifications to enhance HDR and reduce degradation. | Serves as the repair template for high-efficiency knock-in [23] [19]. |
| S-25 MMEJ Donor | dsDNA donor with a single sgRNA cut site and 25-bp microhomology arms. | Optimized for high knock-in efficiency and seamless integration [57]. |
| Base Editor Systems (e.g., ABE-Ultramax) | Engineered fusion proteins for direct conversion of A•T to G•C base pairs without DSBs. | Reversible functional validation by introducing point mutations to inactivate sequences [61]. |
| Self-Cleaving Peptides (P2A, T2A) | Sequences inserted between the endogenous gene and reporter to enable co-translational cleavage. | Ensures the native protein and reporter are functional without fusion artifacts [23]. |
| T3H48 Self-Cleaving Ribozyme | Engineered hammerhead ribozyme for targeted knockdown of mRNA. | Validation of phenotype specificity through conditional gene knockdown [61]. |
Detailed Experimental Protocols
Protocol 1: Genotypic Validation of Knock-In Alleles
Purpose: To confirm the precise integration of the transgene at the intended genomic locus and verify the correct junction sequences.
Materials:
- Genomic DNA from fin clips or embryos.
- PCR primers: One binding within the endogenous gene (outside the homology arm) and one binding within the inserted transgene.
- High-fidelity DNA polymerase.
- Sequencing reagents.
Procedure:
- Design Primers: Design two sets of primers for bidirectional junction PCR.
- 5' Junction: Forward primer in the upstream endogenous region and reverse primer in the 5' part of the inserted sequence.
- 3' Junction: Forward primer in the 3' part of the inserted sequence and reverse primer in the downstream endogenous region.
- PCR Amplification: Perform PCR on genomic DNA from potential founders (F0) or their progeny (F1). Include a wild-type control.
- Gel Electrophoresis: Analyze PCR products by agarose gel electrophoresis. The correct knock-in will yield products of the expected size, absent in the wild-type control.
- Sanger Sequencing: Purify the PCR products and perform Sanger sequencing using the same primers to confirm the precise, in-frame fusion and the absence of unwanted mutations at the junctions [57] [23].
Protocol 2: Expression Fidelity Analysis by Imaging and In Situ Hybridization
Purpose: To compare the spatial and temporal expression pattern of the knock-in reporter with the endogenous gene's known or expected expression profile.
Materials:
- Fixed zebrafish embryos/larvae (knock-in and wild-type).
- Antibodies against the endogenous protein (if available).
- RNA in situ hybridization (ISH) probe for the endogenous gene.
- Fluorescence microscope.
Procedure:
- Live Imaging: Anesthetize and image live knock-in embryos/larvae at various developmental stages using a fluorescence microscope. Document the expression pattern of the reporter.
- Immunofluorescence (IF): If a specific antibody is available, perform IF on fixed knock-in and wild-type samples. The signal from the antibody should co-localize perfectly with the reporter signal in the knock-in, confirming the reporter faithfully labels the cells producing the endogenous protein.
- RNA In Situ Hybridization (ISH): Perform ISH on wild-type siblings using a probe for the endogenous gene. The ISH staining pattern should match the reporter fluorescence pattern in the knock-in animals. This is a powerful confirmation that the reporter is subject to the same transcriptional regulation as the native gene [23].
Protocol 3: Functional Validation via Phenotype Rescue or Knockdown
Purpose: To provide the strongest evidence that the knock-in allele is functional and that observed phenotypes are specific to the targeted gene.
Materials:
- Established mutant line for the gene of interest (loss-of-function allele).
- Base editor system (e.g., ABE-Ultramax) or ribozyme tool (e.g., T3H48).
- Microinjection setup.
Procedure - Phenotype Rescue:
- Cross the knock-in line into the background of a known mutant line that exhibits a clear phenotype (e.g., pigmentation defect, morphological abnormality).
- Analyze Progeny: If the knock-in allele is functional, it should rescue the mutant phenotype in the offspring, demonstrating that the tagged protein retains biological activity.
Procedure - Reversible Knockdown (Base Editing):
- Design a gRNA targeting the ribozyme sequence integrated into the gene's intron to inactivate it via a single base change (e.g., using ABE-Ultramax).
- Microinject the base editor components into embryos carrying the active ribozyme allele, which should exhibit a mutant phenotype (e.g., albinism).
- Score for Phenotype Reversion: Efficient base editing should reverse the ribozyme-induced knockdown, leading to a wild-type phenotype (e.g., rescued pigmentation). This confirms that the phenotype is specifically due to the ribozyme's action on that gene's mRNA and not off-target effects [61].
Data Interpretation and Troubleshooting
- Mosaic Expression in F0: Expected due to delayed integration. Focus on founders with widespread fluorescence for germline screening [57].
- Mismatched Expression Patterns: If the reporter pattern does not match ISH, consider positional effects from residual vector backbone or incomplete regulatory elements. Re-target using linear dsDNA donors with 5' modifications to avoid backbone integration [57] [19].
- Low Germline Transmission: Optimize by using chemically modified dsDNA templates and Cas9 RNP complexes. Confirm high somatic efficiency before screening for founders [45] [19].
The precise modeling of human genetic diseases in experimental organisms is crucial for understanding disease mechanisms and developing therapeutic interventions. Within the context of zebrafish research, homology-independent knock-in strategies have emerged as powerful techniques for introducing patient-specific disease-associated variants into the zebrafish genome. Unlike homology-directed repair (HDR), which remains challenging in zebrafish due to low efficiency, homology-independent approaches leverage alternative DNA repair pathways to enable efficient integration of genetic elements without extensive homology arms [10]. These methods have revolutionized our ability to create accurate zebrafish models that recapitulate human genetic conditions, providing valuable platforms for functional genomics and drug discovery.
The fundamental principle underlying homology-independent knock-in involves the concurrent cleavage of both the chromosomal target site and a donor plasmid using CRISPR/Cas9, followed by integration via non-homologous end joining (NHEJ) repair pathways [14]. This approach circumvents the major limitations of HDR in zebrafish, including variable efficiency and technical complexity, while enabling the precise insertion of various genetic elements ranging from single nucleotide changes to larger DNA cassettes exceeding 5.7 kb [14]. As such, homology-independent knock-in represents a versatile and robust methodology for incorporating human disease-associated variants into the zebrafish genome, facilitating the generation of models that more accurately mirror human genetic diseases.
Technical Approaches for Variant Insertion
CRISPR/Cas9-Mediated Homology-Independent Integration
The CRISPR/Cas9 system has dramatically simplified genome editing in zebrafish, enabling efficient introduction of double-strand breaks at specific genomic loci. For homology-independent knock-in, the approach involves co-injecting a donor plasmid containing the desired genetic modification along with CRISPR/Cas9 components (sgRNA and Cas9 mRNA or protein) into zebrafish embryos [14]. The donor plasmid is engineered to include target sequences ("bait" sequences) for the same sgRNA that directs cleavage at the chromosomal locus, ensuring concurrent cleavage of both the donor plasmid and the genomic target site.
The procedural workflow begins with the design and synthesis of sgRNAs targeting both the genomic locus of interest and the donor plasmid. High-efficiency sgRNAs are essential, with successful implementations typically using sgRNAs that induce indel mutations at rates above 66% [14]. The donor plasmid is constructed to contain the genetic element of interest (e.g., point mutations, epitope tags, or reporter cassettes) flanked by the sgRNA target sequences. When co-injected into zebrafish embryos, the simultaneous cleavage of the genomic DNA and donor plasmid activates the highly active NHEJ pathway in early zebrafish development, leading to integration of the donor sequence at the target locus [14].
This method has demonstrated remarkable efficiency, with one study reporting successful targeted integration in >75% of injected embryos when converting an eGFP transgene to Gal4 [14]. The approach is particularly valuable for creating reporter lines and introducing patient-specific mutations, as it bypasses the need for extensive homology arms and can accommodate large DNA inserts. Furthermore, the same donor plasmids and sgRNAs can be applied across different species where similar genetic tools are available, enhancing the method's versatility [14].
Optimized Point Mutation Knock-In Using ssODNs
For introducing precise point mutations that recapitulate human disease variants, single-stranded oligodeoxynucleotides (ssODNs) serve as effective repair templates when combined with CRISPR/Cas9. This approach has been systematically optimized through several key parameters that significantly impact knock-in efficiency [62].
The design of ssODNs follows specific principles to maximize knock-in rates. Asymmetric anti-sense ssODNs with homology arms of 36 and 90 nucleotides have been demonstrated to be substantially more efficient (3- to 10-fold) compared to symmetric designs [62]. These oligos are designed to be anti-sense to the PAM-containing (non-target) strand, which facilitates HDR by allowing portions of the strand to separate from the Cas9-sgRNA ribonucleoprotein complex and become available for binding to the homology arms of the oligo [62]. Additionally, the distance between the Cas9 cut site and the intended modification critically influences efficiency, with optimal placement within 10-15 nucleotides of the cut site [62].
Chemical modifications of ssODNs, particularly phosphorothioate (PS) linkages that replace phosphate oxygen atoms with sulfur at the ends of ssODNs, have shown significant improvements in knock-in efficiency by protecting against exonuclease degradation [62]. This modification, independent of oligo size, enhances the stability of the repair template and increases the likelihood of successful integration.
Table 1: Optimization Parameters for ssODN-Based Point Mutation Knock-In
| Parameter | Optimal Configuration | Impact on Efficiency | Experimental Evidence |
|---|---|---|---|
| Oligo Design | Asymmetric anti-sense (36nt/90nt arms) | 3- to 10-fold improvement | [62] |
| Cut Site Distance | <15 nucleotides (ideal: <10nt) | 70-80% reduction at 20nt distance | [62] |
| Chemical Modification | Phosphorothioate (PS) linkages at ends | Significant improvement | [62] |
| Strand Orientation | Anti-sense to PAM-containing strand | Substantial efficiency gain | [62] |
Experimental Protocol for Precise Knock-In
sgRNA Design and Validation
The initial step in precise knock-in involves the design and validation of highly efficient sgRNAs. Target sites are selected using bioinformatic tools such as SSC for efficiency prediction and CC-Top for off-target prediction [62]. The target site should be located in close proximity to the intended modification site (within 10-15 nucleotides for point mutations) to maximize knock-in efficiency [62]. For homology-independent approaches using plasmid donors, the sgRNA should target both the genomic locus and the donor plasmid "bait" sequence [14].
sgRNA synthesis typically involves an overlap-extension PCR of sense sgRNA oligos combined with a reverse sgRNA-scaffold oligo, followed by in vitro transcription using systems such as the HiScribe T7 Quick High Yield RNA Synthesis kit [63]. The synthesized sgRNA is then purified using RNA cleanup kits, with careful attention to maintaining RNA integrity.
Validation of sgRNA efficiency is performed by injecting the sgRNA with Cas9 mRNA or protein into wild-type zebrafish embryos and assessing indel formation rates. This is typically done by pooling injected embryos, extracting genomic DNA, performing locus-specific PCR amplification, and analyzing mutation rates through sequencing of individual PCR clones or using fluorescent PCR methods like CRISPR-STAT [63]. Only sgRNAs demonstrating high efficiency (typically >50% indel formation) should be proceeded with for knock-in experiments.
Donor Template Design and Preparation
For point mutations, asymmetric anti-sense ssODNs are designed with 36-nucleotide and 90-nucleotide homology arms, with the modification positioned close to the cut site [62]. These ssODNs can be ordered as ultramers from commercial suppliers and resuspended in TE buffer to 100 μM concentration [63]. For larger insertions, donor plasmids are constructed to include the genetic element of interest flanked by sgRNA target sequences, with the entire cassette typically inserted between the bait sequences [14].
When designing knock-in templates, incorporating silent mutations to modify the PAM site or introduce restriction sites can facilitate subsequent screening and genotyping [62]. These modifications prevent re-cleavage of successfully edited alleles and provide convenient markers for validation.
Microinjection and Embryo Handling
Zebrafish embryos are collected immediately after spawning and maintained in E3 embryo medium. Injection mixtures are prepared containing:
- sgRNA (50-100 ng/μL)
- Cas9 mRNA (150-300 ng/μL) or Cas9 protein (300-500 ng/μL)
- Donor template: ssODN (10-100 ng/μL) or plasmid DNA (25-50 ng/μL)
The components are mixed in nuclease-free water with appropriate buffers and injected into the cell of one-cell stage embryos using standard microinjection techniques [63]. Injected embryos are incubated at 28°C in E3 medium and monitored for development.
Screening and Validation of Knock-In Events
A critical challenge in knock-in generation is the identification of precise integration events amid predominantly mosaic editing. Fluorescent PCR-based screening methods provide a robust and sensitive approach for detecting knock-in events [63]. This protocol involves several key steps:
DNA Extraction: Genomic DNA is extracted from pooled embryos or fin clips of potential founders using extraction solutions such as Tissue Preparation Solution followed by Neutralization Solution B [63].
Fluorescent PCR Amplification: Target regions are amplified using M13F-tailed forward primers and PIG-tailed reverse primers with fluorescent labeling. The PCR products are then subjected to capillary electrophoresis, which allows precise sizing of amplification products and detection of knock-in alleles based on size differences [63].
Restriction Digest Validation: For point mutations that introduce or alter restriction sites, PCR products can be digested with appropriate restriction enzymes and analyzed by capillary electrophoresis to confirm precise editing [63].
Sequencing Confirmation: Potential knock-in events identified through fluorescent PCR are confirmed by Sanger sequencing to verify the precise integration and rule out unintended modifications.
This screening approach allows researchers to distinguish knock-in alleles from wild-type and NHEJ-induced indels with high sensitivity, even in mosaic founders [63]. The method is scalable and can be adapted for high-throughput screening of multiple founders.
Diagram 1: Homology-Independent Knock-In Workflow for Zebrafish Disease Modeling
Validation and Functional Analysis
Germline Transmission and Stable Line Establishment
Following the identification of somatic knock-in events in injected embryos (G0), potential founders are raised to adulthood and outcrossed with wild-type fish to assess germline transmission. progeny (F1) from these crosses are screened using the same fluorescent PCR methods to identify individuals carrying the precise knock-in allele [63]. Typically, multiple founders should be screened, as germline transmission rates can vary significantly.
For established knock-in lines, comprehensive molecular characterization is essential. This includes:
- Sequencing of both alleles to confirm the precise integration and identify any unintended modifications
- Quantitative analysis of transcript levels using RT-PCR to assess potential effects on gene expression
- Immunohistochemistry or Western blotting to verify protein expression and localization for tagged genes
- Functional assays specific to the targeted gene to validate physiological relevance
Phenotypic Characterization of Disease Models
The phenotypic characterization of knock-in models should be tailored to the specific human disease being modeled. For monogenic disorders, this typically involves:
- Comprehensive morphological assessment during development
- Behavioral analyses relevant to the neurological, muscular, or other systems affected
- Physiological measurements (e.g., cardiac function, metabolic parameters)
- Histopathological examination of relevant tissues
- Response to established therapies or chemical modulators
Long-term studies may be necessary for late-onset disorders, requiring careful monitoring of age-related phenotypic progression. Comparative analyses with existing models (e.g., knockout lines) can help distinguish gain-of-function from loss-of-function mechanisms.
Research Reagent Solutions
Table 2: Essential Research Reagents for Homology-Independent Knock-In in Zebrafish
| Reagent Category | Specific Examples | Function and Application | Key Considerations |
|---|---|---|---|
| CRISPR/Cas9 Components | Cas9 mRNA, Cas9 protein, sgRNA | Introduction of site-specific double-strand breaks | High-purity synthesis critical for efficiency; protein can reduce mosaicism |
| Donor Templates | ssODNs (Ultramers), Plasmid donors | Provide template for desired genetic modification | Asymmetric design with phosphorothioate modifications enhances efficiency |
| Screening Reagents | Fluorescent primers, Restriction enzymes, Capillary electrophoresis standards | Detection and validation of precise knock-in events | Fluorescent PCR enables sensitive detection in mosaic founders |
| Microinjection Supplies | Injection needles, Micromanipulators, E3 embryo medium | Delivery of components to zebrafish embryos | Needle calibration critical for embryo viability |
| Bioinformatics Tools | SSC, CC-Top, CRISPOR | sgRNA design and off-target prediction | Multiple tools recommended for comprehensive design |
Advanced Applications and Integration with Variant Effect Mapping
The application of homology-independent knock-in in zebrafish disease modeling is significantly enhanced when integrated with emerging technologies for variant effect mapping. Multiplex assays of variant effect (MAVEs), including deep mutational scanning (DMS) and massively parallel reporter assays (MPRAs), provide high-throughput functional data on thousands of variants simultaneously [64]. These approaches can prioritize variants for in vivo modeling in zebrafish based on their functional impact.
Recent advances in machine learning approaches, such as DeepRVAT (deep rare variant association testing), demonstrate how variant annotations can be integrated to improve the identification of pathogenic variants [65]. These models use deep set networks to learn trait-agnostic gene impairment scores from diverse variant annotations, including missense impact scores (SIFT, PolyPhen2, AlphaMissense), deleteriousness scores (CADD, ConDel), and functional predictions (PrimateAI, SpliceAI) [65]. The resulting scores can guide the selection of variants most likely to have phenotypic consequences when introduced into zebrafish models.
The combination of multiplex functional data and optimized knock-in techniques creates a powerful pipeline for systematic disease variant characterization. This integrated approach moves beyond single-variant modeling toward comprehensive functional annotation of human genetic variation, with zebrafish providing the crucial in vivo validation platform within a vertebrate system.
Diagram 2: Integrated Pipeline for Functional Variant Characterization in Zebrafish
The adult zebrafish brain exhibits remarkable regenerative and neurogenic capabilities, making it a powerful model for studying brain development, disease, and repair. A significant challenge in this field has been the long-term tracking of specific neural cell populations from embryonic stages into adulthood. This application note details how homology-independent knock-in strategies, combined with the Zebrabow system, provide a robust methodological framework for heritable, multicolor labeling of cells, enabling precise lineage tracing and population dynamics analysis in the adult zebrafish brain.
Homology-independent knock-in is a CRISPR/Cas9-mediated genome editing technique that facilitates the targeted integration of large DNA cassettes into specific genomic loci without the need for homologous recombination. This method leverages the cell's non-homologous end joining (NHEJ) repair pathway, which is highly active in zebrafish [14]. The process involves the co-injection of a Cas9 nuclease, a guide RNA (sgRNA) targeting the genomic locus of interest, and a "bait" donor plasmid containing the same sgRNA target sites flanking the insert cassette [14]. Concurrent cleavage of both the genome and the donor plasmid leads to the integration of the entire cassette or fragments thereof at the target site. This approach has been used successfully to convert existing eGFP lines into Gal4 driver lines and to knock-in reporter genes like Venus and turboRFP under the control of endogenous promoters, such as otx2 and pax2a, faithfully recapitulating native expression patterns [16]. For studies of the adult brain, this strategy allows for the precise, heritable labeling of neural stem cells and specific neuronal populations from the earliest stages of development.
Key Experimental Protocols
Protocol: Homology-Independent Knock-in for Endogenous Gene Tagging
This protocol is adapted from Auer et al. (2014) and Kizil et al. (2017) for labeling genes expressed in the Midbrain-Hindbrain Boundary (MHB), a region pertinent to adult brain structure [14] [16].
- sgRNA Design and Synthesis: Design sgRNAs to target a sequence approximately 500 base pairs upstream of the ATG start codon of your gene of interest (e.g., otx2 or pax2a) using web tools like CHOP-CHOP. The target site should be unique in the genome to minimize off-target effects. Synthesize sgRNA using T7 polymerase in vitro transcription [16].
- Donor Plasmid Construction: Clone the sgRNA "bait" target sequence into a plasmid vector upstream of a promoter-less reporter gene, such as a fast-maturing fluorescent protein (e.g., Venus, turboRFP). The bait sequence must be identical to the genomic target. The final donor plasmid should have the structure:
[Bait Sequence]-[Reporter Gene][14] [16]. - Microinjection into Zebrafish Embryos: Co-inject the following into the cytoplasm of one-cell stage zebrafish embryos:
- Cas9 mRNA (e.g., 100-300 pg)
- sgRNA (e.g., 25-100 pg)
- Donor plasmid (e.g., 25-100 pg)
- Screening and Establishment of Stable Lines: Raise injected embryos (F0) and screen for reporter expression at the expected developmental stages and anatomical locations. Outcross founder (F0) fish exhibiting correct expression patterns to wild-type fish to establish stable F1 transgenic lines. Outcross for multiple generations (e.g., >6) to dilute potential off-target mutations [16].
Protocol: Multicolor Labeling with the Zebrabow System for Clonal Analysis
This protocol, based on the Zebrabow (Zebrafish Brainbow) system, enables the stochastic multicolor labeling of cells for lineage tracing [66].
- Transgenic Line Selection: Utilize existing transgenic lines such as
ubi:Zebrabowfor ubiquitous expression orUAS:Zebrabowin combination with tissue-specific Gal4 drivers. The Zebrabow construct contains a promoter followed by cassettes for fluorescent proteins (e.g., RFP, YFP, CFP) flanked by incompatible lox sites [66]. - Induction of Cre Recombinase: Cross the Zebrabow reporter line to a line expressing Cre recombinase under a tissue-specific or inducible promoter (e.g.,
pax2a:CreERT2). To induce recombination, treat larvae or juvenile fish with 4-Hydroxytamoxifen (4-OHT). The concentration and duration of treatment can be optimized to modulate the number of colors generated. A typical working concentration is 5 µM 4-OHT for 24-48 hours [66]. - Imaging and Color Analysis in Adult Brain: In adult fish, the resulting stochastic recombination events lead to combinatorial expression of the fluorescent proteins, granting each progenitor cell and its clonal progeny a unique spectral signature. Analyze fish using confocal microscopy. The colors are stable over time and through cell divisions, allowing long-term clonal analysis in brain structures like the tectum and telencephalon [66].
The Scientist's Toolkit: Essential Research Reagents
| Research Reagent | Function & Application in Adult Zebrafish Brain Research |
|---|---|
| Zebrabow Transgenes | Provides a multicolor palette for stochastic labeling of adjacent cells and long-term lineage tracing in neurogenic zones [66]. |
| CRISPR/Cas9 System | Enables homology-independent knock-in for precise, heritable labeling of endogenous genes (e.g., otx2, pax2a) [14] [16]. |
| Tissue-Specific Cre/Lines | Drives recombination (e.g., in Zebrabow) or reporter expression in specific neural cell types or brain regions (e.g., telencephalon) [66]. |
| 4-Hydroxytamoxifen (4-OHT) | Induces CreER[T2] activity for temporal control of genetic recombination, allowing labeling at specific developmental timepoints [66]. |
| Pan-neuronal GCamp6s | Enables in vivo calcium imaging and functional analysis of neuronal activity in freely behaving adult zebrafish [67]. |
Quantitative Data from Key Methodologies
Table 1: Performance Metrics of Homology-Independent Knock-in and Zebrabow Labeling
| Method / Parameter | Typical Efficiency / Value | Key Quantitative Findings / Outcome |
|---|---|---|
| CRISPR Knock-in Efficiency | Varies by target; high efficiency reported for some loci. | Successful conversion of eGFP to KalTA4 in >75% of injected embryos; ~22% showed widespread RFP expression recapitulating the original pattern [14]. |
| Zebrabow Color Diversity | Optimized by modulating Cre activity. | The broadly expressed ubi:Zebrabow line provides diverse color profiles. Colors remain stable throughout embryonic and larval stages and are inherited equally by daughter cells [66]. |
| Spatial Information from Labeled Cells | Decoding precision from population code. | The activity of telencephalic place cells can be used to decode the animal's spatial location to within a median of 6.69 ± 2.34 mm [67]. |
| Neuronal Yield in Telencephalon | Enrichment of functionally defined cells. | The telencephalon contains the highest fraction of spatially specific cells (69 ± 14%), despite comprising only ~8% of recorded neurons [67]. |
Visualizing the Workflows
Homology-Independent Knock-In
Zebrabow Lineage Tracing Strategy
Conclusion
Homology-independent knock-in has emerged as a simple, flexible, and highly efficient method for precise genome engineering in zebrafish, effectively overcoming the historical barriers of low HDR efficiency. By leveraging the cell's endogenous NHEJ repair pathway and optimized parameters such as chemical inhibition of NHEJ and the use of modified templates, researchers can now achieve high rates of germline transmission for large DNA insertions. This robust strategy has profoundly expanded the zebrafish genetic toolbox, enabling the creation of sophisticated reporter lines, accurate disease models, and conditional alleles with unprecedented reliability. As the field advances, the continued refinement of this technology promises to further accelerate functional genomics and the in vivo modeling of human diseases, solidifying the zebrafish's role as an indispensable model in biomedical research and therapeutic discovery.
References
- 1. Homology-Directed Repair in Zebrafish: Witchcraft and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7793982/]
- 2. CRISPR-Cas9-mediated homology-directed repair for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11531618/]
- 3. HDR vs. NHEJ: CRISPR Gene Editing Techniques [https://www.zeclinics.com/blog/hdr-vs-nhej-in-crispr-gene]
- 4. Efficient high-precision homology-directed repair ... [https://www.nature.com/articles/s41592-023-01949-1]
- 5. Prime editing outperforms homology-directed repair as a ... [https://pubmed.ncbi.nlm.nih.gov/40437234/]
- 6. Homology-directed repair FAQs [https://www.takarabio.com/learning-centers/gene-function/gene-editing/crisprcas9-knockins/homology-directed-repair-faqs?srsltid=AfmBOoo_x9XMgcrvyagcXjA-kqY_0_Qc08hygEWvhZ8vQaMdFCg2m5ZC]
- 7. Chemical reprogramming enhances homology-directed ... [https://www.nature.com/articles/s42003-019-0444-0]
- 8. Optimised genome editing for precise DNA insertion and ... [https://elifesciences.org/reviewed-preprints/107475]
- 9. Base editors in zebrafish: a new era for functional ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1598887/full]
- 10. Genome editing using CRISPR/Cas9-based knock-in ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202316302833]
- 11. CRISPR-Cas9-mediated homology-directed repair for ... [https://www.sciencedirect.com/science/article/pii/S2162253124002312]
- 12. DNA repair by nonhomologous end joining and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2754209/]
- 13. Inhibition of non-homologous end joining increases the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4618510/]
- 14. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3875856/]
- 15. Precise in-frame integration of exogenous DNA mediated ... [https://www.nature.com/articles/srep08841]
- 16. CRISPR/Cas9-Mediated Zebrafish Knock-in as a Novel ... [https://www.frontiersin.org/journals/neuroanatomy/articles/10.3389/fnana.2017.00052/full]
- 17. Improving CRISPR/Cas9 mutagenesis efficiency by delaying the early development of zebrafish embryos | Scientific Reports [https://www.nature.com/articles/s41598-020-77677-9]
- 18. Exogenous gene integration mediated by genome editing technologies in zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5470516/]
- 19. Identifying optimal conditions for precise knock-in of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12211562/]
- 20. Zebrafishology, study design guidelines for rigorous and ... [https://www.nature.com/articles/s42003-025-07496-z]
- 21. Knock-in of large reporter genes in human cells via ... [https://pubmed.ncbi.nlm.nih.gov/26850641/]
- 22. Efficient generation of knock-in transgenic zebrafish ... [https://www.nature.com/articles/srep06545]
- 23. Efficient knock-in method enabling lineage tracing ... [https://www.life-science-alliance.org/content/6/5/e202301944]
- 24. Highly efficient generation of knock-in transgenic medaka by ... [https://zoologicalletters.biomedcentral.com/articles/10.1186/s40851-017-0086-3]
- 25. CRISPR Transfection Protocols Guide: How To Select The ... [https://www.synthego.com/guide/how-to-use-crispr/transfection-protocols/]
- 26. A robust pipeline for efficient knock-in of point mutations and ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08971-1]
- 27. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12282252/]
- 28. Enhanced RNA-targeting CRISPR-Cas technology in ... [https://www.nature.com/articles/s41467-025-57792-9]
- 29. Effects of concentration of CRISPR/Cas9 components on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584178/]
- 30. Improved selection of zebrafish CRISPR editing by early ... [https://www.nature.com/articles/s41598-023-27503-9]
- 31. Precise and efficient genome editing in zebrafish using the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4299274/]
- 32. CRISPR-based functional genomics tools in vertebrate ... [https://www.nature.com/articles/s12276-025-01514-0]
- 33. Efficient targeted integration directed by short homology in ... [https://elifesciences.org/articles/53968]
- 34. Generation of a zebrafish knock-in line expressing MYC- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9050874/]
- 35. Generation of a zebrafish knock-in line expressing MYC ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X22004429]
- 36. Endogenous zebrafish proneural Cre drivers generated by ... [https://www.nature.com/articles/s41598-021-81239-y]
- 37. Cre/lox regulated conditional rescue and inactivation with ... [https://elifesciences.org/articles/71478]
- 38. CRISPR/Cas9-Mediated Knockin and Knockout in Zebrafish [https://www.ncbi.nlm.nih.gov/books/NBK536362/]
- 39. CRISPR/Cas9-mediated Conversion of eGFP- Into Gal4- ... [https://pubmed.ncbi.nlm.nih.gov/25393779/]
- 40. Functional visualization and disruption of targeted genes ... [https://www.nature.com/articles/srep34991]
- 41. Long-read sequencing for fast and robust identification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10954187/]
- 42. CRISPR/Cas9-mediated precise genome modification by a ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6493-4]
- 43. Pharmacological inhibition of DNA-PK stimulates Cas9- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4550049/]
- 44. Refined DNA repair manipulation enables a universal ... [https://www.nature.com/articles/s41467-025-61696-z]
- 45. Fast, precise and cloning-free knock-in of reporter sequences ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10323249/]
- 46. Homology-Directed Repair in Zebrafish: Witchcraft and ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2020.595474/full]
- 47. Homology-independent targeted insertion (HITI) enables ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01799-7]
- 48. T cell gene targeting and selection using non-viral intron ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12261302/]
- 49. One-step efficient generation of dual-function conditional ... [https://elifesciences.org/articles/48081]
- 50. In vivo genome editing via CRISPR/Cas9-mediated ... [https://www.nature.com/articles/s41467-024-48092-9]
- 51. Activities and specificities of CRISPR/Cas9 and Cas12a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6320322/]
- 52. Identifying optimal conditions for precise knock-in of ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/40446205/]
- 53. Comparison of DNA targeting CRISPR editors in human cells [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-023-00958-z]
- 54. Comparison of Cas12a and Cas9-mediated mutagenesis ... [https://www.nature.com/articles/s41598-024-55088-4]
- 55. CRISPR-Cas9/Cas12a systems for efficient genome ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1452496/full]
- 56. Increasing Cas12a- and Cas9-mediated deletion sizes with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12581816/]
- 57. Optimal tagging strategies for illuminating expression ... [https://www.nature.com/articles/s42003-023-05686-1]
- 58. Highly efficient CRISPR/Cas9-mediated knock-in ... [https://pubmed.ncbi.nlm.nih.gov/24179142/]
- 59. In vivo genome editing via CRISPR/Cas9 mediated ... [https://www.nature.com/articles/nature20565]
- 60. CRISPR-Cas9 homology-independent targeted integration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10495553/]
- 61. A recombinase-activated ribozyme to knock down ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11856399/]
- 62. Optimized knock-in of point mutations in zebrafish using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6158492/]
- 63. Fluorescent PCR–based Screening Methods for Precise ... [https://bio-protocol.org/en/bpdetail?id=4732&type=0]
- 64. Linking genome variants to disease: scalable approaches ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8490018/]
- 65. Integration of variant annotations using deep set networks ... [https://www.nature.com/articles/s41588-024-01919-z]
- 66. Zebrabow: multispectral cell labeling for cell tracing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3678346/]
- 67. A population code for spatial representation in the ... [https://www.nature.com/articles/s41586-024-07867-2]