Low-Throughput scRNA-seq for Embryonic Research: A Detailed Workflow Guide from Cell Isolation to Validation
This article provides a comprehensive guide for researchers and drug development professionals on implementing low-throughput single-cell RNA sequencing (scRNA-seq) for embryonic studies. It covers the foundational principles of why low-throughput methods are uniquely suited for precious embryo samples, details step-by-step methodological protocols from single-cell isolation to library preparation, offers solutions for common troubleshooting and optimization challenges, and outlines rigorous validation and comparative analysis techniques. By integrating the latest advancements and best practices, this resource aims to empower scientists to effectively leverage low-throughput scRNA-seq to unravel cellular heterogeneity and lineage trajectories in embryonic development.
Low-Throughput scRNA-seq for Embryonic Research: A Detailed Workflow Guide from Cell Isolation to Validation
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing low-throughput single-cell RNA sequencing (scRNA-seq) for embryonic studies. It covers the foundational principles of why low-throughput methods are uniquely suited for precious embryo samples, details step-by-step methodological protocols from single-cell isolation to library preparation, offers solutions for common troubleshooting and optimization challenges, and outlines rigorous validation and comparative analysis techniques. By integrating the latest advancements and best practices, this resource aims to empower scientists to effectively leverage low-throughput scRNA-seq to unravel cellular heterogeneity and lineage trajectories in embryonic development.
Why Low-Throughput scRNA-seq is Indispensable for Embryo Research
Single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology by enabling the resolution of cellular heterogeneity during embryogenesis. For embryonic studies, where starting material is often extremely limited, low-throughput scRNA-seq methods provide an essential toolset for high-resolution transcriptomic profiling. These approaches, typically processing dozens to a few hundred cells per experiment [1], stand in contrast to high-throughput methods that analyze thousands to millions of cells. The strategic application of low-throughput scRNA-seq is particularly valuable for investigating rare embryonic cell types, characterizing lineage specification events, and validating stem cell-derived embryo models with enhanced sensitivity and analytical depth [2] [3].
In the context of human embryonic development, research faces significant challenges due to ethical considerations, technical limitations, and scarce biological material [3]. Low-throughput scRNA-seq methodologies address these constraints by maximizing information yield from minimal input, sometimes even at the level of individual cells. This capability has proven fundamental for creating comprehensive reference atlases of human development [4] and for elucidating transcriptional dynamics during critical developmental windows such as preimplantation stages and gastrulation [3]. As the field progresses toward more sophisticated embryo models, low-throughput scRNA-seq remains indispensable for authenticating these systems against in vivo reference data [4].
Defining Scope and Scale: Key Characteristics of Low-Throughput scRNA-seq
Operational Parameters and Technical Specifications
Low-throughput scRNA-seq methods are distinctly characterized by their cell processing capacity, which generally ranges from dozens to a few hundred cells per experiment [1]. This stands in stark contrast to high-throughput microdroplet systems, which can profile hundreds of thousands to millions of cells in a single run [5] [1]. The defining feature of low-throughput approaches is their emphasis on analytical depth over cell volume, often achieving more comprehensive transcriptome coverage per cell through more extensive RNA sequencing [6].
The operational boundaries of low-throughput scRNA-seq can be delineated by several key parameters, as summarized in Table 1:
Table 1: Key Parameters Defining Low-Throughput scRNA-seq for Embryonic Studies
| Parameter | Low-Throughput Scope | Representative Technologies | Embryonic Study Applications |
|---|---|---|---|
| Cell Throughput | Dozens to few hundred cells per experiment [1] | Fluidigm C1, SMART-seq2, Plate-based methods [6] [7] | Analysis of rare embryonic cell populations, limited embryo samples |
| Sequencing Depth | High coverage per cell (full-length transcript preferred) | Smart-seq2 [6] | Alternative splicing analysis, allele-specific expression, comprehensive transcriptome characterization |
| Cell Isolation Approach | Mechanical manipulation, FACS, manual picking [1] [6] | Micromanipulation, FACS, limiting dilution [6] | Precise selection of specific embryonic cell types based on morphology or markers |
| mRNA Capture Efficiency | 10-20% of transcripts reverse transcribed [6] | Poly(dT) primers with template switching [6] | Critical for detecting low-abundance transcripts in early embryos |
| Amplification Method | PCR or in vitro transcription [6] | SMARTer technology [2] | Linear amplification for minimal bias in precious samples |
| Unique Molecular Identifiers | Optional implementation [6] | Barcoded reverse transcription primers [6] | Quantitative molecular counting for transcriptional bursting studies |
Comparative Analysis of Low- vs. High-Throughput Approaches
The strategic selection between low- and high-throughput scRNA-seq methodologies involves careful consideration of their complementary strengths and limitations. Low-throughput methods excel in scenarios requiring deep transcriptional profiling, full-length transcript coverage, and maximized mRNA recovery from limited cell numbers - all common requirements in embryonic research [6] [2]. These platforms typically employ microfluidic chambers (e.g., Fluidigm C1) or plate-based setups that provide superior control over reaction conditions and enable more efficient mRNA capture compared to high-throughput droplet systems [5].
High-throughput methods, in contrast, prioritize cell number scalability and cost efficiency at the expense of transcriptome completeness per cell [5]. They typically sequence only the 5' or 3' ends of transcripts and have lower mRNA capture rates, making them better suited for comprehensive atlas-building of heterogeneous tissues where identifying all cell types takes precedence over deep molecular characterization of each cell [6]. For embryonic studies, the choice between these approaches often depends on the specific research question: high-throughput for comprehensive cellular census across developmental stages, and low-throughput for deep mechanistic investigation of specific lineage decisions or rare cell populations.
Experimental Protocols for Embryonic Studies
Sample Preparation and Cell Isolation
The initial phase of low-throughput scRNA-seq for embryonic material requires meticulous sample preparation to preserve RNA integrity and ensure representative cell capture. For preimplantation embryos, careful zymogen removal and zona pellucida dissolution are critical first steps, followed by gentle dissociation to individual blastomeres using enzymatic treatments (e.g., Trypsin-EDTA) tailored to embryonic stage [3]. Cell isolation represents perhaps the most critical step, with several approaches available:
- Limiting Dilution: A straightforward method where cell suspensions are diluted to approximately 0.5 cells per aliquot and manually dispensed into multi-well plates [6]. While technically simple, this approach is inefficient, with only about one-third of prepared wells typically containing a single cell.
- Micromanipulation: Using microscope-guided capillary pipettes to extract individual cells from embryo suspensions [6]. This method provides visual confirmation of cell integrity and is particularly valuable for targeting specific blastomeres based on spatial position or morphological criteria.
- Fluorescence-Activated Cell Sorting (FACS): The most commonly used method for obtaining highly purified single cells [6]. FACS enables selection based on fluorescent markers (e.g., lineage-specific reporters) or physical parameters (size, granularity). This method requires careful optimization of pressure and nozzle size to maintain embryonic cell viability.
- Laser Capture Microdissection: Utilizes a laser system to isolate specific cells from tissue sections [6], particularly relevant for later embryonic stages or specific anatomical regions.
Table 2: Critical Reagents for Embryonic scRNA-seq Sample Preparation
| Reagent Category | Specific Examples | Function in Protocol | Considerations for Embryonic Samples |
|---|---|---|---|
| Dissociation Reagents | Trypsin-EDTA, Accutase | Breakdown of embryonic cell-cell junctions | Concentration and exposure time must be optimized for developmental stage |
| Cell Viability Dyes | Propidium iodide, DAPI, Calcein AM | Distinguish live/dead cells during sorting | Potential toxicity requires minimal exposure |
| Surface Marker Antibodies | CD34, CD133, CD45 [8] | FACS isolation of specific progenitor populations | Validated clones with demonstrated specificity for embryonic epitopes |
| Nuclease Inhibitors | RNaseOUT, RiboLock | Preserve RNA integrity during processing | Essential given extended processing times of low-throughput methods |
| Cell Culture Media | KSOM, DMEM/F12 with supplements | Maintain cell viability during processing | Stage-specific formulations to minimize transcriptional stress responses |
Following isolation, cells are immediately lysed in hypotonic buffers containing denaturants (e.g., guanidine thiocyanate) and nuclease inhibitors to preserve RNA integrity and prevent degradation [6]. The inclusion of spike-in RNA controls (e.g., ERCC RNA Spike-In Mix) is strongly recommended at this stage to enable subsequent quality control and normalization steps [7].
Library Preparation and Sequencing
Library construction for low-throughput scRNA-seq emphasizes transcript completeness and detection sensitivity. The SMART-seq2 protocol has emerged as a gold standard for embryonic studies due to its full-length transcript coverage and enhanced sensitivity for low-input samples [6] [7]. The key steps include:
- Reverse Transcription: Using engineered Moloney murine leukemia virus reverse transcriptase with low RNase H activity and increased thermostability [6]. Template-switching oligonucleotides are incorporated to ensure uniform coverage while maintaining strand specificity.
- cDNA Amplification: Employing limited-cycle PCR (typically 18-22 cycles) to amplify cDNA without introducing substantial bias [6]. Alternatively, in vitro transcription methods can provide linear amplification but require additional steps and time.
- Library Construction: Fragmenting amplified cDNA, followed by adapter ligation and additional PCR amplification to incorporate sequencing compatibilities [6]. Unique dual indices should be used to enable sample multiplexing.
For embryonic studies, special consideration must be given to the high proportion of ribosomal RNA and the relatively low mRNA content in individual blastomeres. Poly(A) selection using poly(dT) primers remains the standard approach for enriching mRNA, although methods for capturing non-polyadenylated transcripts are available for specific applications [2]. The incorporation of Unique Molecular Identifiers (UMIs) - random 4-8 bp sequences included in the reverse transcription primers - enables precise quantification by correcting for PCR amplification bias, though this typically comes at the cost of full-length transcript information [6].
Sequencing depth requirements depend on the specific biological questions. For comprehensive transcriptome characterization, a minimum of 1-2 million reads per cell is recommended, while targeted analyses may require less depth [6]. The use of spike-in controls enables accurate normalization across cells and experimental batches, which is particularly important when comparing across developmental stages or experimental conditions [7].
Data Analysis Workflow for Embryonic scRNA-seq
The analysis of low-throughput scRNA-seq data from embryonic samples requires specialized computational approaches that address the unique characteristics of these datasets. The following workflow outlines the key steps from raw data processing to biological interpretation:
Quality Control and Normalization
Quality control represents a critical first step in scRNA-seq analysis, particularly for embryonic data where sample quality can be highly variable. Key metrics include:
- Library Size: The total sum of counts across all features per cell. Cells with unusually small library sizes indicate poor RNA capture efficiency [7].
- Number of Expressed Features: The count of genes with non-zero counts per cell. Cells with very few detected genes suggest poor quality or incomplete capture [7].
- Spike-in Proportions: The percentage of reads mapping to exogenous spike-in transcripts. Deviations from expected proportions indicate technical artifacts [7].
- Mitochondrial RNA Content: The fraction of reads mapping to mitochondrial genes. Elevated levels often indicate cellular stress or apoptosis [7].
Following quality control, normalization addresses technical variations between cells. For data with spike-in controls, methods like BASiCS use these exogenous standards to separate technical noise from biological heterogeneity [7]. For data without spike-ins, approaches like scran implement pooling-based normalization to stabilize variance estimates [7]. For embryonic studies, special consideration should be given to cell cycle effects, which can be pronounced in rapidly dividing embryonic cells. Computational correction methods (e.g., scran's cyclone classifier) can identify cell cycle phase and regress out these effects [7].
Dimensionality Reduction and Cell Type Identification
The high-dimensional nature of scRNA-seq data necessitates dimensionality reduction for visualization and interpretation. Principal component analysis (PCA) typically serves as the initial step, followed by non-linear methods such as t-distributed Stochastic Neighbor Embedding (t-SNE) or Uniform Manifold Approximation and Projection (UMAP) [4]. For embryonic studies, where developmental trajectories are continuous, UMAP often provides superior visualization of lineage relationships [4].
Clustering algorithms (e.g., Louvain, Leiden) group cells based on transcriptional similarity, enabling identification of distinct cell types or states [7]. The resolution parameters of these algorithms should be carefully tuned to match the biological context - higher resolution for fine-grained separation of closely related progenitors, lower resolution for broad lineage classification. Following clustering, marker gene identification algorithms (e.g., Wilcoxon rank-sum test, MAST) statistically compare gene expression between clusters to identify defining transcriptional signatures [7].
For embryonic development studies, trajectory inference methods (e.g., Slingshot, Monocle) can reconstruct temporal ordering of cells along differentiation pathways, even from snapshot data [4]. These methods model the transcriptional dynamics of lineage specification, identifying genes associated with fate decisions and branch points in developmental trajectories.
Applications in Embryonic Development Research
Lineage Specification and Cell Fate Decisions
Low-throughput scRNA-seq has dramatically advanced our understanding of lineage specification during embryonic development. By profiling individual cells from preimplantation embryos, researchers have delineated the transcriptional programs underlying the first lineage decisions - segregation of the inner cell mass (ICM) from the trophectoderm (TE) [4] [3]. These studies have identified key transcription factors (e.g., NANOG and GATA4 for ICM; GATA2 and GATA3 for TE) and revealed the existence of intermediate cell states that were previously unrecognized [3].
In later developmental stages, low-throughput scRNA-seq has enabled the deconstruction of complex processes such as gastrulation, where the three germ layers (ectoderm, mesoderm, and endoderm) are established. Studies of human gastrula stages have identified distinct subtypes within the primitive streak, mesoderm, and endoderm lineages, revealing unexpected heterogeneity in these foundational populations [4]. The high sensitivity of full-length transcript protocols has been particularly valuable for detecting low-abundance transcription factors that drive these fate decisions.
Validation of Embryo Models
The emergence of stem cell-derived embryo models (e.g., blastoids, gastruloids) represents a promising approach for studying early human development while addressing ethical and technical limitations [4] [3]. Low-throughput scRNA-seq serves as a critical validation tool for assessing the fidelity of these models by comparing their transcriptional profiles to in vivo reference data [4].
Integrated analysis pipelines, such as the stabilized UMAP projection method described by Chen et al., enable quantitative assessment of transcriptional similarity between model systems and natural embryos [4]. These approaches can identify subtle deviations in gene expression patterns that may reflect functional deficiencies in the models. The comprehensive human embryo reference tool integrating six published datasets provides an essential benchmark for such validation studies [4].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of low-throughput scRNA-seq for embryonic studies requires careful selection of reagents and tools optimized for minimal input samples. The following table summarizes key solutions and their applications:
Table 3: Essential Research Reagent Solutions for Embryonic scRNA-seq
| Reagent Category | Specific Product Examples | Application in Workflow | Performance Considerations |
|---|---|---|---|
| Cell Isolation Kits | Fluidigm C1 Reagents, FACS antibodies | Single-cell isolation and capture | Embryo-validated protocols preserve cell viability |
| Whole Transcriptome Amplification | SMARTer Ultra Low Input RNA Kit, Smart-seq2 reagents [6] | cDNA synthesis and amplification | High sensitivity for low-input samples; maintains full-length transcripts |
| Library Preparation | Illumina Nextera XT, Nextera Flex | Sequencing library construction | Compatibility with low DNA input; minimized PCR bias |
| RNA Spike-in Controls | ERCC RNA Spike-In Mix [7] | Quality control and normalization | Accurate quantification of technical variation |
| Cell Lysis Buffers | Takara Lysis Buffer, Single Cell Lysis Kit | RNA release and stabilization | Effective lysis while preserving RNA integrity |
| Nuclease-Free Reagents | Ambion RNase Zap, DEPC-treated water | Contamination prevention | Essential for maintaining RNA quality in low-biomass samples |
| Sequence Capture Beads | Dynabeads MyOne Streptavidin, AMPure XP | Nucleic acid purification | High recovery efficiency for precious samples |
| Rauvovertine B | Rauvovertine B, MF:C19H22N2O3, MW:326.4 g/mol | Chemical Reagent | Bench Chemicals |
| Schizolaenone C | Schizolaenone C, MF:C25H28O6, MW:424.5 g/mol | Chemical Reagent | Bench Chemicals |
Low-throughput scRNA-seq methods provide an essential methodological framework for embryonic development research, offering deep transcriptional profiling of limited cell numbers with high sensitivity and analytical completeness. The strategic application of these approaches has illuminated fundamental mechanisms of lineage specification, revealed previously unrecognized cellular heterogeneity in developing embryos, and provided critical validation tools for emerging embryo model systems. As single-cell technologies continue to evolve, low-throughput scRNA-seq will remain indispensable for extracting maximum biological insight from minimal embryonic material, particularly as the field advances toward more comprehensive integration of multimodal single-cell data and spatial transcriptomic approaches.
Single-cell RNA sequencing (scRNA-seq) has redefined the landscape of developmental biology by enabling the resolution of cellular heterogeneity with unprecedented precision. For research on embryonic development, where starting materials are often limited to dozens or hundreds of cells, specific low-to-mid-throughput scRNA-seq workflows offer distinct advantages. These protocols balance high sensitivity with cost-effectiveness, making them particularly suitable for precious samples like human embryos and stem cell-derived embryo models [3]. This application note details the specialized methodologies, key advantages, and practical implementation of these tailored scRNA-seq approaches within a low-throughput workflow for embryo research.
The power of scRNA-seq in embryology stems from its ability to overcome the limitations of bulk RNA sequencing, which obscures critical heterogeneity within biological systems [9]. While bulk methods provide population averages, they inevitably mask the nuanced differences between individual cells that drive developmental lineages and cell fate decisions [10]. This resolution gap is particularly crucial in early development, where a limited number of cells undergo rapid specialization events [11].
Key Advantages for Embryo Analysis
High Sensitivity for Limited Cell Numbers
Low-throughput scRNA-seq platforms excel in detecting genes from small cell populations, a fundamental requirement in embryo research where sample availability is constrained by both biological and ethical considerations [3].
- Comprehensive Gene Detection: Full-length transcript protocols such as Smart-Seq2 and MATQ-Seq demonstrate enhanced sensitivity for detecting low-abundance genes, which is critical for identifying key developmental regulators present in limited quantities within small cell populations [12].
- Optimized for Miniature Samples: These methods are specifically designed to handle the challenges of limited input material, enabling researchers to obtain meaningful data from samples containing as few as dozens to hundreds of cells without pre-amplification steps that could introduce bias [11].
Cost-Effectiveness for Focused Studies
While high-throughput droplet methods excel for large cell atlas projects, low-throughput approaches provide significant economic advantages for studies with limited sample sizes.
- Reduced Per-Sample Costs: By focusing on smaller cell numbers, these methods avoid the premium costs associated with high-throughput commercial platforms, making scRNA-seq accessible for labs with constrained budgets or focused research questions [13].
- Efficient Resource Allocation: Researchers can strategically allocate sequencing depth to fewer cells, achieving higher quality data per cell without the financial burden of processing thousands of cells simultaneously [12].
Resolution of Cellular Heterogeneity
The unparalleled ability to resolve cellular heterogeneity within seemingly homogeneous populations makes scRNA-seq particularly valuable for understanding embryonic development.
- Rare Cell Identification: scRNA-seq can identify rare subpopulations within embryonic tissues, such as primordial germ cells or specific progenitor cells, which would be undetectable using bulk sequencing approaches [9] [10].
- Lineage Specification Mapping: During critical developmental transitions, such as the formation of the inner cell mass (ICM), trophectoderm (TE), and primitive endoderm, scRNA-seq reveals subtle transcriptional differences that precede morphological changes [4] [3].
Table 1: Technical Performance Metrics of scRNA-seq Platforms Suitable for Embryo Research
| Platform/ Method | Cell Throughput Range | Transcript Coverage | Sensitivity (Genes/Cell) | Cost Per Cell | Ideal Embryonic Applications |
|---|---|---|---|---|---|
| Smart-Seq2 [12] | Dozens to hundreds | Full-length | 1,000-5,000 | Moderate | Preimplantation embryos, rare cell types |
| MATQ-Seq [12] | Dozens to hundreds | Full-length | 1,500-6,000 | Moderate | Low-abundance transcript detection |
| Quartz-Seq2 [12] | Dozens to hundreds | Full-length | 1,000-4,500 | Moderate | Lineage tracing, developmental kinetics |
| Fluidigm C1 [12] | Dozens to hundreds | Full-length | 1,200-5,000 | High | Integrated workflow, automated processing |
| Drop-seq [9] [12] | Thousands to millions | 3'-end | 500-2,000 | Low | Larger embryo samples, atlas building |
Dynamic Trajectory Reconstruction
Beyond static snapshots, scRNA-seq enables the reconstruction of dynamic developmental processes through computational trajectory inference.
- Pseudotime Analysis: Algorithms like Monocle and Slingshot can order individual cells along pseudotemporal trajectories, reconstructing developmental pathways without the need for physical time-series experiments [13].
- Transition State Identification: These analyses reveal intermediate cellular states during critical developmental transitions, such as the maternal-to-zygotic transition (MZT) or the emergence of the three primary lineages in the blastocyst [3].
Experimental Protocols for Embryo scRNA-seq
Sample Preparation and Quality Control
The initial stage of performing scRNA-seq on embryonic samples involves careful extraction of viable individual cells while preserving RNA integrity.
- Gentle Dissociation Protocols: Embryo dissociation requires optimized enzymatic combinations and temperature conditions (often 4°C) to minimize artificial stress responses that can alter transcriptional profiles [14].
- Rigorous Quality Assessment: Cell viability (>85%) and integrity must be confirmed before processing, with particular attention to avoiding RNA degradation in delicate embryonic cells [9].
- Adapted Isolation Techniques: For challenging embryonic tissues or frozen samples, single-nucleus RNA sequencing (snRNA-seq) provides an alternative approach that minimizes dissociation artifacts and preserves spatial information about nuclear RNA [14].
Library Preparation Methodologies
Different scRNA-seq protocols offer distinct advantages depending on the specific research question and embryonic stage being studied.
- Full-Length Transcript Protocols: Methods such as Smart-Seq2 provide comprehensive transcript coverage, enabling detection of alternative splicing, allele-specific expression, and RNA editing events crucial for understanding regulatory mechanisms in development [12].
- 3'-End focused Methods: Protocols like Drop-seq offer higher throughput at lower cost per cell, making them suitable for larger-scale studies where detecting novel cell types rather than isoform-level resolution is the primary goal [9] [12].
Table 2: Key Research Reagent Solutions for Embryo scRNA-seq
| Reagent/Chemical | Function in Workflow | Specific Application in Embryo Research |
|---|---|---|
| Poly(T) Primers [12] | mRNA capture via polyA tail binding | Selective analysis of polyadenylated mRNA while minimizing ribosomal RNA capture |
| Unique Molecular Identifiers (UMIs) [9] [14] | Barcode individual mRNA molecules | Account for amplification biases through molecular counting; essential for quantitative analysis |
| Template-Switch Oligos (TSO) [9] | Enable cDNA synthesis independent of poly(A) tails | Improve cDNA yield from partially degraded RNA in delicate embryonic samples |
| Barcoded Beads [9] [13] | Uniquely label cellular mRNA during capture | Trace transcripts to individual cells in droplet-based systems |
| 4-Thiouridine (4sU) [15] | Metabolic RNA labeling for nascent transcript detection | Track newly synthesized RNA during rapid developmental transitions like zygotic genome activation |
Specialized Methodologies for Developmental Studies
Advanced applications of scRNA-seq in embryo research incorporate specialized techniques to address specific biological questions.
- Metabolic Labeling Integration: Techniques such as scNT-seq incorporate nucleoside analogs (e.g., 4-Thiouridine) to label newly synthesized RNA, enabling precise measurement of RNA kinetics during critical developmental transitions like the maternal-to-zygotic transition [15].
- Multi-Omic Approaches: Combining scRNA-seq with epigenetic profiling methods allows for integrated analysis of gene expression and regulatory landscape changes throughout development [9].
Data Analysis and Interpretation
Computational Workflow for Embryonic Data
The analysis of scRNA-seq data from embryonic samples requires specialized computational approaches tailored to the unique characteristics of developing systems.
- Quality Control and Normalization: Rigorous filtering to remove low-quality cells, doublets, and background noise, followed by normalization methods specifically designed for single-cell data to address technical variability [12].
- Batch Effect Correction: Integration of multiple datasets using algorithms like fastMNN or Harmony to enable comparisons across different embryonic stages, individuals, or experimental conditions [4].
- Reference-Based Annotation: Projection of query datasets onto established embryonic references, such as the integrated human embryo atlas containing 3,304 cells from zygote to gastrula stages, for consistent cell type identification [4].
Specialized Analytical Approaches for Development
Embryonic scRNA-seq data enables specific analytical approaches that leverage the unique properties of developing systems.
- Trajectory Inference: Algorithms such as Slingshot utilize the continuous nature of developmental processes to reconstruct lineage relationships and temporal ordering of cells, revealing the sequence of molecular events driving cell fate decisions [4] [13].
- Regulatory Network Analysis: SCENIC (Single-Cell Regulatory Network Inference and Clustering) identifies key transcription factors driving lineage specification, such as DUXA in morula, VENTX in epiblast, and OVOL2 in trophectoderm [4].
- Cross-Species Comparison: Integration of human embryonic data with model organism datasets identifies conserved and species-specific aspects of development, providing evolutionary context to developmental mechanisms [4].
Applications in Embryo Research
Preimplantation Development
scRNA-seq has revolutionized our understanding of the earliest stages of human development, from zygote to blastocyst formation.
- Maternal-to-Zygotic Transition: Single-cell analyses have revealed the highly dynamic transcriptome during preimplantation, identifying the timing and magnitude of zygotic genome activation (ZGA) and the degradation of maternal mRNAs [3].
- Lineage Specification: Comprehensive transcriptomic atlases have defined the molecular signatures of the three primary lineages (epiblast, primitive endoderm, and trophectoderm) and identified key regulators of these fate decisions [4] [3].
Gastrulation and Organogenesis
Beyond implantation, scRNA-seq enables the exploration of later developmental events despite technical and ethical challenges.
- Germ Layer Formation: Analysis of gastrulating embryos has revealed the transcriptional programs driving the emergence of definitive endoderm, mesoderm, and ectoderm from the epiblast [4].
- Cell Atlas Construction: Integrated datasets spanning multiple developmental stages provide reference frameworks for identifying the origin of specific cell types and understanding the progression of normal and abnormal development [4].
Embryo Model Validation
scRNA-seq serves as a critical tool for validating stem cell-derived embryo models, which provide ethically accessible systems for studying human development.
- Transcriptomic Benchmarking: Comparison of embryo models (e.g., blastoids, gastruloids) with in vivo references assesses the fidelity of these models and identifies areas for improvement [4] [3].
- Lineage Annotation: Projection of model-derived cells onto reference atlases ensures accurate cell type identification and prevents misannotation of lineages [4].
Low-throughput scRNA-seq workflows tailored for dozens to hundreds of cells provide an optimal balance of sensitivity, cost-effectiveness, and analytical power for embryonic research. The strategic implementation of these methods enables researchers to overcome the fundamental challenges of limited starting material while generating comprehensive insights into developmental mechanisms. As the field advances, integration with spatial transcriptomics, multi-omics approaches, and artificial intelligence-driven analysis will further enhance the resolution at which we can study embryonic development [9]. These specialized scRNA-seq protocols continue to drive discoveries in basic developmental biology while simultaneously providing critical tools for understanding developmental disorders and improving regenerative medicine applications.
Application Note: Scientific Rationale and Value
Understanding the initial steps of cell fate decision-making is fundamental to developmental biology and regenerative medicine. For embryo research, single-cell RNA sequencing (scRNA-seq) provides an unbiased method to deconstruct cellular heterogeneity and map the precise transcriptional trajectories that guide a single zygote into a complex organism. Low-throughput scRNA-seq workflows are particularly critical for embryonic studies, where starting material is often limited, and high-resolution, deep sequencing of individual cells is required to capture the full complexity of early lineage decisions and identify rare, transient progenitor populations [16] [17].
Key biological questions addressable with this approach include:
- Mapping Lineage Bifurcations: Precisely defining the points at which homogeneous cell populations diverge into distinct lineages, such as the separation of musculoskeletal precursors into cartilage, bone, and muscle lineages in the developing limb [16].
- Identifying Rare Progenitors: Discovering and characterizing rare, transcriptionally distinct cell populations that serve as key intermediates in developmental pathways but are too scarce for bulk sequencing methods to detect [18].
- Deciphering Molecular Drivers: Uncovering the core transcription factors and signaling pathways that initiate and maintain cell type specification, moving beyond a priori selection of marker genes [19].
Experimental Protocols
Low-Throughput scRNA-seq Wet-Lab Protocol (SMART-seq Technology)
This protocol is optimized for maximum transcript coverage and sensitivity from low cell inputs, such as those obtained from embryonic tissues [12] [17].
Sample Preparation and Cell Isolation:
- Tissue Dissociation: Gently dissociate embryonic tissue into a single-cell suspension using enzymatic and/or mechanical methods. Preserve cell viability.
- Cell Sorting and Lysis: Using a micromanipulator or fluorescence-activated cell sorting (FACS), individually collect 1 to 100 target cells into separate 0.2 mL tubes containing lysis buffer. Immediately freeze samples at -80°C until library preparation [17].
Library Construction (SMART-seq2):
- Reverse Transcription: Thaw lysed cells and perform reverse transcription using an oligo(dT) primer and a template-switching oligonucleotide (TSO). This ensures full-length cDNA synthesis and incorporates a universal adapter sequence.
- cDNA Amplification: Amplify the full-length cDNA by PCR using primers that bind to the universal adapter.
- Tagmentation and Indexing: Fragment the amplified cDNA and add sequencing adapters with unique sample indices using a tagmentation-based library construction kit (e.g., Nextera XD).
- Library Purification and QC: Purify the final libraries and assess quality using a Bioanalyzer or TapeStation [12].
Sequencing:
- Sequence libraries on an Illumina platform using paired-end 50 bp reads.
- Target a sequencing depth of 20-25 million reads per cell for mammalian samples to ensure robust gene detection [17].
Computational Protocol for Rare Cell Population Identification
This two-step protocol leverages CellSIUS (Cell Subtype Identification from Upregulated gene Sets) for sensitive detection of rare cell types from scRNA-seq data [18].
Pre-processing and Coarse Clustering:
- Quality Control: Filter out low-quality cells based on thresholds for count depth, number of genes detected, and mitochondrial read fraction [20].
- Normalization and Feature Selection: Normalize the count data and select highly variable genes for downstream analysis.
- Initial Clustering: Perform an initial, coarse-grained clustering using a standard method (e.g., Seurat) to identify major cell populations. This step defines the primary clusters
C1...Cmwithin which CellSIUS will search for rare subtypes [18].
Rare Cell Population Detection with CellSIUS:
- Input: Provide the normalized expression values of
Ncells grouped intoMcoarse clusters from the previous step. - Identify Candidate Marker Genes: For each coarse cluster
Cm, perform a Wilcoxon rank-sum test to find genes significantly upregulated in a small subset of cells withinCmcompared to the rest of the cluster. - Form Gene Sets: For each upregulated gene, define a "gene set" comprising the gene itself and its highly correlated genes.
- Score Cells and Cluster: Score all cells in
Cmbased on their aggregate expression of each gene set. Use these scores to perform a new round of clustering, specifically withinCm, to identify a potential rare subpopulation. - Filter and Output: Apply specificity filters to ensure the identified subpopulation is distinct. The output is a list of rare cell populations and their transcriptomic signature genes [18].
Quantitative Data and Performance Metrics
Table 1: Key scRNA-seq Performance Metrics from Benchmarking Studies
| Metric Category | Specific Metric | Reported Performance / Typical Range | Context / Method |
|---|---|---|---|
| Library Complexity | Genes detected per cell | ~2,700 genes/cell | Mouse hindlimb development (10x Genomics) [16] |
| >20,000 reads/cell | Recommended sequencing depth for 10x Genomics [17] | ||
| Rare Cell Detection | Adjusted Rand Index (ARI) | 0.76 (Seurat) to 0.99 (DBSCAN as outlier) | Performance on a rare population (0.15% of cells) [18] |
| CellSIUS Performance | Outperforms other methods in specificity/selectivity | Identification of rare cell types in complex data [18] | |
| Metabolic Labeling* | T-to-C substitution rate | 8.40% (mean) | mCPBA/TFEA pH 7.4 chemistry on Drop-seq [15] |
| Labeled mRNA UMIs per cell | 36.87% - 45.98% of total mRNAs | On-beads IAA and mCPBA/TFEA methods [15] |
Note: Metabolic labeling enables the study of RNA dynamics, crucial for understanding cell state transitions during embryogenesis [15].
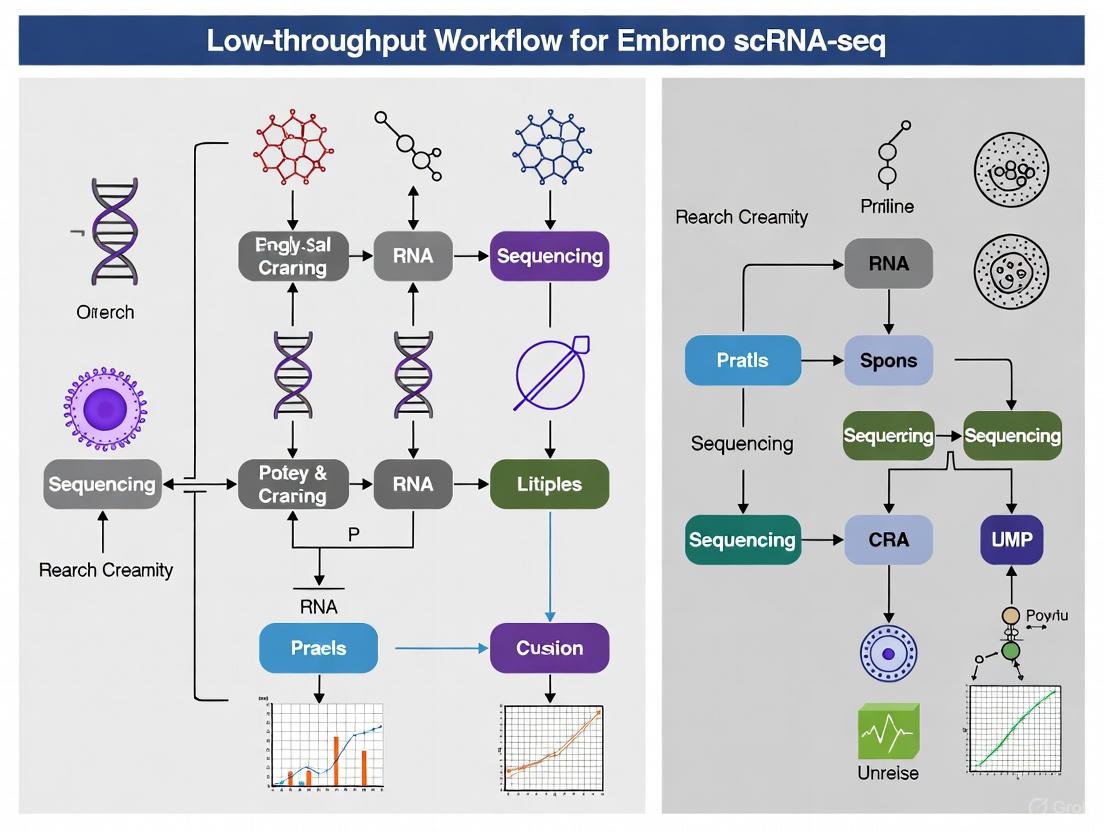
Visualizing Experimental Workflows
Low-Throughput Embryo scRNA-seq Workflow
Computational Detection of Rare Cell Populations
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for Embryo scRNA-seq Studies
| Item | Function / Application | Example / Note |
|---|---|---|
| SMART-seq2 Reagents | Full-length cDNA synthesis and amplification from single cells. Maximizes gene detection from low-input samples. | Template Switching Oligo (TSO), SMARTScribe Reverse Transcriptase [12]. |
| Unique Molecular Identifiers (UMIs) | Tags individual mRNA molecules during reverse transcription to correct for PCR amplification bias and enable absolute transcript counting. | Essential for droplet-based protocols (e.g., 10x Genomics) [20] [12]. |
| Nucleoside Analogs (4sU, 5-EU) | Metabolic RNA labeling. Incorporated into newly synthesized RNA, allowing for the study of transcriptional dynamics during cell state transitions. | Critical for studying RNA kinetics in embryogenesis [15]. |
| CellSIUS Software | Computational tool for sensitive and specific identification of rare cell populations and their transcriptomic signatures from complex scRNA-seq data. | R package; used after initial coarse clustering [18]. |
| InferCNV | Computational method to identify large-scale chromosomal copy number alterations (CNVs) from scRNA-seq data. Helps distinguish malignant from normal cells in studies of cancer ontogeny. | Used to confirm somatic CNVs in AT2-like cells during lung adenocarcinoma progression [19]. |
| 6-O-Acetylcoriatin | 6-O-Acetylcoriatin, MF:C17H22O7, MW:338.4 g/mol | Chemical Reagent |
| Tinosporoside A | Tinosporoside A | Tinosporoside A stimulates glucose uptake via PI3K/AMPK pathways. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
Selecting the appropriate single-cell RNA sequencing (scRNA-seq) platform is a critical first step in embryonic development research. The fundamental choice between full-length transcript and 3'/5'-end counting methods directly impacts transcriptome coverage, detection capability, and experimental outcomes [12]. For embryo research, where cell numbers are often limited and transcriptomic dynamics are rapid, this platform selection must balance comprehensive biological insight with practical experimental constraints [4].
Full-length transcript sequencing provides complete coverage across mRNA transcripts, enabling isoform resolution, variant detection, and comprehensive transcriptome annotation [12]. In contrast, 3'-end counting methods focus sequencing on transcript termini, providing digital gene expression quantification with reduced sequencing depth requirements [21] [22]. Understanding the technical and practical distinctions between these approaches ensures appropriate technology selection for specific embryological research questions within low-throughput workflows.
Technical Foundations and Methodological Comparisons
Core Technological Principles
Full-length transcript sequencing employs random priming during reverse transcription, generating sequencing reads distributed across the entire transcript length [21]. This approach requires effective ribosomal RNA depletion or polyadenylated RNA selection prior to library preparation to prevent capture of unwanted RNA species [21]. Protocols such as Smart-Seq2, MATQ-Seq, and Fluidigm C1 utilize this principle, with some demonstrating enhanced sensitivity for detecting low-abundance genes and comprehensive transcript variant analysis [12].
3'-end counting methods initiate cDNA synthesis from the transcript's 3'-end using oligo(dT) primers, localizing sequencing reads to the 3'-untranslated region (UTR) [21] [22]. Each transcript generates approximately one sequencing fragment, simplifying quantification by directly relating read counts to transcript abundance [21]. Techniques implementing this approach include Lexogen QuantSeq, Drop-Seq, inDrop, and 10x Genomics Chromium systems [21] [12].
Table 1: Fundamental Methodological Differences Between Sequencing Approaches
| Parameter | Full-Length Sequencing | 3'/5'-End Counting Methods |
|---|---|---|
| Priming Strategy | Random primers | Oligo(dT) primers targeting poly(A) tail |
| Transcript Coverage | Distributed across entire transcript | Localized to 3' or 5' end |
| Reads per Transcript | Proportional to transcript length | Approximately one fragment per transcript |
| rRNA Depletion | Required (poly(A) selection or rRNA depletion) | Built-in through poly(A) selection |
| Protocol Examples | Smart-Seq2, MATQ-Seq, Fluidigm C1 | QuantSeq, Drop-Seq, inDrop, 10x Genomics |
Experimental Workflows and Protocol Details
The experimental workflow diverges significantly after RNA extraction. For full-length methods, the protocol involves: (1) ribosomal RNA depletion or mRNA enrichment, (2) random primed reverse transcription, (3) cDNA amplification, and (4) library preparation [21]. This workflow typically requires more processing steps and time compared to end-counting methods [21].
For 3'-end counting methods, the streamlined protocol includes: (1) oligo(dT) primed reverse transcription, (2) template switching, and (3) PCR amplification with barcoding [21] [22]. The simplified workflow reduces hands-on time and is more robust for challenging sample types, including degraded RNA and FFPE material [21].
Diagram 1: Experimental workflow comparison between full-length and 3'-end sequencing methods. Yellow indicates initial sample processing, green represents RNA selection steps, blue shows full-length protocol steps, and red indicates 3'-end method steps.
Performance Characteristics and Analytical Capabilities
Quantitative Performance in Embryonic Systems
Sequencing technology selection significantly impacts detection capability in embryonic environments characterized by rapid transcriptional changes and diverse isoform expression. Performance evaluations reveal method-specific advantages under different experimental conditions.
Table 2: Performance Comparison for Embryo Research Applications
| Performance Metric | Full-Length Sequencing | 3'/5'-End Counting Methods |
|---|---|---|
| Genes Detected per Cell | Higher for full-length transcripts [12] | Lower but sufficient for major cell types |
| Short Transcript Detection | Reduced sensitivity [22] | Enhanced detection capability [22] |
| Differentially Expressed Genes | Detects more DEGs [21] [23] | Fewer DEGs but consistent biological conclusions [21] |
| Transcript Length Bias | Favors longer transcripts [22] | Minimal length bias [22] |
| Sequencing Depth Requirement | Higher (typically >20M reads/sample) [21] | Lower (1-5M reads/sample) [21] |
| Isoform Resolution | Excellent for splice variants and novel isoforms [21] | Limited to gene-level quantification |
| Rare Cell Type Detection | Enhanced sensitivity for rare transcripts [12] | Requires specialized computational methods [24] |
Full-length sequencing demonstrates superior detection of differentially expressed genes (DEGs) regardless of sequencing depth, with one study identifying approximately 30% more DEGs compared to 3'-end methods [21] [23]. However, 3'-end counting methods show particular advantage in detecting short transcripts, especially under conditions of sparse data or reduced sequencing depth [22]. At sequencing depths of 2.5 million reads, 3'-end methods detected approximately 400 more transcripts shorter than 1,000 base pairs compared to full-length approaches [22].
For pathway analysis, full-length sequencing identifies more functionally enriched pathways through DEG analysis, though both methods provide highly similar biological conclusions when employing gene set enrichment analysis of all genes [23]. The reproducibility between biological replicates is similar for both approaches, making 3'-end methods suitable for large-scale screening experiments where cost efficiency is paramount [21] [22].
Applications in Embryonic Development Research
The selection between full-length and end-sequencing methods should align with specific research objectives in embryo research. Full-length transcript sequencing is indispensable for investigations requiring isoform-level resolution, such as characterizing alternative splicing during lineage specification [12], identifying novel embryonic transcripts [21], and detecting allelic expression patterns in early development [12].
3'-end counting methods provide optimal solutions for quantitative gene expression profiling across large sample sets [21], lineage tracing through barcoding approaches [4], and experiments utilizing challenging sample types including fixed embryos or low-quality RNA [21]. These methods also excel in time-series studies of embryonic development where numerous time points require processing [21].
For constructing comprehensive embryonic reference atlases, full-length methods offer more complete transcriptome annotation, as demonstrated in integrated human embryo datasets covering development from zygote to gastrula stages [4]. These references enable precise benchmarking of stem cell-derived embryo models through unbiased transcriptional comparison [4].
Implementation Considerations for Low-Throughput Embryo Workflows
Experimental Design and Practical Considerations
Low-throughput embryo research necessitates careful consideration of several practical aspects. Sample availability often limits experimental design, with embryo studies typically processing fewer than 100 cells per condition [4]. For such limited samples, full-length methods maximize biological information capture per cell, while 3'-end methods enable more experimental conditions with the same sequencing budget.
Cell dissociation and viability present particular challenges for embryonic tissues. Enzymatic dissociation can trigger stress responses altering transcriptional profiles [12]. Single-nuclei RNA-seq (snRNA-seq) provides an alternative when tissue dissociation is problematic, especially for frozen samples or fragile embryonic cells [12]. Split-pooling techniques with combinatorial indexing accommodate minute sample sizes while eliminating need for specialized microfluidic equipment [12].
Sequencing depth requirements vary significantly between approaches. Full-length methods typically require 20-50 million reads per sample for comprehensive transcriptome coverage, while 3'-end methods provide quantitative expression data with just 1-5 million reads per sample [21]. This substantial difference directly impacts per-sample costs and should inform technology selection based on available sequencing resources.
Data Analysis and Computational Requirements
Data analysis approaches differ substantially between sequencing methods. Full-length data supports sophisticated analyses including isoform quantification, splicing analysis, and RNA editing detection [12]. The computational pipeline involves alignment, transcript assembly, and isoform quantification, requiring specialized tools and significant processing resources.
3'-end counting data analysis focuses on digital gene expression matrices, simplifying preprocessing to alignment and unique molecular identifier (UMI) counting [21]. The reduced data complexity enables faster processing and simpler statistical analysis for differential expression [21].
Feature selection represents a critical step in scRNA-seq analysis, particularly for identifying subtle cell-type differences in embryonic development [24]. While standard highly variable gene selection performs adequately for abundant, well-separated cell types, specialized feature selection methods significantly improve rare cell type identification [24]. For embryo research where transitional states are common, careful feature selection enhances detection of developing lineages.
Diagram 2: Decision framework for selecting between full-length and 3'-end sequencing methods in embryo research. Yellow indicates input considerations, green represents decision points, and blue/red show method selection outcomes.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for Embryo scRNA-seq
| Reagent/Platform | Function | Application Context |
|---|---|---|
| Lexogen QuantSeq 3' mRNA-Seq Kit | 3'-end library preparation | Cost-effective gene expression quantification; degraded RNA samples [21] |
| KAPA Stranded mRNA-Seq Kit | Full-length library preparation | Traditional whole transcriptome analysis; isoform detection [22] |
| Smart-Seq2 | Full-length protocol | Enhanced sensitivity for low-abundance transcripts; single-cell resolution [12] |
| 10x Genomics Chromium | 3'-end counting with droplet microfluidics | High-throughput single-cell profiling; large cell numbers [12] |
| Fluidigm C1 | Full-length automated platform | Microfluidics-based single-cell capture; precise cell handling [12] |
| MATQ-Seq | Full-length protocol | Increased accuracy in quantifying transcripts; efficient variant detection [12] |
| Drop-Seq | 3'-end droplet method | High-throughput, low cost per cell; scalable to thousands of cells [12] |
| Karaviloside X | Karaviloside X, MF:C42H68O14, MW:797.0 g/mol | Chemical Reagent |
| Aspergillon A | Aspergillon A|AbMole | Aspergillon A is a natural product for research applications. This product is for research use only, not for human consumption. |
Platform selection between full-length and 3'/5'-end sequencing methods represents a fundamental strategic decision in embryo research. Full-length transcript sequencing provides comprehensive biological insight through complete transcriptome characterization, making it ideal for discovery-phase research, isoform-level analysis, and rare transcript detection. Conversely, 3'-end counting methods offer practical advantages in cost efficiency, sample throughput, and analytical simplicity, suitable for quantitative screening studies and large-scale comparative analyses.
The emerging paradigm in embryonic research leverages both approaches strategically: employing 3'-end methods for large-scale screening to identify conditions of interest, followed by focused full-length sequencing for mechanistic investigation [21]. This integrated approach maximizes both throughput and biological depth, advancing our understanding of embryonic development through appropriate technological implementation.
A Step-by-Step Low-Throughput scRNA-seq Workflow for Embryonic Cells
Single-cell RNA sequencing (scRNA-seq) has revolutionized transcriptomics by enabling the resolution of gene expression to the level of individual cells, thereby uncovering cellular heterogeneity that is averaged out in bulk sequencing approaches [13] [25]. The foundation of any successful scRNA-seq experiment, particularly in the context of low-throughput workflows for precious samples like embryos, is the effective and reliable isolation of viable single cells. Cell capture strategies determine the scale, precision, and ultimate quality of the resulting transcriptomic data.
For embryo research, where cell numbers are inherently limited and each sample is of immense scientific value, the choice of cell capture method is paramount. These strategies must balance the need for high-quality data with the practical constraints of working with low cell inputs. The three primary platforms—Fluorescence-Activated Cell Sorting (FACS), Micromanipulation, and Microfluidic Systems—each offer distinct advantages and limitations for specific embryonic research applications. This application note details these methodologies within the context of establishing a robust, low-throughput scRNA-seq workflow for embryonic development studies.
Comparative Analysis of Cell Capture Platforms
The selection of a cell isolation method dictates the scale, cost, and type of biological questions that can be addressed. The table below provides a systematic comparison of the three core platforms.
Table 1: Comparative Analysis of Single-Cell Isolation Methods for Embryonic Research
| Method | Throughput | Key Advantage | Primary Limitation | Ideal Application in Embryo Research |
|---|---|---|---|---|
| FACS | Medium | Enables selection based on specific surface markers (e.g., CD34, CD133) [8]; high versatility. | Requires large input volume and cell number (>10,000 cells); dependent on antibody availability [13]. | Isolation of specific, marker-defined progenitor populations (e.g., hematopoietic stem cells) from dissociated embryonic tissues [8]. |
| Micromanipulation | Very Low | Ultimate precision for hand-picking individual cells; minimal equipment requirements. | Extremely time-consuming and low-throughput [13]; high technical skill requirement. | Targeting specific, morphologically distinct blastomeres in preimplantation embryos [26]. |
| Microfluidics | High (Droplet) / Low (IFC) | Low sample consumption; cost-effective per cell; precise fluid control [13] [27]. | Requires >1,000 cells; can be restricted by homogeneous cell size requirements [13]. | High-throughput profiling of thousands of cells from dissociated embryonic organs [27] [12]. |
| Microdroplet | Very High | Capable of processing thousands to millions of cells in parallel; very low cost per cell [13] [27]. | Lower sensitivity in gene expression detection; only sequences the 3' or 5' end of transcripts [13] [12]. | Large-scale atlas projects aiming to capture full cellular heterogeneity of a complex embryonic tissue. |
| Microwell | High | Cost-effective and high-throughput; portable systems available (e.g., Seq-Well) [13] [27]. | Cell loading is governed by Poisson distribution statistics, which can lead to multiple cells per well [13]. | High-throughput profiling when cost is a primary constraint and equipment access is limited. |
Detailed Experimental Protocols for Low-Throughput Workflows
Protocol 1: Targeted Cell Isolation via FACS for Hematopoietic Stem/Progenitor Cells (HSPCs)
This protocol is adapted from studies on human umbilical cord blood-derived HSPCs, demonstrating a workflow for isolating rare cell populations from a mixed sample, a common requirement in embryonic research [8].
A. Sample Preparation and Staining
- Dissociation: Obtain a single-cell suspension from the embryonic tissue of interest using enzymatic (e.g., trypsin) or mechanical dissociation. For sensitive tissues, consider a cold dissociation technique using cryophilic proteases to maintain high cell viability and reduce artifacts [28].
- Staining: Resuspend the cell pellet in a cold buffer (e.g., RPMI-1640 with 2% FBS).
- Prepare a cocktail of antibodies. For HSPCs, this includes:
- Lineage (Lin) depletion cocktail: FITC-conjugated antibodies against differentiation markers (e.g., CD235a, CD2, CD3, CD14, CD16, CD19, CD24, CD56, CD66b).
- Positive selection antibodies: PE-conjugated anti-CD34 and APC-conjugated anti-CD133.
- Viability marker: A dye to exclude dead cells (e.g., DAPI or Propidium Iodide).
- Incubate the cell suspension with the antibody cocktail in the dark at 4°C for 30 minutes.
- Wash cells with buffer to remove unbound antibodies [8].
- Prepare a cocktail of antibodies. For HSPCs, this includes:
B. Fluorescence-Activated Cell Sorting
- Gating Strategy:
- P1 Gate: Select single cells based on forward-scatter height (FSC-H) vs. forward-scatter area (FSC-A) to exclude doublets.
- P2 Gate: From P1, select viable cells by excluding DAPI-positive events.
- P3 Gate: From P2, select Lin-negative (FITC-negative) cells to exclude differentiated lineages.
- P4/P5 Gates: From the Lin-negative population, sort the target populations—specifically, CD34+CD45+ and/or CD133+CD45+ HSPCs [8].
- Collection: Sort directly into a tube containing a compatible cell culture medium or lysis buffer, depending on the immediate downstream application. For scRNA-seq, ensure sorted cells are kept cold and processed promptly for library preparation.
Protocol 2: Manual Selection of Preimplantation Blastomeres via Micromanipulation
This protocol outlines the precise isolation of individual cells from early-stage embryos for full-length transcriptome analysis, as used in co-sequencing studies of mRNA and small non-coding RNAs [26].
A. Embryo Handling and Preparation
- Embryo Collection: Obtain preimplantation embryos (e.g., mouse or human) following standard assisted reproductive protocols and ethical guidelines.
- Zona Pellucida Removal: Briefly expose embryos to an acidic Tyrode's solution or protease to remove the zona pellucida, facilitating access to individual blastomeres.
- Transfer: Wash and transfer the denuded embryos into a drop of Ca2+/Mg2+-free buffer on a microscope slide or dish to weaken cell-cell adhesions.
B. Micromanipulation and Cell Picking
- Setup: Install a sharp, clean glass capillary needle on a micromanipulator attached to an inverted microscope.
- Dissociation: Use the needle to gently tease apart individual blastomeres from the embryo. Avoid excessive mechanical stress.
- Aspiration: Carefully aspirate a single, intact blastomere into the capillary needle.
- Transfer and Lysis: Expel the isolated blastomere into a small volume of a specific lysis buffer. For protocols like Smart-seq2, this buffer should contain detergents and RNase inhibitors to immediately lyse the cell and stabilize RNA [26] [12]. The lysate can then be used for full-length transcriptome library construction.
Protocol 3: High-Throughput Profiling using Microfluidic Droplet Systems
This protocol leverages commercial platforms like the 10x Genomics Chromium system for high-cell-throughput studies of later-stage embryonic organs [27] [12].
A. Sample and Reagent Preparation
- Single-Cell Suspension: Prepare a high-viability (>90%) single-cell suspension from the embryonic tissue at a recommended concentration of 700-1,200 cells/µL.
- Reagent Setup: Thaw and prepare the required reagents from a commercial kit (e.g., Chromium Next GEM Chip G Single Cell Kit, Library & Gel Bead Kit v3.1).
B. Microfluidic Workflow on 10x Genomics Chromium
- Loading: Load the cell suspension, gel beads, and partitioning oil into the designated wells of a microfluidic "GEM Chip."
- Droplet Generation: Run the chip on the Chromium Controller. The system partitions each cell with a uniquely barcoded gel bead into a nanoliter-scale droplet, creating Gel Bead-in-Emulsions (GEMs). This process achieves high cell-throughput, encapsulating thousands of cells per run [27].
- Reverse Transcription: Inside each droplet, the cell is lysed, and mRNA transcripts are captured by the poly(dT) primers on the barcoded beads. Reverse transcription occurs, creating cDNA tagged with the cell-specific barcode and a Unique Molecular Identifier (UMI).
- Library Preparation: Break the droplets, purify the barcoded cDNA, and amplify it via PCR. Construct a sequencing library following the manufacturer's instructions (e.g., using the Chromium Single Cell 3' Kit) [8].
- Sequencing: The final libraries are sequenced on an Illumina platform (e.g., NextSeq 1000/2000), typically aiming for 20,000-50,000 reads per cell [8].
Visualizing the scRNA-seq Workflow from Cell to Data
The following diagram illustrates the logical and experimental workflow for single-cell RNA sequencing, integrating the cell capture methods described above.
Diagram 1: scRNA-seq Workflow from Cell Capture to Data Analysis.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of the protocols above requires specific reagents and tools. The following table lists key solutions and their functions.
Table 2: Essential Research Reagent Solutions for Embryonic scRNA-seq
| Item | Function/Description | Example Use Case |
|---|---|---|
| Fluorescence-Activated Cell Sorter | Instrument that sorts individual cells from a suspension based on light scattering and fluorescent characteristics. | Isolation of CD34+/CD133+ HSPCs from a heterogeneous suspension of umbilical cord blood cells [8]. |
| Micromanipulation System | A setup with fine-control hydraulic or mechanical manipulators and an inverted microscope for precise handling of single cells. | Manual picking of specific blastomeres from an 8-cell stage embryo for single-cell multi-omics [26]. |
| Chromium Controller & Kits (10x Genomics) | Integrated microfluidic system and reagent kits for automated, high-throughput single-cell library preparation. | Generating barcoded scRNA-seq libraries from thousands of cells dissociated from an embryonic organ [8]. |
| Lineage Depletion Cocktail | A mixture of antibodies against lineage-specific markers (e.g., CD2, CD3, CD14, etc.) for negative selection. | Enriching for primitive hematopoietic stem cells by removing differentiated cell types during FACS [8]. |
| Cold-Active Protease | Enzyme (e.g., from Bacillus species) that remains highly active at low temperatures (4-10°C) for tissue dissociation. | Generating high-viability single-cell suspensions from sensitive embryonic tissues while minimizing stress-induced artifacts [28]. |
| Smart-seq2 Lysis Buffer | A specialized buffer containing detergents, dNTPs, oligo-dT primers, and RNase inhibitors for immediate cell lysis and RNA capture. | Lysing a single, micromanipulated blastomere to initiate full-length transcriptome sequencing [26] [12]. |
| Tannagine | Tannagine | Tannagine is a high-purity tannin reagent for research on protein binding, antioxidants, and antimicrobials. For Research Use Only. Not for diagnostic or therapeutic use. |
| Cephalandole B | Cephalandole B, MF:C17H14N2O3, MW:294.30 g/mol | Chemical Reagent |
The strategic selection of a cell capture platform—FACS, Micromanipulation, or Microfluidics—is the cornerstone of a successful low-throughput scRNA-seq workflow in embryo research. The choice is not one of superiority but of application-specific suitability. FACS provides antibody-based precision for isolating defined populations, Micromanipulation offers unparalleled manual control for the most precious samples, and Microfluidic platforms deliver scalability for capturing complex heterogeneity. By understanding the capabilities and limitations of each method, as detailed in these application notes and protocols, researchers can robustly leverage scRNA-seq to unravel the intricate transcriptional landscapes of embryonic development.
Library preparation for low-input RNA is a critical step in single-cell RNA sequencing (scRNA-seq), enabling the detailed exploration of cellular heterogeneity. Within embryo research, where starting material is often extremely limited, optimized protocols for reverse transcription, cDNA amplification, and barcoding are essential for obtaining meaningful transcriptomic data. These methods have completely transformed our understanding of human embryonic development by allowing researchers to systematically investigate lineage specification and cellular differentiation events during preimplantation stages and beyond [3] [4]. This application note details established methodologies and considerations for implementing a low-throughput scRNA-seq workflow specifically tailored for embryonic research applications.
Key Methodological Approaches
Current technologies for scRNA-seq library preparation employ distinct strategies for partitioning individual cells and barcoding their transcripts.
Platform Selection for Low-Throughput Workflows
For projects not requiring ultra-high throughput, such as studies on precious embryo samples, several platforms offer flexible cell number accommodation. The table below compares relevant technologies suitable for lower-throughput applications.
Table 1: scRNA-seq Platform Comparison for Low- to Mid-Throughput Applications
| Commercial Solution | Capture Platform | Throughput (Cells/Run) | Capture Efficiency (%) | Max Cell Size | Fixed Cell Support |
|---|---|---|---|---|---|
| 10× Genomics Chromium | Microfluidic oil partitioning | 500–20,000 | 70–95 | 30 µm | Yes |
| BD Rhapsody | Microwell partitioning | 100–20,000 | 50–80 | 30 µm | Yes |
| Singleron SCOPE-seq | Microwell partitioning | 500–30,000 | 70–90 | < 100 µm | Yes |
| Plate-based Combinatorial Barcoding (e.g., Parse, Scale) | Multiwell-plate | 1,000–1M+ | > 85 | – | Yes [29] [30] |
The general workflow for scRNA-seq library preparation involves sequential molecular biology steps to convert RNA from single cells into a sequencer-ready library.
Experimental Protocols
Detailed Protocol: cDNA-PCR Barcoding for Low-Input RNA
This protocol is adapted from the Oxford Nanopore cDNA-PCR Sequencing V14 Barcoding kit, suitable for full-length cDNA sequencing from low-input samples [31].
Sample Preparation and Input Requirements
- Input Material: 10 ng enriched Poly(A)+ RNA or 500 ng total RNA per sample.
- Quality Control: Assess RNA length, quantity, and purity using appropriate methods (e.g., Qubit RNA HS Assay Kit, Bioanalyzer).
- Cell Preparation: For embryonic tissues, gentle dissociation protocols are critical. Enzymatic digestion on ice can help mediate transcriptomic stress responses. Fixed cell methods (e.g., ACME methanol fixation) may be employed to preserve transcriptomic states [29].
Reverse Transcription and Strand-Switching (170 minutes)
- Primer Annealing: Combine RNA with RT Primer (provided in kit) and dNTPs.
- Reverse Transcription: Add Maxima H Minus Reverse Transcriptase with 5× RT Buffer, RNaseOUT, and incubate.
- Reaction conditions: 42°C for 90 minutes, followed by 85°C for 5 minutes.
- Strand-Switching: Add cDNA RT Adapter and Strand Switching Primer II (SSPII).
- This step incorporates a Unique Molecular Identifier (UMI) for downstream quantification.
- Stopping Point: cDNA can be stored at -20°C overnight if needed [31].
cDNA Amplification and Barcoding (40 minutes)
- PCR Setup: Combine strand-switched cDNA with LongAmp Hot Start Taq 2X Master Mix and Barcode Primers.
- Thermal Cycling:
- Initial denaturation: 95°C for 3 minutes
- Cycling (12-15 cycles): 95°C for 15s, 62°C for 15s, 65°C for 10 minutes
- Final extension: 65°C for 5 minutes
- Product Cleanup: Use Agencourt AMPure XP beads to purify amplified cDNA.
- Quality Assessment: Check cDNA size distribution (300-400 bp to 9,000-10,000 bp range expected) using fragment analyzers [31] [30].
Library Preparation and Sequencing
- Adapter Ligation: Pool barcoded samples and add Rapid Sequencing Adapters (5 minutes).
- Priming and Loading: Prime flow cell and load prepared cDNA library.
- Sequencing: Perform on R10.4.1 flow cells for optimal results [31].
Alternative Protocol: Droplet-Based scRNA-seq
For platforms like 10× Genomics, the workflow differs in its initial partitioning approach [32]:
- GEM Generation: Co-encapsulate single cells with barcoded gel beads in oil-emulsion droplets.
- Reverse Transcription: Within each droplet, mRNA binds to beads via poly(dT) sequences, and reverse transcription occurs.
- Library Construction: Break droplets, pool barcoded cDNA, and proceed with library preparation through fragmentation, adapter ligation, and PCR amplification.
- Sequencing: Utilize paired-end sequencing on Illumina platforms with recommended read depths of 20,000-50,000 reads per cell [30] [32].
Technical Considerations and Optimization
Addressing Common Challenges in Low-Input scRNA-seq
Table 2: Troubleshooting Common Issues in Embryo scRNA-seq
| Challenge | Impact on Data | Mitigation Strategies |
|---|---|---|
| Multiplets | Two or more cells share same barcode; inflated expression values | - Accurate cell counting and dilution- Proper sample dissociation to prevent clumps- Add DNase to reduce genomic DNA-mediated stickiness [30] |
| Ambient RNA | Background RNA from damaged cells misattributed to cells | - Optimize tissue dissociation to minimize cell death- Include wash steps in combinatorial barcoding protocols- Computational background correction [30] |
| Low Capture Efficiency | Reduced gene detection sensitivity | - Use fresh enzymes and quality-controlled reagents- Optimize input RNA quantity and quality- Consider nuclear sequencing for difficult-to-dissociate tissues [29] |
| Batch Effects | Technical variability obscuring biological signals | - Process all samples for a comparative study simultaneously- Use multiplexing with sample barcoding- Employ standardized protocols across samples [29] [4] |
Quality Control Checkpoints
- Post-Amplification cDNA: Size distribution should show a gradual rise from 300-400 bp to 9,000-10,000 bp.
- Final Library: Ideal size distribution between 400-500 bp for Illumina sequencing.
- Sequencing QC: Use FastQC/MultiQC to evaluate per-base sequence quality, sequence diversity, and GC content [30].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Low-Input RNA Library Preparation
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Reverse Transcriptase | Maxima H Minus Reverse Transcriptase | Synthesizes cDNA from mRNA templates; high processivity needed for low-input samples [31] |
| Barcoding Primers | cDNA-PCR Barcoding Kit 24 V14 (Oxford Nanopore) | Enable sample multiplexing; contain unique barcodes and UMIs for cell and molecule identification [31] |
| Amplification Master Mix | LongAmp Hot Start Taq 2X Master Mix | Amplifies cDNA with high fidelity and processivity for full-length transcript coverage [31] |
| Cleanup Beads | Agencourt AMPure XP beads | Size selection and purification of nucleic acids between reaction steps [31] |
| Transposase Enzyme | Tn5 Transposase (for multiomics) | Simultaneously fragments DNA and adds adapters in scATAC-seq workflows [32] |
| Viability Stains | DAPI, Propidium Iodide | Assess cell membrane integrity and identify live cells for sorting [32] |
| Rauvoyunine B | Rauvoyunine B, MF:C23H26N2O6, MW:426.5 g/mol | Chemical Reagent |
Robust library preparation for low-input RNA is fundamental to successful embryo scRNA-seq research. The choice between droplet-based, microwell, and combinatorial barcoding approaches depends on specific experimental needs, including cell number, desired throughput, and available resources. By implementing the detailed protocols and quality control measures outlined in this document, researchers can reliably generate high-quality transcriptomic data from precious embryonic materials, ultimately advancing our understanding of early development, cell fate decisions, and the molecular basis of developmental disorders.
For researchers investigating embryonic development, single-cell RNA sequencing (scRNA-seq) provides unprecedented resolution to explore cell fate decisions, lineage specification, and transcriptional heterogeneity. However, the successful application of this technology, particularly within low-throughput workflows designed for precious embryonic samples, demands careful optimization of key sequencing parameters. Among these, sequencing depth and coverage are fundamentally critical yet distinct considerations that directly impact data quality, interpretive power, and cost-efficiency [33] [34].
Sequencing depth (or read depth) refers to the average number of times a specific nucleotide is read during the sequencing process, typically expressed as a multiple (e.g., 50,000 reads per cell). It is a key determinant of data accuracy and the sensitivity for detecting lowly-expressed transcripts [34]. In contrast, sequencing coverage describes the percentage of the transcriptome that is successfully sequenced at least once, ensuring comprehensive representation of all expressed genes [33] [34]. For embryonic studies, where cell numbers are often limited and each sample is invaluable, striking the optimal balance between these two parameters is paramount to maximize biological insights while conserving resources.
This application note outlines detailed protocols and evidence-based recommendations for balancing sequencing depth and coverage within a low-throughput scRNA-seq workflow for embryonic research.
Key Concepts and Definitions
Distinguishing Depth from Coverage
- Sequencing Depth: The average number of times a given nucleotide is sequenced. Deeper sequencing increases confidence in base calling and facilitates the detection of rare variants and low-abundance transcripts [33] [34].
- Sequencing Coverage: The proportion of the genome or transcriptome that has been sequenced at least once. High coverage ensures that the entirety of the target region is represented, minimizing gaps in the data [33] [34].
The Interplay in Embryonic Transcriptomes
In embryonic scRNA-seq, sufficient coverage ensures that transcripts from all genes, including those specific to rare or transient cell populations, are captured. Adequate depth is then necessary to quantify the expression of these genes accurately, especially critical transcription factors that may be expressed at low levels but have pivotal biological roles [35] [4]. A failure to achieve adequate coverage risks missing key genes entirely, while insufficient depth leads to noisy, unreliable quantification and an inability to distinguish biological variation from technical noise.
Optimal Sequencing Parameters for Embryonic Transcriptomes
Determining the appropriate sequencing depth is influenced by the specific biological question, the complexity of the embryonic sample, and the scRNA-seq protocol employed. The following recommendations synthesize findings from recent studies.
Recommended Sequencing Depth
Table 1: Recommended Sequencing Depth for Embryonic Transcriptome Analysis
| Application / Context | Recommended Sequencing Depth | Key Rationale |
|---|---|---|
| Standard Embryo Profiling (Cell-type identification, primary lineage specification) | 20,000 - 50,000 reads per cell | Provides a robust balance for detecting a majority of expressed genes and defining major cell populations [35] [36]. |
| Detection of Low-Abundance Transcripts (Rare transcription factors, signaling molecules) | 50,000 - 100,000 reads per cell | Increased depth enhances sensitivity for quantifying weakly expressed but biologically critical genes [35] [34]. |
| Comprehensive Gene Detection (Near-complete transcriptome cataloguing) | >100,000 reads per cell | Required to detect >90% of annotated genes, as demonstrated in chicken embryo studies where 28.7-29.6 million reads achieved this goal [35]. |
| De Novo Transcriptome Assembly (Whole-animal samples) | ~30 million total reads | A cross-phyla comparison suggested this depth provides a good balance between gene discovery and noise for whole-animal assemblies [36]. |
| De Novo Transcriptome Assembly (Single-tissue samples) | ~20 million total reads | The same study found that single-tissue assemblies require slightly lower depth for representative assembly [36]. |
Impact of Depth on Transcript Discovery
The relationship between sequencing depth and gene detection is non-linear. A study on chicken embryos demonstrated that while increasing depth from 1.6 million to 10 million reads significantly boosted the proportion of detected genes from 68% to about 80%, the marginal gain diminished beyond 10-20 million reads [35]. This highlights that for many applications, a depth of 10-20 million reads (or its per-cell equivalent) can be a cost-effective point of saturation for gene detection.
Detailed Experimental Protocols
A Low-Throughput scRNA-Seq Workflow for Embryonic Samples
The following diagram illustrates a generalized low-throughput scRNA-seq workflow tailored for embryonic samples. Key decision points for depth and coverage are integrated into the process.
Diagram 1: Low-throughput scRNA-seq workflow for embryonic samples, highlighting key steps where sequencing parameters are determined.
Protocol: Sample Preparation and Library Construction
Sample Preparation and Cell Isolation:
- Isolate viable individual cells or nuclei from the embryo. For fragile or frozen samples, single-nucleus RNA-seq (snRNA-seq) is a suitable alternative [12].
- Use gentle dissociation protocols to minimize stress-induced transcriptional changes.
- Assess cell viability and integrity using a cell counter or flow cytometry. Aim for >80% viability [12].
Library Preparation:
- Select a scRNA-seq protocol appropriate for the study goals. Full-length protocols (e.g., SMART-Seq2) are ideal for detecting splice variants and have enhanced sensitivity for low-abundance genes, while 3'-end counting protocols (e.g., 10x Genomics) enable higher cell throughput at a lower cost per cell [12].
- Follow manufacturer instructions for cDNA synthesis and amplification. Incorporate Unique Molecular Identifiers (UMIs) to correct for PCR amplification bias and enable accurate digital quantification of transcripts [37] [12].
- Perform quality control on the final libraries using a Fragment Analyzer or Bioanalyzer and quantify via qPCR.
Protocol: Sequencing Depth Optimization via Pilot Study
For novel embryonic systems, a pilot study is highly recommended to empirically determine the optimal sequencing depth.
- Sequencing Run: Sequence a subset of libraries to a high depth (e.g., >100,000 reads per cell).
- In Silico Down-Sampling: Use computational tools to randomly sub-sample the sequenced reads to lower depths (e.g., 10,000, 20,000, 50,000 reads per cell) from the original high-depth data [35].
- Saturation Analysis: For each down-sampled dataset, calculate the number of genes detected. Plot the number of genes detected against sequencing depth.
- Determine Optimal Depth: Identify the point where the gene detection curve begins to plateau. The depth just before this plateau is a cost-effective optimal depth for subsequent experiments.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Embryonic scRNA-seq
| Reagent / Kit | Function | Considerations for Embryonic Work |
|---|---|---|
| Gentle Cell Dissociation Kit | Liberates individual cells from embryonic tissues while preserving viability and RNA integrity. | Critical for minimizing transcriptional stress responses. Enzymatic blends (e.g., collagenase) are often preferred for early embryos. |
| scRNA-seq Library Prep Kit(e.g., SMART-Seq2, 10x Chromium) | Converts mRNA from single cells into a sequencer-ready cDNA library. | Full-length (SMART-Seq2): Best for isoform analysis, lowly-expressed genes. 3'-end (10x): Best for cell throughput, population heterogeneity [12]. |
| Unique Molecular Identifiers (UMIs) | Short random barcodes that tag individual mRNA molecules, allowing for accurate transcript counting by correcting for PCR duplicates. | Essential for precise quantification of gene expression levels [37] [12]. |
| rRNA Depletion Kit | Removes abundant ribosomal RNA (rRNA) to increase the sequencing power dedicated to mRNA. | Increases the effective coverage of the transcriptome. Useful when total RNA input is limited. |
| Viability Staining Dye(e.g., Propidium Iodide, DAPI) | Distinguishes live cells from dead cells or debris during cell sorting. | Essential for ensuring high-quality input material, as RNA from dead cells contributes to background noise. |
| Benchmarking Reference Atlas(e.g., Integrated Human Embryo Data) | A curated scRNA-seq dataset serving as a universal reference for authenticating embryo models and annotating cell identities. | Enables unbiased comparison of in-house data against a gold-standard in vivo reference, preventing misannotation [4]. |
Technology Selection and Strategic Considerations
The choice of scRNA-seq technology directly influences the required sequencing depth and the overall experimental strategy. The following diagram outlines the decision-making process for selecting the appropriate protocol in the context of a low-throughput embryonic research workflow.
Diagram 2: A decision tree for selecting an scRNA-seq protocol and corresponding sequencing depth for embryonic research.
Applications in Drug Discovery and Development
The principles of optimizing scRNA-seq for embryos extend directly into drug discovery. This technology can reveal the cellular heterogeneity of diseases, identify key therapeutic targets, and evaluate the fidelity of stem cell-derived embryo models used for drug testing [3] [37] [38]. A well-sequenced embryonic transcriptome serves as a critical benchmark for assessing whether in vitro models accurately recapitulate in vivo development, thereby validating their use in preclinical screens [4].
Embryonic scRNA-seq is a powerful tool that demands careful experimental planning. There is no universal "best" depth or coverage; rather, the optimal parameters must be tailored to the biological question, the embryonic system under study, and the chosen technology. A pilot study with in-silico down-sampling is a highly effective strategy for determining the most efficient sequencing depth. By adhering to the protocols and considerations outlined in this document, researchers can design robust, cost-effective low-throughput workflows that maximize the scientific return from precious embryonic samples.
Overcoming Technical Challenges in Embryo-Derived scRNA-seq
Addressing Low RNA Input and Amplification Bias in Embryonic Cells
Single-cell RNA sequencing (scRNA-seq) of embryonic tissues presents unique challenges, primarily due to the naturally low amounts of RNA in individual cells and the amplification biases introduced during library preparation [12] [3]. These challenges are particularly pronounced in early human development studies, where sample availability is often restricted by ethical considerations and technical limitations [4] [3]. Overcoming these obstacles is critical for obtaining accurate transcriptional profiles that can reveal novel insights into cellular heterogeneity, lineage specification, and developmental disorders [39] [10]. This application note outlines optimized low-throughput workflows and protocols specifically designed for embryonic scRNA-seq research, focusing on strategies to mitigate technical artifacts while preserving biological fidelity.
Technical Challenges in Embryonic scRNA-seq
Embryonic cells typically contain limited RNA material, with vertebrate cells generally estimated to contain approximately 10âµâ€“10ⶠmRNA molecules [10]. This scarcity is compounded during early embryonic development stages, where rapid cell divisions and compact transcriptional programs further reduce RNA complexity [3]. The minute RNA quantities necessitate amplification steps that can introduce significant technical artifacts, including:
- Amplification Bias: Non-linear amplification during cDNA synthesis and PCR preferentially enriches certain transcripts over others, distorting true expression ratios [12] [39].
- 3' Bias: Protocols relying on poly(A) selection often exhibit coverage skewing toward the 3' end of transcripts, compromising isoform-level analysis [12] [40].
- Molecular Loss: Inefficient reverse transcription and capture leads to undersampling of the transcriptome, particularly affecting low-abundance genes [12] [40].
These technical variabilities can obscure crucial biological signals in embryonic development, such as the subtle transcriptional differences driving early lineage specification [4] [3].
Table 1: Comparison of scRNA-seq Protocols for Embryonic Research
| Protocol | Amplification Method | Transcript Coverage | UMI Implementation | Unique Advantages for Embryonic Cells |
|---|---|---|---|---|
| Smart-Seq2 [12] | PCR-based | Full-length | No | Enhanced sensitivity for low-abundance transcripts; ideal for detecting rare regulatory RNAs in early embryos |
| CEL-Seq2 [12] | IVT-based | 3'-only | Yes | Linear amplification reduces bias; suitable for quantifying expression levels in preimplantation embryos |
| Quartz-Seq2 [12] | PCR-based | Full-length | No | Optimized reaction conditions improve sensitivity for limited embryonic RNA input |
| MATQ-Seq [12] | PCR-based | Full-length | Yes | Increased accuracy in quantifying transcripts; efficient detection of transcript variants in developing lineages |
| Drop-Seq [12] [40] | PCR-based | 3'-end | Yes | High-throughput capability for profiling heterogeneous embryonic cell populations |
Optimized Low-Throughput Workflow for Embryonic Cells
The following workflow has been specifically optimized for low-throughput studies of embryonic development, balancing sensitivity with technical accuracy for precious embryo samples.
Sample Preparation and Quality Control
Begin with rigorous sample preparation to preserve RNA integrity and ensure single-cell suspension quality:
- Embryo Dissociation: Use gentle enzymatic cocktails (e.g., Accutase or TrypLE) with minimal mechanical disruption to preserve RNA integrity [3]. For fragile embryonic cells, consider single-nucleus RNA-seq (snRNA-seq) approaches to overcome dissociation challenges [12].
- Viability Assessment: Employ fluorescence-activated cell sorting (FACS) with viability dyes (e.g., propidium iodide) to ensure >85% cell viability, critical for reducing ambient RNA contamination [40].
- Cell Concentration Optimization: Adjust cell suspension to 700–1,200 cells/μL to minimize doublet formation while maintaining capture efficiency [40].
Protocol Selection for Embryonic Applications
Based on the specific research question and embryonic stage, select an appropriate protocol:
- For Preimplantation Embryos (Limited Cell Numbers): Smart-Seq2 is recommended for its enhanced sensitivity in detecting low-abundance transcripts and capability for full-length transcript coverage, enabling isoform analysis during zygotic genome activation [12] [3].
- For Lineage Tracing Studies: Droplet-based methods like Drop-Seq provide the throughput needed to capture rare transitional states during gastrulation, with UMIs enabling accurate molecular counting [12] [40].
- For Quantitative Expression Analysis: CEL-Seq2's linear amplification (IVT-based) reduces PCR biases, providing more accurate quantification of expression levels in embryonic cell subtypes [12].
Molecular Strategies for Bias Mitigation
Implement these molecular biology strategies to address amplification bias and low RNA input:
- Unique Molecular Identifiers (UMIs): Incorporate UMIs during reverse transcription to correct for amplification biases and enable absolute molecular counting [12] [40]. This is particularly valuable for accurately quantifying transcriptional bursting in embryonic cells.
- Template-Switch Oligos (TSOs): Utilize TSOs in Smart-Seq2 and related protocols to enable cDNA synthesis independent of poly(A) tails, improving coverage of potentially degraded transcripts from embryonic samples [40].
- Spike-in Controls: Add exogenous RNA controls (e.g., ERCC RNA Spike-In Mix) in known quantities to monitor technical variability and enable normalization between samples [12].
- Reduced Amplification Cycles: Optimize PCR cycle numbers to maintain representation of low-abundance transcripts while minimizing duplication rates [12] [40].
Table 2: Research Reagent Solutions for Embryonic scRNA-seq
| Reagent/Category | Specific Examples | Function in Embryonic scRNA-seq |
|---|---|---|
| Cell Viability Dyes | Propidium iodide, DAPI | Distinguish viable cells in embryonic dissociations; critical for reducing background RNA |
| Amplification Kits | Smart-Seq2 kit, CEL-Seq2 reagents | Optimized chemistry for limited embryonic RNA; maintain representation of rare transcripts |
| Barcoding Systems | 10x Barcoded Gel Beads, Custom UMIs | Enable multiplexing of precious embryonic samples; correct for amplification biases |
| Spike-in Controls | ERCC RNA Spike-In Mix, SIRVs | Monitor technical variability; enable cross-sample normalization for comparative embryology |
| Reverse Transcriptase | Maxima H-minus, Template-switching RT | High-efficiency cDNA synthesis from limited embryonic RNA; reduce 3' bias |
Experimental Protocol: Smart-Seq2 Optimization for Embryonic Cells
The following detailed protocol adapts Smart-Seq2 specifically for embryonic cell applications, incorporating modifications to address low RNA input and amplification bias:
Cell Lysis and Reverse Transcription
Cell Lysis Buffer Preparation:
- Prepare lysis buffer containing: 0.5% Triton X-100, 2 U/μL RNase inhibitor, 2.5 mM dNTPs, and 1:120,000 ERCC RNA Spike-In Mix.
- Distribute 4 μL lysis buffer into individual PCR tubes.
Single-Cell Capture and Lysis:
- Manually pick individual embryonic cells using a micromanipulator and transfer into lysis buffer.
- Immediately freeze samples at -80°C for 10 minutes, then thaw on ice to ensure complete lysis.
- Centrifuge briefly (500 × g, 1 minute) to collect contents.
Reverse Transcription with Template Switching:
- Prepare RT mix (per reaction): 1× Maxima RT buffer, 2 U/μL RNase inhibitor, 2.5 μM TSO, 5 mM DTT, 4 U/μL Maxima H-minus Reverse Transcriptase.
- Add 6 μL RT mix to each lysed cell, mixing gently.
- Incubate: 42°C for 60 minutes, 50°C for 10 minutes, then 70°C for 15 minutes.
- Store cDNA at -20°C or proceed immediately to preamplification.
cDNA Preamplification and Quality Control
PCR Preamplification:
- Prepare PCR mix (per reaction): 1× KAPA HiFi HotStart ReadyMix, 0.5 μM ISPCR primer.
- Add 15 μL PCR mix to 10 μL RT reaction.
- Amplify with cycling conditions: 98°C for 3 minutes; 21-24 cycles (optimized for embryonic cells) of 98°C for 20 seconds, 65°C for 15 seconds, 72°C for 4 minutes; final extension at 72°C for 5 minutes.
cDNA Quality Control:
- Analyze 1 μL amplified cDNA on High Sensitivity DNA Chip (Bioanalyzer) or Fragment Analyzer.
- Expected profile: Broad distribution from 0.5-6 kb, with peak around 1.5-2 kb.
- Quantify cDNA using fluorometric methods (e.g., Qubit dsDNA HS Assay).
Tagmentation and Library Construction
Tagmentation Reaction:
- Dilute high-quality cDNA to 0.2-0.5 ng/μL in 10 mM Tris-HCl (pH 8.0).
- Prepare tagmentation mix (per reaction): 1× TD buffer, 0.5-1.0 μL TDE1 enzyme (Nextera Th5 Transposase).
- Combine 5 μL diluted cDNA with 5 μL tagmentation mix, incubate at 55°C for 10 minutes.
- Immediately add 5 μL Neutralization Buffer and incubate at room temperature for 5 minutes.
Library Amplification:
- Prepare PCR mix (per reaction): 1× KAPA HiFi HotStart ReadyMix, 5 μM each i5 and i7 indexing primers.
- Combine 15 μL PCR mix with 5 μL tagmented cDNA.
- Amplify with cycling conditions: 72°C for 3 minutes; 98°C for 30 seconds; 10-12 cycles of 98°C for 10 seconds, 63°C for 30 seconds, 72°C for 1 minute; final extension at 72°C for 5 minutes.
Library Cleanup and Quality Control:
- Purify libraries using 0.8× SPRIselect beads.
- Assess library quality on Fragment Analyzer; expected size distribution: 200-1,000 bp.
- Quantify libraries by qPCR (KAPA Library Quantification Kit) before sequencing.
Data Analysis Considerations for Embryonic Applications
Computational analysis of embryonic scRNA-seq data requires special considerations to address technical artifacts:
- Quality Control Metrics: Establish embryo-specific QC thresholds: genes/cell >1,000, mitochondrial percentage <10% (higher thresholds may indicate stressed embryonic cells) [4].
- Doublet Detection: Employ computational doublet detection tools (e.g., Scrublet, DoubletFinder) adapted for embryonic datasets, as embryonic cells have distinct size and RNA content compared to somatic cells [40].
- Batch Effect Correction: Apply mutual nearest neighbors (MNN) or similar methods to correct for technical variability between experimental batches, particularly crucial when integrating data from multiple embryo donations [4].
- Trajectory Inference: Utilize pseudotime algorithms (e.g., Slingshot, Monocle3) to reconstruct developmental trajectories, accounting for the rapid transcriptional shifts characteristic of embryonic development [4].
Successful scRNA-seq of embryonic cells requires careful optimization at every step, from sample preparation through data analysis, to overcome the inherent challenges of low RNA input and amplification bias. The protocols and strategies outlined here provide a foundation for obtaining high-quality transcriptional data from precious embryonic samples, enabling researchers to explore the complex regulatory landscapes of early development with unprecedented resolution. As the field advances, integration of these approaches with emerging multi-omics technologies promises to further illuminate the molecular mechanisms governing human embryogenesis.
Mitigating Dropout Events and Managing Technical Noise in Sparse Data
Single-cell RNA sequencing (scRNA-seq) of embryonic tissues presents unique challenges, primarily due to the inherently low starting mRNA quantities and the critical nature of rare cell populations driving development. A predominant issue is the "dropout" phenomenon, where a gene is expressed at a moderate level in one cell but not detected in another cell of the same type [41]. These dropout events, stemming from the stochastic capture of limited mRNA molecules during library preparation, result in highly sparse data matrices that can obscure genuine biological signals [42] [41]. In the context of low-throughput embryo research, where every cell is valuable and sample sizes are often smaller, effectively mitigating technical noise and dropouts is not merely a preprocessing step but a fundamental necessity for achieving biological fidelity. This protocol outlines a streamlined, robust workflow designed to address these challenges, enabling researchers to distinguish true biological zeros from technical artifacts and thereby uncover the subtle transcriptional dynamics that underpin embryonic development.
Computational Strategies for Dropout Mitigation and Noise Reduction
A range of computational methods has been developed to address sparsity in scRNA-seq data. These can be broadly categorized into imputation methods, which aim to fill in missing values, and noise reduction models, which seek to stabilize the data. The choice of method should be guided by the specific research question and the need to preserve biological heterogeneity, a key concern in embryonic development studies.
The table below summarizes the core strategies, their representative tools, and key considerations for their application in a low-throughput embryo research workflow.
Table 1: Computational Strategies for Managing Dropouts and Noise
| Category | Representative Methods | Underlying Principle | Considerations for Embryo scRNA-seq |
|---|---|---|---|
| Model-Based & Probabilistic Imputation | scRecover [43], scImpute [43], SAVER [43] | Employs statistical models (e.g., Zero-Inflated Negative Binomial) to distinguish technical dropouts from biological zeros. | High transparency; preserves true biological zeros; may scale poorly with very large datasets. |
| Smoothing & Low-Rank Reconstruction | MAGIC [41] [43], KNN-smoothing [43], ALRA [43] | Diffuses information across similar cells or enforces global structural constraints to denoise data. | Efficient scaling; risk of over-smoothing and blurring rare cell-type signals. |
| Deep Neural Models | scVI [44] [43], DCA [43], DeepImpute [43] | Uses nonlinear embeddings (e.g., variational autoencoders) to capture complex data structures and impute values. | Effective for complex dependencies; training can be less stable and models less interpretable. |
| High-Dimensional Statistical Denoising | RECODE/iRECODE [42] | Uses high-dimensional statistics and eigenvalue modification to reduce technical noise without dimensionality reduction. | Preserves full-dimensional data; effectively mitigates both technical and batch noise. |
| Leveraging Dropout Patterns | Co-occurrence Clustering [41] | Treats the binary dropout pattern as a useful biological signal for clustering cells, rather than a problem. | Identifies cell populations based on gene pathways beyond highly variable genes. |
The following diagram illustrates how these methods can be integrated into a cohesive analytical workflow for embryo scRNA-seq data, from raw data processing to downstream biological interpretation.
Protocol: Implementing iRECODE for Dual Noise Reduction
For embryo studies that may involve integrating data from multiple batches or donors, the iRECODE algorithm provides a powerful solution for simultaneous technical and batch noise reduction while preserving the full dimensionality of the data [42]. The following is a detailed application protocol.
Principle: iRECODE synergizes the high-dimensional statistical approach of RECODE with established batch correction methods. It first maps gene expression data to an essential space using noise variance-stabilizing normalization (NVSN) and singular value decomposition. The key innovation is that batch correction is integrated within this essential space, bypassing high-dimensional calculations that typically reduce accuracy and increase computational cost [42].
Experimental Procedure:
- Input Data Preparation: Begin with a raw UMI count matrix from your embryo scRNA-seq experiment, where cells are columns and genes are rows. Ensure that batch metadata (e.g., sequencing run, sample donor) is accurately recorded.
- Platform and Dependency Setup: The RECODE platform is available as an R package. Install it from the designated repository (e.g., Bioconductor or GitHub). Ensure all dependencies, such as Harmony [42], are installed.
- Parameter Configuration: iRECODE is designed to be largely parameter-free. The primary choice is the batch correction method. Based on benchmarking, using Harmony within the iRECODE framework is recommended for optimal performance [42].
- Execution Code:
- Output and Quality Assessment: The output is a denoised and batch-corrected expression matrix. Evaluate success using two key metrics:
- Batch Mixing: Visualize the data using UMAP, coloring points by batch. Successful integration will show cells from different batches well-mixed within cell type clusters [42].
- Biological Conservation: Cell-type identities should remain distinct. Calculate the local inverse Simpson's index (iLISI) for batch mixing and cell-type LISI (cLISI) for biological conservation. iRECODE should yield high iLISI scores while maintaining low cLISI scores, comparable to state-of-the-art batch correction methods [42].
Protocol: Leveraging Dropout Patterns with Co-occurrence Clustering
As an alternative to imputation, this protocol uses the binary dropout pattern itself as a biological signal for identifying cell populations in embryo data [41].
Principle: Instead of treating dropouts as noise, this method hypothesizes that genes within the same functional pathway tend to exhibit similar dropout patterns across different cell types. Binarizing the count matrix (0 for non-detection, 1 for detection) and analyzing co-occurrence can reveal these pathways and define cell populations [41].
Experimental Procedure:
- Input Data: Start with a normalized scRNA-seq count matrix from embryo cells.
- Data Binarization: Convert the count matrix into a binary matrix where any non-zero value is set to 1, representing gene detection.
- Algorithm Execution: Implement the co-occurrence clustering algorithm, which operates iteratively:
- Step A - Gene-Gene Graph: For all cells in a cluster, compute a statistical measure of co-occurrence for each pair of genes. Construct a weighted gene-gene graph and partition it into gene clusters (pathways) using community detection (e.g., the Louvain algorithm) [41].
- Step B - Pathway Activity: For each identified gene pathway, calculate the percentage of detected genes for every cell. This creates a low-dimensional "pathway activity" representation.
- Step C - Cell-Cell Graph: Build a cell-cell graph using Euclidean distances in the pathway activity space. Apply community detection to partition the cells into clusters.
- Step D - Cluster Merging: Merge cell clusters if none of the gene pathways show differential activity between them, based on pre-defined thresholds for signal-to-noise ratio, mean difference, and mean ratio [41].
- Step E - Iteration: Repeat steps A-D on each new cell cluster until no further subdivisions are possible, resulting in a hierarchical tree of cell types.
- Software and Code: While a complete packaged implementation may not be universally available, the analytical steps can be executed using standard single-cell analysis toolkits like Scanpy or Seurat for graph construction and clustering, following the described logic.
- Interpretation: The final output is a set of cell clusters defined by the coordinated presence or absence of gene modules. This can reveal cell subtypes in the developing embryo that might be missed by analyses relying solely on highly variable genes.
The Scientist's Toolkit: Essential Reagents and Computational Tools
Successful execution of the protocols above relies on a combination of wet-lab reagents and computational tools. The following table details the key components of the toolkit for managing technical noise in embryo scRNA-seq.
Table 2: Research Reagent and Computational Solutions for Embryo scRNA-seq
| Item Name | Function/Application | Specifications & Alternatives |
|---|---|---|
| Chromium Single Cell 3' Reagent Kits (10x Genomics) | A droplet-based system for high-throughput barcoding and library preparation of single-cell transcriptomes. | Enables the generation of UMI-based count matrices from embryonic cell suspensions. |
| Cell Ranger (10x Genomics) | A standardized pipeline for processing raw sequencing data (FASTQ) from 10x assays. | Aligns reads, generates feature-barcode matrices, and performs initial QC. Crucial for consistent raw data processing [45]. |
| scRecover | An R package for accurate dropout imputation that distinguishes technical zeros from biological zeros using a ZINB model. | Particularly useful for preserving true biological absences, critical for interpreting signaling in development [43]. |
| RECODE/iRECODE | A platform for technical noise reduction and batch correction based on high-dimensional statistics. | Ideal for complex embryo studies involving multiple samples or data integration, as it preserves full-dimensional data [42]. |
| Harmony | A robust algorithm for integrating scRNA-seq data across multiple batches or experiments. | Can be used standalone or integrated within the iRECODE framework to correct for batch effects while preserving biological variation [42] [44]. |
| Seurat / Scanpy | Comprehensive R/Python-based toolkits for end-to-end analysis of scRNA-seq data. | Provide the foundational environment for QC, normalization, clustering, visualization, and the implementation of many advanced protocols [46] [44]. |
The sparse nature of scRNA-seq data demands rigorous and thoughtful analytical strategies, especially in the low-throughput, high-value context of embryo research. The protocols detailed herein—from the dual-noise reduction capability of iRECODE to the innovative signal extraction from dropout patterns—provide a robust framework for confronting technical variability. By carefully selecting and implementing these methods, researchers can significantly enhance the biological fidelity of their data, paving the way for groundbreaking discoveries in embryonic development, cell fate decisions, and the mechanistic underpinnings of developmental disorders.
In single-cell RNA sequencing (scRNA-seq) of embryonic samples, quality control (QC) is a critical first step in data analysis. Low-quality cells, if not properly identified and removed, can lead to erroneous biological interpretations by obscuring genuine cellular heterogeneity or creating artifactual cell populations [46]. This is particularly crucial in low-throughput embryo research, where the biological material is often scarce and the identification of rare cell populations is a primary goal. This application note details a robust QC workflow focusing on three fundamental metrics: cell viability, doublet detection, and mitochondrial gene content, providing embryology researchers with standardized protocols to ensure data integrity.
Key Quality Control Metrics and Thresholding
The initial QC stage involves calculating key metrics and setting appropriate thresholds to filter out low-quality cells. Table 1 summarizes the core QC metrics and recommended thresholding strategies for embryo scRNA-seq studies.
Table 1: Core QC Metrics and Thresholding Strategies for scRNA-seq Data
| QC Metric | Biological Significance | Recommended Thresholding Method | Notes for Embryo Research |
|---|---|---|---|
| Count Depth (total counts/cell) | Low counts may indicate poorly captured or dying cells [46]. | Median Absolute Deviation (MAD) [46]. | Be permissive to avoid losing rare embryonic cell types. |
| Detected Genes (genes/cell) | Low gene numbers can indicate broken cells or low-quality libraries [46]. | Median Absolute Deviation (MAD) [46]. | |
| Mitochondrial Proportion (mtDNA%) | High proportions often indicate cells undergoing apoptosis or stress [46] [47]. | Tissue-specific reference values; 5% is often too stringent for human samples [47]. | Human embryos generally show higher mtDNA% than mouse; avoid uniform 5% threshold [47]. |
A critical consideration is the mitochondrial proportion (mtDNA%). A systematic analysis of over 5 million cells revealed that the commonly used default threshold of 5% is frequently unsuitable, particularly for human tissues. Human cells exhibit significantly higher average mtDNA% than mouse cells, and a 5% threshold fails to accurately discriminate between healthy and low-quality cells in 29.5% of human tissues analyzed [47]. Therefore, researchers should consult tissue-specific reference values or use data-driven methods instead of relying on a universal 5% cutoff.
The following workflow diagram outlines the sequential steps for the quality control process.
Assessing Cell Viability and Sample Quality
High cell viability is a prerequisite for successful single-cell sequencing, especially for sensitive embryonic tissues. Viability is typically assessed prior to library preparation using membrane integrity assays.
Protocol: Cell Viability Assessment via Dye Exclusion
This protocol is adapted from optimized tissue dissociation methods for scRNA-seq [48] [49].
- Preparation: After tissue dissociation into a single-cell suspension, obtain a representative sample of cells.
- Staining: Mix 10-20 µL of the cell suspension with an equal volume of 0.4% Trypan Blue solution. Incubate for 1-2 minutes at room temperature [49].
- Counting: Load the mixture onto a hemocytometer. Under a bright-field microscope, count both unstained (viable) and blue-stained (non-viable) cells.
- Calculation: Calculate viability as follows: Viability (%) = [Number of viable cells / Total number of cells] × 100
- Quality Standard: Proceed with library preparation only if cell viability exceeds 70% [17]. For lower viability, consider repeating the dissociation protocol or performing additional dead cell removal.
Detection and Removal of Doublets
Doublets are artifactual libraries generated from two cells that were incorrectly encapsulated together. They can be mistaken for novel cell types or intermediate states, posing a significant risk to data interpretation [50]. In embryonic development, where cells transition through transient states, this is a major concern. While experimental techniques exist, computational detection is a widely accessible and effective approach.
Protocol: Computational Doublet Detection with DoubletFinder
DoubletFinder is a benchmarked method that demonstrates high detection accuracy [51]. The following protocol is implemented in R.
- Preprocessing: Begin with a high-quality gene-barcode matrix that has undergone initial QC (filtering on count depth and mtDNA%) and standard normalization.
- Parameter Estimation:
- Run Principal Component Analysis (PCA) on the preprocessed data.
- Use the
paramSweepfunction to simulate artificial doublets and test a range of pK (proportion of artificial nearest neighbors) values. - Identify the optimal pK value that minimizes the variance between real and artificial doublet classifications.
- Doublet Calling:
- Execute the
doubletFinderfunction, providing the preprocessed data, the estimated pK value, and the expected doublet rate. The expected doublet rate depends on the number of cells loaded and should be estimated based on the platform's specifications (e.g., ~1% per 1000 cells recovered for 10x Genomics) [51].
- Execute the
- Result Interpretation: The function returns a new metadata column classifying each cell as "Singlet" or "Doublet." Remove all cells classified as "Doublets" before proceeding with downstream analysis.
The diagram below illustrates how doublets are computationally identified by comparing real cells to simulated artificial doublets.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for scRNA-seq QC in Embryo Research
| Item | Function / Application | Example |
|---|---|---|
| Collagenase Type II | Enzymatic dissociation of complex embryonic tissues into single-cell suspensions. | Used in the dissociation of the mouse female reproductive tract for scRNA-seq [48]. |
| Trypan Blue Solution | A dye exclusion assay for assessing cell viability prior to library construction. | A standard, widely used method to determine the proportion of live cells in a suspension [49]. |
| Chromium Single Cell 3' Kit | A high-throughput, droplet-based library preparation system for single-cell RNA sequencing. | A popular commercial solution for generating barcoded scRNA-seq libraries from single-cell suspensions [17]. |
| Illumina Single Cell 3' RNA Prep Kit | A flexible, vortexing-based library preparation method that does not require microfluidic equipment. | An alternative to droplet-based methods, suitable for 100 to 100,000 cells [1] [17]. |
| scDblFinder / DoubletFinder R Packages | Computational tools for identifying and removing doublets from scRNA-seq data post-sequencing. | Benchmarking studies show DoubletFinder has high detection accuracy for identifying heterotypic doublets [50] [51]. |
Benchmarking and Validating Embryonic scRNA-seq Findings
The study of early human development is fundamental for understanding congenital diseases, infertility, and early pregnancy loss. However, research using human embryos faces significant challenges, including scarcity of donated embryos and ethical/legal constraints such as the "14-day rule." Stem cell-based embryo models have emerged as transformative tools that offer unprecedented experimental access to mimic early human development. The usefulness of these in vitro models hinges entirely on their fidelity to in vivo embryonic processes. Without rigorous benchmarking against natural embryonic development, findings from these models may lead to inaccurate biological conclusions.
Molecular characterization of embryo models has traditionally relied on examining individual lineage markers. However, this approach has limitations as many co-developing cell lineages share molecular markers. Unbiased transcriptional profiling through single-cell RNA sequencing (scRNA-seq) has therefore become the gold standard for validating embryo models. This protocol details a comprehensive framework for benchmarking in vitro embryo models against in vivo embryonic references using scRNA-seq data, with particular emphasis on low-throughput workflows suitable for laboratories with limited computational resources or smaller sample sizes.
Establishing the Reference: An Integrated Human Embryo Transcriptome Atlas
Reference Dataset Composition and Integration
A high-quality, integrated reference dataset serves as the foundation for rigorous benchmarking. This protocol utilizes an integrated human embryogenesis transcriptome reference compiled from six published scRNA-seq datasets covering developmental stages from zygote to gastrula (Carnegie Stage 7). The integration process employs fast mutual nearest neighbor (fastMNN) methods to minimize batch effects while preserving biological variance [4].
Table: Composition of Integrated Human Embryo Reference Atlas
| Developmental Stage | Key Lineages Present | Technology | Notable Features |
|---|---|---|---|
| Preimplantation Embryos | Trophectoderm (TE), Inner Cell Mass (ICM), Epiblast, Hypoblast | scRNA-seq | Covers lineage bifurcation events |
| Postimplantation Blastocysts (3D cultured) | Cytotrophoblast (CTB), Syncytiotrophoblast (STB), Extravillous Trophoblast (EVT) | scRNA-seq | Maturing trophoblast lineages |
| Carnegie Stage 7 Gastrula | Primitive Streak, Definitive Endoderm, Mesoderm, Amnion, Extraembryonic Mesoderm | scRNA-seq | Captures gastrulation events |
The resulting reference encompasses 3,304 early human embryonic cells embedded in a unified computational space using Uniform Manifold Approximation and Projection (UMAP), displaying continuous developmental progression with temporal and lineage specification. The first lineage branch point occurs as ICM and TE cells diverge around embryonic day 5 (E5), followed by ICM bifurcation into epiblast and hypoblast lineages [4].
Lineage Annotation and Validation
The reference atlas employs multiple validation approaches to ensure accurate lineage annotation:
- Cross-species comparison: Contrasting annotations with available non-human primate datasets
- Transcription factor activity: Using SCENIC analysis to identify known lineage-specific transcription factors
- Pseudotime analysis: Applying Slingshot trajectory inference to identify transcription factors with modulated expression across developmental trajectories
Key lineage markers validated in the reference include:
- Epiblast: TDGF1, POU5F1
- Trophectoderm: CDX2, NR2F2 (early); GATA2, GATA3, PPARG (mature CTB)
- Hypoblast: GATA4, SOX17 (early); FOXA2, HMGN3 (late)
- Primitive Streak: TBXT
- Amnion: ISL1, GABRP
Low-Throughput scRNA-seq Experimental Workflow
Sample Preparation and Single-Cell Isolation
For low-throughput studies focusing on specific developmental stages or lineage commitments, careful sample preparation is critical:
A. Tissue Dissociation Protocol
- Perform mechanical and enzymatic dissociation at 4°C to minimize artificial stress responses
- Use validated enzyme cocktails appropriate for embryonic tissues (e.g., Accutase for delicate embryonic cells)
- Limit dissociation time to 20-30 minutes with gentle agitation
- Include RNA stabilizers in dissociation buffers to preserve transcriptome integrity
B. Single-Cell Capture Methods for Low-Throughput Studies
- Fluorescence-Activated Cell Sorting (FACS): Enables selection of specific progenitor populations using surface markers
- Magnetic-Activated Cell Sorting (MACS): Suitable for enriching or depleting specific cell types
- Limiting Dilution: Simple and cost-effective for small cell numbers (<100 cells)
- Microfluidic Systems: Provide high-quality libraries for moderate throughput (96-384 cells)
C. Alternative Nuclear RNA-seq Considerations For tissues difficult to dissociate or when working with archived frozen samples, single-nucleus RNA sequencing (snRNA-seq) offers advantages:
- Minimizes dissociation-induced stress genes
- Applicable to frozen samples
- Particularly useful for brain, muscle, and other complex tissues
Library Preparation and Sequencing
A. cDNA Synthesis and Amplification Low-throughput studies benefit from full-length transcript protocols that provide better gene coverage:
- Smart-seq2 Protocol: Provides superior detection of splice variants and single-nucleotide polymorphisms
- Modifications for Embryonic Cells:
- Increase reverse transcriptase concentration due to high ribosomal RNA content
- Include ERCC spike-in controls for quality assessment
- Use reduced cycle numbers in PCR amplification to minimize bias
B. Unique Molecular Identifiers (UMIs) Incorporate UMIs during reverse transcription to:
- Correct for PCR amplification biases
- Enable accurate transcript counting
- Improve quantitative accuracy, especially for low-abundance transcripts
C. Sequencing Parameters
- Depth: 50,000-100,000 reads per cell sufficient for cell type identification
- Platform: Any standard sequencing platform (Illumina recommended for consistency with reference)
Workflow for Low-Throughput Embryo scRNA-seq Benchmarking (Max Width: 760px)
Computational Analysis Pipeline for Low-Throughput Studies
Data Preprocessing and Quality Control
A. Read Alignment and Quantification
- Align to GRCh38 human genome reference using Spliced Transcripts Alignment to a Reference (STAR)
- Generate gene count matrices with featureCounts or similar tools
- Process all samples with identical parameters to minimize technical variation
B. Quality Control Metrics
- Cell-level QC: Remove cells with <500 genes or >10% mitochondrial reads
- Gene-level QC: Filter genes detected in <10 cells
- Contamination Screening: Check for unexpected cell types using lineage markers
- Stress Gene Assessment: Monitor dissociation-induced stress genes (FOS, JUN, heat shock proteins)
Reference Mapping and Cell Identity Prediction
The integrated embryo reference provides an Early Embryogenesis Prediction Tool that enables:
A. Data Projection
- Project query datasets onto the reference UMAP using stabilized projection methods
- Transfer cell type labels based on nearest neighbors in reference space
- Calculate confidence scores for each cell type assignment
B. Lineage Validation
- Verify transferred labels with expression of known lineage markers
- Identify mismatches that may indicate model-specific deviations
- Detect novel cell states not present in the reference
Table: Key Transcription Factors for Lineage Validation
| Lineage | Early Markers | Late Markers | Functional Significance |
|---|---|---|---|
| Epiblast | POU5F1, NANOG | VENTX, HMGN3 | Pluripotency establishment |
| Trophectoderm | CDX2, NR2F2 | GATA3, PPARG | Trophoblast differentiation |
| Hypoblast | GATA4, SOX17 | FOXA2, HMGN3 | Primitive endoderm formation |
| Primitive Streak | TBXT, MESP2 | EOMES, MIXL1 | Mesendodermal specification |
Differential Expression and Trajectory Analysis
A. Lineage-Specific Gene Expression
- Identify genes differentially expressed between model and reference
- Focus on key developmental pathways (WNT, TGF-β, NOTCH signaling)
- Calculate effect sizes to distinguish biological from technical variation
B. Pseudotime Analysis
- Construct developmental trajectories using Slingshot or Monocle3
- Compare timing of lineage specification events between model and reference
- Identify genes with divergent expression dynamics along developmental trajectories
Computational Analysis Pipeline for Embryo Model Benchmarking (Max Width: 760px)
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for Embryo scRNA-seq Workflows
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Gentle Cell Dissociation Reagent | Tissue dissociation while preserving viability | Use at 4°C; include RNA stabilizers |
| FACS Antibody Panels | Isolation of specific progenitor populations | Validate specificity for embryonic antigens |
| Smart-seq2 Reagent Kit | Full-length scRNA-seq library preparation | Optimize cycle number for embryonic cells |
| UMI Barcoded Primers | Unique molecular identifiers for quantification | Essential for accurate transcript counting |
| ERCC RNA Spike-In Mix | Technical quality control | Add during cell lysis for normalization |
| Chromium Single Cell Kit (10x) | High-throughput library preparation | Alternative for larger-scale studies |
| SCENIC Analysis Pipeline | Transcription factor regulatory network inference | Identify key lineage-determining factors |
| Slingshot R Package | Pseudotime and trajectory analysis | Map developmental trajectories in models |
Quality Assessment and Troubleshooting
Common Technical Challenges and Solutions
A. High Mitochondrial RNA Content
- Cause: Cell stress or apoptosis
- Solution: Optimize dissociation protocol; include viability dyes during cell sorting
B. Batch Effects Between Model and Reference
- Cause: Technical variation in library preparation
- Solution: Process reference and model data through identical pipelines; use batch correction methods
C. Low Alignment Rates
- Cause: RNA degradation or adapter contamination
- Solution: Implement rigorous RNA quality control; use fresh fragmentation reagents
Interpretation Guidelines
A. Assessing Developmental Fidelity
- Evaluate presence/absence of expected lineages using marker expression
- Compare timing of developmental milestones using pseudotime analysis
- Quantify proportion of "off-target" cell types not present in natural embryos
B. Reporting Standards
- Document percentage of cells confidently mapping to reference lineages
- Report lineage-specific correlation coefficients with reference data
- Include negative controls (undifferentiated cells) to establish baseline
This protocol establishes a comprehensive framework for benchmarking in vitro embryo models against in vivo references using scRNA-seq. The integrated human embryo reference dataset and associated analysis tools provide an essential resource for validating model fidelity. For low-throughput workflows, focusing on specific developmental windows and employing full-length transcript methods maximizes biological insights while maintaining practical feasibility. Standardized benchmarking following these guidelines will enhance reproducibility and biological relevance in the rapidly advancing field of synthetic embryology.
Cell Type Annotation and Lineage Validation Using Marker Genes and Trajectory Inference
Single-cell RNA sequencing (scRNA-seq) has revolutionized transcriptomic studies by enabling the analysis of gene expression at the individual cell level, thereby uncovering cellular heterogeneity in complex biological systems [12]. This technological advancement is particularly transformative in embryo research, where it enables the tracking of dynamic cell differentiation events and lineage decisions during early development. In low-throughput scRNA-seq workflows designed for embryonic studies, where processing dozens to a few hundred cells is common, two analytical pillars form the foundation for biological interpretation: cell type annotation using marker genes and lineage validation through trajectory inference [1] [52].
Cell type annotation provides the essential identity cards for individual cells, allowing researchers to decipher the cellular composition of embryonic tissues. Simultaneously, trajectory inference reconstructs the developmental pathways these cells follow, mapping their journey from progenitor states to fully differentiated fates. When integrated together, these approaches form a powerful framework for validating lineage relationships and understanding the molecular dynamics driving embryonic development [52] [13]. This application note details standardized protocols and analytical frameworks for implementing these methods within low-throughput embryo scRNA-seq research, providing researchers with practical tools for uncovering the complexities of developmental biology.
Background
scRNA-seq in Embryo Research
The application of scRNA-seq to embryonic development has fundamentally changed our understanding of early cell fate decisions. Unlike bulk RNA-seq, which averages gene expression across cell populations, scRNA-seq captures the transcriptional heterogeneity between individual cells, making it ideal for studying the rapidly changing cellular landscape of developing embryos [13]. Low-throughput workflows are particularly suited to embryonic research where cell numbers may be limited, such as studies focusing on specific embryonic structures or time points, and where deeper sequencing per cell is desirable to capture more transcripts [1].
Key Concepts and Definitions
Cell Type Annotation: The process of identifying and labeling cell types within scRNA-seq data based on characteristic gene expression patterns, particularly using marker genes [53].
Marker Genes: Genes that exhibit specific expression in particular cell types or states, enabling their distinction from other cells. Canonical markers are frequently used for cell identification, while differentially expressed genes (DEGs) provide additional discriminatory power [54] [53].
Trajectory Inference: A computational method that reconstructs developmental or differentiation pathways by ordering cells along a pseudotemporal continuum based on transcriptional similarities [52].
Pseudotime: A quantitative measure that represents a cell's progression along a reconstructed developmental trajectory, with values reflecting relative positions rather than actual chronological time [52].
Methods and Experimental Design
Low-Throughput scRNA-seq Workflow for Embryo Research
Low-throughput scRNA-seq approaches are characterized by their focus on processing smaller numbers of cells (typically dozens to a few hundred) while often providing more comprehensive transcript coverage. For embryonic research, this balance is particularly advantageous as it allows for deeper sequencing per cell while maintaining manageable experimental scale [1]. The following diagram illustrates the complete workflow from sample preparation through final validation:
Sample Preparation and Single-Cell Isolation
For embryo research, careful sample preparation is critical. Embryonic tissues must be gently dissociated into single-cell suspensions while preserving cell viability and RNA integrity. Low-throughput methods particularly suited for embryonic studies include:
- Fluorescence-Activated Cell Sorting (FACS): Enables selection of specific cell types based on surface markers or fluorescent reporters, allowing researchers to focus on particular embryonic lineages [12] [25].
- Microfluidic Platforms (e.g., Fluidigm C1): Provide integrated circuits for cell capture, lysis, and library preparation, offering full-length transcript coverage which is advantageous for detecting isoform usage during development [25] [12].
- Micromanipulation: Manual selection of individual cells under microscopic visualization, particularly useful when working with very early embryonic stages where cell numbers are extremely limited [13].
Each method offers distinct advantages for embryonic research, with the choice depending on specific experimental needs regarding cell throughput, transcript coverage, and required equipment [12].
Research Reagent Solutions
Table 1: Essential Research Reagents and Platforms for Low-Throughput Embryo scRNA-seq
| Category | Specific Examples | Function in Workflow | Considerations for Embryo Research |
|---|---|---|---|
| Single-Cell Isolation | Fluidigm C1, FACS | Individual cell capture and partitioning | FACS enables pre-selection of specific embryonic cell populations; microfluidics offers integrated processing |
| Library Preparation | Smart-Seq2, Smart-Seq3 | Full-length cDNA amplification and library construction | Superior for detecting isoform usage and allelic expression in developing embryos [25] |
| Cell Type Annotation | ACT server, LICT, Seurat | Cell identity assignment using marker databases and algorithms | ACT provides embryonic development-specific marker references; LICT leverages AI for annotation [53] [55] |
| Trajectory Inference | Monocle3, Slingshot | Reconstruction of developmental paths from scRNA-seq data | Monocle3 is particularly effective for complex branching trajectories common in embryogenesis [52] |
| Data Analysis | Seurat, Scanpy, scViewer | Comprehensive analysis environment for scRNA-seq data | scViewer provides interactive visualization specifically useful for exploring embryonic datasets [56] |
Cell Type Annotation Using Marker Genes
Annotation Strategies and Tools
Cell type annotation in embryonic scRNA-seq data can be approached through multiple strategies, each with distinct advantages for developmental studies:
Knowledge-Based Annotation with ACT: The Annotation of Cell Types (ACT) web server provides a convenient platform that utilizes a hierarchically organized marker map curated from thousands of publications [53]. For embryonic research, this resource is particularly valuable as it integrates tissue-specific cellular hierarchies and employs a Weighted and Integrated gene Set Enrichment (WISE) method. The platform requires only a list of upregulated genes from cell clusters and returns comprehensive annotation suggestions with statistical support.
Automated Annotation with LICT: For more complex embryonic datasets where manual annotation becomes challenging, the Large Language Model-based Identifier for Cell Types (LICT) tool offers an automated alternative. LICT integrates multiple large language models in a "talk-to-machine" approach that iteratively refines annotations based on marker gene expression patterns [55]. This approach has demonstrated particular strength in annotating cell populations with low heterogeneity, which is common in early embryonic development.
Marker Gene Selection Methods: The foundation of accurate cell type annotation lies in selecting robust marker genes. Recent benchmarking studies have evaluated 59 computational methods for marker gene selection and found that simple methods, particularly the Wilcoxon rank-sum test, Student's t-test, and logistic regression, often outperform more complex approaches [54]. These methods excel at identifying genes with the specific expression patterns needed for distinguishing embryonic cell types – characterized by strong up-regulation in the cell type of interest with minimal expression in others.
Practical Annotation Protocol
For researchers implementing cell type annotation in embryonic scRNA-seq studies, the following step-by-step protocol provides a robust framework:
Cluster Identification: After quality control and normalization, perform cell clustering using Seurat or Scanpy pipelines. Use UMAP visualization to assess cluster separation and identify potential subpopulations [52] [57].
Differential Expression Analysis: For each cluster, perform differential expression analysis using a "one-vs-rest" approach to identify upregulated genes. The Wilcoxon rank-sum test implemented in Seurat provides a reliable starting point [54].
Marker Gene Selection: Select the top 10-20 marker genes per cluster based on statistical significance (adjusted p-value) and effect size (log fold-change). Prioritize genes with known biological relevance to embryonic development when available [54].
Multi-Method Annotation: Submit the marker gene lists to both ACT and LICT platforms. For ACT, utilize the embryonic development-specific hierarchies. For LICT, employ the multi-model integration strategy to leverage complementary strengths of different AI models [53] [55].
Annotation Validation: Use the objective credibility evaluation strategy from LICT, which assesses whether more than four marker genes are expressed in at least 80% of cells within the cluster. This provides a quantitative measure of annotation reliability [55].
Visualization and Interpretation: Generate visualization plots including UMAPs with cluster annotations, violin plots showing marker gene expression across clusters, and dot plots displaying expression strength and prevalence [57].
The following diagram illustrates the logical workflow and decision points in the cell type annotation process:
Lineage Validation Through Trajectory Inference
Trajectory Inference Concepts and Tools
Trajectory inference (TI) methods computationally reconstruct developmental trajectories by ordering cells along pseudotemporal progressions based on transcriptional similarity [52]. In embryonic research, TI enables researchers to trace lineage relationships between cell populations, identify branching points where cell fate decisions occur, and characterize the transcriptional dynamics driving differentiation.
For low-throughput embryo scRNA-seq studies, Monocle3 has emerged as a particularly effective tool [52]. It excels at learning complex trajectory structures with multiple branches, which is common in embryonic development where progenitor cells give rise to diverse differentiated descendants. The method works by modeling transcriptional changes as a stochastic process and projecting cells into a reduced-dimensional space where progress along developmental paths can be quantified as "pseudotime" – a continuous value representing each cell's relative position in the differentiation process [52].
Integrated Trajectory Analysis Protocol
Implementing trajectory inference for lineage validation in embryonic development involves a multi-step process that integrates with cell type annotation:
Data Preparation: Begin with an annotated Seurat object containing cell type identities and normalized expression counts. Convert the object to a CellDataSet format compatible with Monocle3 [52].
Dimension Reduction: Perform dimension reduction specifically for trajectory inference using UMAP or DDRTree algorithms. These methods preserve the continuous relationships between cells that reflect developmental progressions [52].
Cell Ordering: Define the trajectory starting point based on biological knowledge (e.g., pluripotent stem cells in embryonic datasets) or computational identification of root cells. Monocle3 will then order all cells along the trajectory based on transcriptional similarity [52].
Branch Analysis: Identify branch points where cells diverge into different lineages. For each branch, perform differential expression testing to identify genes that are significantly associated with the lineage decision [52].
Pseudotime-Based DEG Analysis: Conduct pseudotime-series analysis using a pseudo-bulk approach with edgeR to identify genes significantly associated with developmental progression [52]. This involves:
- Creating pseudo-bulk samples by aggregating cells with similar pseudotime values
- Fitting a negative binomial generalized linear model with pseudotime as a covariate
- Testing for genes exhibiting significant expression changes along pseudotime
Lineage Validation: Integrate trajectory results with cell type annotations to validate lineage relationships. Cells of related lineages should position along connected trajectories, while distinct cell types should separate into different branches [52].
Table 2: Trajectory Inference Tools for Embryonic Lineage Analysis
| Tool | Algorithm Type | Strengths | Embryonic Application Examples |
|---|---|---|---|
| Monocle3 | Reversed graph embedding | Handles complex branching trajectories; integrates with Seurat | Mammary gland development; embryonic cell fate mapping [52] |
| Slingshot | Minimum spanning trees | Identifies lineage paths from cluster centers; works with any clustering | Early embryonic lineage specification [13] |
| Pseudo-time | |||
| SCORPIUS | Principal curves | Ordering of cells without requiring branch detection | Linear differentiation pathways in embryo development |
The power of trajectory inference extends beyond merely ordering cells – it enables the identification of genes dynamically regulated along developmental paths, providing mechanistic insights into embryonic lineage specification.
Case Study: Integrated Analysis in Mouse Mammary Gland Development
A comprehensive workflow demonstrating the integration of cell type annotation and trajectory inference was applied to scRNA-seq data from mouse mammary gland epithelium across five developmental stages: embryonic, early postnatal, pre-puberty, puberty, and adult [52]. This study exemplifies the practical application of the methods described in this application note.
The analysis began with quality control and data integration using Seurat to harmonize data from multiple developmental stages. Cell type annotation was performed using marker-based methods, identifying distinct epithelial subpopulations. Trajectory inference using Monocle3 successfully reconstructed the developmental path from embryonic to adult stages, positioning cells along a pseudotemporal continuum that represented biological progression rather than chronological age [52].
Pseudotime-based differential expression analysis using edgeR's quasi-likelihood framework identified numerous genes significantly associated with developmental progression. The analysis employed a sophisticated design matrix that incorporated both pseudotime and sample effects, substantially increasing statistical power to detect dynamically regulated genes [52]. This approach successfully captured the transcriptional dynamics driving mammary gland maturation, demonstrating how integrated cell type annotation and trajectory inference can unravel developmental processes.
Cell type annotation and trajectory inference represent complementary pillars of scRNA-seq analysis in embryonic development research. When implemented within low-throughput workflows optimized for embryonic studies, these methods provide a powerful framework for deciphering lineage relationships and validating developmental pathways. The protocols and tools detailed in this application note – from knowledge-based annotation with ACT to trajectory reconstruction with Monocle3 – offer researchers standardized approaches for extracting meaningful biological insights from complex embryonic scRNA-seq datasets.
As single-cell technologies continue to evolve, the integration of these methods with emerging multi-omics approaches – including spatial transcriptomics, single-cell ATAC-seq, and computational prediction of cell-cell communication – will further enhance our ability to reconstruct embryonic development with unprecedented resolution. The standardized workflows presented here provide a foundation for these advanced applications, enabling researchers to consistently validate lineage relationships and uncover the molecular mechanisms governing embryogenesis.
Single-cell RNA sequencing (scRNA-seq) has revolutionized the study of embryonic development by enabling the unbiased transcriptional profiling of individual cells, thereby revealing cellular heterogeneity and dynamic state transitions that are fundamental to understanding how complex organisms are built. Unlike bulk RNA-seq, which averages gene expression across thousands of cells, scRNA-seq can identify rare cell types, define novel lineages, and reconstruct developmental trajectories at unprecedented resolution [10] [2]. This capability is particularly critical for studying early human development, where cellular diversity arises rapidly, and the molecular fidelity of in vitro embryo models must be rigorously validated against their in vivo counterparts [4] [58].
In low-throughput embryo research, the analytical journey from a count matrix to meaningful biological insights involves a series of critical steps: robust quality control, accurate cell clustering, precise cluster annotation, and insightful trajectory inference. The complexity of this workflow, combined with the unique challenges of working with precious and often limited embryonic material, necessitates a carefully considered and well-executed analytical strategy [12] [2]. This guide provides a detailed protocol for navigating this journey, focusing on the specific context of low-throughput scRNA-seq studies in embryonic development.
Experimental Design and Wet-Lab Protocols
Selecting an Appropriate scRNA-seq Protocol
The choice of scRNA-seq protocol is a primary determinant of data quality and should be aligned with the specific biological questions and experimental constraints. For low-throughput embryo studies, the trade-off between the number of cells profiled and the depth of transcriptional information per cell is a key consideration. The table below summarizes common protocols, highlighting their suitability for embryonic research.
Table 1: Comparison of scRNA-seq Protocols Relevant to Embryo Research
| Protocol | Isolation Strategy | Transcript Coverage | UMI | Amplification Method | Unique Features & Suitability |
|---|---|---|---|---|---|
| Smart-Seq2 [12] | FACS | Full-length | No | PCR | High sensitivity for lowly-expressed transcripts; ideal for detecting isoforms and allelic expression in embryos. |
| Drop-Seq [12] | Droplet-based | 3'-end | Yes | PCR | High-throughput, lower cost per cell; suitable for profiling larger, heterogeneous cell populations. |
| inDrop [12] | Droplet-based | 3'-end | Yes | IVT | Uses hydrogel beads; efficient barcode capture. |
| CEL-Seq2 [12] | FACS | 3'-only | Yes | IVT | Linear amplification reduces bias. |
| SPLiT-Seq [12] | Not required | 3'-only | Yes | PCR | Fixed cells, combinatorial indexing; no complex equipment needed, ideal for difficult-to-dissociate tissues. |
For studies where tissue dissociation is challenging or samples are frozen, single-nucleus RNA-seq (snRNA-seq) or split-pooling techniques like SPLiT-Seq offer viable alternatives, as they eliminate the need for isolating intact single cells [12] [2].
A Low-Throughput Workflow for Embryo Dissociation and Library Preparation
This protocol is adapted for a low-throughput, high-quality data generation approach, suitable for processing a limited number of embryonic cells.
Materials & Reagents
- Pronase or Accutase: For gentle enzymatic dissociation of embryonic tissues.
- Phosphate-Buffered Saline (PBS): With or without bovine serum albumin (BSA).
- Viability Stain: Such as propidium iodide or DAPI for flow cytometry.
- Lysis Buffer: A buffer containing a detergent (e.g., Triton X-100) and RNase inhibitors.
- Poly(T) Primers with UMIs and Barcodes: For reverse transcription and mRNA capture.
- Reverse Transcriptase Enzyme: For cDNA synthesis.
- PCR Amplification Reagents: Including a high-fidelity polymerase.
- Library Preparation Kit: For NGS, such as Illumina's Nextera.
Procedure
- Tissue Dissociation: Isolate the embryo or embryonic tissue. Gently wash with PBS. Incubate in a pre-warmed Pronase or Accutase solution for 5-15 minutes at 37°C to dissociate into a single-cell suspension. Gently pipette periodically to aid dissociation. Quench the enzyme activity with a serum-containing medium or PBS-BSA.
- Cell Washing and Viability Assessment: Pellet the cells by gentle centrifugation. Resuspend in PBS-BSA and pass through a 40 μm cell strainer to remove clumps. Assess cell viability and count using a hemocytometer and viability stain. A viability of >90% is recommended.
- Single-Cell Isolation (FACS): Dilute the cell suspension to a concentration of approximately 500-1,000 cells/μL. Use a Fluorescence-Activated Cell Sorter (FACS) to sort single, viable cells into the individual wells of a 96-well or 384-well plate containing lysis buffer and poly(T) primers. This method provides precise control over cell quality and number, which is crucial for low-throughput studies.
- mRNA Capture and Reverse Transcription: Immediately seal the plate after sorting and centrifuge briefly. Incubate the plate to lyse cells and allow poly(A)+ mRNA to hybridize to the poly(T) primers. Perform reverse transcription to generate cDNA. The primers should include Unique Molecular Identifiers (UMIs) to correct for amplification bias and cell barcodes to mark the cellular origin of each transcript [2].
- cDNA Amplification and Library Prep: Amplify the cDNA using PCR. The number of PCR cycles should be optimized to minimize over-amplification. Quality-check the amplified cDNA using a bioanalyzer. Proceed to tagment the cDNA and add sequencing adaptors using a commercial library preparation kit.
- Library QC and Sequencing: Quantify the final libraries using a fluorometric method and assess their size distribution. Pool libraries at equimolar ratios for sequencing on an appropriate Illumina platform. Aim for a sequencing depth of 50,000-100,000 reads per cell as a starting point.
Computational Analysis: From Raw Data to Clusters
Quality Control and Preprocessing
After sequencing, raw reads are processed through a pipeline like Cell Ranger (10x Genomics) or an in-house workflow to generate a gene-by-cell count matrix [45]. The first critical analytical step is quality control (QC) to remove low-quality cells and technical artifacts.
Table 2: Key Metrics for scRNA-Seq Quality Control
| Metric | Description | Acceptable Range (Example) | Indication of Low Quality |
|---|---|---|---|
| UMI Counts per Cell | Total number of transcripts detected. | Dataset-dependent (e.g., 1,000-30,000 for embryos). | Too low: empty droplet; Too high: multiplets. |
| Genes Detected per Cell | Number of unique genes detected. | Correlates with UMI count. | Low values suggest poor cell capture or RNA degradation. |
| Mitochondrial Read Fraction | Percentage of reads mapping to mt-DNA. | Typically <5-10% [45]. | Elevated levels indicate apoptotic or stressed cells. |
| Ribosomal Read Fraction | Percentage of reads mapping to rRNA. | Dataset-dependent. | Unusually high levels can indicate incomplete rRNA depletion. |
Procedure:
- Filter Cells: Remove cells with UMI counts or genes detected significantly below (empty droplets) or above (multiplets) the population median. Exclude cells with a high percentage of mitochondrial reads (e.g., >10% in most embryonic cells) [45].
- Filter Genes: Remove genes that are detected in only a very small number of cells (e.g., less than 10).
- Normalize Data: Normalize the UMI counts for each cell by the total counts for that cell and multiply by a scaling factor (e.g., 10,000), followed by a log transformation. This corrects for differences in sequencing depth per cell.
Dimensionality Reduction and Clustering
The normalized data, containing expression values for thousands of genes per cell, is inherently high-dimensional. To group cells with similar expression profiles, a multi-step process is used.
- Feature Selection: Identify a subset of highly variable genes (HVGs) that drive heterogeneity across the population. These genes are used for downstream analysis.
- Principal Component Analysis (PCA): Perform linear dimensionality reduction on the HVGs. This projects the data into a lower-dimensional space defined by the principal components (PCs), which capture the major axes of variation.
- Graph-Based Clustering: Construct a graph where cells are nodes connected to their nearest neighbors in PCA space. Use a community detection algorithm, such as the Louvain or Leiden algorithm, to identify clusters of cells—the fundamental units for initial biological interpretation [59].
- Visualization with UMAP: Use non-linear dimensionality reduction techniques like Uniform Manifold Approximation and Projection (UMAP) to visualize the cells in 2D or 3D space. Cells that are close together in the UMAP plot have similar transcriptomes.
The following diagram illustrates the core computational workflow from raw data to clustered cells.
Core Interpretation: Annotating Clusters and Inferring Trajectories
Strategies for Cell Cluster Annotation
Assigning biological identities to computational clusters is a pivotal step. A multi-faceted approach that combines computational tools with biological knowledge yields the most reliable annotations [59].
Practical Annotation Strategies:
- Biology-First Manual Annotation: Start by identifying the top differentially expressed genes (DEGs) for each cluster. Compare these marker genes to established databases (e.g., CellMarker 2.0) and literature on embryonic development. For example, the expression of POU5F1 (OCT4) and NANOG suggests an epiblast identity, while ISL1 and GABRP are associated with amnion [4] [58].
- Reference-Based Annotation with Label Transfer: Tools like SingleR or Azimuth can automatically annotate clusters by comparing their transcriptomes to a curated reference atlas. For human embryonic studies, the integrated human embryo reference spanning zygote to gastrula stages is an invaluable resource [4]. This method can project query data onto the reference UMAP and predict cell identities.
- AI-Driven Classification: Tools like CellTypist use machine learning models trained on large collections of annotated datasets to classify cells rapidly. These are particularly useful for standard cell types but may require validation for novel populations [59].
Table 3: Key Tools for scRNA-seq Cluster Annotation
| Tool | Method | Key Feature | Application in Embryology |
|---|---|---|---|
| SingleR [59] | Reference-based | Fast cell-type recognition by pairwise comparison. | Label transfer from human embryo atlases. |
| Garnett [59] | Marker-based | Uses a pre-trained classifier based on marker genes. | Classifying canonical lineages (e.g., trophectoderm). |
| CellTypist [59] | AI-driven | Automated model matching for large datasets. | Rapid initial annotation of common cell types. |
| Manual Curation | Biology-first | Expert knowledge and literature-based validation. | Essential for validating novel or rare cell types. |
Trajectory Inference and RNA Velocity Analysis
To move beyond static cell types and understand developmental processes, trajectory inference (pseudotime analysis) is used to reconstruct the dynamic transitions cells undergo.
- Choosing a Trajectory Tool: Tools like Monocle, Slingshot, or PAGA can order cells along a pseudotemporal trajectory based on their transcriptional similarity [59] [4]. This reveals the sequence of gene expression changes as cells differentiate.
- Interpreting Pseudotime: The inferred trajectory can model key developmental bifurcations, such as the divergence of the epiblast, hypoblast, and trophoblast lineages in early embryogenesis [4]. Analyzing genes that are differentially expressed along pseudotime can identify drivers of these fate decisions.
- RNA Velocity: This technique leverages the ratio of unspliced to spliced mRNA for each gene to predict the future state of individual cells. RNA velocity analysis can confirm inferred trajectories and reveal the directionality of cell-state transitions, such as the progression from epiblast-like cells (EpiLCs) to amniotic ectoderm-like cells (AMLCs) and mesoderm-like cells (MeLCs) in embryoid models [58].
The diagram below illustrates how trajectory inference and RNA velocity are used to derive dynamic insights from static snapshot data.
Validating Biological Insights
Computational predictions must be validated to ensure biological relevance. This is especially critical when working with embryo models to authenticate their fidelity to in vivo development [4].
Validation Strategies:
- Orthogonal Molecular Assays: Perform fluorescence in situ hybridization (FISH) on the original embryo or model to confirm the spatial expression of key marker genes identified in your analysis. Immunostaining for proteins encoded by these genes provides an additional layer of confirmation [59].
- Functional Experiments: Perturb key signaling pathways predicted to regulate lineage specification. For instance, the role of NODAL signaling in human mesoderm and primordial germ cell specification was identified through scRNA-seq comparative analysis and subsequently validated through functional experiments in embryoid models [58].
- Cross-Species and Cross-Study Comparison: Compare your annotated dataset with published scRNA-seq data from similar stages in human or non-human primate embryos. This can reveal conserved genetic programs and highlight species-specific differences, while also serving as a benchmark for your annotations [4] [58].
Table 4: Key Research Reagent Solutions for Embryo scRNA-Seq
| Item | Function | Example & Notes |
|---|---|---|
| Commercial scRNA-seq Kit | All-in-one reagent solution for library prep. | 10x Genomics Chromium Single Cell 3' Kit (droplet-based); SMART-Seq v4 (plate-based). |
| Cell Strainer | Removal of cell clumps post-dissociation. | 40 μm nylon mesh strainer. Critical for preventing channel/ droplet clogging. |
| Viability Stain | Distinguishing live from dead cells. | Propidium Iodide (PI) or DAPI for FACS sorting. |
| RNase Inhibitor | Prevention of RNA degradation during processing. | Added to lysis and reaction buffers to maintain RNA integrity. |
| UMI & Cell Barcode Primers | mRNA capture, reverse transcription, and cellular/ molecular indexing. | Found in commercial kits; essential for accurate quantification and multiplexing. |
| Curated Reference Atlas | Benchmarking and annotating embryonic cell clusters. | Integrated human embryo reference (Zygote to Gastrula) [4]. |
| Batch Effect Correction Tool | Harmonizing data from multiple experiments or samples. | Harmony or Seurat's CCA to integrate datasets without altering biological variance [59]. |
Conclusion
Low-throughput scRNA-seq emerges as a powerful and essential methodology for embryonic research, offering the sensitivity and resolution required to decode the complex processes of early development. By mastering the foundational principles, optimized workflows, and robust validation frameworks outlined in this guide, researchers can reliably profile precious embryonic samples, from initial cell isolation to final data interpretation. The future of this field points toward deeper integration with multi-omics approaches, the development of more sophisticated computational tools for data analysis, and the expanded use of embryo reference atlases. These advancements will undoubtedly accelerate discoveries in developmental biology, illuminate the mechanisms of developmental diseases, and pave the way for innovations in regenerative medicine and therapeutic development.
References
- 1. Low-input and single-cell RNA sequencing [https://www.illumina.com/techniques/sequencing/rna-sequencing/ultra-low-input-single-cell-rna-seq.html]
- 2. A practical guide to single-cell RNA-sequencing for ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0467-4]
- 3. Single Cell RNA Sequencing and Its Impact on ... [https://www.mdpi.com/1422-0067/26/16/7741]
- 4. A comprehensive human embryo reference tool using ... [https://www.nature.com/articles/s41592-024-02493-2]
- 5. Single-cell RNA sequencing for the study of development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6070143/]
- 6. Single-cell RNA sequencing technologies and ... [https://www.nature.com/articles/s12276-018-0071-8]
- 7. A step-by-step workflow for low-level analysis of single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5112579/]
- 8. Optimizing single cell RNA sequencing of stem ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1590889/full]
- 9. Droplet-based single-cell RNA sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522672/]
- 10. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 11. Application of single-cell RNA sequencing in embryonic ... [https://www.sciencedirect.com/science/article/pii/S0888754320302251]
- 12. Navigating single-cell RNA-sequencing: protocols, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 13. scRNA-seq Explained: Workflow, Tools, and Applications [https://www.metwarebio.com/single-cell-rna-seq-workflow-tools-applications/]
- 14. Singleâ€cell RNA sequencing technologies and applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 15. Benchmarking metabolic RNA labeling techniques for high ... [https://www.nature.com/articles/s41467-025-61375-z]
- 16. Single cell RNA-sequencing reveals cellular heterogeneity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7282974/]
- 17. Single-Cell Sequencing - Center for Genetic Medicine [https://www.cgm.northwestern.edu/cores/nuseq/services/next-generation-sequencing/single-cell-seq.html]
- 18. CellSIUS provides sensitive and specific detection of rare cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1739-7]
- 19. Deciphering cell lineage specification of human lung ... [https://www.nature.com/articles/s41467-021-26770-2]
- 20. Current best practices in singleâ€cell RNAâ€seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 21. Whole Transcriptome vs 3' mRNA-Seq: How to Choose ... [https://www.lexogen.com/blog/choosing-between-whole-transcriptome-total-rna-seq-and-3-mrna-seq/]
- 22. A comparison between whole transcript and 3' RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5393-3]
- 23. 3′ RNA-seq is superior to standard RNA-seq in cases of ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2023.1234218/full]
- 24. Statistically principled feature selection for single cell ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06240-y]
- 25. The Evolution of Single- Cell RNA Sequencing Technology and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9918030/]
- 26. - Single mRNA-sncRNA Co- Cell of... | SpringerLink sequencing [https://link.springer.com/protocol/10.1007/7651_2023_487]
- 27. Advances in Microfluidic Single-Cell RNA Sequencing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029715/]
- 28. Preparation of Cells from Embryonic Organs for Single - Cell ... RNA [https://pubmed.ncbi.nlm.nih.gov/30957983/]
- 29. Establishing single cell RNA transcriptomics: a brief guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403637/]
- 30. Getting Started with scRNA-seq: Library Preparation, QC ... [https://www.parsebiosciences.com/blog/getting-started-with-scrna-seq-library-preparation-qc-and-sequencing/]
- 31. cDNA-PCR Sequencing V14 - Barcoding (SQK-PCB114.24) [https://nanoporetech.com/document/pcr-cdna-sequencing-v14-barcoding-sqk-pcb114-24]
- 32. A drop-based workflow for fast and robust joint profiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599661/]
- 33. vs. Sequencing : Key Metrics in NGS Explained Depth Coverage [https://3billion.io/blog/sequencing-depth-vs-coverage]
- 34. Decoding Sequencing and Depth - CD Genomics Coverage [https://www.cd-genomics.com/resource-sequencing-depth-and-coverage.html]
- 35. Evaluation of the coverage and depth of transcriptome by RNA-Seq... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-S10-S5]
- 36. A comparison across non-model animals suggests an... : Internet Archive [https://archive.org/details/pubmed-PMC3655071]
- 37. Applications of single-cell RNA sequencing in drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141847/]
- 38. Single-cell technology for drug discovery and development [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2024.1459962/full]
- 39. Single-cell RNA Sequencing: Current Progresses and ... [https://openbiotechnologyjournal.com/VOLUME/18/ELOCATOR/e18740707311249/FULLTEXT/]
- 40. Droplet-based single-cell RNA sequencing [https://link.springer.com/article/10.1186/s12967-025-06996-0]
- 41. Embracing the dropouts in single-cell RNA-seq analysis [https://www.nature.com/articles/s41467-020-14976-9]
- 42. Comprehensive noise reduction in single-cell data with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570323/]
- 43. A Hybrid Computational Intelligence Framework for scRNA ... [https://arxiv.org/html/2511.16923v1]
- 44. Best practices for single-cell analysis across modalities [https://pmc.ncbi.nlm.nih.gov/articles/PMC10066026/]
- 45. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 46. 6. Quality Control [https://www.sc-best-practices.org/preprocessing_visualization/quality_control.html]
- 47. Systematic determination of the mitochondrial proportion in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8599307/]
- 48. Optimized protocol for isolation of high-quality single cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8605478/]
- 49. From dye exclusion to high-throughput screening: A review ... [https://www.sciencedirect.com/science/article/pii/S0734975025002502]
- 50. Chapter 8 Doublet detection | Advanced Single-Cell ... [https://bioconductor.org/books/3.15/OSCA.advanced/doublet-detection.html]
- 51. Benchmarking computational doublet-detection methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7897250/]
- 52. Unraveling the timeline of gene expression: A pseudotemporal... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10663991/]
- 53. Annotation of cell types (ACT): a convenient web server for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10623726/]
- 54. A comparison of marker gene selection methods for single-cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03183-0]
- 55. Evaluation of cell type annotation reliability using a large ... [https://www.nature.com/articles/s42003-025-08745-x]
- 56. scViewer: An Interactive Single-Cell Gene Expression ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10253102/]
- 57. The Beginner's Guide to Single-Cell RNA-seq Data Analysis [https://www.pythiabio.com/post/the-beginners-guide-to-single-cell-rna-seq-data-analysis-essential-plot-types]
- 58. Single-cell analysis of embryoids reveals lineage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9499422/]
- 59. A Practical Guide to Single-Cell RNA-Seq Cluster Annotation [https://www.nygen.io/resources/blog/scrna-seq-cluster-annotation]