A Complete Guide to Preventing Embryo Drying for Flawless Immunofluorescence in Pre-Implantation Research
This protocol provides a detailed, step-by-step guide for researchers and drug development professionals on preventing embryo desiccation during immunofluorescence staining.
A Complete Guide to Preventing Embryo Drying for Flawless Immunofluorescence in Pre-Implantation Research
Abstract
This protocol provides a detailed, step-by-step guide for researchers and drug development professionals on preventing embryo desiccation during immunofluorescence staining. Covering foundational principles, a robust methodological workflow, advanced troubleshooting, and rigorous validation techniques, this article synthesizes current best practices to ensure the preservation of embryo morphology and antigen integrity. By implementing these strategies, scientists can significantly improve the reliability of data obtained from precious pre-implantation human and mammalian embryos, thereby enhancing research on early embryonic development and cell lineage specification.
Understanding Embryo Vulnerability: Why Pre-Implantation Embryos Are Prone to Desiccation
FAQs: Blastocyst Handling and Drying Risks
Q1: Why are blastocysts particularly susceptible to drying during immunofluorescence protocols?
Blastocysts are uniquely vulnerable to drying due to their fundamental structure. They are composed of a thin, fluid-filled trophectoderm (TE) layer surrounding a blastocoel cavity. This large fluid-filled cavity and the minimal cytoplasmic volume in the TE cells mean that even minor volume loss from evaporation can cause catastrophic structural collapse, compromising the embryo's integrity and the experiment's success [1].
Q2: What specific structural parameters of a blastocyst correlate with better outcomes and potentially lower susceptibility to handling stress?
Research on 3D morphological parameters has identified several metrics associated with positive clinical outcomes. Blastocysts with these characteristics are generally more robust. The table below summarizes key parameters from a large-scale study.
Table 1: 3D Blastocyst Morphological Parameters Associated with Positive Outcomes
| Parameter | Description | Association with Clinical Pregnancy & Live Birth |
|---|---|---|
| Blastocyst Surface Area | Total external surface area of the blastocyst. | Larger values significantly associated with higher success rates (P < 0.001) [1]. |
| Blastocyst Volume | Total volume of the blastocyst. | Larger values significantly associated with higher success rates (P < 0.001) [1]. |
| Blastocyst Diameter | Diameter of the blastocyst. | Larger values significantly associated with higher success rates (P < 0.001) [1]. |
| TE Surface Area | Surface area of the trophectoderm facing the blastocyst cavity. | Larger values significantly associated with higher success rates (P < 0.001) [1]. |
| TE Cell Number | Number of cells in the trophectoderm. | Larger values significantly associated with higher success rates (P < 0.001) [1]. |
| ICM Shape Factor | Measure of how spherical the Inner Cell Mass (ICM) is. | A smaller value (shape closer to a sphere) correlated with higher success rates (P < 0.05) [1]. |
Q3: During which specific steps of the immunofluorescence protocol is the drying risk highest?
The primary risk is during solution changes, particularly between the fixation, permeabilization, and washing steps. A critical finding is that air-drying of unfixed or fixed cryosections, even briefly, causes significant loss and diffusion of soluble proteins like GFP due to damaged cell membranes [2]. The dehydration steps in traditional paraffin embedding, which use ethyl alcohol, are also a major cause of fluorescence quenching and structural damage [3].
Q4: How can I quickly check if my blastocyst has undergone drying damage during processing?
Signs of drying damage under the microscope include:
- Visible shrinkage or wrinkling of the trophectoderm.
- Granular appearance of the cytoplasm.
- Compromised structural integrity, making the blastocyst difficult to handle.
- Dim or anomalous fluorescence signal in immunofluorescence.
Troubleshooting Guides
Problem: Low or Lost Fluorescence Signal After Staining
Potential Cause 1: Fluorescent protein leaching during drying steps.
- Solution: Eliminate all air-drying steps for unfixed samples. For cryosections, use a Direct Fixation (DF) protocol where sections are immediately immersed in pre-warmed (30-37°C) 4% Paraformaldehyde (PFA) after mounting, omitting any drying step [2].
Potential Cause 2: Fluorescence quenching during dehydration.
- Solution: For paraffin embedding, replace the traditional ethyl alcohol dehydration series with a tertiary butanol (TBA) series. This modification has been shown to increase fluorescence intensity by over 12-fold compared to the traditional method [3].
Potential Cause 3: Antibody penetration issues due to incomplete permeabilization.
- Solution: Ensure the permeabilization buffer (e.g., containing Triton X-100) is fresh and the incubation time is optimized for the blastocyst's tough outer layer. A control stain with a nuclear dye (e.g., Hoechst) can help verify permeabilization efficiency.
Problem: Poor Structural Preservation of Blastocyst Morphology
Potential Cause: Osmotic or physical stress during fluid handling.
- Solution:
- Use pre-equilibrated media: Ensure all buffers and media are at 37°C and properly pH-balanced before use.
- Minimize transfer: Use plate-based protocols (e.g., in an 8-well IBIDI plate) to reduce the need for physically moving the blastocyst between droplets [4].
- Gentle solution exchange: Never fully aspirate the well. Always leave a small volume of liquid covering the blastocyst before gently adding the next solution.
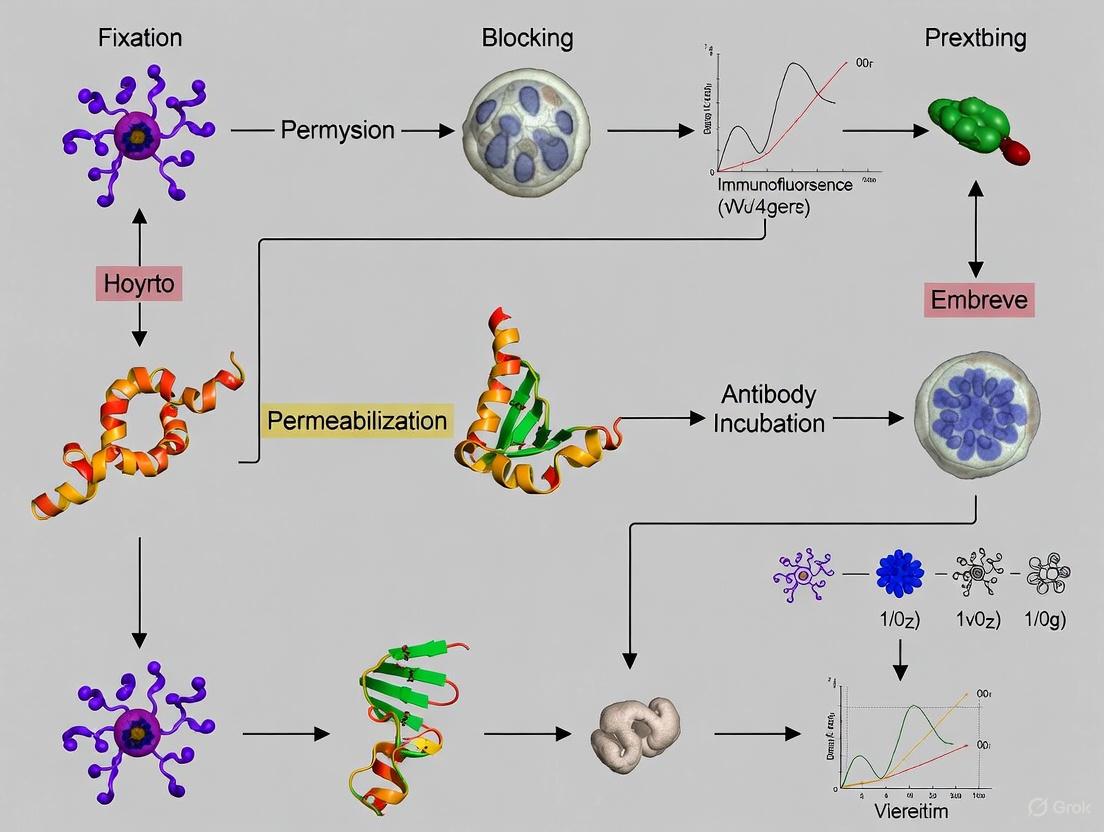
Experimental Protocol: Immunofluorescence for Mouse Blastocysts with Drying Prevention
This protocol is adapted for maximum retention of fluorescence and structure [4] [2] [3].
Workflow: Immunofluorescence with Drying Prevention
Materials & Reagents
- Culture/Handling: Pre-equilibrated embryo culture media.
- Fixation: 4% Paraformaldehyde (PFA) in PBS.
- Permeabilization/Blocking Buffer: PBS with 0.1-0.5% Triton X-100 and 1-5% Bovine Serum Albumin (BSA).
- Antibody Dilution Buffer: PBS with 1% BSA.
- Primary & Secondary Antibodies: Target-specific.
- Nuclear Stain: e.g., Hoechst 33342.
- Mounting Medium: Anti-fade fluorescent mounting medium.
- Equipment: 8-well glass-bottom chambered slides (e.g., IBIDI), fine-tipped transfer pipettes, humidified chamber.
Step-by-Step Procedure
- Sample Preparation: Transfer blastocysts into an 8-well glass-bottom plate pre-filled with culture medium. Using chambered slides minimizes subsequent handling.
- Fixation: Gently remove half the medium and add an equal volume of pre-warmed (30-37°C) 4% PFA to achieve a final concentration of ~2-4%. Incubate for 15-20 minutes at room temperature. Critical Step: Never allow the blastocysts to be exposed to air.
- Washing: Gently remove the fixative and add washing buffer (PBS). Repeat 3 times, ensuring the blastocysts remain submerged at all times.
- Permeabilization and Blocking: Incubate with permeabilization/blocking buffer for 30-60 minutes at room temperature.
- Primary Antibody Incubation: Dilute the primary antibody in antibody dilution buffer. Gently replace the blocking buffer with the primary antibody solution. Incubate overnight at 4°C in a humidified chamber to prevent evaporation.
- Washing: Carefully remove the primary antibody and wash 3 times with washing buffer, allowing 5-10 minutes per wash.
- Secondary Antibody and Nuclear Stain Incubation: Prepare a solution containing the fluorophore-conjugated secondary antibody and nuclear stain in antibody dilution buffer. Incubate for 1-2 hours at room temperature, protected from light.
- Final Washes: Wash 3 times with PBS, allowing 10 minutes per wash.
- Mounting: For chambered slides, the blastocysts are already positioned for imaging. Gently add a drop of anti-fade mounting medium and carefully lower a coverslip if required, avoiding air bubbles. For traditional slides, transfer the blastocysts in a minimal volume to a drop of mountant on a slide before coverslipping. Proceed to imaging as soon as possible.
Research Reagent Solutions
Table 2: Essential Reagents for Blastocyst Immunofluorescence
| Reagent | Function | Key Consideration for Drying Prevention |
|---|---|---|
| 8-well Glass-bottom Plates | Provides a stable, contained environment for processing. | Eliminates the need for multiple transfers between droplets, drastically reducing drying risk [4]. |
| Pre-warmed Fixative (PFA) | Cross-links and preserves cellular structures. | Pre-warming to 30-37°C accelerates fixation, rapidly stabilizing proteins before they can leak from compromised membranes [2]. |
| Tertiary Butanol (TBA) | Dehydrating agent for embedding. | A less-denaturing alternative to ethanol for dehydration, preserving fluorescent protein signal significantly better [3]. |
| Anti-fade Mounting Medium | Presves fluorescence during imaging. | Seals the specimen and reduces photobleaching. Essential for preserving signal after careful processing. |
| Bovine Serum Albumin (BSA) | Blocking agent to reduce non-specific antibody binding. | Used in buffers to stabilize antibodies and block non-specific sites, improving signal-to-noise ratio. |
A technical support resource for researchers combating drying artifacts in immunofluorescence
Frequently Asked Questions
1. What are the immediate consequences of letting my sample dry out during an immunofluorescence protocol?
Air-drying of a tissue section, whether partial or complete, during immunostaining negatively affects both the staining of tissue antigens and the ability to remove previously deposited antibody layers in sequential staining rounds. The core issue is a progressive loss of protein-associated water, which can lead to irreversible, high-energy misfolding of proteins. This results in antigen masking (the epitope becomes inaccessible to antibodies) and erratic, unpredictable staining artifacts that compromise the reproducibility and diagnostic reliability of your experiment [5].
2. Why does drying make it impossible to strip antibodies for sequential staining?
Drying causes irreversible changes to the bound antibody layers themselves. When a section dries, the antibody-protein complexes appear to undergo a conformational shift that makes them resistant to removal by standard chemical stripping buffers. The presence of a disaccharide like lactose during critical steps can prevent this, permitting the complete removal of bound antibodies and allowing for multiple rounds of staining and destaining on the same sample [5].
3. I work with embryos. How critical is drying during cryopreservation?
Drying is a significant risk during cryopreservation, but the principles are managed through vitrification. The goal of rapid-cooling vitrification is to solidify the cell so quickly that the remaining intracellular water does not have time to form damaging ice crystals. The warming rate is at least as important as the cooling rate; a slow warming rate allows lethal intracellular ice crystals to form through recrystallization. Successful protocols use a combination of cryoprotectants and ultra-rapid temperature changes to avoid both ice crystal formation and the damaging effects of excessive dehydration [6].
4. How can I visually detect or quantify drying in my samples?
Advanced techniques like Fluorescence Lifetime Imaging (FLIM) can be used to investigate drying processes. For instance, the fluorescence lifetime of certain dyes (e.g., ATTO 655) is quenched by water molecules. By measuring the fluorescence lifetime, researchers can access the local water concentration within a sample, such as a hydrogel microgel, and track its decrease over time during drying under ambient lab conditions [7].
Troubleshooting Guides
Problem: High Background and Non-Specific Staining
Potential Cause: Incomplete blocking or non-specific antibody binding, potentially exacerbated by sample drying that alters protein conformation.
Solutions:
- Optimize Blocking: Use a 2–10% solution of serum from the secondary antibody host species or Bovine Serum Albumin (BSA) in PBS. Incubate for 1–2 hours at room temperature. Ensure the blocking serum does not come from the same species as your primary antibody [8].
- Improve Washing: After antibody incubations, wash slides several times with PBS containing a detergent like 0.05% Tween 20 (PBS-T) to reduce background. Let the washing solution sit on the sample for about 5 minutes per wash and repeat 3-4 times [9] [10].
- Include Protective Sugars: Add 10% lactose or sucrose to your antibody dilution buffer. This helps stabilize proteins against drying-induced aggregation and non-specific binding that can contribute to background [5].
Problem: Weak or Absent Specific Signal
Potential Cause: Antigen masking due to accidental drying during protocol steps or over-fixation.
Solutions:
- Prevent Drying Meticulously: Never let sections dry out after rehydration. Always keep them submerged in buffer or in a humidified chamber during antibody incubations. When removing excess fluid between steps, be careful not to dry the section completely [5] [9].
- Employ Antigen Retrieval: For formalin-fixed paraffin-embedded (FFPE) samples, a heat-induced antigen retrieval step is often essential. Heat slides in a citrate-based (pH 6.0) or EDTA-based retrieval buffer using a microwave or pressure cooker [10].
- Use Disaccharides in Retrieval: Supplement your antigen retrieval buffer with 10% lactose or sucrose. This protects epitopes from re-masking that can occur when sections are extracted from the hot buffer and briefly exposed to air [5].
Problem: Inconsistent Staining Between Repeated Experiments
Potential Cause: Uncontrolled variations in humidity and drying times during staining, leading to sporadic antigen masking.
Solutions:
- Standardize with Humidified Chambers: Perform all antibody incubation steps in a properly sealed humidified chamber to ensure uniform hydration across the sample and between experiments [9].
- Add a Hydration Safeguard: Incorporate a disaccharide like lactose (10%) into your washing buffers or as a post-retrieval soak. This provides a buffer against minor, inadvertent drying events, leading to more homogeneous and consistent staining [5].
Quantitative Data on Desiccation Effects
The following table summarizes key experimental findings on how desiccation impacts immunoassay components and how disaccharides can mitigate this damage [5].
Table 1: Quantified Effects of Air-Drying on Immunoassays and Protective Efficacy of Disaccharides
| Experimental Condition | Impact on Antigen Immunoavailability | Impact on Antibody Stripping | Protective Effect of 10% Disaccharide (e.g., Lactose, Sucrose) |
|---|---|---|---|
| Oven Drying (60°C, 1 hr) | Severe loss | Bound antibodies become irremovable | Prevents antigen masking; enables complete antibody removal after drying |
| Freeze Drying (-53°C, 24-48 hr) | Severe loss | Not tested | Preserves antigenicity |
| Desiccation (Room Temp, 24-48 hr) | Severe loss | Not tested | Preserves antigenicity |
| "Accidental" Drying During Staining | Erratic and unpredictable staining artifacts | Inability to strip deposited layers | Prevents artifacts and allows homogeneous staining |
| Long-Term Storage (6 months, dry) | Progressive antigen masking over time | Not tested | Protects against antigen masking during dry storage; a second AR step can restore staining |
Experimental Protocol: Using Disaccharides to Prevent Drying-Induced Damage
This protocol is adapted from a study demonstrating that disaccharides prevent air-drying artifacts in FFPE tissue sections [5].
Objective: To protect tissue-bound antigens and antibodies from air-drying-induced damage during immunohistochemistry or immunofluorescence, enabling consistent staining and multiple rounds of re-staining.
Materials Needed:
- Dewaxed and rehydrated FFPE tissue sections.
- Antigen Retrieval Buffer (e.g., 10 mM EDTA, pH 8, or citrate buffer, pH 6.0).
- D-Lactose monohydrate or Sucrose.
- Tris-Buffered Saline (TBS) or Phosphate-Buffered Saline (PBS).
- Primary and secondary antibodies.
- Standard IHC/IF detection reagents.
Method:
- Antigen Retrieval with Disaccharide Supplement:
- Perform heat-induced antigen retrieval as you normally would.
- Critical Step: Supplement the antigen retrieval buffer with 10% (w/v) lactose or sucrose.
- After retrieval, allow the slides to cool in the buffer to avoid re-masking upon exposure to air.
Application of Protective Solution:
- Following antigen retrieval and cooling, immerse the slides in a 10% solution of lactose or sucrose in TBS or distilled water for at least 1 hour at room temperature in a humidified chamber. This step can also be performed after primary antibody incubation.
Controlled Drying (if necessary):
- If a drying step is unavoidable in your protocol (e.g., to apply a specific reagent), drain excess fluid and allow the slide to dry in the presence of the disaccharide.
- Control Experiment: Always include a paired section that undergoes the same drying process but without the protective disaccharide in any step.
Immunostaining:
- Proceed with your standard blocking, primary antibody incubation, washing, and secondary antibody incubation steps.
- Optional: For added protection, the primary antibody can also be diluted in a buffer containing 10% lactose or sucrose.
Stripping and Re-staining (for sequential staining):
- If performing multiple rounds of staining, the presence of lactose during the previous steps will allow for complete removal of bound antibodies using standard chemical stripping buffers (e.g., SDS, 2-mercaptoethanol).
Mechanism of Disaccharide Protection Against Desiccation in IF
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Preventing Desiccation Artifacts
| Reagent / Material | Function / Explanation | Example Application in Protocol |
|---|---|---|
| Disaccharides (Lactose, Sucrose) | Stabilize proteins by replacing water molecules in the hydration shell, preventing irreversible misfolding during dehydration. | Add at 10% (w/v) to antigen retrieval buffers, antibody dilution buffers, or as a post-retrieval soak [5]. |
| Humidified Chamber | A sealed container with a saturated atmosphere prevents evaporation of aqueous solutions from the sample during long incubations. | Use for all antibody incubation steps [9]. |
| Bovine Serum Albumin (BSA) | A common blocking agent that reduces non-specific binding of antibodies to the tissue, lowering background. | Use at 1-5% in PBS or TBS for blocking and antibody dilution [10] [8]. |
| Normal Serum | A blocking agent containing a mixture of proteins that bind non-specific sites. Should be from the same species as the secondary antibody. | Use at 2-10% for blocking. More effective than BSA for some applications [8]. |
| Cryoprotectants (e.g., EG, DMSO, Sucrose) | Permeable agents that protect cells from ice crystal formation during cryopreservation by reducing the freezing point and promoting vitrification. | Used in specific cocktails for oocyte and embryo vitrification [11] [6]. |
| Fluorescence-Compatible Mounting Medium | A solution that preserves fluorescence, often with anti-fade agents, and provides the correct refractive index for high-resolution microscopy. | Use after staining is complete to protect your sample for imaging [9]. |
Frequently Asked Questions (FAQs)
Q1: Why is humidity control critical in embryo culture systems? Humidity control is primarily essential to prevent the evaporation of culture medium. Even when covered with oil, media can evaporate over time in dry incubator conditions. This evaporation increases the osmolality (solute concentration) and can alter the pH of the medium, creating a suboptimal environment that can impair embryonic development and reduce pregnancy rates [12].
Q2: What is the direct link between osmolality and developmental arrest? Preimplantation embryos are exquisitely sensitive to increases in osmolality. Research on porcine embryos, which are a strong model for human development, shows that even a minor increase in osmolarity to 330 mOsm under organic osmolyte deficiency disrupts cell volume homeostasis. This disruption triggers a cascade of issues, including metabolic reprogramming and compromised zygotic genome activation (ZGA), ultimately leading to developmental arrest at the 4-cell stage [13].
Q3: How do organic osmolytes like glycine and betaine help embryos? Organic osmolytes are uncharged molecules that embryos accumulate to regulate their cell volume. When external osmolality increases, embryos take in inorganic ions as an initial response. High levels of these ions can disrupt cellular biochemistry. Organic osmolytes like glycine and betaine replace a portion of these inorganic ions, allowing the cell to maintain osmotic balance without the detrimental effects of high ionic strength, thereby supporting continued development [13] [14].
Q4: Can covering culture media with oil eliminate the need for humidified incubators? While using a sufficient volume of high-quality mineral oil is a crucial first line of defense against evaporation, evidence suggests it may not always be fully sufficient. Studies comparing dry and humidified time-lapse incubator chambers have shown that even under oil, hyperosmotic changes can occur in dry conditions, potentially affecting development. Therefore, for optimal results, using oil overlay in a properly humidified incubator is recommended [12].
Troubleshooting Common Problems
Problem: Decreased Blastocyst Formation Rates Under Hyperosmotic Stress
Potential Cause: Cell volume dysregulation leading to metabolic-epigenetic disruption. Solution:
- Supplement with organic osmolytes: Add 1 mM glycine or betaine to the culture medium. This has been shown to completely reverse hyperosmotic stress-induced developmental arrest in porcine embryos by restoring cell volume homeostasis [13].
- Verify medium osmolality: Regularly calibrate instruments used to measure osmolality. Ensure that the base osmolality of the medium is appropriate for the species (e.g., ~288 mOsm for porcine embryos [13]).
Problem: Increased Embryo Fragmentation and Developmental Delay
Potential Cause: Evaporative loss in culture medium leading to increased osmolality and pH shifts. Solution:
- Ensure proper humidification: Confirm that your incubator's humidity reservoir is filled with sterile water and that the chamber maintains a saturated humidity environment [12].
- Check oil quality: Use fresh, high-quality embryo-tested mineral oil. Ensure a sufficient overlay (e.g., 4 mL over 80 µL of medium) to create an effective vapor barrier [12].
- Review dish handling: Minimize the time culture dishes spend outside the incubator. When they are out, use a pre-warmed stage top or chamber to reduce condensation and temperature fluctuations.
Problem: Developmental Block During Maternal-to-Zygotic Transition
Potential Cause: Osmotic stress disrupting mitochondrial function and epigenetic remodeling. Solution:
- Consider stage-specific osmolality: For oocyte maturation, a two-stage system with lower osmolality (290 mOsm) for the first 22 hours, followed by higher osmolality (320 mOsm), in the presence of 1 mM glycine, has been shown to improve outcomes in porcine oocytes [14].
- Pharmacological intervention: In cases of stress-induced PDH inactivation (a key metabolic enzyme), treatment with Dichloroacetate (DCA) can reactivate PDH and rescue development, though organic osmolyte supplementation is a more physiological approach [13].
Table 1: Impact of Humidity on Embryo Culture Outcomes in a Time-Lapse System
| Parameter | Dry Conditions (DC) | Humidified Conditions (HC) | Significance |
|---|---|---|---|
| Ongoing Pregnancy Rate | No significant difference | No significant difference | Not significant [12] |
| Blastulation Rate | No significant difference | No significant difference | Not significant [12] |
| Morphokinetics (t2, t4) | Earlier and more synchronous | Slightly delayed | Significant [12] |
| Medium Osmolality/pH | Increased over time | More stable | Significant [12] |
Table 2: Strategies to Overcome Osmotic Stress in Different Species
| Intervention | Experimental Model | Effect on Development | Key Findings |
|---|---|---|---|
| Glycine/Betaine Supplementation | Porcine 2-cell embryos | Rescues developmental arrest | Restores cell volume, corrects metabolic-epigenetic dysregulation, enables ZGA [13] |
| Stage-Adjusted Osmolality | Porcine oocyte IVM | Improves maturation quality | 290 mOsm (first 22h) → 320 mOsm (last 22h) with 1mM Glycine improves mitochondrial function and embryo yield [14] |
| Reduced Base Osmolality | Mouse embryos | Prevents 2-cell block | Culture in KSOM/CZB media at 250-275 mOsm overcomes developmental arrest [14] |
Experimental Protocol: Immunofluorescence in Human Blastocysts with Osmotic Protection
This protocol is adapted for the detection of nuclear transcription factors and phosphorylated proteins, with critical steps to prevent osmotic stress and embryo drying [15].
Key Resources for Osmotic Protection:
- Phosphate-buffered saline (PBS) with Ca2+ and Mg2+: Essential for maintaining membrane integrity during washes.
- 4% Paraformaldehyde (PFA) in PBS with Ca2+ and Mg2+: Freshly prepared and stored at 4°C for no more than 7 days to ensure optimal fixation and minimal osmotic shock.
- Triton X-100 in PBS (without Ca2+ and Mg2+): Prepared fresh on the day of use for effective and consistent permeabilization.
Detailed Steps:
- Handling and Fixation:
- Use a glass capillary with a smooth, fire-polished opening (>300 µm diameter) for all manual handling of human blastocysts to minimize shear stress.
- Fix embryos in 4% PFA (prepared in PBS with Ca2+/Mg2+) for 30 minutes at room temperature on a rocking platform.
- Critical Step: Perform all washes and incubation steps in solutions with correct ion composition. Perform all incubations in 4-well dishes to minimize embryo handling and transfer stress.
Permeabilization and Blocking:
- Wash embryos 3 times in PBS with Ca2+/Mg2+.
- Permeabilize with 0.1% Triton X-100 in PBS (without Ca2+/Mg2+) for 30 minutes at room temperature.
- Block in a solution containing a serum (e.g., normal donkey serum) and PBS with Ca2+/Mg2+ for 1 hour to reduce non-specific antibody binding.
Antibody Staining:
- Incubate with primary antibody (e.g., anti-phospho-SMAD2) diluted in blocking solution overnight at 4°C on a rocker.
- Wash 3 times in PBS with Ca2+/Mg2+ over 2 hours.
- Incubate with fluorescently-conjugated secondary antibody and DAPI (for DNA stain) diluted in blocking solution for 2 hours at room temperature, protected from light.
- Perform final 3 washes in PBS with Ca2+/Mg2+ before mounting.
Mounting:
- Mount embryos in a small drop of DAPI-containing mounting medium on a glass slide. Ensure the embryo is completely surrounded by mounting medium to prevent drying during imaging.
Visual Workflows
Workflow for Embryo Handling
Osmotic Stress and Rescue Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Managing Humidity and Osmolarity
| Reagent/Material | Function | Technical Notes |
|---|---|---|
| Embryo-Tested Mineral Oil | Prevents evaporation of culture medium by creating a vapor barrier. | Use a sufficient volume (e.g., 4 mL over 80 µL medium). Quality is critical [12]. |
| Organic Osmolytes (Glycine, Betaine) | Protect against hyperosmotic stress by enabling cell volume regulation. | Supplement at 1 mM concentration in culture medium to prevent developmental arrest [13] [14]. |
| PBS with Ca²⁺ and Mg²⁺ | Maintains membrane integrity and fluid balance during immunofluorescence washes. | Essential for all washing and fixation steps to avoid osmotic shock [15]. |
| D-(+)-Raffinose | A non-metabolizable sugar used to precisely adjust medium osmolality in experimental models. | Useful for creating hyperosmotic conditions without introducing variable metabolites [13] [14]. |
| Sterile Water for Humidification | Maintains saturated humidity in incubator chambers to minimize medium evaporation. | Use in water reservoirs of time-lapse or standard incubators [12]. |
A Robust Step-by-Step Protocol: Hydration-Centric Immunofluorescence for Human Blastocysts
Why is maintaining a hydrated workspace critical for embryo research?
A consistently hydrated environment is fundamental to successful embryo culture and immunofluorescence (IF) research. It prevents osmotic stress and physical damage that can occur if embryos dry out, even briefly. Furthermore, for IF protocols, proper hydration is essential for preserving delicate cellular structures, ensuring antibody binding efficiency, and achieving reproducible, high-quality results.
Step-by-Step Experimental Protocols
Protocol 1: Standard Preparation of a Hydrated Workspace for Immunofluorescence
This protocol details the setup for culturing cells on glass-bottom dishes, a common requirement for high-resolution imaging, using pre-equilibrated media and oil overlays to maintain hydration [4].
Materials Required:
- Glass-bottom culture dishes (e.g., 8-well IBIDI plate)
- Pre-equilibrated cell culture media (e.g., 2i/LIF for naïve mouse embryonic stem cells, mESCs)
- Embryo-tested mineral oil (light or heavy)
- Coating agent (e.g., 0.1% gelatin or laminin)
- DPBS (Dulbecco's Phosphate Buffered Saline)
- Sterile pipettes and tips
Procedure:
- Coating the Substrate: For mESCs, coat the wells of the glass-bottom plate with 130 µL of 0.1% gelatin for at least 15 minutes at room temperature. Alternatively, for a 2D culture, coat with laminin and incubate at 37°C overnight or for a minimum of 4 hours [4].
- Preparing the Workspace: Aseptically remove the coating solution from the wells.
- Adding Pre-equilibrated Media: Add enough pre-equilibrated culture media (approximately 200 µL per well) to cover the bottom of the wells [4].
- Seeding Cells: Plate your cells at the required concentration. For an 8-well plate with a 48-hour culture, plate 30,000 cells per well [4].
- Applying the Oil Overlay: Carefully overlay the media drop with the appropriate volume of embryo-tested mineral oil to create a "closed" system. In a standard 35 mm dish, 2 mL of oil might be used to cover 20 µL media drops (a 1:100 media-to-oil ratio) [16].
- Incubation: Incubate the culture dish at 37°C and the appropriate CO2 concentration until the cells are ready for fixation and immunostaining.
Protocol 2: Warming and Recovery of Vitrified Oocytes/Embryos in a Hydrated System
This protocol highlights the critical use of pre-equilibrated media for the recovery of cryopreserved specimens, ensuring proper hydration and osmotic balance after warming [17].
Materials Required:
- Vitrified oocytes/embryos (e.g., in a CryoTip or HSV device)
- Pre-equilibrated culture medium supplemented with protein (e.g., 20% v/v SSS or 12 mg/mL HSA)
- Warming solutions (TS, DS, WS1, WS2) as per vitrification kit instructions
- Sterile Petri dishes
- Mineral oil
- 37°C waterbath (minimum volume 500 mL)
Procedure:
- Preparation: Do not begin warming until you have a dish of pre-equilibrated culture medium ready for the final recovery of specimens [17].
- Setup Warming Dish: Aseptically dispense the required warming solutions (e.g., 50 µL drops of TS, DS, and WS) on a sterile Petri dish. For oocytes, dispense a minimum of 100 µL of TS [17].
- Warming: Quickly remove the vitrification device from liquid nitrogen and immerse it in a 37°C waterbath for 3 seconds, swirling gently [17].
- Specimen Recovery: Dispense the contents of the device into the TS drop and leave for 1 minute. Subsequently, transfer the specimens through DS, WS1, and WS2, following the specified timings (e.g., 4 minutes each) [17].
- Final Recovery in Pre-equilibrated Media: Transfer the warmed oocytes or embryos into the pre-equilibrated culture medium with protein supplement.
Note: To further prevent evaporation during the warming steps, the DS and WS drops may be overlaid with 8.0-8.5 mL of equilibrated mineral oil at least 45 minutes prior to starting the procedure [17].
Table 1: Effect of Media-to-Oil Ratio on Small Molecule Inhibitor Efficacy
This quantitative data demonstrates how the physical setup of your culture workspace can profoundly alter the effective concentration of small molecules, a critical consideration for drug studies [16].
| Media-to-Oil Ratio | Dish Setup Description | Effective [Nocodazole] for 100% PBE Block | Experimental Implication |
|---|---|---|---|
| No Oil | 2 mL media in humidified incubator | ~100 nM | Baseline activity in an open, humidified system. |
| 1:4 | 1 × 500 µL media under 2 mL oil | >100 nM | Slight reduction in effective drug concentration. |
| 1:10 | 10 × 20 µL media under 2 mL oil | >100 nM | Noticeable shift in EC50. |
| 1:100 | 1 × 20 µL media under 2 mL oil | >>100 nM (EC50 = 88.4 nM) | Standard culture setup shows significant drug partitioning into oil. |
| 1:1000 | 1 × 2 µL media under 2 mL oil | >>1000 nM (EC50 = 785 nM) | Severe loss of drug activity; common in live imaging/micromanipulation. |
Abbreviations: PBE, Polar Body Extrusion; EC50, Half Maximal Effective Concentration. Data adapted from [16].
Troubleshooting Guides & FAQs
Why is my small molecule inhibitor not working in my embryo culture assay?
This is a common issue often linked to the culture setup. Small, lipophilic molecules can partition into the oil overlay, reducing their effective concentration in the culture media [16]. The lower the media-to-oil volume ratio, the more pronounced this effect becomes.
- Solution: Avoid oil-covered culture for drug/inhibitor experiments wherever possible. If oil is necessary, use the largest practical media volume and the smallest necessary oil volume to reduce the media-to-oil ratio. Always report the media-to-oil ratio used in your methods section to improve reproducibility [16].
I am observing high background in my immunofluorescence staining. Could this be related to my culture conditions?
While culture conditions are not a direct cause, improper handling during subsequent IF steps can lead to high background. However, the principles of a controlled environment are key.
- Solution: Ensure you are using an appropriate blocking buffer (e.g., 5% normal serum from the same species as your secondary antibody, or 1-5% BSA) for a sufficient time (30-60 minutes) to prevent non-specific antibody binding [18] [19]. Always include a no-primary-antibody control to check for non-specific binding of your secondary antibody [20].
How long can I pre-equilibrate my culture media before use?
The stability of pre-equilibrated media is finite.
- Solution: Refer to the stability of your specific media formulations. For example, some specialized media like 2i/LIF for mESCs can be used for up to 2 weeks, while N2B27 may be stable for about 3 weeks once prepared [4]. Always follow manufacturer guidelines and validate media performance for critical applications.
My cells are detaching during the immunofluorescence washing steps. What can I do?
This can occur with loosely adherent cells, such as embryonic stem cells grown in 3D colonies [4].
- Solution:
- Optimize Coating: Ensure your culture substrate (e.g., gelatin, laminin) is fresh and applied correctly.
- Gentle Handling: Perform all wash steps gently. Avoid direct pipetting onto the cell layer; instead, add and remove solutions from the side of the well.
- Fixation Check: Verify that your fixation method (e.g., 4% PFA for 15 minutes) is adequate for your cell type [4] [19].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Maintaining a Hydrated Workspace
This table lists key reagents used in preparing and maintaining a hydrated culture environment for embryo and cell research.
| Reagent | Function | Example Application |
|---|---|---|
| Embryo-Tested Mineral Oil | Creates a physical overlay to prevent evaporation and microbial contamination; maintains a stable pH and osmotic environment. | Used to cover micro-drop cultures of oocytes and embryos in Petri dishes [16] [17]. |
| Pre-equilibrated Culture Media | Provides nutrients and a stable, pH-balanced (via CO2/bicarbonate buffer) environment for cell growth and maintenance. | Essential for all cell culture procedures, including post-thaw recovery and long-term embryo culture [4] [17]. |
| Protein Supplement (e.g., SSS, HSA) | Acts as a surfactant, reducing surface tension and preventing cell stickiness and damage. Provides carriers for lipids and hormones. | Added to handling and culture media for oocytes/embryos (e.g., at 20% v/v or 12 mg/mL) [17]. |
| Gelatin/Laminin | Coats culture surfaces to enhance cell attachment and growth, which is especially important for sensitive stem cells [4]. | Used as a substrate for plating mouse embryonic stem cells in 2D or 3D formats prior to IF [4]. |
| Blocking Buffer (e.g., BSA, Normal Serum) | Reduces non-specific binding of antibodies to the sample, thereby lowering background noise in immunofluorescence [18] [19]. | Applied after fixation and permeabilization, just before primary antibody incubation [18] [19]. |
Workflow and Conceptual Diagrams
Diagram 1: Experimental Workflow for Hydrated IF Preparation
Diagram 2: Impact of Media-to-Oil Ratio on Drug Availability
In immunofluorescence protocols for embryonic research, the fixation and permeabilization steps are critical for preserving cellular architecture and enabling antibody access. However, these very steps introduce significant risk of sample drying through extended air exposure. For delicate samples such as embryos, even minimal drying causes irreversible morphological damage, loss of antigenicity, and increased autofluorescence, compromising experimental validity. This guide details the mechanisms behind these artifacts and provides optimized, timed protocols to maintain sample hydration throughout processing.
FAQs: Addressing Critical Concerns
Q1: Why is preventing air exposure specifically critical during fixation and permeabilization?
Air exposure during these steps leads to rapid dehydration. In fixation, aldehydes like formaldehyde work by crosslinking proteins; if samples dry during this process, the crosslinking occurs in a distorted physical state, altering epitope structure and often masking antigen binding sites. During permeabilization, which follows fixation and often involves alcohol or detergent treatments, drying can create inconsistent "pores" in membranes, leading to uneven antibody penetration and high background noise. For embryos, which have complex three-dimensional structures, this results in non-representative staining and unreliable data [21].
Q2: What are the observable consequences of embryo drying, and how do I distinguish them from other artifacts?
The consequences can be observed under microscopy:
- Irreversible Morphological Collapse: The embryo will appear shrunken, flattened, or distorted rather than maintaining its spherical, blastocoel-filled structure.
- High Background Noise: Drying concentrates salts and cellular components, leading to nonspecific antibody binding and a "speckled" or generally high fluorescent background.
- Loss of Specific Signal: Critical antigens may fail to stain, not due to antibody failure, but because the epitopes have been denatured by drying during fixation.
You can distinguish drying artifacts from poor fixation or inadequate permeabilization by the physical collapse of the structure. Other issues might preserve morphology but yield no signal or high background, respectively.
Q3: My protocol requires multiple solution changes. What is the safest way to perform washes without air exposure?
The gold standard is to never remove all liquid from the sample dish. A timed wash protocol should be employed:
- Never Fully Decant: When exchanging solutions, always leave a small residual volume (e.g., 50-100 µL) covering the samples.
- Gently Add Fresh Solution: Add your fresh wash buffer or reagent down the side of the well or dish to avoid direct stream on samples. The residual volume prevents air contact.
- Time Your Incubations: For a standard 5-minute wash, ensure the sample is fully submerged for the entire duration. Using a volumetric pipette to add and remove solutions consistently can help maintain timing and minimize turbulence.
- For Non-Adherent Cells/Embryos: Consider using centrifugal filter devices. These devices allow you to perform solution exchanges via brief centrifugation, which keeps the cells captured on a membrane and submerged in buffer until the spin, drastically minimizing air exposure [22].
Troubleshooting Guide: Fixation and Permeabilization Errors
| Problem | Primary Symptom | Root Cause | Preventive Solution |
|---|---|---|---|
| Incomplete Fixation | Cellular autofluorescence, poor morphology, loss of signal. | Fixative solution evaporated during incubation; insufficient fixative volume. | Always use adequate volume; ensure container is sealed during incubation; use 4% formaldehyde for most targets [21]. |
| Over-Permeabilization | Loss of intracellular structure, diffuse signal, high background. | Overly long exposure to detergent; drying during permeabilization concentrates detergent. | Strictly time permeabilization steps (e.g., 15 min for Triton X-100); never let sample dry [23] [21]. |
| Uneven Staining | Patchy or variable signal intensity across the embryo. | Sample partially dried, creating inconsistent antibody access; insufficient washing. | Maintain liquid cover at all times; follow timed wash protocols with sufficient buffer volumes. |
| Altered Scatter Profile | Abnormal cell size (FSC) and granularity (SSC) in flow cytometry. | Use of high-concentration alcohol-based fixatives, which can be exacerbated by drying. | For flow, test and standardize fixation/permeabilization buffers; avoid protocol variations [24]. |
Table 1: Common issues arising from improper handling during fixation and permeabilization.
Optimized Protocols with Timed Washes
Protocol 1: Standard Immunofluorescence for Adherent Cultures
This protocol, adapted from commercial best practices, is designed for cells but outlines the timed, non-drying principles essential for any sample [23].
Workflow: Standard IF with Timed Washes
Materials:
- Image-iT Fixation/Permeabilization Kit (or equivalent: 4% formaldehyde, Triton X-100, blocking buffer) [23]
- Wash Buffer (e.g., PBS)
- Primary and secondary antibodies
Method:
- Fixation: Aspirate culture media to a residual volume of ~100 µL. Gently add 1 mL of 4% Formaldehyde Fixative Solution. Incubate for 15 minutes at room temperature in a sealed container to prevent evaporation.
- Wash 1: Aspirate fixative, leaving a residual volume. Gently add 2 mL Wash Buffer. Incubate with agitation for 2-5 minutes. Aspirate and repeat twice more for a total of three timed washes.
- Permeabilization: Remove wash buffer, leaving residual volume. Add 1 mL Permeabilization Solution (e.g., 0.1% Triton X-100). Incubate for 15 minutes at room temperature.
- Wash 2: Aspirate permeabilization solution, leaving a residual volume. Perform three timed washes with 2 mL Wash Buffer, each for 2-5 minutes, as before.
- Blocking and Staining: Proceed to blocking and antibody incubation steps, maintaining the "no-dry" principle throughout.
Protocol 2: Centrifugal Filter-Based Staining for Non-Adherent Embryos and Cells
This protocol is ideal for delicate, non-adherent samples like embryos or organoids, as it physically prevents air exposure during solution exchanges [22].
Workflow: Centrifugal Filter-Based Staining
Materials:
- Centrifugal Filter Device with PVDF membrane
- Microcentrifuge
- Fixative (e.g., 4% PFA) and Permeabilization Buffer (e.g., Methanol or detergent-based) [22]
Method:
- Load: Transfer your sample (e.g., embryos in suspension) to the centrifugal filter device.
- Fix: Add the appropriate fixative (e.g., 4% PFA) directly to the device. Incubate for the required time without centrifuging.
- Wash 1: Place the device in the centrifuge and spin briefly at low speed to remove the fixative. Add wash buffer to the device and centrifuge again. Repeat for a total of three washes. The sample remains captured on the moist membrane.
- Permeabilize: Add permeabilization buffer (e.g., methanol) to the device. Incubate for the required time without centrifuging.
- Wash 2: Centrifuge to remove the perm buffer. Add wash buffer and centrifuge again. Repeat for three washes.
- Elute: After the final wash, reverse the device and centrifuge into a fresh tube to recover the stained sample for mounting.
Research Reagent Solutions
Table 2: Essential reagents for fixation and permeabilization protocols, highlighting their function and considerations for preventing drying artifacts.
| Reagent | Function | Key Considerations |
|---|---|---|
| 4% Formaldehyde | Crosslinking fixative. Preserves cellular structure by creating protein crosslinks. | The gold standard for most targets. Drying during fixation causes crosslinking in a distorted state, ruining epitopes [21]. |
| Methanol | Denaturing fixative & permeabilizer. Precipitates proteins and dissolves lipids. | Can be used cold (-20°C) as a combined fix/perm. Highly volatile; requires sealed containers to prevent evaporation and concentration [21]. |
| Triton X-100 | Detergent-based permeabilization. Creates pores in lipid membranes for antibody entry. | Strict timing is required (e.g., 15 min). Drying concentrates the detergent locally, leading to over-permeabilization and loss of subcellular structures [23] [21]. |
| Saponin | Mild detergent permeabilization. Binds cholesterol to create reversible pores. | Often used for intracellular organelle staining. Must be included in all subsequent antibody and wash steps to maintain permeability [21]. |
| Paraformaldehyde (PFA) | Purified, non-polymerized form of formaldehyde. Used for making fresh fixative. | Crosslinks proteins, reducing surface stickiness of cells and organoids, which can be a factor during washes [25]. |
| BD Pharmingen FoxP3 Buffer Set | Commercial fix/perm buffer set. | Optimized for transcription factors. Testing is key, as different commercial kits can dramatically impact surface marker fluorescence and scatter profiles [24]. |
VECTASHIELD Mounting Media: Troubleshooting Guides and FAQs
Frequently Asked Questions
Q: I am using VECTASHIELD HardSet Mounting Medium with DAPI and see bubble formation under the coverslip after some time. How can this be remedied? A: For hardening formulations like HardSet, ensure you are using fixed, thin-cut sections (<10 µm). Using unfixed material, thicker sections, or chamber slides can increase bubble formation. Apply an adequate volume of medium to spread evenly under the coverslip. Storage of mounted slides at -20°C may also help. For existing bubbles, you can remove the coverslip by soaking the slide in buffer and remount with fresh media [26].
Q: Do I need to seal the coverslip after applying VECTASHIELD? A: It depends on the formulation. For non-setting formulations (e.g., H-1000, H-1200), sealing with nail polish or a plastic sealant is recommended for long-term storage beyond a day or so. For hardening/setting formulations (e.g., HardSet, Vibrance), sealing is generally not required when using thin sections or cell monolayers [27] [28] [26].
Q: Do I need to dehydrate tissue sections before applying VECTASHIELD? A: No. Dehydration (e.g., air drying or ethanol exposure) is not required or recommended. For optimal antifade performance, the preparation should be removed from the final buffer/water rinse, kept slightly wet/moist, and then coverslipped with a small volume (25-50 µL) of VECTASHIELD [27] [28] [26].
Q: How quickly can I view sections after mounting with a hardening medium like VECTASHIELD Vibrance? A: Slides mounted with VECTASHIELD Vibrance can be viewed as soon as one hour after mounting, with two hours recommended for optimal signal intensity. The medium cures enough to hold the coverslip in place within one hour, with complete curing taking 4–24 hours at room temperature [27].
Q: How long can I store slides mounted with VECTASHIELD? A: Slides mounted with VECTASHIELD Vibrance can be stored at room temperature for several months without media retraction, bubble formation, or loss of signal intensity. Storage at 4°C is also effective [27]. For the non-setting VECTASHIELD formulation with DAPI, mounted slides should be stored at 4°C, protected from light [28].
Troubleshooting Common Issues
| Problem | Possible Cause | Solution |
|---|---|---|
| Bubble Formation | Using thick sections, chamber slides, or insufficient mounting medium [26]. | Use thin sections (<10 µm), ensure adequate volume of medium is applied, store slides at -20°C [26]. |
| Media Retraction | Use of non-setting formulations over long storage periods [27]. | Use a hardening formulation (e.g., Vibrance, HardSet) for long-term storage [27]. |
| Signal Fading (Photobleaching) | Use of a mounting medium with insufficient antifade properties. | All VECTASHIELD formulations are specifically designed to inhibit photobleaching [28] [26]. |
| Specimen Drying | Failure to properly coverslip or use a sealed medium. | Use a hardening formulation that does not require sealing [27]. Ensure specimen is moist before applying medium [27]. |
Research Reagent Solutions
The following table details key reagents for secure mounting in immunofluorescence, particularly in the context of embryo research.
| Item | Function in the Protocol | Example from Search Results |
|---|---|---|
| VECTASHIELD Vibrance with DAPI [27] | Aqueous, hardening mounting medium with antifade properties and nuclear counterstain. Prevents drying and photobleaching. | Sets at room temperature; viewable in 1 hour; room temperature storage; minimal bubbles [27]. |
| VECTASHIELD with DAPI [28] | Non-hardening mounting medium with antifade properties and DAPI. Requires sealing for long-term storage. | Liquid on slide; ideal refractive index; stable formula; store at 4°C [28]. |
| VECTASHIELD HardSet with DAPI [26] | Hardening mounting medium with antifade properties and DAPI. Does not require sealing. | Hardens in ~15 min; ideal refractive index; ready-to-use [26]. |
| DAPI Stock Solution [29] | Nucleic acid stain for nuclear counterstaining. Used independently of mounting media. | Preferentially stains dsDNA; excitation/emission at ~358/461 nm; prepare as 5 mg/mL stock [29]. |
| Formaldehyde [4] | Fixative for preserving cell structure and antigenicity before staining and mounting. | 16% formaldehyde solution used in mESC immunofluorescence protocol [4]. |
| Triton X-100 [4] | Detergent for permeabilizing cell membranes to allow antibodies to enter. | Used in permeabilization buffer for mESC protocol [4]. |
Mounting Media Comparison and Selection Guide
The table below summarizes the key characteristics of different VECTASHIELD formulations that contain DAPI to aid in selection.
| Product Name | Formulation Type | Counterstain | Refractive Index (Cured) | Recommended Viewing Time | Long-Term Storage |
|---|---|---|---|---|---|
| VECTASHIELD Vibrance [27] | Aqueous (Hardening) | DAPI | 1.47 [27] | 1-2 hours [27] | Room temperature for weeks/months [27] |
| VECTASHIELD HardSet [26] | Aqueous (Hardening) | DAPI | 1.46 [26] | ~15 minutes for immobilization [26] | No sealing required [26] |
| VECTASHIELD [28] | Aqueous (Non-Hardening) | DAPI | 1.45 [28] | Not specified | Store at 4°C; sealing recommended [28] |
Workflow for Selecting and Using Mounting Media
The following diagram illustrates the decision pathway for choosing the appropriate VECTASHIELD formulation and the subsequent steps for secure mounting to prevent drying.
Experimental Protocols for Secure Mounting
Detailed Protocol: Using a Hardening Mounting Medium (e.g., VECTASHIELD Vibrance)
This protocol is designed for secure mounting that prevents drying and is suitable for long-term archiving, critical for precious embryo samples [27].
- Sample Preparation: After the final immunofluorescence staining step and final buffer rinse, ensure the specimen is kept slightly wet or moist. Do not allow the sample to air-dry completely, and do not dehydrate the tissue with ethanol [27].
- Apply Mounting Medium: Pipette a small volume (25-50 µL) of VECTASHIELD Vibrance with DAPI onto the specimen [27].
- Apply Coverslip: Gently lower a clean coverslip onto the slide, avoiding the introduction of air bubbles.
- Curing: Allow the slide to sit at room temperature, protected from light. The coverslip will become immobilized in about one hour, and the slide can be imaged. Complete curing will occur within 4 to 24 hours [27].
- Storage: Once cured, slides can be stored at room temperature in a standard slide box for several months without media retraction, bubble formation, or loss of signal intensity. Sealing the coverslip is not required [27].
Alternative Protocol: Using a Non-Hardening Mounting Medium (e.g., VECTASHIELD with DAPI)
This protocol is for non-hardening media, which remain liquid and require sealing for long-term storage [28].
- Sample Preparation: As with the hardening protocol, ensure the specimen is moist after the final buffer rinse. Do not dehydrate [28].
- Apply Mounting Medium and Coverslip: Apply 25-50 µL of VECTASHIELD with DAPI and lower the coverslip.
- Seal the Coverslip: If you intend to keep the slides beyond a day or so, seal the perimeter of the coverslip with nail polish or a plastic sealant to prevent evaporation and drying [28].
- Storage: Store the sealed slides at 4°C, protected from light, to preserve fluorescence [28].
Independent DAPI Staining Protocol
If using a mounting medium without DAPI, a separate counterstaining step is required. The following protocol can be used for adherent cells prior to mounting [29].
- Stain Preparation: Dilute a 5 mg/mL DAPI stock solution to a working concentration of 300 nM in Phosphate-Buffered Saline (PBS) [29].
- Staining: After all other staining steps and a final PBS equilibration, add approximately 300 µL of the dilute DAPI staining solution to the coverslip, ensuring the cells are covered.
- Incubation: Incubate for 1 to 5 minutes at room temperature [29].
- Rinsing: Rinse the sample several times with PBS to remove unbound dye [29].
- Mounting: Drain excess buffer and proceed to mount the sample with your chosen antifade mounting medium [29].
Technical Support Center
Frequently Asked Questions (FAQs)
FAQ 1: What is fire polishing, and how does it protect my embryonic samples? Fire polishing is a process where the tip of a glass capillary is briefly exposed to heat, smoothing sharp edges and creating a uniform, polished finish. This is critical for preventing the pipette from scratching or damaging delicate cell membranes [30]. A smooth tip minimizes fluid turbulence during dispensing or aspiration, directly reducing the shear stress forces that can compromise embryo viability.
FAQ 2: I am struggling with embryo drying during extended immunofluorescence procedures. How can fire-polished capillaries help? Fluid loss and embryo drying are often exacerbated by poor seals or rough handling. Fire-polished capillaries facilitate the formation of a tighter seal with cells or other surfaces during microinjection or fluid handling [31]. This superior seal minimizes unintended fluid loss from the sample environment, helping to maintain the hydration of embryos throughout multi-step immunofluorescence protocols [4].
FAQ 3: Why does my sample experience high shear stress during fluid handling, and how can I reduce it? Shear stress is frequently caused by rapid fluid flow through narrow, unpolished openings. The internal surface roughness of a standard pipette can create turbulent flow. Using fire-polished capillaries, which have a smoother internal bore, promotes laminar flow and reduces turbulence [32]. This is especially important when working with sensitive cells like neurons or embryos, as it preserves membrane integrity and function.
FAQ 4: Can I use any type of glass capillary for fire polishing? No, the type of glass is important. Borosilicate glass is the most common and versatile choice for electrophysiology and microinjection [31]. It has suitable thermal properties for pulling and fire polishing. Other types, such as quartz or aluminosilicate glass, have different characteristics and may require specialized equipment. Always ensure the glass capillaries you select are compatible with your puller and intended fire-polishing setup.
Troubleshooting Guide
| Problem | Possible Cause | Solution |
|---|---|---|
| Poor Seal Formation | Rough or irregular pipette tip damaging the cell membrane. | Implement a consistent fire-polishing step. Use a microforge or heated filament to smooth the tip under microscopic observation [31]. |
| Excessive Fluid Loss | Poor tip geometry leading to turbulent flow and drips. | Use fire-polished capillaries with a consistent, smooth bore. Ensure the tip diameter is appropriate for your sample size and fluid viscosity. |
| High Sample Mortality | Shear stress from rough internal capillary surfaces. | Switch to fire-polished capillaries to minimize turbulent flow. Optimize aspiration and dispensing pressures to use the gentlest possible flow rates [32]. |
| Clogged Capillary Tips | Debris accumulation on internal rough surfaces. | Use fire-polished capillaries to reduce internal friction. Filter all buffers and solutions using a 0.2 µm filter before loading. |
Research Reagent Solutions
The following table details essential materials for experiments utilizing fire-polished glass capillaries.
| Item | Function & Explanation |
|---|---|
| Borosilicate Glass Capillaries | The primary material for creating micropipettes. Its thermal properties allow for consistent pulling and fire polishing, and it offers a good balance of low electrical noise and mechanical strength [30] [31]. |
| Micropipette Puller | An instrument that uses heat and mechanical force to stretch glass capillaries into two fine-tipped micropipettes. Puller parameters (heat, force, velocity) determine the final tip geometry [33]. |
| Microforge | A specialized instrument that combines a microscope with a fine, heatable filament. It is used for fire polishing pipette tips to smooth them and for precisely breaking the tip to a desired diameter [31]. |
| High-Purity Water | Used for rinsing and preparing solutions. Essential for preventing particulate contamination that can clog fine capillary tips. |
| 0.2 µm Filters | Used for sterilizing and removing particulates from all buffers and solutions before loading them into capillaries to prevent clogging [4]. |
Experimental Workflow and Parameter Optimization
The diagram below illustrates the key decision points and parameter adjustments in the process of preparing an ideal fire-polished capillary.
Quantitative Data for Capillary Selection
The table below summarizes the physical properties of common glass types used for capillaries, which influence their performance during pulling and fire polishing.
| Glass Type | Softening Point (°C) | Annealing Point (°C) | Thermal Expansion (0–300°C) | Common Usage |
|---|---|---|---|---|
| Duran (Schott 8250) | 720 | 500 | 5.0 x 10⁻⁶/K | Patch clamp, general use |
| Schott AR-GLAS | 720 | 530 | 9.1 x 10⁻⁶/K | Not specified |
| Schott BORO-8330 | 825 | 560 | 33 x 10⁻⁷ cm/cm/°C | Thin-wall capillaries |
| Corning 7800 | 789 | 565 | 55 x 10⁻⁷ cm/cm/°C | Multi-barrel, septum theta |
| Kimble N51A | 785 | 570 | 55 x 10⁻⁷ cm/cm/°C | Single barrel tubing |
Detailed Protocol: Fire Polishing a Glass Capillary
This protocol describes the process of fire polishing a pulled glass capillary using a microforge.
Materials and Equipment:
- Pulled borosilicate glass capillary
- Microforge
- High-magnification microscope
Procedure:
- Secure the Capillary: Mount the pulled capillary securely in the holder of the microforge, positioning the tip within the field of view under high magnification.
- Position the Heat Source: Bring the heat source (typically a platinum-iridium filament) near the tip of the capillary. Ensure it does not contact the glass directly.
- Apply Brief Heat: Briefly activate the heat source. The intensity and duration of the heat pulse must be empirically determined. The goal is to see the very end of the tip begin to melt and smooth out, without causing the tip to close up entirely.
- Inspect the Tip: Deactivate the heat and visually inspect the tip. A properly fire-polished tip will appear smooth and rounded under the microscope, with no sharp or jagged edges [31].
- Test the Capillary: Before use with precious embryonic samples, test the capillary with a buffer solution to ensure flow characteristics are as expected and the tip is not obstructed.
Troubleshooting the Protocol:
- Tip Closed Entirely: Used too much heat or held it for too long. Reduce the heat intensity or duration for the next attempt.
- No Visible Change: The heat was insufficient or the tip was too far from the heat source. Increase heat slightly or reposition the capillary.
- Tip Became Deformed: The heat was applied unevenly or was excessive. Ensure the filament is clean and straight, and reduce the heat setting.
Troubleshooting Embryo Integrity: Solving Common Drying Artifacts and Staining Failures
FAQ: How do I identify drying artifacts in my immunofluorescence samples?
Drying artifacts manifest through two primary types of observable defects in your samples: morphological changes and staining abnormalities.
- Shriveled or Distorted Morphology: Cells or tissues that have dried out often appear shrunken, puckered, or creased. This physical distortion occurs because the loss of water compromises cellular structures.
- High, Uniform Background Staining: A pervasive, speckled, or uniformly high signal across your sample, obscuring specific staining, is a classic sign. When the sample dries, antibodies become trapped and bind non-specifically throughout the tissue, not just to the target antigen [34].
FAQ: What is the direct link between sample drying and high background?
The connection is direct and mechanical. When the aqueous buffer on your sample evaporates, the following sequence of events occurs [34] [35] [36]:
- Antibody Concentration: As the liquid evaporates, the concentration of antibodies and other proteins in the solution increases dramatically on the tissue surface.
- Non-specific Trapping: The drying process physically traps these concentrated antibodies onto the tissue matrix and cellular components in a non-specific manner.
- Irreversible Binding: Once dried onto the tissue, these antibodies bind irreversibly. Subsequent washing steps are ineffective at removing them, leading to a high, uniform background that can mask your specific signal.
FAQ: My embryos look shriveled and show high background. What should I do?
A combination of immediate corrective actions and long-term protocol adjustments is required to address this issue.
Immediate Corrective Actions
- Re-prepare your samples: Unfortunately, the drying damage is often irreversible [34]. The most reliable solution is to start a new sample preparation, meticulously ensuring the slides do not dry out at any step.
- Review your protocol: Immediately check and confirm that all incubation and washing steps are performed in a humidified chamber and that sufficient volume of liquid is used to fully cover the sample at all times [37] [36].
Long-Term Protocol Adjustments
- Use a Humidified Chamber: For all antibody incubation steps, which can last several hours or overnight, use a sealed container lined with moist paper towels or use commercially available humidified chambers. This is non-negotiable for delicate samples like embryos [15] [35].
- Avoid Edge Effects: When applying solutions to slides, ensure the liquid forms a continuous film without air bubbles. Be careful not to let the meniscus of the liquid cross over the tissue section, as this will cause localized drying [34].
- Segment Your Work: If processing multiple slides, handle them in small batches to minimize the time each slide spends outside of a liquid buffer or humidified environment.
The following diagram illustrates the cause-and-effect relationship that leads to drying artifacts and the key preventive measures to break this cycle.
FAQ: How can I distinguish drying-induced background from other types of background staining?
Different causes of high background often present with distinct characteristics. The table below summarizes key differentiators to aid in diagnosis.
Table: Differentiating Drying Artifacts from Other Common Background Issues
| Cause of Background | Characteristic Appearance | How to Confirm |
|---|---|---|
| Sample Drying [34] [36] | High, often uniform background; possible "edge effects" where staining is stronger at the sample periphery; shriveled morphology. | Check if background is consistent with areas that may have dried. Re-prepare samples with strict humidity control. |
| Primary Antibody Concentration Too High [34] [38] | Speckled or granular staining pattern across the entire sample, including non-cellular areas. | Perform an antibody titration experiment. The background should decrease at lower concentrations without loss of specific signal. |
| Insufficient Blocking [34] [37] | General, diffuse background across the tissue section. | Increase blocking incubation time or try a different blocking agent (e.g., normal serum from the secondary antibody host). |
| Insufficient Washing [37] [36] | A cloudy or hazy appearance with precipitate-like artifacts. | Increase the number, duration, and volume of washes between steps. Include gentle agitation. |
| Autofluorescence [39] [38] | Present even in an unstained control sample. Often uniform but can be granular (e.g., from lipofuscin in aged tissue). | Image an unstained section with the same settings. If positive, use quenching reagents (e.g., Sudan Black B) or choose a far-red fluorophore. |
The Scientist's Toolkit: Essential Materials for Preventing Drying Artifacts
The following table lists key reagents and tools crucial for maintaining sample hydration and integrity during immunofluorescence protocols, especially for sensitive embryos.
Table: Essential Reagents and Tools for Hydration Control
| Item | Function in Preventing Drying |
|---|---|
| Humidified Chamber | A sealed container that maintains a saturated environment, preventing evaporation during long incubation steps [35] [36]. |
| PBS or TBS Buffer | Standard physiological buffers used for washing and diluting reagents. Keeping samples submerged in these is critical [15]. |
| Triton X-100 or Tween-20 | Detergents added to wash buffers (typically 0.05-0.1%) to reduce surface tension and help ensure even coverage, minimizing hydrophobic interactions that can cause non-specific binding [34] [15]. |
| Normal Serum or BSA | Common blocking agents. Adequate blocking is part of a robust protocol that, when combined with hydration control, ensures clean staining [34] [15]. |
| Glass Capillaries (Hand-pulled) | For handling delicate embryos, capillaries with smooth, fire-polished edges are essential to avoid damaging structures and potentially causing leaks or drying [15]. |
| Four-Well Dishes | Ideal for processing small numbers of embryos or tissue sections, as they allow for easy medium changes while keeping samples submerged and organized [15]. |
Optimization of Wash Buffer Composition and Incubation Times for Enhanced Viability
Frequently Asked Questions
What is the most critical component to include in a wash buffer to prevent cell loss during centrifugation? Incorporating albumin is crucial for protecting cells from mechanical stress during washing. Data demonstrates that wash buffers containing 1% albumin can maintain cell retention rates greater than 80% and prevent significant drops in viability. The protective effect is due to albumin's ability to shield cells from oxidation and shear stress [40].
How does buffer osmolarity and ion composition affect delicate embryonic cells? Using a physiologically balanced base solution, such as PBS or Plasma-Lyte, is essential. The base solution provides the correct osmotic pressure to prevent cell swelling or shrinkage. Furthermore, the specific ionic composition can influence cell size and stress marker expression, underscoring the importance of using a well-established, isotonic solution [41] [40].
What is the maximum recommended hold time for cells in a wash buffer? Experimental data supports a hold time of up to 2 hours in a properly formulated wash buffer without significant loss of viability. For longer procedures, it is critical to validate the buffer composition and hold time for your specific cell type [40].
Why is it necessary to include serum or BSA in the antibody incubation buffer? Bovine Serum Albumin (BSA) or serum is used as a blocking agent in the incubation buffer to minimize non-specific background staining by occupying reactive sites on the sample. A common incubation buffer formulation includes 1% BSA, 1% normal serum, and 0.3% Triton X-100 in PBS [42].
Troubleshooting Guides
Problem: Low Cell Viability After Washing
| Potential Cause | Recommended Solution | Underlying Principle |
|---|---|---|
| Harsh centrifugation | Reduce centrifugal force to 500 × g and ensure consistent time (e.g., 5 minutes) [40]. | Excessive g-forces damage cell membranes and internal structures. |
| Prolonged hold time | Limit hold time in wash buffer to less than 2 hours and keep samples on ice if possible [40]. | Depletes nutrients and increases exposure to metabolic byproducts. |
| Improper buffer composition | Use a balanced salt solution (PBS/Plasma-Lyte) with 1-5% recombinant albumin [40]. | Albumin provides a protective colloid effect against shear stress and oxidation. |
Problem: High Background Signal in Immunofluorescence
| Potential Cause | Recommended Solution | Underlying Principle |
|---|---|---|
| Insufficient blocking | Extend blocking to 30-60 minutes using the serum of the secondary antibody host or 1% BSA [43]. | Blocks Fc receptors and non-specific binding sites on the sample. |
| Inadequate washing | Perform three washes, 5-15 minutes each, with ample PBS after each antibody step [43] [42]. | Removes unbound and loosely-associated antibodies. |
| Antibody concentration too high | Titrate the primary and secondary antibodies to find the optimal dilution [43]. | High antibody concentrations increase non-specific binding. |
Experimental Data and Protocols
Table 1: Impact of Wash Buffer Composition on Cell Retention and Viability
Data derived from primary human T cell wash experiments, providing quantitative metrics for buffer optimization [40].
| Buffer Composition | Cell Retention (%) | Viability Change (Post-Wash) |
|---|---|---|
| Plasma-Lyte only | ~82% | -4% |
| Plasma-Lyte + 1% Optibumin | ~92% | -1% |
| Plasma-Lyte + 1% HSA | ~85% | -2% |
| PBS only | ~81% | -3% |
| PBS + 1% Optibumin | ~88% | -1% |
| PBS + 1% HSA | ~89% | -1% |
Protocol: Optimized Wash and Staining Steps for Sensitive Cells
This protocol integrates best practices for maintaining viability during immunofluorescence [4] [43].
- Fixation: Fix cells for 10 minutes at room temperature with 4% formaldehyde in PBS.
- Permeabilization and Blocking: Permeabilize and block simultaneously by incubating for 30-60 minutes in a buffer containing 1% BSA and 0.1% Triton X-100 in PBS.
- Antibody Incubation:
- Incubate with primary antibody diluted in incubation buffer (e.g., with 1% BSA).
- Wash 3 times for 5 minutes each with PBS containing 0.1% Tween-20 (PBST).
- Incubate with fluorophore-conjugated secondary antibody in incubation buffer, protected from light.
- Wash 3 times for 5 minutes each with PBST.
- Final Wash and Mounting: Perform a final rinse with PBS, apply an anti-fade mounting medium with DAPI, and image promptly [43] [42].
IF Protocol with Critical Wash Steps
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Cell Washing and Staining
Key materials and their functions for robust and reproducible immunofluorescence experiments.
| Reagent | Function | Example Application |
|---|---|---|
| Recombinant Albumin (Optibumin) | Animal-free origin, consistent quality; protects cells from shear stress during washing steps [40]. | Used at 1-5% in PBS or Plasma-Lyte as a wash buffer component. |
| Bovine Serum Albumin (BSA) | Blocking agent; reduces non-specific antibody binding [43] [42]. | Used at 1% in PBS, often with serum, for antibody dilution and blocking. |
| Triton X-100 | Detergent for cell permeabilization; allows antibodies to access intracellular targets [43]. | Used at 0.1-0.3% in PBS for permeabilization. |
| Tween-20 | Mild detergent; reduces surface tension for effective washing without excessive cell disruption [43]. | Used at 0.1% in PBS (PBST) for wash steps between antibody incubations. |
| ibidi μ-Slides | Coverslip-bottom dishes; optimized for high-resolution microscopy; prevent sample drying [4] [43]. | All staining steps can be performed directly in the slide, minimizing cell loss. |
| Anti-Fade Mounting Medium | Preserves fluorescence signal; reduces photobleaching during imaging and storage [43]. | Applied after final wash before placing a coverslip for microscopy. |
Why is a humidified chamber non-negotiable for long incubations?
During extended antibody incubations, particularly the common overnight step at 4°C, the primary risk is evaporation. Even minor evaporation can have catastrophic effects on your experiment:
- Increased Antibody Concentration: As water evaporates, the remaining antibody solution becomes more concentrated. This leads to non-specific binding and high background staining, obscuring your true signal [34].
- Sample Drying: If the section dries out completely, it causes irreversible, non-specific antibody binding. This creates severe artifacts and often renders the sample useless [34] [35].
- Osmotic Stress: Changes in solute concentration can stress or damage delicate samples, such as embryos or tissues.
A humidified chamber creates a saturated environment that prevents the evaporation of your precious antibody droplets, ensuring consistent concentration and specific binding throughout the incubation.
How do I set up a simple humidified chamber?
You do not need expensive equipment to create an effective humidified chamber. A reliable setup can be assembled quickly with common lab materials.
Basic Protocol for Assembling a Humidified Chamber:
- Select a Container: Use a sealed plastic box, pipette tip box lid, or a glass Petri dish with a tight-fitting lid.
- Add Humidifying Material: Line the bottom of the container with a damp paper towel, gauze, or laboratory wipes moistened with distilled water. Alternatively, you can use a small reservoir of water.
- Create a Platform: Place a support frame (e.g., a slide rack, cut pipette tip box lid, or several wooden or plastic applicator sticks) over the moist material. This platform keeps your slides above direct contact with the water, preventing cross-contamination and ensuring even humidity.
- Apply Samples: Pipette your antibody solution onto the tissue sections or cells on your slides. Carefully place the slides on the platform.
- Seal and Incubate: Close the lid securely to create a sealed, humid environment. Place the entire chamber in a refrigerator or incubator at the required temperature for the duration of the incubation.
The diagram below illustrates this simple and effective setup.
What are the most common problems and their solutions?
Even with a humidified chamber, issues can arise. The table below summarizes frequent problems and their evidence-based solutions, drawing from general IHC/IF best practices [34] [35].
| Problem | Possible Cause | Solution |
|---|---|---|
| Evaporation Still Occurs | Leaky or ill-fitting chamber lid | Ensure the lid seals completely. Use parafilm to seal the edges if necessary. |
| High Background Staining | Antibody concentration increased due to evaporation; insufficient blocking. | Confirm chamber is sealed. Re-titrate antibody concentration. Extend blocking time to 1 hour using serum from the secondary antibody host species [34] [35]. |
| Uneven or Patchy Staining | Antibody solution did not fully cover the tissue section. | Ensure the antibody solution forms a continuous bubble over the entire section. Use a pap pen to create a hydrophobic barrier around the section. |
| Weak or No Signal | Inactive antibody; epitope masking. | Include a positive control tissue. Optimize antigen retrieval methods (e.g., test citrate vs. Tris-EDTA buffer at different pH levels) [34]. |
| Tissue Detachment | Sections dried out before incubation; insufficient slide coating. | Never let sections air dry after deparaffinization/hydration. Use positively charged or poly-L-lysine coated slides. |
Frequently Asked Questions (FAQs)
Q1: Can I stack slides in the humidified chamber? Yes, but with caution. Use a proper slide rack that ensures slides are level and separated. Stacking slides directly on top of each other can cause uneven antibody distribution and pressure on the samples.
Q2: How long can I store a prepared humidified chamber before use? It is best practice to prepare the chamber immediately before use to prevent microbial growth. For long-term storage of the chamber itself, ensure it is completely dry to avoid mold.
Q3: My fluorescent signal is weak. Could the humidified chamber be a factor? The humidified chamber itself should not cause weak signal. However, if evaporation occurred, the effective antibody concentration would be higher, typically leading to increased background, not weaker specific signal. Focus troubleshooting on primary antibody viability, antigen retrieval efficiency, and fluorophore protection from light [35].
Q4: Is there a difference between incubation at 4°C vs. room temperature in a humidified chamber? The key principle of preventing evaporation applies to both. Overnight incubation at 4°C often yields lower background and is standard for many primary antibodies. For room temperature incubations, ensure the chamber is kept in a dark, stable location away from heat sources.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key materials and reagents critical for successfully implementing humidified chambers and achieving high-quality immunofluorescence results.
| Item | Function & Rationale |
|---|---|
| Sealed Container | Creates the physical chamber to maintain a saturated atmosphere and prevent evaporation. |
| Normal Serum | Used for blocking non-specific binding sites. Should be from the same species as the host of the secondary antibody [35]. |
| Antibody Diluent | A buffered solution (e.g., PBS) often containing a gentle detergent (e.g., 0.05% Tween-20) to reduce hydrophobic interactions and non-specific binding [34]. |
| Positively Charged Slides | Prevents tissue sections from detaching during long incubation and washing steps. |
| Sodium Borohydride (NaBH4) | Quenches autofluorescence caused by aldehyde fixatives. A 1% solution in PBS can be used after fixation [35]. |
| Protease & Phosphatase Inhibitors | Crucial for preserving labile epitopes, especially phosphorylated targets, during sample preparation and storage [35]. |
Workflow & Decision-Making for Successful Incubations
To achieve optimal results, follow a systematic workflow from setup to troubleshooting. The diagram below outlines the key steps and decision points for implementing a humidified chamber in your immunofluorescence protocol.
This technical support guide provides targeted troubleshooting advice for researchers, particularly those working within the context of embryo immunofluorescence (IF) protocols, to prevent sample drying and other common issues.
Frequently Asked Questions (FAQs)
Q: At which steps is my embryo sample most at risk of drying, and how can I prevent it?
- A: The risk is highest during antibody incubations and washing steps. To prevent drying, always perform these procedures in a humidified chamber (e.g., a sealed container with damp paper towels) and avoid using excessive wash volumes that can lead to uneven drying at the edges of your sample [44].
Q: My immunofluorescence signal is weak or absent. What are the primary causes?
- A: Weak signal can result from several factors, including inadequate permeabilization (preventing antibody access), insufficient antibody concentration or incubation time, or over-fixation which can destroy antigen epitopes. Ensure you are using a strong enough permeabilization agent like Triton X-100 for intracellular targets and optimize your antibody dilutions [45].
Q: I am seeing high background fluorescence. How can I reduce it?
- A: High background is often due to non-specific antibody binding or incomplete blocking. Ensure your blocking serum is from a different species than your primary antibody and extend your blocking time to 30-60 minutes. Additionally, perform thorough washing after each antibody incubation step to remove unbound antibodies [45].
Q: What is the purpose of a hydrophobic barrier, and when should I use one?
- A: A hydrophobic barrier, created with a special pen, forms a well on the slide to contain small volumes of antibody solutions, preventing them from spreading and evaporating. This is crucial for embryo samples and when working with limited reagent volumes to ensure the sample stays submerged and does not dry out [44].
Troubleshooting Guide
| Problem | Possible Cause | Solution |
|---|---|---|
| Sample Drying | Incubation outside a humidified chamber; insufficient liquid coverage. | Always use a humidified chamber during all incubation steps. Use a hydrophobic barrier to contain liquid [44]. |
| Weak or No Specific Signal | Inefficient permeabilization; low antibody titer; epitope damaged by fixation. | For intracellular targets, use 0.1-0.2% Triton X-100 for permeabilization. Titrate antibodies for optimal concentration. For aldehyde fixation, avoid over-fixating [45]. |
| High Background Signal | Inadequate blocking; non-specific antibody binding; insufficient washing. | Block with 1-5% BSA or serum from an unrelated species. Increase wash times and volume after antibody incubations [45]. |
| Cell or Tissue Detachment | Overly harsh washing; slide not properly coated. | Use gentle washing techniques. For delicate samples like embryos, use poly-L-lysine coated slides to improve adhesion [44] [45]. |
| Non-specific Nuclear Staining | Contamination or over-staining with counterstain. | Optimize the concentration and incubation time of nuclear dyes like DAPI (0.1-1 µg/mL for 5 minutes) and wash thoroughly [45]. |
Experimental Protocol: Key Stages for Embryo Integrity
The following workflow outlines a generalized indirect immunofluorescence protocol, highlighting critical checkpoints for visual inspection to preemptively catch issues, with special considerations for embryo samples.
Sample Preparation and Fixation
- Methodology: Fix embryo samples by incubating in 2-4% Paraformaldehyde (PFA) for 10-20 minutes at room temperature. Aldehyde fixatives like PFA are recommended for preserving cellular architecture [45].
- Quality Checkpoint: Visually confirm that the sample remains fully submerged and that fixation time is not exceeded, as over-fixation can mask antigen epitopes.
Permeabilization and Blocking
- Methodology: To allow antibody access to intracellular targets, permeabilize fixed embryos by incubating with 0.1-0.2% Triton X-100 in PBS for 10 minutes. Subsequently, block samples by incubating with a 1-5% solution of Bovine Serum Albumin (BSA) or normal serum from an unrelated species for 30-60 minutes to prevent non-specific antibody binding [45].
- Quality Checkpoint: Ensure the blocking solution fully covers the sample. The sample should not appear dry or sticky at this stage.
Antibody Incubation and Washing
- Methodology: Incubate samples with the primary antibody diluted in blocking buffer, typically for 1-2 hours at room temperature or overnight at 4°C. After thorough washing, incubate with the fluorophore-conjugated secondary antibody, also diluted in blocking buffer, for about 1 hour at room temperature [46] [45]. All incubations must be performed in a humidified chamber to prevent evaporation [44].
- Quality Checkpoint: Before and after each incubation, verify that the sample is still covered with liquid and has not dried out. After washes, check for smooth, even tissue appearance without salt crystals or residue.
Mounting and Imaging
- Methodology: Apply an antifade mounting medium and gently lower a coverslip, avoiding air bubbles. For stability, the coverslip edges can be sealed with clear nail polish. Store the slides in the dark at 4°C until imaging [44] [45].
- Quality Checkpoint: Visually inspect under a microscope for large air bubbles, sample detachment, or signs of drying at the edges before proceeding with image acquisition.
The Scientist's Toolkit: Essential Research Reagents
| Reagent | Function in the Protocol |
|---|---|
| Paraformaldehyde (PFA) | An aldehyde fixative that cross-links proteins, preserving cellular morphology and immobilizing antigens [45]. |
| Triton X-100 | A non-ionic detergent used for permeabilizing cell membranes, allowing antibodies to access intracellular targets [45]. |
| Bovine Serum Albumin (BSA) | A blocking agent used to cover non-specific binding sites on the tissue, reducing background signal [45]. |
| Primary Antibody | The antibody that specifically recognizes and binds to the target antigen of interest [46]. |
| Fluorophore-conjugated Secondary Antibody | The antibody that recognizes the primary antibody and is coupled to a fluorescent dye, providing signal amplification and detection [46]. |
| Poly-L-lysine | A slide coating that provides a charged surface to enhance adhesion of cells or tissues, preventing wash-off during processing [44] [45]. |
| DAPI | A fluorescent dye that binds strongly to DNA, used as a nuclear counterstain to visualize all nuclei in a sample [46] [45]. |
| Antifade Mounting Medium | A reagent used to preserve fluorescence and prevent photobleaching during microscopy and storage [44] [45]. |
Validating Sample Integrity: Confirming Successful Staining and Nuclear Segmentation
In the study of early mammalian development, specific transcription factors serve as critical markers for identifying and isolating distinct cell lineages. The first cell fate choice in the mammalian embryo is the segregation of the inner cell mass (ICM), which gives rise to the fetus, and the trophectoderm (TE), which forms the placenta. This process, and the subsequent specification of the primitive endoderm (PE), is regulated by a core set of transcription factors, including NANOG, CDX2, and GATA4, which are often mutually antagonistic [47] [48]. Accurately assessing the expression of these markers via immunofluorescence (IF) is fundamental to developmental biology research. However, the success of these experiments is highly dependent on optimized protocols, with a critical factor being the consistent prevention of embryo drying throughout the staining procedure [49] [50].
The following diagram illustrates the core antagonistic relationships between these key transcription factors that govern early cell fate decisions:
Troubleshooting Guide: FAQs on Staining Key Markers
This section addresses common challenges researchers face when detecting lineage specification markers.
FAQ 1: I am observing weak or no staining for NANOG in my inner cell mass (ICM) samples. What could be the cause?
Weak or absent signal is a frequent issue in IF. The causes and solutions are multifaceted, but ensuring your samples do not dry out at any point is paramount, as this can destroy antigenicity [49] [50].
- Possible Cause: Sample Drying. If samples dry out during the staining procedure, the antigen can become denatured and inaccessible to the antibody [49] [50].
- Recommendation: It is vital that the sample remains covered in liquid throughout the entire staining procedure [49]. Carefully manage buffer volumes and container seals.
- Possible Cause: Inadequate Fixation or Permeabilization. Improper fixation may not preserve the antigen, while insufficient permeabilization will prevent antibody access to intracellular targets like NANOG.
- Possible Cause: Low Abundance of Target.
- Possible Cause: Antibody Incubation.
- Recommendation: Ensure you are using the correct antibody dilution and incubating for the recommended time. Many antibodies require incubation at 4°C overnight for optimal results [49].
FAQ 2: My immunofluorescence results for CDX2 and GATA4 show high background. How can I improve the signal-to-noise ratio?
High background can obscure specific signal and make interpretation difficult.
- Possible Cause: Sample Autofluorescence. This is a common pitfall, especially in embryonic tissues.
- Possible Cause: Insufficient Blocking. Inadequate blocking leaves non-specific sites available for antibody binding.
- Possible Cause: Antibody Concentration Too High.
- Possible Cause: Non-specific Binding of Secondary Antibody.
FAQ 3: I need to confirm the specificity of my GATA4 antibody. What is the best experimental control?
Demonstrating antibody specificity is crucial for publishing robust data.
- Recommendation:
- Knockout/Knockdown Control: The gold standard is to use knockout cells or tissue (where the gene has been deleted) or knockdown cells (e.g., via siRNA). The absence of staining in this control confirms specificity [49].
- Isotype Control: Use an antibody of the same isotype (e.g., IgG) that targets an irrelevant antigen to identify non-specific binding of the antibody itself [49].
- Biological Validation: Ensure the staining pattern matches the expected biological expression. For example, GATA4 should be restricted to the primitive endoderm lineage in the blastocyst, not the epiblast or trophectoderm [47].
Protocol for Preventing Embryo Drying During Immunofluorescence
Preventing embryo drying is a non-negotiable aspect of a successful IF protocol. The following workflow is designed to minimize this risk.
Detailed Critical Steps:
- Liquid Handling: When removing or adding solutions, use a vacuum aspiration system with a fine tip (e.g., a rubber tube capped by a yellow pipette tip). This allows for precise removal of most liquid without disturbing or exposing the delicate embryos to air [53]. Never leave embryos exposed to air.
- Continuous Liquid Cover: The most critical rule is that samples must be kept covered in liquid at all times during the staining process [49] [50]. Check samples frequently to ensure they remain fully submerged.
- Humidified Chamber: During all antibody incubation steps, which can last for hours or overnight, place the slides or vials in a humidified chamber. This can be a sealed container with a damp paper towel at the bottom to saturate the atmosphere and prevent evaporation.
- Mounting: After the final wash, mount the samples immediately in an anti-fade mounting medium to preserve fluorescence and image them as soon as possible for the best results [49].
Experimental Protocol: Validating Marker Expression
The following table provides a generalized protocol for co-staining blastocyst embryos for NANOG, CDX2, and GATA4, incorporating the critical step of preventing drying.
Table 1: Immunofluorescence Protocol for Key Lineage Markers in Blastocysts
| Step | Procedure | Critical Parameters | Purpose & Rationale |
|---|---|---|---|
| 1. Fixation | Fix embryos in 4% PFA for 20-30 minutes at room temperature. | Use freshly prepared PFA. Time varies by embryo stage. | Preserves cellular architecture and immobilizes antigens. |
| 2. Permeabilization & Blocking | Permeabilize with 0.5% Triton X-100 for 20 min. Block with 10% normal serum for 1 hour. | Serum should match secondary antibody species. | Allows antibody entry and blocks non-specific binding sites. |
| 3. Primary Antibody Incubation | Incubate with anti-NANOG, -CDX2, -GATA4 in blocking buffer. | Incubate at 4°C overnight in a humidified dark chamber. | Ensures specific, high-affinity binding to target antigens. |
| 4. Washing | Wash 3x 15 min with PBS containing 0.1% Tween-20 (PBSw). | Ensure samples are fully submerged during and between washes. | Removes unbound and loosely-bound antibodies to reduce background. |
| 5. Secondary Antibody Incubation | Incubate with species-matched fluorophore-conjugated secondary antibodies. | Protect from light. Incubate for 1-2 hours at room temp. | Binds to primary antibody, providing a detectable signal. |
| 6. Final Wash & Mounting | Wash 3x 15 min with PBSw. Mount on slides with anti-fade reagent. | Perform final washes carefully. Image immediately or store at 4°C in dark. | Removes excess secondary antibody. Preserves signal for microscopy. |
Research Reagent Solutions
Selecting and validating the right reagents is fundamental to achieving specific and reproducible staining results.
Table 2: Essential Reagents for Lineage Marker Immunofluorescence
| Reagent | Function | Key Considerations |
|---|---|---|
| Primary Antibodies (e.g., anti-NANOG, anti-CDX2, anti-GATA4) | Specifically bind to the target lineage specification marker. | Must be validated for IF. Confirm species reactivity. Use knockout controls to verify specificity [49]. |
| Fluorophore-Conjugated Secondary Antibodies | Bind to the primary antibody, providing a detectable fluorescent signal. | Must be raised against the host species of the primary antibody. Select bright fluorophores (e.g., Alexa Fluor 488, 594, 647) with minimal spectral overlap for multiplexing [51] [52]. |
| Blocking Serum | Contains antibodies that bind to non-specific sites to reduce background. | Should be from the same species as the secondary antibody. Increase incubation time if background is high [49] [52]. |
| Permeabilization Agent (e.g., Triton X-100, Tween-20) | Creates pores in the cell membrane to allow antibody entry for intracellular targets. | Concentration and time must be optimized. Methanol fixation also permeabilizes [50]. |
| Antifade Mounting Medium | Preserves fluorescence by reducing photobleaching during microscopy. | Essential for long-term storage and for capturing clear images. Use a product containing antifading agents [49]. |
Quantitative Data and Marker Profiles
Understanding the expected expression patterns and the quantitative relationships between these markers is key to accurate analysis.
Table 3: Quantitative Expression Profiles of Key Lineage Markers in the Mouse Blastocyst
| Marker | Primary Lineage | Key Functional Role | Expression Level (Relative) | Mutually Antagonistic With |
|---|---|---|---|---|
| NANOG | Epiblast (EPI) | Sustains pluripotency and specifies the epiblast [48]. | High in EPI, absent in TE/PE | CDX2, GATA4 [48] |
| CDX2 | Trophectoderm (TE) | Required for correct cell fate specification and differentiation of trophectoderm [48]. | High in TE, absent in ICM | NANOG, OCT4 [48] |
| GATA4 | Primitive Endoderm (PE) | Critical for differentiation and function of the primitive endoderm. | High in PE, absent in EPI/TE | NANOG [47] |
| OCT4 | Inner Cell Mass (ICM) | Master regulator of pluripotency; essential for ICM formation. | High in ICM, downregulated in TE | CDX2 [48] |
Frequently Asked Questions (FAQs)
Q1: What makes StarDist particularly suitable for nuclear segmentation in sensitive samples like embryos? StarDist uses deep learning with star-convex shape priors, making it exceptionally good at separating touching nuclei without manual intervention. This is crucial for embryonic tissues where preserving 3D architecture is critical to prevent drying artifacts. Unlike classical methods that require parameter tuning for each image, StarDist's pre-trained models provide consistent results across different tissue types and imaging conditions [54]. Its ability to handle varying nuclear sizes and densities with minimal user input makes it ideal for high-throughput analysis of delicate samples [55] [56].
Q2: How can I prevent embryo drying during immunofluorescence sample preparation for imaging? Preventing embryo drying requires careful experimental planning and specific laboratory techniques. Always perform immunofluorescence staining in chambers with coverslip bottoms that eliminate air exposure, such as ibidi μ-Slides. Maintain adequate liquid volume throughout all washing and incubation steps, and never let samples dry between solution changes. Using mounting medium with low autofluorescence immediately after staining creates a protective barrier against dehydration [4] [43]. For embryonic stem cells specifically, plate cells in small-well chambers like 8-well IBIDI plates with appropriate coating (gelatin for 3D structure preservation or laminin for 2D imaging) to maintain hydration and 3D architecture [4].
Q3: My StarDist segmentation results include too many false positives. How can I improve this? Adjust the Probability/Score Threshold to a higher value (e.g., 0.5-0.7) to reduce false detections, particularly in images with background debris or noise. For the Versatile (fluorescent nuclei) model, the default probability threshold is 0.479071 [55] [57]. You can also increase the Overlap Threshold to prevent excessive segmentation of overlapping nuclei. For cleanup of small debris in the final output, apply post-processing size filters to remove objects smaller than 500 pixels, which typically represent noise rather than true nuclei [57].
Q4: What are the most common causes of poor nuclear segmentation in embryonic tissues? The primary issues include: (1) over-fixation causing nuclear morphology alterations, (2) incomplete permeabilization preventing antibody penetration, (3) excessive nuclear clustering in 3D colonies making individual segmentation difficult, and (4) non-specific DAPI staining creating background noise. For embryonic stem cells, optimize fixation to 10 minutes with 4% formaldehyde and use appropriate detergent concentrations (Triton X-100) for permeabilization based on your specific cell type [4] [43].
Q5: Can I use StarDist for batch processing multiple images? Yes, StarDist supports batch processing through both the Fiji plugin interface and scripting. For the Fiji GUI, process multiple images using the built-in batch mode. For advanced workflows, use Jython or Python scripts to automate processing of entire folders. The Multi-Channel Nuclear Analysis plugin provides a user-friendly interface for batch processing with StarDist while maintaining all segmentation parameters across images [55] [56].
Troubleshooting Guides
Poor Nuclear Segmentation Quality
Problem: StarDist fails to properly segment nuclei, either missing valid nuclei or including excessive background.
Solutions:
- Adjust probability threshold: Increase the value (range 0.3-0.8) to reduce false positives in noisy images [55]
- Modify overlap threshold: For dense nuclear regions, decrease the overlap threshold (e.g., 0.2-0.4) to better separate touching nuclei [55]
- Pre-process images: Apply background subtraction and adjust brightness/contrast before segmentation using ImageJ's Process > Subtract Background and Image > Adjust > Brightness/Contrast tools [58] [59]
- Tile large images: For memory issues with large images, increase the number of tiles in StarDist's advanced options [55]
- Filter results: Remove small objects post-segmentation using size-based filtering (typically 500-10,000 pixels for nuclei) [57]
Sample Drying During Immunofluorescence
Problem: Embryonic samples dry out during staining procedures, compromising morphology and experimental results.
Solutions:
- Use chambered slides: Process samples in ibidi μ-Slides or similar chambers with coverslip bottoms to minimize air exposure [43]
- Optimize liquid volumes: For 8-well plates, use 130-200μL per well to maintain hydration without dilution [4]
- Reduce processing time: Streamline protocol to half-day instead of multi-day procedures [4]
- Humidity control: Perform antibody incubations in humidified chambers
- Rapid transitions: Have all solutions pre-prepared for quick changes between steps
Inconsistent Intensity Measurements
Problem: Fluorescence intensity measurements vary inconsistently between samples or experimental batches.
Solutions:
- Standardize imaging parameters: Use identical exposure times, laser powers, and gain settings across all samples
- Implement background subtraction: Subtract background fluorescence from empty regions for each channel [56]
- Use consistent fixation: Fix all samples for exactly the same duration (10 minutes for 4% formaldehyde) [43]
- Include controls: Process positive and negative control samples with each batch
- Normalize data: Express intensities relative to internal controls or reference standards
Performance Comparison of Nuclear Segmentation Tools
The table below summarizes key segmentation tools and their performance characteristics based on recent benchmarking studies:
| Segmentation Platform | Segmentation Method | Accuracy (F1-score) | Best Use Case | Tissue Type Performance |
|---|---|---|---|---|
| StarDist | Deep learning (star-convex polygons) | 0.67 F1-score at IoU 0.5 [54] | Limited computational resources, fast processing | Struggles with very dense nuclear regions [54] |
| Mesmer | Deep learning (centroid + boundary) | Highest overall accuracy (0.67 F1-score) [54] | Maximum accuracy requirements | Consistent across tissue types [54] |
| Cellpose | Deep learning (gradient flows) | Varies by tissue type [54] | Tonsil tissue with non-specific staining | Best for tonsil, less robust with high variance data [54] |
| QuPath | Classical image processing | Lower than deep learning methods [54] | When GUI preference overrides accuracy needs | Moderate across tissue types [54] |
| Fiji/CellProfiler | Classical thresholding + watershed | Limited accuracy [54] | Basic segmentation needs | Lower performance across tissue types [54] |
Experimental Protocols
Immunofluorescence Protocol for Embryonic Stem Cells with Drying Prevention
Timing: 4-6 hours
Materials:
- Pre-coated 8-well ibidi μ-Slides (gelatin or laminin coated)
- Freshly prepared 4% formaldehyde in PBS
- Permeabilization buffer (0.1% Triton X-100 in PBS)
- Blocking buffer (5% BSA or serum in PBS)
- Primary and secondary antibodies
- Mounting medium with DAPI
Procedure:
- Sample Preparation: Plate mouse embryonic stem cells at 30,000 cells per well in 8-well ibidi plates 48 hours before staining [4]
- Fixation: Aspirate media and add 130μL of 4% formaldehyde for 10 minutes at room temperature
- Washing: Wash 3× with PBS (5 minutes each) without letting wells dry between changes
- Permeabilization: Incubate with 0.1% Triton X-100 for 10 minutes
- Blocking: Block with 5% BSA for 30-60 minutes
- Primary Antibody: Incubate with primary antibody in blocking buffer for 1 hour
- Washing: Wash 3× with PBS (5 minutes each)
- Secondary Antibody: Incubate with fluorophore-conjugated secondary antibody for 1 hour in the dark
- Final Wash: Wash 3× with PBS (5 minutes each)
- Mounting: Add mounting medium with DAPI and store in dark at 4°C
Critical Steps for Drying Prevention:
- Always have next solution ready before removing current solution
- Never leave wells uncovered for more than 10-15 seconds
- Maintain consistent liquid volumes (130-200μL) across all wells
- Process samples in batches of ≤8 to minimize processing time
StarDist Nuclear Segmentation Workflow
Timing: 10-30 minutes depending on image size
Procedure:
- Install StarDist in Fiji via Help > Update > Manage Update Sites > Enable StarDist, CSBDeep, and TensorFlow [55]
- Open Image in Fiji (2D or 2D+time only)
- Pre-process Image:
- Split channels (Image > Color > Split Channels)
- Apply background subtraction if needed
- Adjust brightness/contrast for optimal visualization [58]
- Run StarDist:
- Plugins > StarDist > StarDist 2D
- Select appropriate model (Versatile for fluorescent nuclei)
- Adjust parameters:
- Probability Threshold: 0.4-0.7
- Overlap Threshold: 0.3-0.5
- Output to: Label Image or ROI Manager
- Post-processing:
- Apply size filter to remove small objects
- Validate segmentation against original image
- Export measurements (Analyze > Analyze Particles)
Research Reagent Solutions
The table below outlines essential materials for successful immunofluorescence and nuclear segmentation workflows:
| Reagent/Equipment | Function | Specific Application |
|---|---|---|
| ibidi μ-Slide 8-well | Prevents sample drying during processing | Maintains hydration for embryonic samples [4] [43] |
| 0.1% Gelatin Coating | Preserves 3D colony architecture | Mouse embryonic stem cell culture [4] |
| Laminin Coating | Promotes 2D cell growth | Easier imaging of cortical proteins [4] |
| 4% Formaldehyde | Protein cross-linking fixation | Standard fixation for most antigens [43] |
| Triton X-100 | Cell membrane permeabilization | Antibody access to intracellular targets [43] |
| BSA Blocking Solution | Reduces non-specific binding | Decreases background fluorescence [4] |
| DAPI Mounting Medium | Nuclear counterstain and preservation | Final mounting for imaging stability [43] |
| StarDist Pre-trained Models | Automated nuclear segmentation | Accurate nuclear identification without training [55] |
Frequently Asked Questions
Why is my embryo sample drying out during long staining procedures? Drying is often caused by insufficient humidity control during long incubation or washing steps. For whole-mount embryos, ensure the sample is fully submerged in buffer at all times. For sections, perform incubations in a humidified chamber (a sealed box containing damp paper towels) to prevent evaporation [60].
How can I tell if my immunofluorescence signal is genuine or an artifact from a dried sample? Drying artifacts often manifest as unnaturally high, speckled background fluorescence across the entire sample, which can obscure specific staining. A genuine signal should be localized to expected cellular or subcellular structures. Including a no-primary-antibody control is essential to identify this issue [60].
My negative control shows high background. Is this due to drying? Possibly. Inadequate blocking or washing can cause high background, and these issues can be exacerbated if the sample partially dries, concentrating reagents non-specifically. Ensure your blocking buffer is fresh and increase wash volumes and times, all while maintaining a humidified environment [60].
Which published dataset should I use for benchmarking my embryo imaging data? The choice depends on your organism and research focus. Reputable sources for published embryo data include institutional repositories and published benchmarking studies. Select a dataset generated using a similar model (e.g., mouse, zebrafish), developmental stage, and imaging technology (e.g., confocal, spatial transcriptomics) for a valid comparison [61].
What are the key metrics for benchmarking my nuclear segmentation results? The F1-score is a key metric that balances precision and recall, often evaluated at a standard Intersection over Union (IoU) threshold of 0.5. A higher F1-score indicates better accuracy in identifying and outlining nuclei. Benchmarking studies consistently show that pre-trained deep learning models like Mesmer and Cellpose outperform classical algorithms [54].
Troubleshooting Guide
This guide addresses common issues encountered during embryo immunofluorescence protocols, with a focus on preventing drying and achieving results robust enough for benchmarking.
| Problem | Possible Cause | Solution | Prevention Tip |
|---|---|---|---|
| High background fluorescence | Sample dried during incubation [60]; Inadequate blocking or washing. | Re-optimize protocol ensuring sample never dries; Increase blocking time; Use more thorough washing steps. | Always use a humidified chamber; Keep samples fully submerged in buffer. |
| Weak or absent specific signal | Over-fixation leading to epitope masking; Antibody cannot penetrate whole-mount tissue [60]. | For whole-mounts, try alternative fixatives like methanol; Increase permeabilization time and antibody incubation times. | For whole-mount IHC, validate antibody on cryosections first; Optimize fixation time [60]. |
| Inconsistent nuclear segmentation | Using a segmentation tool not optimized for your tissue density or type [54]. | Benchmark segmentation tools on a small part of your data. For dense tissues, use Mesmer; for speed, use StarDist; for challenging staining, try Cellpose [54]. | Consult benchmarking studies to select the most appropriate pre-trained model for your specific data characteristics [54]. |
| Poor agreement with published datasets | Technological differences in platform sensitivity or gene panels; Biological variability [61]. | When comparing spatial transcriptomics data, focus on trends of marker genes rather than absolute counts; Use datasets from the same technology platform if possible [61]. | Plan experiments with benchmarking in mind, and use the same analysis pipelines as the published study for a fair comparison. |
Experimental Protocols for Robust Results
Whole-Mount Immunofluorescence Staining with Anti-Drying Measures
This protocol is adapted for preventing embryo drying, based on established whole-mount IHC methods [60].
- Fixation: Immerse embryos in 4% Paraformaldehyde (PFA) at 4°C overnight. Ensure volume is at least 20x the tissue volume.
- Permeabilization and Blocking: Wash 3x with PBS. Incubate with permeabilization/blocking buffer (e.g., PBS with 0.5% Triton X-100 and 5% serum) for 24-48 hours at 4°C on a rocking platform. Critical Step: Perform all subsequent incubations in a humidified chamber.
- Primary Antibody Incubation: Incubate with primary antibody diluted in blocking buffer for 24-48 hours at 4°C.
- Washing: Wash 6x over 24 hours with PBS containing 0.1% Tween-20 (PBTw) to reduce background.
- Secondary Antibody Incubation: Incubate with fluorophore-conjugated secondary antibody and DAPI (for nuclei) in blocking buffer for 24 hours at 4°C, protected from light.
- Final Washing and Mounting: Wash 6x over 24 hours with PBTw. Clear and mount the embryo in an appropriate mounting medium (e.g., glycerol-based), ensuring the sample is fully covered by the medium and the coverslip is sealed to prevent drying.
Workflow for Benchmarking Nuclear Segmentation
Accurate nuclear segmentation is the foundation for many downstream analyses in embryo imaging. Follow this workflow to validate your results [54].
Protocol for Embedding and Sectioning Organoids to Prevent Drying
This protocol for handling delicate inner ear organoids exemplifies practices that prevent drying and morphological damage in 3D samples [62].
- Fixation: Fix organoids following your standard protocol (e.g., with 4% PFA).
- Agarose Embedding:
- Prepare a solution of low-melting-point agarose.
- Carefully transfer the fixed organoids into the agarose solution.
- Let the agarose solidify completely at room temperature or 4°C. This step is critical for preserving the 3D structure during sectioning.
- Vibratome Sectioning:
- Use a vibratome to section the agarose-embedded organoids into thick slices (e.g., 50-200 μm).
- Keep the sections submerged in PBS or a protective buffer at all times.
- Immunofluorescence:
- Perform immunostaining on the free-floating sections.
- Conduct all antibody incubations on a shaker to ensure even exposure and prevent settling.
- Maintain sections in buffer throughout the process.
Benchmarking Data for Segmentation Tools
When analyzing embryo images, selecting the right nuclear segmentation tool is critical. Errors at this stage propagate to all downstream analyses. The following table summarizes a quantitative benchmark of common tools across different tissue types, which can guide your selection [54].
| Segmentation Platform | Technology Type | Recommended Use Case | Key Strength | Reported F1-Score (Sample) |
|---|---|---|---|---|
| Mesmer [54] | Deep Learning | General purpose; Dense tissue (e.g., Skin) | Highest overall accuracy on composite dataset [54] | 0.67 (Composite Dataset) |
| Cellpose [54] | Deep Learning | Tonsil tissue; Non-specific staining | Robustness to challenging staining patterns [54] | Top Performer (Tonsil) |
| StarDist [54] | Deep Learning | Limited computational resources | ~12x faster runtime with CPU than Mesmer [54] | Good Performance (Skin) |
| QuPath [54] | Classical Algorithm | Free, GUI-based software | Better or similar to expensive licensed software [54] | Varies with manual optimization |
The Scientist's Toolkit
This table lists essential reagents and materials for executing the embryo immunofluorescence and benchmarking protocols detailed above.
| Item | Function/Benefit |
|---|---|
| 4% Paraformaldehyde (PFA) | A standard cross-linking fixative that preserves tissue architecture and antigenicity [60]. |
| Methanol | An alternative precipitating fixative used when PFA causes epitope masking, particularly in whole-mount staining [60]. |
| Humidified Chamber | A sealed container with moistened paper towels to prevent evaporation and sample drying during antibody incubations [60]. |
| Triton X-100 / Tween-20 | Detergents used to permeabilize cell membranes, allowing antibodies to access intracellular targets, and to reduce background in wash buffers [60]. |
| DAPI (6-diamidino-2-phenylindole) | A fluorescent dye that binds strongly to DNA, used to stain nuclei and visualize nuclear morphology and location [62] [60]. |
| Low-Melting-Point Agarose | Used for embedding delicate 3D samples like organoids to provide structural support during vibratome sectioning, preventing collapse and damage [62]. |
| Validated Primary Antibodies | Antibies that have been confirmed to work in IHC on cryosections (IHC-Fr) are strong candidates for success in whole-mount staining protocols [60]. |
| Pre-trained Segmentation Models (e.g., Mesmer) | Deep learning models that provide high-accuracy, generalizable nuclear segmentation without the need for extensive manual parameter tuning [54]. |
Leveraging Tools like CellProfiler for Automated Tracking and Population-Level Analysis
Technical Support Center: FAQs & Troubleshooting Guides
Installation and Startup
Q: CellProfiler fails to start on Windows 10/11, showing only a brief terminal flash. A: This is a known issue with some versions. If you encounter this with version 4.2.8, it is recommended to uninstall it and install the stable version 4.2.7 instead [63]. Antivirus software, particularly Sentinel One, can also block CellProfiler from launching; temporarily disabling the software for installation may resolve the issue [63].
Q: I encounter permission errors during installation on a Linux system.
A: This can often be fixed by using the sudo command to grant administrative privileges during installation (e.g., sudo pip2.7 install -e .). Ensure you are using the correct version of pip (pip2.7) for CellProfiler's Python version [64].
Image Processing and Analysis
Q: How can I mimic Fiji/ImageJ's 'Auto Brightness/Contrast' function within CellProfiler? A: You can use the RescaleIntensity module. This module performs a similar function by adjusting the image contrast, much like the 'Auto' button in ImageJ [65].
Q: What is the best way to correct for uneven illumination in my images? A: Use the CorrectIlluminationCalculate module to generate an illumination function from your images, and then apply it using the CorrectIlluminationApply module. This retrospective, multi-image method is more robust for quantitative profiling than single-image methods [66] [67].
Q: My images are in color, but I need grayscale for analysis. How do I convert them? A: The ColorToGray module is designed for this. You can either combine all color channels into one grayscale image or split each channel into a separate grayscale image for individual analysis [68] [67].
Object Classification and Visualization
Q: Can I assign specific colors to objects based on their measurements? A: While the ClassifyObjects module can classify objects, its color assignment can be random. A reliable workaround is to use the FilterObjects module to create sub-populations based on measurements. You can then use ConvertObjectsToImage and OverlayOutlines to visualize these populations with your chosen colors [69].
Q: The colors in my output images and plots are hard to distinguish. Can I change them? A: Yes. You can select a more suitable color palette in the CellProfiler Preferences dialog. This is particularly helpful for improving accessibility for colorblind users [70].
Tracking and Population Analysis
Q: What is a general workflow for tracking objects over time, such as nuclei in an embryo? A: A standard tracking workflow in CellProfiler involves several key steps [68]:
- Read Images & Metadata: Use the
StartingModulestool (which combinesImages,Metadata,NamesAndTypes, andGroups) to load your images and organize them into temporal sequences. - Preprocess Images: Convert images to grayscale using
ColorToGrayif necessary. - Segment Objects: Identify the primary objects (e.g., nuclei) in each frame using
IdentifyPrimaryObjects. - Track Objects: Use the
TrackObjectsmodule to link the segmented objects across frames.
The diagram below illustrates this workflow and the key settings for tracking dividing nuclei, based on a tutorial using a Drosophila embryo sample [68].
Q: After tracking, how can I analyze population-level heterogeneity instead of just using averages? A: To preserve information on population heterogeneity, you can use external tools like PopulationProfiler. This software imports per-cell measurements from CellProfiler, visualizes the distribution of measurements for each well as histograms, and allows for gating to define and quantify sub-populations. This is crucial for analyzing processes like cell cycle disruption [71].
Key Research Reagent Solutions
The following table details essential materials used in the featured experiment for tracking nuclei in a Drosophila embryo [68] [72].
| Item | Function in the Experiment |
|---|---|
| Drosophila Embryo | A biological model system for studying early development and cell division. |
| GFP-Histone | A fluorescent marker that labels chromatin, allowing visualization of nuclei and their behavior over time. |
| CellProfiler | Open-source software for automated image analysis, including segmentation and tracking of objects. |
| CellProfiler Analyst with Tracer | A data visualization tool for exploring and assessing the quality of cellular trajectories from time-lapse data [72]. |
Troubleshooting Common Experimental Issues
Q: My segmented objects are not tracking correctly across frames. A: This can often be due to poor segmentation. Use the quality assessment metrics in CellProfiler Tracer (such as "Singletons" for transient objects or "Crossings" for potential merge errors) to visually diagnose and assess tracking quality. This helps identify frames where segmentation may have failed [72].
Q: My intensity measurements seem inconsistent. What could be wrong?
A: Inhomogeneous illumination can corrupt intensity measurements by 10-30% [66]. Always perform illumination correction (see above) for quantitative intensity profiling. Also, ensure you are not using the ImageMath module in a way that rescales your intensity data outside the 0-1 range expected by some CellProfiler modules [67].
Conclusion
Preventing embryo drying is not a single step but a critical principle that underpins every stage of a successful immunofluorescence protocol, from handling to mounting. By integrating the foundational understanding of embryo vulnerability, a meticulous hydration-centric methodology, proactive troubleshooting, and rigorous validation, researchers can reliably preserve the structural and molecular integrity of pre-implantation embryos. Mastering these techniques is paramount for generating high-quality, reproducible data that can accelerate discoveries in early human development, improve assisted reproductive technologies, and inform the creation of more accurate in vitro models. Future directions will likely involve the development of even more refined culture-medium-based mounting media and automated handling systems to further minimize manual manipulation risks.
References
- 1. Timelapse-based 3D reconstruction of blastocysts reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12623974/]
- 2. A novel protocol to detect green fluorescent protein in unfixed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7474079/]
- 3. Maintenance of Fluorescence During Paraffin Embedding ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2019.00752/full]
- 4. A quick, cheap, and reliable protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9876944/]
- 5. Disaccharides Protect Antigens from Drying-Induced Damage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4810794/]
- 6. A review of best practices of rapid-cooling vitrification for ... [https://www.asrm.org/practice-guidance/practice-committee-documents/a-review-of-best-practices-of-rapid-cooling-vitrication-for-oocytes-and-embryos-a-committee-opinion-2021/]
- 7. Moisture changes inside hydrogel particles during their ... [https://pubs.rsc.org/en/content/articlehtml/2024/cp/d4cp02684e]
- 8. Immunocytochemistry protocol [https://www.abcam.com/en-us/technical-resources/protocols/icc-protocol]
- 9. Introduction to Sample Preparation: Immunofluorescence [https://microscopy.duke.edu/guides/intro-sample-prep-immunofluorescence]
- 10. Immunofluorescence Staining of Paraffin Sections Step by ... [https://www.frontiersin.org/journals/neuroanatomy/articles/10.3389/fnana.2020.582218/full]
- 11. Optimized protocol for cryopreservation of human eggs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3577428/]
- 12. Humidified atmosphere in a time-lapse embryo culture system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10310598/]
- 13. Cellular osmoregulation enhances porcine embryo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616923/]
- 14. Stage-dependent changes in culture medium osmolality ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1524749/full]
- 15. Protocol for immunofluorescence detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12156156/]
- 16. Oocyte and embryo culture under oil profoundly alters ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1337937/full]
- 17. Simplified Embryo and Oocyte Warming Protocols [https://fujifilmbiosciences.fujifilm.com/us/simplified-embryo-and-oocyte-warming-protocols]
- 18. ICC/IF Protocol [https://www.antibodies.com/applications/immunofluorescence/immunofluorescence-protocol]
- 19. Comprehensive Immunofluorescence Protocols: IF Protocol Hub [https://ifprotocol.org/]
- 20. 5 Controls for Immunofluorescence: An Easy Guide [https://bitesizebio.com/44370/controls-for-immunofluorescence-a-beginners-guide/]
- 21. Successful Immunofluorescence : Fixation & Permeabilization [https://blog.cellsignal.com/successful-immunofluorescence-fixation-and-permeabilization]
- 22. Protocol for immunostaining of non-adherent cells and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272898/]
- 23. Image-iT® Fixation Kit | Thermo Fisher Scientific - US Permeabilization [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/immunofluorescence-protocol/immunofluorescence-image-it-fixation-permeabilization-kit.html]
- 24. Comparison of Different Fix/Permeabilization Buffers for use in ... [https://blog.td2inc.com/comparison-of-different-fix-permeabilization-buffers]
- 25. A microfluidic platform for culturing and high-content ... [https://www.nature.com/articles/s41598-025-23883-2]
- 26. VECTASHIELD HardSet Antifade Mounting Medium With ... [https://vectorlabs.com/products/vectashield-hardset-with-dapi/?srsltid=AfmBOopZ3hQAN2EZ8DZbFo6FZBkGXH-FatWA-K_HFHMPLa4BCCPlGBlN]
- 27. VECTASHIELD Vibrance® Antifade Mounting Medium With ... [https://vectorlabs.com/products/vectashield-vibrance-antifade-dapi/?srsltid=AfmBOoo4gUNhKtgFm3LT3B4us5o9Q0wAUywLNoshVFpWCVLyzVj1OaP1]
- 28. VECTASHIELD Antifade Mounting Medium With DAPI [https://vectorlabs.com/products/vectashield-with-dapi/?srsltid=AfmBOoqzZBbTilFhBsZRsxL-CAMi5TKmTPsAZwncVCoYCxzLtDcpwCDy]
- 29. DAPI Counterstaining Protocols [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/dapi-protocol/basic-dapi-counterstaining-protocols.html]
- 30. Glass Capillaries - Quality Glass for Microinjection [https://www.wpiinc.com/glass-capillaries?srsltid=AfmBOoorymFT-KUeN1MjrTlGBhB3Bvx7Zz6vawHtmkwx9CAUF826eIVM]
- 31. Becoming a Patch Clamping Pipette Wizard [https://bitesizebio.com/49075/patch-clamping-pipette/]
- 32. Fire-Shaped Nozzles to Produce a Stress Peak for ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9321844/]
- 33. Science and Art of Pulling Micropipettes [https://www.wpiinc.com/blog/post/Science-and-Art-of-Pulling-Micropipettes?srsltid=AfmBOoo6UzrK9clviDZYa74ZJBuJwrjgo4AiMb4paxaGl3XsTJjphuM5]
- 34. Ultimate IHC Troubleshooting Guide: Fix Weak Staining & ... [https://www.atlasantibodies.com/knowledge-hub/blog/the-ultimate-ihc-troubleshooting-guide-from-weak-staining-to-high-background/]
- 35. ICC/IF Troubleshooting [https://www.antibodies.com/applications/immunofluorescence/immunofluorescence-troubleshooting]
- 36. Troubleshooting - Immunofluorescence Assays [https://ibidi.com/content/366--troubleshooting]
- 37. Immunofluorescence (IF) Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/if-troubleshooting-guide?srsltid=AfmBOorPcje3nu1qr3O3fdlgTqLYagSt-kFAnIyidz-LjZW6wE1208zr]
- 38. Immunofluorescence Troubleshooting | Tips & Tricks [https://www.stressmarq.com/support/technical-support/troubleshooting/immunofluorescence-troubleshooting/?srsltid=AfmBOopCHLyEDwP0MjWn-h14BieRdwkCIKUI3J5vUaRjHMvD5xSrWJIL]
- 39. Background in Fluorescence Imaging [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/molecular-probes-school-of-fluorescence/imaging-basics/protocols-troubleshooting/troubleshooting/background-fluorescence.html]
- 40. Maintaining Viable Cell Count in Primary T Cells During ... [https://invitria.com/resources/optibumin-tcell-washing/]
- 41. Impact of buffer composition on biochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9764254/]
- 42. Frozen Tissue Preparation & IHC Cryosection Staining ... [https://www.rndsystems.com/resources/protocols/protocol-preparation-and-fluorescent-ihc-staining-frozen-tissue-sections]
- 43. Immunofluorescence Staining | A Typical Workflow [https://ibidi.com/content/365-immunofluorescence-staining-a-typical-workflow]
- 44. Immunofluorescence Staining of Bacterial Samples [https://www.protocols.io/view/immunofluorescence-staining-of-bacterial-samples-frrbbm52p.html]
- 45. 9 Tips to optimize your IF experiments [https://www.ptglab.com/news/blog/9-tips-to-optimize-your-if-experiments/?srsltid=AfmBOorK1rvTBhGYWwgu-NnZ-2fJAhkV-6zmAr7G2TmygUCBCJKtV387]
- 46. Comprehensive Guide to Immunofluorescence (IF) Protocols [https://www.clyte.tech/post/comprehensive-guide-to-immunofluorescence-if-protocols?srsltid=AfmBOoqPxxHKPJZaO75wmAl-0DXuXjGQCoidRSesoWfzJIknuKp48Hz3]
- 47. Cell-Surface Proteomics Identifies Lineage-Specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3405530/]
- 48. Cross-regulation of the Nanog and Cdx2 promoters [https://www.nature.com/articles/cr200979]
- 49. Immunofluorescence (IF) Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/if-troubleshooting-guide?srsltid=AfmBOoo4kuabD4GdUqq_lytcSgHbnwpvBNVlf8WbBqrr0JDopBf3tFGm]
- 50. Immunofluorescence Troubleshooting | Tips & Tricks [https://www.stressmarq.com/support/technical-support/troubleshooting/immunofluorescence-troubleshooting/?srsltid=AfmBOootX8ySjAOySKXjObijOXCsDTiO2b3SqPL6osgUgLxG5xOvJLuy]
- 51. Designing a multicolor flow cytometry protocol [https://www.abcam.com/en-us/technical-resources/guides/flow-cytometry-guide/designing-a-multicolor-protocol]
- 52. Immunofluorescence Troubleshooting [https://stjohnslabs.com/blog/immunofluorescence-troubleshooting?srsltid=AfmBOop9zTyJDa5iW2TvKkvJVthoEplK-34e8u123OIUQR72drLglsS9]
- 53. An Immunofluorescence Method to Analyze the Proliferation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3425951/]
- 54. Quantitative benchmarking of nuclear segmentation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12125308/]
- 55. StarDist [https://imagej.net/plugins/stardist]
- 56. Multi-Channel Nuclear Analysis: An ImageJ/FIJI Plugin for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12181773/]
- 57. Cellpose/StarDist Segmentation — PyImageJ documentation [https://py.imagej.net/en/latest/Cellpose-StarDist-Segmentation.html]
- 58. Brightness and Contrast [https://imagej.net/learn/brightness-and-contrast]
- 59. Display and Contrast Enhancement [https://imagej.net/nih-image/more-docs/Tutorial/Enhancement.html]
- 60. IHC staining protocol for whole mount samples [https://www.abcam.com/en-us/technical-resources/protocols/wholemount-staining-for-ihc]
- 61. Systematic benchmarking of high-throughput subcellular ... [https://www.nature.com/articles/s41467-025-64292-3]
- 62. Protocol for vibratome sectioning, immunofluorescence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392772/]
- 63. Cellprofiler 4.2.8 does not start on Windows 10-11 [https://forum.image.sc/t/cellprofiler-4-2-8-does-not-start-on-windows-10-11/102073]
- 64. Troubleshooting CellProfiler Installation - GitHub Pages [https://drk-lo.github.io/lotterhoslabprotocols/code_TroubleshootingCellprofiler.html]
- 65. CellProfiler Adjust Brightness/Contrast to mimic FIJI ImageJ [https://forum.image.sc/t/cellprofiler-adjust-brightness-contrast-to-mimic-fiji-imagej/22875]
- 66. Data-analysis strategies for image-based cell profiling [https://www.nature.com/articles/nmeth.4397]
- 67. Image Processing — CellProfiler 4.0.7 documentation [https://cellprofiler-manual.s3.amazonaws.com/CellProfiler-4.0.7/modules/imageprocessing.html]
- 68. Imaging / Object tracking using CellProfiler / Hands-on [https://training.galaxyproject.org/training-material/topics/imaging/tutorials/object-tracking-using-cell-profiler/tutorial.html]
- 69. How to assign specific colors to Objects based on ... [https://forum.image.sc/t/how-to-assign-specific-colors-to-objects-based-on-meanintensity-measurements/24797]
- 70. Set and modify color palette for output of ClassifyObjects ... [https://forum.image.sc/t/set-and-modify-color-palette-for-output-of-classifyobjects-module-colorblind/55228]
- 71. PopulationProfiler: A Tool for Population Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4795740/]
- 72. Tracer [https://cellprofiler.org/tracer]