A Comprehensive Guide to Digoxigenin-Labeled RNA Probes: Protocol, Optimization, and Applications
This article provides a complete resource for researchers and drug development professionals utilizing digoxigenin (DIG)-labeled RNA probes.
A Comprehensive Guide to Digoxigenin-Labeled RNA Probes: Protocol, Optimization, and Applications
Abstract
This article provides a complete resource for researchers and drug development professionals utilizing digoxigenin (DIG)-labeled RNA probes. It covers foundational principles of the non-radioactive DIG system and its advantages over other labeling techniques. A detailed, step-by-step protocol for in vitro transcription and probe generation is presented, alongside specialized applications in techniques like in situ hybridization and EMSA. Critical troubleshooting guidance for common issues such as low yield and high background is included, along with methods for validating probe sensitivity and specificity. The content synthesizes current best practices to enable robust, reproducible results in nucleic acid detection.
Understanding Digoxigenin-Labeled RNA Probes: Principles and Advantages
The DIG (Digoxigenin) System represents a cornerstone non-radioactive technology for the sensitive and specific detection of nucleic acids in molecular biology, histology, and diagnostic applications. As a safer and more versatile alternative to radioactive isotopes, the system utilizes digoxigenin, a plant-derived steroid molecule, to label DNA, RNA, or oligonucleotide probes. This guide provides an in-depth technical overview of the DIG system, detailing its fundamental principles, key advantages, experimental protocols, and essential reagents, framed within the context of advancing digoxigenin-labeled RNA probe research.
Principles of the DIG System
The core principle of the DIG system involves the covalent attachment of digoxigenin, a hapten isolated from Digitalis plants, into nucleic acid probes [1]. This labeled probe hybridizes with its complementary target sequence (DNA or RNA) in a sample. Detection is achieved through an enzyme-conjugated antibody specific for the digoxigenin molecule, followed by a colorimetric, fluorescent, or chemiluminescent substrate reaction [1].
- High Specificity: The anti-DIG antibody exhibits exceptional specificity for the digoxigenin molecule because digoxigenin does not naturally occur in biological tissues, resulting in minimal non-specific background binding [1].
- Versatility: DIG-labeled probes are suitable for multiple applications, including Northern and Southern blotting, in situ hybridization (ISH), and microarray analysis [2] [1].
- Safety and Convenience: The system eliminates the hazards, regulatory hurdles, and short half-lives associated with radioactive isotopes like 32P, allowing for longer probe storage and safer laboratory handling [2] [1].
Advantages Over Other Labeling Methods
The following table summarizes the key characteristics of the DIG system in the context of the broader non-radioactive nucleic acid labeling product market.
Table 1: Comparison of Key Non-Radioactive Nucleic Acid Labeling Technologies
| Characteristic | DIG System | Biotin-Based | Fluorescent |
|---|---|---|---|
| Label Molecule | Digoxigenin (plant steroid) | Biotin (Vitamin) | Fluorescent Dyes (e.g., Cy3, FITC) |
| Detection Principle | Anti-DIG Antibody | Streptavidin/Avidin | Direct Fluorescence |
| Sensitivity | High [3] | High | Variable (technology-dependent) |
| Specificity | Very High (low background) [1] | High (endogenous biotin can cause background) | High |
| Primary Applications | Filter hybridization, ISH, Northern/Southern blotting [1] | Various blotting and detection techniques | Real-time PCR, microscopy, microarrays [2] |
| Key Market Players | Roche (via Merck Millipore) [1] | Various suppliers | Thermo Fisher, Promega [2] |
The global market for non-radioactive nucleic acid labeling products, valued at an estimated $550.6 million, is driven by increasing demand for molecular diagnostics and personalized medicine [2]. The DIG system holds a significant position within this market, distinguished by its proven track record, with thousands of publications attesting to its performance and reliability [1].
Detailed Experimental Workflow
A standard workflow for using a DIG-labeled RNA probe, for example in in situ hybridization, involves several critical stages from probe preparation to final detection. The diagram below outlines this comprehensive process.
Diagram 1: DIG-Labeled RNA Probe Workflow
Critical Protocol Steps and Methodologies
A. Probe Selection and Design For optimal results in ISH, RNA probes should be 250–1,500 bases in length, with probes of approximately 800 bases exhibiting the highest sensitivity and specificity [4]. The probe must be complementary to the target mRNA (an "antisense" probe) to ensure specific hybridization. A "sense" strand probe should always be synthesized and used in parallel as a negative control [4].
B. Tissue Preparation and Pre-Treatment Proper sample fixation and storage are critical for preserving nucleic acid integrity and preventing RNA degradation by RNases [4].
- Deparaffinization and Rehydration: For formalin-fixed paraffin-embedded (FFPE) tissues, slides must be treated with xylene and a graded ethanol series to remove paraffin completely [4].
- Antigen Retrieval: Treatment with Proteinase K (e.g., 20 µg/mL for 10-20 minutes at 37°C) is essential to permeabilize the tissue, making the target nucleic acid accessible to the probe. The concentration and incubation time must be optimized; insufficient digestion reduces signal, while over-digestion damages tissue morphology [4].
C. Hybridization and Stringency Washes The probe is diluted in a hybridization buffer containing formamide (which lowers the required hybridization temperature) and denatured before application [4].
- Hybridization Temperature: Typically occurs between 55°C and 65°C overnight [4].
- Stringency Washes: Post-hybridization, slides are washed with solutions like 50% formamide in 2x SSC to remove excess and non-specifically bound probe. The temperature and salt concentration (e.g., 0.1-2x SSC) of these washes are critical for removing background without dissatching specific hybrids [4].
D. Immunological Detection After hybridization and washing, the DIG label is detected.
- Blocking: Tissues are incubated with a blocking buffer (e.g., containing 2% BSA, milk, or serum) to prevent non-specific antibody binding [4].
- Antibody Incubation: An anti-DIG antibody conjugated to an enzyme (Alkaline Phosphatase or Horseradish Peroxidase) is applied [1].
- Substrate Addition: A chromogenic or chemiluminescent substrate is added. For colorimetric detection, the enzyme catalyzes a reaction that produces a colored precipitate at the site of the probe [1].
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of the DIG system requires a set of core reagents. The following table catalogs the essential components for a typical experiment.
Table 2: Essential Research Reagent Solutions for DIG Labeling and Detection
| Reagent/Material | Function/Description | Key Considerations |
|---|---|---|
| Template DNA | A linearized plasmid or PCR product containing the target sequence and an RNA polymerase promoter. | Must be linearized with a restriction enzyme that creates a 5'-overhang for efficient in vitro transcription [3]. |
| RNA Polymerase (SP6, T7, T3) | Drives the in vitro transcription reaction to synthesize the RNA probe. | Choice of polymerase depends on the promoter sequence in the template. |
| DIG RNA Labeling Mix | Contains nucleotide precursors (e.g., DIG-UTP) for incorporation into the nascent RNA probe. | A standardized mix ensures consistent and efficient labeling [1]. |
| Anti-DIG-AP Antibody | Polyclonal antibody conjugated to Alkaline Phosphatase (AP) that binds specifically to the DIG hapten. | This is the primary detection reagent. Must be diluted in blocking buffer prior to use [4]. |
| Hybridization Buffer | A solution containing formamide, salts, and blocking agents to facilitate specific probe binding. | The high formamide concentration allows hybridization to occur at a lower, less destructive temperature [4]. |
| Wash Buffers (SSC, MABT) | Used for post-hybridization stringency washes to remove unbound probe. | MABT (Maleic Acid Buffer with Tween) is gentler than PBS for nucleic acid detection steps [4]. |
| Blocking Reagent | (e.g., BSA, skim milk, or serum) Prevents non-specific binding of the antibody to the tissue sample. | Critical for achieving a low background signal [4]. |
| Detection Substrate (NBT/BCIP or CDP-Star) | For colorimetric (NBT/BCIP) or chemiluminescent (CDP-Star) detection. The enzyme catalyzes a color change or light emission. | Chemiluminescent substrates offer higher sensitivity for low-abundance targets [1]. |
The DIG system remains a robust, sensitive, and safe standard for non-radioactive nucleic acid detection. Its high specificity, proven reliability, and adaptability to various detection modalities make it an indispensable tool for researchers in genomics, disease research, and drug development. As the field of molecular biology continues to advance, with a growing emphasis on safety and high-throughput applications, the principles and protocols of the DIG system provide a solid foundation for current and future research utilizing digoxigenin-labeled probes.
In molecular biology and diagnostic research, the need for precise, safe, and robust nucleic acid detection methods is paramount. For decades, radioisotopic labeling was the gold standard for techniques like Northern blotting and in situ hybridization due to its high sensitivity. However, the significant safety hazards, regulatory burdens, and instability of radioactive probes have driven the scientific community to seek superior alternatives. Among these, digoxigenin (DIG)-labeled RNA probes have emerged as a leading technology, combining the critical advantages of high sensitivity and specificity with an excellent safety profile. This whitepaper details the technical foundations of DIG-based labeling, provides a quantitative comparison with traditional methods, and outlines detailed protocols that empower researchers to leverage this powerful technology within a modern drug development and research framework.
Digoxigenin is a steroid hapten derived from plants of the Digitalis species [5]. Its fundamental application in molecular biology involves conjugating digoxigenin to nucleotide triphosphates (e.g., DIG-11-UTP for RNA probes), which are then enzymatically incorporated into nucleic acid probes [5]. Post-hybridization, these probes are detected via an enzyme-linked immunoassay using an antibody conjugate (e.g., anti-DIG-alkaline phosphatase) and subsequent colorimetric or chemiluminescent substrate incubation [5]. This core mechanism provides a versatile and powerful platform for sensitive nucleic acid detection.
Core Advantages: A Technical Comparison
The transition to DIG-labeled probes from radioactive methods is supported by direct, measurable benefits across key performance and operational categories.
Superior Safety and Operational Stability
The most immediate advantage of DIG labeling is the complete elimination of radiation hazards.
- Enhanced Safety Profile: DIG-labeled probes pose no radiation exposure risks, require no specialized shielding, and circumvent the complex disposal procedures and costs associated with radioisotopes like (^{32}\text{P}) [6]. This makes them suitable for educational settings and institutions with restricted radionuclide access.
- Superior Probe Stability: Radioactively labeled probes decay and have short useful lifespans. In contrast, DIG-labeled probes are highly stable and can be stored at –20°C for at least one year without loss of activity, enabling reproducible results over time and reducing reagent waste [5].
High Sensitivity and Specificity
DIG-based detection is not merely a safer alternative; it delivers performance that meets or exceeds radioactive standards.
- Exceptional Sensitivity: Chemiluminescent detection with DIG-labeled probes can achieve femtomole-level sensitivity, capable of detecting small RNAs in Northern blots with exposure times as short as one minute [6]. This level of sensitivity is fully comparable to that achieved with radioactive probes.
- High Specificity with Low Background: The digoxigenin hapten is not endogenous to animal cells, which minimizes non-specific background interference [5]. The high specificity of the anti-DIG antibody conjugate further ensures that the signal is derived exclusively from the hybridized probe, resulting in a high signal-to-noise ratio critical for clear and reliable data interpretation.
Table 1: Quantitative Comparison of DIG-Labeled vs. Radioactive RNA Probes
| Feature | DIG-Labeled RNA Probes | Radioactive Probes (e.g., ³²P) |
|---|---|---|
| Sensitivity | Femtomole-level [6] | Femtomole-level |
| Probe Stability | >1 year at -20°C [5] | Short (depends on isotope half-life) |
| Safety & Handling | No special radiation precautions | Requires shielding, monitoring, and regulated disposal |
| Detection Time | ~1 minute (chemiluminescence) [6] | Hours to days (autoradiography) |
| Spatial Resolution | High, ideal for ISH [7] [8] | Lower, due to radiation scatter |
| Cost & Regulation | Lower long-term cost; minimal regulation | High cost for disposal and regulatory compliance |
Detailed Experimental Protocol for Northern Blotting
The following optimized protocol for nonradioactive Northern analysis using DIG-labeled DNA probes demonstrates the practical application of this technology in a genome-wide screening context [6].
Probe Synthesis and Hybridization
This protocol utilizes a DIG-labeled DNA probe synthesized via random priming.
- Probe Labeling: Synthesize a DIG-labeled DNA probe using a DIG High Prime DNA Labeling and Detection Starter Kit or equivalent. The random priming method incorporates DIG-11-dUTP into the probe. Purify the labeled probe to remove unincorporated nucleotides.
- Membrane Transfer and Fixation: Following standard gel electrophoresis, transfer the RNA onto a positively charged nylon membrane via capillary or electroblotting. Fix the RNA to the membrane by UV crosslinking.
- Pre-hybridization and Hybridization:
- Pre-hybridize the membrane in a suitable volume of DIG Easy Hyb buffer at the determined hybridization temperature (e.g., 42°C) for 30 minutes.
- Denature the DIG-labeled DNA probe by boiling for 5 minutes, then immediately chill on ice.
- Dilute the denatured probe in fresh DIG Easy Hyb buffer and incubate with the membrane for at least 6 hours (or overnight).
Post-Hybridization Washes and Chemiluminescent Detection
- Stringency Washes:
- Wash the membrane twice with 2X SSC, 0.1% SDS at room temperature for 5 minutes per wash.
- Perform two additional stringency washes with 0.5X SSC, 0.1% SDS at a higher temperature (e.g., 68°C) for 15 minutes per wash.
- Immunological Detection:
- Blocking: Briefly rinse the membrane in Wash Buffer. Incubate the membrane in Blocking Solution for 30 minutes.
- Antibody Incubation: Dilute anti-DIG-alkaline phosphatase (AP) conjugate in Blocking Solution. Incubate the membrane in this solution for 30 minutes.
- Washing: Remove unbound antibody by washing the membrane twice with Wash Buffer for 15 minutes per wash.
- Equilibration: Equilibrate the membrane in Detection Buffer for 2-5 minutes.
- Signal Development:
- Place the membrane in a plastic sleeve or hybridization bag.
- Apply a chemiluminescent AP substrate (e.g., CDP-Star or CSPD) directly onto the membrane, ensuring even coverage.
- Incubate for 5 minutes at room temperature, then drain excess liquid.
- Seal the bag and expose the membrane to a digital imager or X-ray film. A positive signal can often be visualized in under one minute [6].
The workflow for this protocol is systematized in the diagram below.
Advanced Applications: mRNA Fluorescence In Situ Hybridization (FISH)
The versatility of DIG labeling is powerfully demonstrated in advanced spatial transcriptomics and multiplexed assays. The following protocol combines mRNA FISH with immunohistochemistry (IHC) for co-detection of RNA and protein in the same tissue section [7].
Tissue Preparation and mRNA FISH
- Tissue Sectioning: Use fresh-frozen or formalin-fixed paraffin-embedded (FFPE) tissue sections (5–7 μm) mounted on slides. For FFPE tissues, perform deparaffinization and rehydration through a graded series of xylenes and ethanol.
- Protease Pretreatment: To expose target mRNA, treat slides with a mild protease solution. Optimization is critical, as over-digestion damages tissue morphology and under-digestion reduces probe accessibility.
- Hybridization with Multiplex Probes: Apply a multiplex probe set, which may include DIG-labeled probes targeting specific mRNAs (e.g., complement components C4b or C1qa) alongside other fluorescently labeled probes for cell-type markers (e.g., Slc1a3 for astrocytes, Tyrobp for microglia) [7]. Co-hybridize in a humidified chamber overnight.
- Signal Amplification and Development: After post-hybridization washes, develop the FISH signal using a tyramide signal amplification (TSA) system. Incubate slides with an anti-DIG antibody conjugated to horseradish peroxidase (HRP), followed by incubation with a fluorophore-labeled tyramide substrate (e.g., Cyanine 5). The HRP enzyme catalyzes the deposition of the fluorescent tyramide, resulting in a highly amplified, localized signal at the site of probe hybridization [7].
Concurrent Immunohistochemistry for Protein Detection
- Blocking and Antibody Incubation: Following the FISH procedure, block the tissue sections with a protein block to minimize non-specific antibody binding. Incubate with a primary antibody against the protein of interest (e.g., anti-β-amyloid for plaques).
- Signal Detection: Detect the primary antibody using an HRP-conjugated secondary antibody and a tyramide substrate conjugated to a fluorophore with a distinct emission spectrum from the FISH channels. This sequential TSA-based detection allows for robust multiplexing without antibody cross-reactivity.
- Imaging and Analysis: After counterstaining with DAPI, apply an antifade mounting medium. Acquire high-resolution images using a fluorescence microscope or a slide scanner. Analyze the spatial relationships between mRNA expression, protein accumulation, and cellular organization.
The integrated workflow for this multiplexed assay is illustrated below.
The Scientist's Toolkit: Essential Research Reagents
Success with DIG-based methodologies relies on a core set of specialized reagents. The following table details these essential components.
Table 2: Key Reagents for DIG-Based Nucleic Acid Detection
| Reagent / Kit | Function / Description | Example Use Case |
|---|---|---|
| DIG-11-UTP | Digoxigenin-labeled UTP for synthesizing RNA probes via in vitro transcription [5]. | Generating high-sensitivity riboprobes for in situ hybridization. |
| DIG Oligonucleotide 3'-End Labeling Kit | Template-independent enzymatic addition of a single DIG-ddUTP to the 3'-end of oligonucleotides using Terminal Transferase (TdT) [9]. | Creating labeled probes for ISH or EMSA with minimal steric hindrance. |
| Anti-Digoxigenin-AP | Alkaline phosphatase-conjugated antibody for chemiluminescent or colorimetric detection [5]. | Standard detection for Northern, Southern, and Western blots. |
| Anti-Digoxigenin-HRP | Horseradish peroxidase-conjugated antibody for use with tyramide signal amplification (TSA) [7]. | High-sensitivity, amplified detection in multiplexed FISH assays. |
| CDP-Star / CSPD | Chemiluminescent substrates for Alkaline Phosphatase. Emit light upon dephosphorylation [6]. | Sensitive detection in blotting applications. |
| Tyramide Signal Amplification (TSA) Kits | Fluorophore-labeled tyramide substrates that are activated by HRP to deposit a localized, amplified signal [7]. | Enabling highly sensitive multiplex RNA/protein co-detection. |
| DIG Easy Hyb Buffer | A standardized, optimized hybridization solution for use with DIG-labeled probes. | Streamlining Northern and Southern blot procedures. |
Digoxigenin-labeled RNA and DNA probes represent a mature, powerful, and indispensable technology in the modern research and drug development arsenal. They successfully address the critical limitations of radioactive methods by offering an unmatched safety profile, superior reagent stability, and operational simplicity, without compromising on the high sensitivity and specificity required for cutting-edge science. As demonstrated in both foundational techniques like Northern blotting and advanced multiplexed spatial genomics, the flexibility and performance of the DIG system make it a cornerstone for reliable nucleic acid detection. Its continued evolution and integration with signal amplification technologies ensure that it will remain a vital tool for researchers and scientists dedicated to precision medicine and molecular discovery.
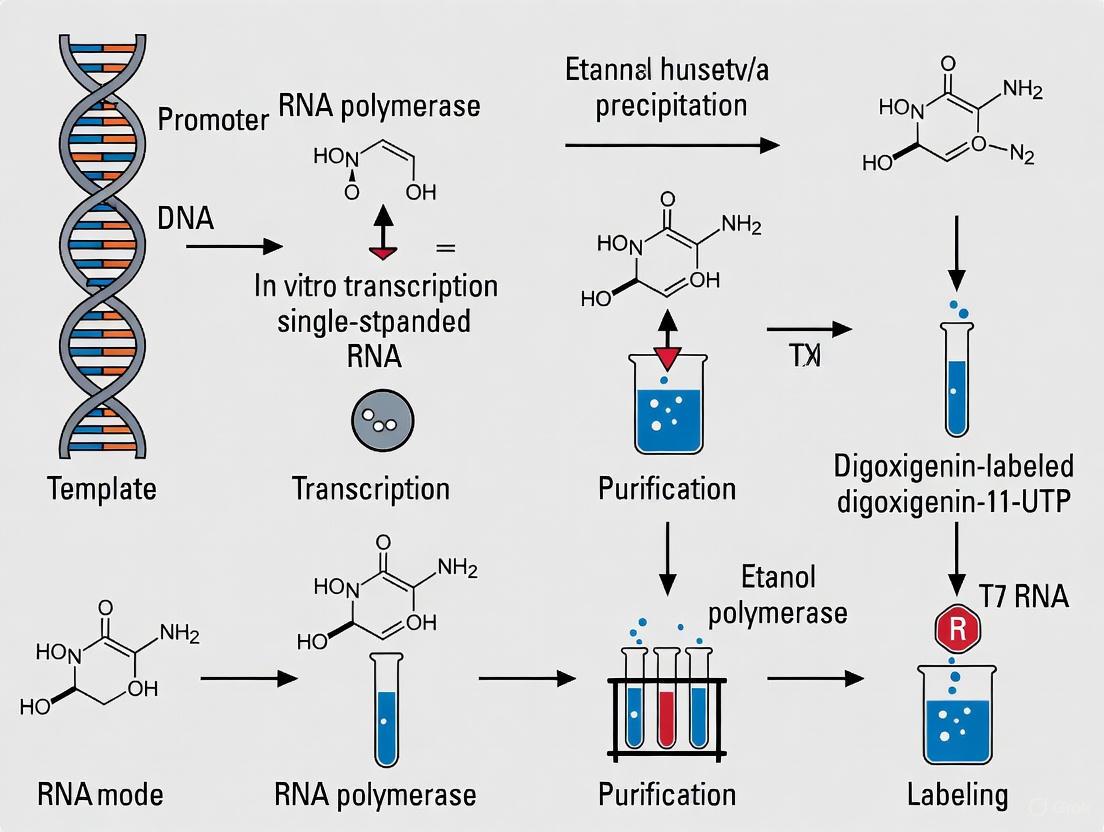
The synthesis of digoxigenin (DIG)-labeled RNA probes represents a cornerstone technology in molecular biology, enabling the sensitive detection of specific nucleic acid sequences in techniques such as in situ hybridization, northern blotting, and microarray analysis. The core principle underlying this methodology involves the enzymatic incorporation of DIG-11-UTP—a uridine triphosphate molecule conjugated to digoxigenin at the 11-position—into RNA transcripts during in vitro transcription. This modification creates stable, highly specific hybridization probes that can be detected immunohistochemically with anti-digoxigenin antibodies conjugated to reporter enzymes such as alkaline phosphatase or horseradish peroxidase. The efficiency of this labeling process is critically dependent on the ability of RNA polymerases to recognize and incorporate the modified nucleotide into growing RNA chains while maintaining transcriptional fidelity and yield. This technical guide examines the fundamental reaction principles governing DIG-11-UTP incorporation by RNA polymerases, with particular emphasis on T7, T3, and SP6 RNA polymerases commonly employed for in vitro transcriptions.
Chemical and Enzymatic Fundamentals
The DIG-11-UTP Molecule: Structure and Properties
DIG-11-UTP consists of a standard uridine triphosphate molecule covalently linked to a digoxigenin hapten via an 11-atom spacer arm attached to the C5 position of the pyrimidine ring. This structural configuration is crucial for its biological function. The digoxigenin moiety is a steroid derivative isolated from Digitalis plants, while the 11-atom spacer provides sufficient distance between the nucleotide base and the hapten to minimize steric interference with polymerase recognition and incorporation. Unlike natural nucleotides, DIG-11-UTP contains a bulky hydrophobic group that can potentially affect enzyme kinetics and incorporation efficiency. The molecule is typically supplied as a lithium salt in aqueous solution and is stable at -20°C for extended periods when protected from light and repeated freeze-thaw cycles.
RNA Polymerase Structure and Function
Bacteriophage-encoded RNA polymerases (T7, T3, and SP6) are single-subunit enzymes that exhibit high promoter specificity and processivity, making them ideal for in vitro transcription applications. These enzymes share a structurally conserved core composed of thumb, palm, and fingers subdomains that form the active site for template-directed RNA synthesis [10]. The palm domain contains the catalytic center responsible for nucleotidyl transfer, while the fingers domain participates in nucleotide recognition and binding. Unlike multi-subunit cellular RNA polymerases, these phage enzymes require no additional protein factors for promoter recognition or transcription initiation, simplifying their use in diagnostic and biotechnology applications. The N-terminal domain of T7 RNA polymerase, for instance, mediates promoter recognition and melting, while accessory modules provide RNA binding and displacement functions [10].
Table 1: Properties of Common Bacteriophage RNA Polymerases Used for DIG-Labeled Probe Synthesis
| Polymerase | Molecular Weight (kDa) | Promoter Specificity | Transcription Rate (nt/sec) | Processivity |
|---|---|---|---|---|
| T7 | 100 | TAATACGACTCACTATAGGGAGA | 230 (at 37°C) | High |
| T3 | 100 | ATTAACCCTCACTAAAGGGAGA | ~200 (at 37°C) | High |
| SP6 | 100 | ATTTAGGTGACACTATAGAAGTG | ~200 (at 37°C) | High |
Mechanism of DIG-11-UTP Incorporation
Template Recognition and Transcription Initiation
The incorporation of DIG-11-UTP begins with promoter recognition and transcription initiation. Bacteriophage RNA polymerases recognize specific promoter sequences of approximately 23 base pairs, with highly conserved regions from -7 to +1 relative to the transcription start site [10]. Upon promoter binding, the enzyme undergoes conformational changes that unwind approximately 8 base pairs of DNA to form the transcription bubble, positioning the template strand in the active site channel. Transcription initiation commences with the formation of the first phosphodiester bond between the initiating nucleotide (typically a purine) and the next complementary nucleotide. During this initiation phase, the enzyme remains promoter-bound and undergoes several abortive cycles before transitioning to the elongation phase.
Nucleotide Recognition and Incorporation
During elongation, the polymerase progresses along the template DNA, recruiting complementary nucleotides to the active site for incorporation into the growing RNA chain. DIG-11-UTP competes with natural UTP for incorporation opposite adenine residues in the template. The enzyme's nucleotide binding pocket accommodates the modified nucleotide through conformational flexibility, though the bulky digoxigenin moiety can affect binding kinetics and incorporation efficiency. Experimental evidence indicates that bacteriophage RNA polymerases can successfully incorporate DIG-11-UTP despite its steric bulk, though at reduced rates compared to unmodified UTP. The incorporation follows the standard mechanism of nucleotidyl transfer, with the 3'-hydroxyl of the growing RNA chain attacking the α-phosphate of the incoming DIG-11-UTP, releasing pyrophosphate and extending the chain by one nucleotide.
Effect on Transcription Elongation and Fidelity
The incorporation of DIG-11-UTP can influence transcription elongation dynamics. The bulky digoxigenin tag may cause transient pausing or reduced elongation rates, particularly when multiple incorporated modifications occur in close proximity. However, the 11-atom spacer arm provides sufficient flexibility to minimize severe steric clashes with polymerase domains. Processivity—the number of nucleotides incorporated per binding event—may be moderately reduced compared to transcription with only natural nucleotides. Despite these potential limitations, the fidelity of base pairing is generally maintained, as the hydrogen-bonding face of uracil remains unmodified and available for specific recognition of adenine residues in the template.
Experimental Protocols and Optimization
Standard In Vitro Transcription with DIG-11-UTP
The following protocol, adapted from established methodologies, details the optimized procedure for synthesizing DIG-labeled RNA probes [11]:
Template Preparation: Linearize 10-20 µg of plasmid DNA containing the gene of interest downstream of a bacteriophage promoter (T7, T3, or SP6) with an appropriate restriction enzyme. Purify the linearized template by phenol/chloroform extraction and ethanol precipitation. Resuspend the DNA in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) at a concentration of approximately 1 µg/µL.
Transcription Reaction Setup: Assemble the reaction at room temperature in the following order:
- 2.5 µL linearized template DNA (1 µg/µL)
- 4.0 µL 5x transcription buffer (supplied with polymerase)
- 6.0 µL 100 mM DTT
- 2.0 µL 10x DIG RNA labeling mix (10 mM ATP, 10 mM CTP, 10 mM GTP, 6.5 mM UTP, 3.5 mM DIG-11-UTP)
- 1.0 µL RNasin (RNase inhibitor, 40 U/µL)
- 20 units of appropriate RNA polymerase (T7, T3, or SP6)
- Nuclease-free water to a final volume of 20 µL
Incubation: Incubate the reaction at 37°C for 2 hours.
Quality Assessment: Analyze 1 µL of the reaction product by agarose gel electrophoresis to verify RNA synthesis. A discrete band of expected size should be visible, though some shorter abortive transcripts may also be present.
Probe Storage: Dilute the labeled probe to 100 µL with 10 mM DTT and store in aliquots at -70°C. Optimal working dilutions for hybridization typically range from 1:500 to 1:2000.
Reaction Optimization Strategies
Several parameters can be optimized to maximize DIG-11-UTP incorporation and probe yield:
Table 2: Optimization Parameters for DIG-Labeled RNA Probe Synthesis
| Parameter | Standard Condition | Optimization Approach | Effect on Yield |
|---|---|---|---|
| DIG-11-UTP:UTP Ratio | 35:65 (3.5 mM DIG-11-UTP:6.5 mM UTP) | Increase to 50:50 for higher labeling density; decrease to 25:75 for longer probes | Higher ratio increases detection sensitivity but may reduce total yield |
| Incubation Time | 2 hours | Extend to 4 hours for increased yield | Moderate improvement (20-50%) in total RNA synthesized |
| Template Concentration | 0.5-1 µg per 20 µL reaction | Increase to 2 µg for high-copy number templates | Increases yield but may exhaust NTPs prematurely |
| NTP Concentration | 1 mM each ATP, CTP, GTP; 0.65 mM UTP; 0.35 mM DIG-11-UTP | Increase to 8.5 mM each NTP for high-yield synthesis | Significantly increases yield but may increase production of short transcripts |
The concentration of monovalent ions significantly affects transcription efficiency. While T7 RNA polymerase is strongly inhibited by NaCl or KCl concentrations above 50 mM, it tolerates potassium glutamate up to at least 100 mM [10]. The inclusion of spermidine in reaction buffers (typically 1-2 mM) enhances template binding and promoter melting, particularly for GC-rich sequences. Magnesium concentration is critical for catalytic activity, with an optimum around 20 mM, though promoter binding occurs optimally at 2-5 mM MgCl₂ [10].
Technical Considerations and Troubleshooting
Labeling Efficiency and Probe Performance
The incorporation rate of DIG-11-UTP directly influences probe sensitivity in detection applications. Under standard conditions with a 35:65 ratio of DIG-11-UTP to UTP, approximately 1 DIG molecule is incorporated every 25-35 nucleotides. This density provides sufficient hapten incorporation for sensitive detection while maintaining acceptable hybridization kinetics and specificity. Higher incorporation rates may be desirable for detecting low-abundance targets but can potentially increase non-specific binding and background signal. The length of the RNA probe also affects performance; optimal probes typically range from 200-1000 nucleotides, balancing penetration efficiency in tissue sections with hybridization specificity.
Common Challenges and Solutions
- Low Yield: Potential causes include template quality, RNase contamination, or suboptimal NTP concentrations. Solution: Repurify template DNA, use fresh RNase-free reagents, and verify NTP concentrations.
- Short Transcripts: Often results from template secondary structure or premature termination. Solution: Increase incubation temperature to 42°C, supplement with pyrophosphatase, or use template-linearizing enzymes that produce 5'-overhangs or blunt ends.
- High Background in Detection: May stem from over-labeling or incomplete purification. Solution: Titrate DIG-11-UTP concentration, implement post-synthesis purification (e.g., LiCl precipitation), and optimize hybridization stringency.
Research Reagent Solutions
Table 3: Essential Reagents for DIG-Labeled RNA Probe Synthesis
| Reagent | Function | Example Specifications |
|---|---|---|
| DIG-11-UTP | Modified nucleotide for probe labeling | 3.5 mM in labeling mix; lithium salt [11] |
| T7/T3/SP6 RNA Polymerase | DNA-dependent RNA polymerase for probe synthesis | 20 U/µL in 50% glycerol [10] |
| 5x Transcription Buffer | Optimal reaction conditions for transcription | 200 mM Tris-HCl (pH 8.0), 40 mM MgCl₂, 10 mM spermidine, 250 mM NaCl [11] |
| RNasin | RNase inhibitor | 40 U/µL; protects RNA transcripts from degradation [11] |
| NTP Mix | Building blocks for RNA synthesis | 10 mM each ATP, CTP, GTP; 6.5 mM UTP [11] |
| Template DNA | Source of target sequence for probe synthesis | Linearized plasmid with phage promoter; 0.5-1 µg/µL [12] |
Applications in Molecular Biology
DIG-labeled RNA probes synthesized through DIG-11-UTP incorporation have enabled numerous applications in molecular biology and diagnostics. In situ hybridization techniques benefit from the high sensitivity and low background afforded by these probes, allowing spatial localization of gene expression in tissues and whole mounts [13] [12]. The high affinity of anti-digoxigenin antibodies (typically conjugated to alkaline phosphatase) enables detection down to single-copy transcripts in optimally prepared samples. Northern blot applications utilize the same principles for detecting specific RNA species separated by electrophoresis, with chemiluminescent or colorimetric detection methods. More recently, these probes have been adapted for high-throughput screening approaches, including microarray-based expression profiling and automated in situ hybridization platforms [13]. The non-radioactive nature of DIG labeling eliminates safety concerns and regulatory hurdles associated with isotopic methods while providing comparable sensitivity for most applications.
Workflow Diagram
Diagram 1: Workflow of DIG-Labeled RNA Probe Synthesis. This diagram illustrates the sequential process from template preparation to final application, highlighting the key enzymatic steps where DIG-11-UTP is incorporated during transcription.
The incorporation of DIG-11-UTP by RNA polymerases represents a robust and well-characterized methodology for generating non-radioactive hybridization probes with sensitivity comparable to radioactive alternatives. The reaction principle leverages the natural substrate flexibility of bacteriophage RNA polymerases to incorporate the modified nucleotide while maintaining transcriptional fidelity. Through optimization of reaction parameters including nucleotide ratios, ionic conditions, and incubation times, researchers can generate high-specificity probes suitable for a wide range of molecular applications. The continued utility of this technology across diverse fields including developmental biology, pathology, and functional genomics underscores its fundamental importance in modern molecular research.
This technical guide details the core components required for the synthesis of digoxigenin (DIG)-labeled RNA probes, a critical methodology in molecular biology for the detection of nucleic acids. Within the broader context of thesis research on DIG-labeled RNA probe protocols, this document provides an in-depth examination of the essential reagents—labeling mixes, polymerases, and templates—their functional mechanisms, and precise experimental requirements. This information is fundamental for researchers and drug development professionals aiming to optimize protocols for techniques such as in situ hybridization, Northern blotting, and other hybridization-based assays.
RNA Labeling Mix: Composition and Function
The RNA labeling mix is a precisely formulated nucleotide solution that enables the incorporation of the hapten digoxigenin into nascent RNA transcripts during in vitro transcription. The core function of the mix is to provide the necessary building blocks for the RNA polymerase while supplying a digoxigenin-tagged nucleotide for label integration.
Core Composition
A standard DIG RNA Labeling Mix, as commercially available, is a solution containing the following components [14]:
- 10x Concentration Solution: The mix is typically supplied as a 10x concentrate, containing:
- 10 mM each of ATP, CTP, and GTP.
- 6.5 mM UTP.
- 3.5 mM DIG-11-UTP (DIG-labeled uridine triphosphate).
- Labeling Ratio: The ratio of DIG-11-UTP to UTP is optimized to facilitate efficient incorporation by bacteriophage RNA polymerases. The formulation inserts a DIG-11-UTP residue at an average interval of every 20 to 25 nucleotides in the transcribed RNA [14].
Table 1: Quantitative Data of a Standard DIG RNA Labeling Mix
| Component | Concentration in 10x Mix | Final Reaction Concentration (1x) | Role in Transcription |
|---|---|---|---|
| ATP, CTP, GTP | 10 mM each | 1 mM each | Unlabeled nucleotide substrates for RNA chain elongation |
| UTP | 6.5 mM | 0.65 mM | Unlabeled uridine triphosphate substrate |
| DIG-11-UTP | 3.5 mM | 0.35 mM | Digoxigenin-labeled nucleotide for probe detection |
| Total Nucleotides | 40 µL package | Sufficient for 20 reactions | N/A |
Characteristics and Handling
- Physical Properties: The solution is typically colorless and miscible with water [14].
- Storage: Must be stored at -20°C to maintain stability [14].
- Labeling Efficiency: Under standard conditions using a linearized template, this system can produce approximately 10 µg of full-length DIG-labeled RNA from 1 µg of template DNA [14].
Polymerases: Enzymatic Drivers of Probe Synthesis
The synthesis of DIG-labeled RNA probes relies on bacteriophage-encoded DNA-dependent RNA polymerases. These enzymes are highly specific for their corresponding promoter sequences and exhibit high processivity, making them ideal for in vitro transcription.
Key Bacteriophage Polymerases
The most commonly used polymerases are derived from bacteriophages and are named accordingly [14] [15] [16]:
- SP6 RNA Polymerase: Isolated from Salmonella typhimurium LT2.
- T7 RNA Polymerase: Isolated from Escherichia coli T7 phage.
- T3 RNA Polymerase: Isolated from Escherichia coli T3 phage.
A critical feature of these enzymes is their high promoter specificity, meaning they demonstrate virtually no cross-activation by each other's promoters. This allows for the targeted transcription of either the "sense" or "antisense" strand from the same DNA template simply by placing different promoters on either side of the insert clonings [16].
Polymerase Mechanism and Fidelity
While the aforementioned enzymes are RNA polymerases, the fundamental mechanism of nucleotide addition is shared with DNA polymerases, which have been more extensively characterized. DNA polymerases catalyze the addition of nucleotides to the 3'-hydroxyl end of a growing DNA strand in a 5' to 3' direction, reading the template strand in the 3' to 5' direction [17]. They act as molecular motors, undergoing conformational changes between "open" and "closed" states upon binding of the correct nucleotide (dNTP), which is crucial for substrate discrimination and fidelity [18]. This induced-fit mechanism ensures that the active site is optimally organized only when a correct Watson-Crick base pair is formed, thereby enhancing the accuracy of nucleic acid synthesis [19] [18].
Table 2: Comparative Analysis of Nucleic Acid Polymerases for Probe Synthesis
| Feature | Bacteriophage RNA Polymerases (T7, T3, SP6) | DNA-Dependent DNA Polymerases |
|---|---|---|
| Primary Role | In vitro transcription of RNA probes | DNA replication and repair in vivo |
| Template | Double-stranded DNA with specific promoter | Primed, single-stranded DNA template |
| Product | Single-stranded RNA | Double-stranded DNA |
| Key Application | Production of labeled RNA probes (riboprobes) | Polymerase Chain Reaction (PCR), cDNA synthesis |
| Promoter Specificity | High (no cross-reactivity between T7, T3, SP6) | Not applicable |
| Proofreading Activity | Generally no 3'→5' exonuclease activity | Present in some high-fidelity enzymes |
Template Requirements: The Blueprint for Transcription
The DNA template is the foundational component that dictates the sequence, length, and specificity of the resulting DIG-labeled RNA probe. The integrity and preparation of the template are paramount to the success of the transcription reaction.
Template Types and Preparation
Two primary types of DNA templates are used for in vitro transcription [14]:
- Linearized Plasmid DNA: The gene of interest is cloned into a transcription vector downstream of a bacteriophage promoter (e.g., pGEM, pBluescript) [15] [16].
- Linearization: The plasmid must be linearized by a restriction enzyme cut downstream of the insert to prevent the transcription of long, uncontrollable concatenated RNAs.
- End Type: Restriction enzymes that generate 5'-overhangs are preferred; those generating 3'-overhangs should be avoided as they can act as spurious transcription initiation sites [14].
- Purification: Following linearization, the DNA should be purified by phenol-chloroform extraction and ethanol precipitation to remove contaminants like RNases and salts [14].
- PCR Products: PCR fragments that have a bacteriophage promoter sequence incorporated into the primer can also serve as templates [14] [20].
- Purification: PCR products should be purified, for instance, using silica-membrane-based columns (e.g., HighPure columns), before use in transcription to remove excess primers, nucleotides, and enzymes [14].
Promoter Sequence Requirements
The promoter sequence is the binding site for the RNA polymerase and is absolutely required for the initiation of transcription. The minimal consensus sequences for common promoters are [21]:
- T7 Class III Phi6.5 Promoter:
5'-TAATACGACTCACTATAGNN...-3' - T7 Class II Phi2.5 Promoter:
5'-TAATACGACTCACTATTANN...-3' - Note: The "NN" represents the first two bases of the transcribed RNA, which are ideally "CG" for the Class III promoter and "GG" for the Class II promoter for optimal efficiency [21].
The following diagram illustrates the complete workflow for synthesizing a DIG-labeled RNA probe, from template preparation to the final product.
Research Reagent Solutions: Essential Materials
The following table catalogs the key reagents and their functions essential for performing DIG-labeled RNA probe synthesis and analysis [14] [21] [20].
Table 3: Essential Research Reagents for DIG-Labeled RNA Probe Protocols
| Reagent / Kit | Function / Description | Key Features / Notes |
|---|---|---|
| DIG RNA Labeling Mix | Provides nucleotides for in vitro transcription, including DIG-11-UTP. | Pre-mixed solution; optimized for SP6, T3, and T7 RNA polymerases; insert DIG every 20-25 nucleotides. |
| SP6, T3, or T7 RNA Polymerase | Enzymatically synthesizes RNA from a DNA template. | High promoter specificity; no cross-reactivity; supplied with optimized transcription buffer. |
| Transcription Vector (e.g., pGEM, pBluescript) | Plasmid DNA containing bacteriophage promoters for cloning the gene of interest. | Flanked by multiple cloning sites and two different RNA polymerase promoters for sense/antisense probe synthesis. |
| RNase Inhibitor | Protects synthesized RNA probes from degradation by RNases. | Critical for maintaining RNA integrity; often included in commercial polymerase mixes. |
| RNase-free DNase I | Degrades the DNA template after transcription is complete. | Removes template DNA to prevent competition during hybridization; must be RNase-free. |
| Anti-Digoxigenin-AP Antibody | Conjugated antibody for detecting the DIG label in hybridized probes. | Used in colorimetric or chemiluminescent detection; conjugated to Alkaline Phosphatase (AP). |
| NBT/BCIP | Colorimetric substrate for Alkaline Phosphatase (AP). | Produces an insoluble purple precipitate for visual detection in situ hybridization. |
Detailed Experimental Protocol: Synthesis of DIG-Labeled RNA Probes
This protocol is adapted from established methods for producing DIG-labeled RNA probes suitable for techniques such as in situ hybridization [14] [20] [22].
Template DNA Linearization and Purification
- Linearize Plasmid DNA: Digest 5-20 µg of plasmid DNA containing your insert and the appropriate promoter using a restriction enzyme that cuts downstream of the insert. Use an enzyme that produces a 5'-overhang or blunt end. Include RNase inhibitor if necessary.
- Verify Digestion: Analyze a small aliquot (e.g., 100 ng) by agarose gel electrophoresis to confirm complete linearization.
- Purify DNA: Purify the linearized DNA by phenol-chloroform extraction followed by ethanol precipitation [14] [22]. Alternatively, use a commercial PCR purification kit or spin column. Resuspend the purified DNA in RNase-free water or TE buffer at a concentration of 0.2-0.5 µg/µL.
In Vitro Transcription Reaction
- Assemble Reaction at Room Temperature: In an RNase-free microcentrifuge tube, combine the following components in order:
- 1 µg purified, linearized DNA template (or 100-200 ng of purified PCR product)
- 2 µL 10x DIG RNA Labeling Mix (from Roche, final concentration 1x) [14]
- 2 µL 10x Transcription Buffer (polymerase-specific)
- 1 µL RNase Inhibitor (e.g., 40 U/µL)
- 2 µL RNA Polymerase (e.g., T7, SP6, or T3, typically 20 U/µL)
- RNase-free water to a final volume of 20 µL
- Mix gently by pipetting and centrifuge briefly to collect the reaction at the bottom of the tube.
- Incubate at 37°C for 2 hours. For longer transcripts, incubation can be extended up to 4 hours.
DNase Treatment and Probe Purification
- Terminate Transcription: Add 2 µL of 0.2 M EDTA (pH 8.0) to stop the reaction [14].
- Degrade Template DNA: Add 1 µL of RNase-free DNase I and incubate at 37°C for 15-30 minutes [20] [16].
- Purify RNA Probe: Purify the probe using a commercial RNA cleanup kit (e.g., Zymo Research RNA Clean & Concentrator) according to the manufacturer's instructions. This method effectively removes proteins, salts, and unincorporated nucleotides. Alternatively, precipitate the RNA with ethanol or LiCl [21].
- Resuspend and Quantify: Resuspend the purified RNA probe in RNase-free water or hybridization buffer. Quantify the concentration by spectrophotometry.
Troubleshooting and Quality Control
- Low Yield: Ensure template DNA is pure, fully linearized, and of high concentration. Verify enzyme activity.
- Background in Hybridization: Include an RNase treatment step post-hybridization to degrade single-stranded, non-hybridized probe [16]. Always use sense strand probes as negative controls [20].
- RNA Degradation: Maintain an RNase-free work environment by using gloves, RNase-free tips and tubes, and dedicated reagents.
In the broader context of optimizing digoxigenin (DIG)-labeled RNA probe protocols, understanding and predicting the expected yield and incorporation efficiency is fundamental to experimental success. These parameters directly influence the sensitivity and specificity of downstream applications like in situ hybridization, northern blotting, and other nucleic acid detection methods [23] [24] [25]. This guide provides a detailed technical overview of the quantitative benchmarks and methodological controls that researchers can expect when synthesizing DIG-labeled RNA probes, serving as a critical resource for scientists and drug development professionals in planning and troubleshooting their experiments.
Quantifying Probe Yield and Incorporation
The yield and incorporation efficiency of a DIG-labeling reaction are primary indicators of its success. These metrics determine the amount of probe available for hybridization and its effective specific activity.
Direct Measurement of Probe Yield
Probe yield can be estimated through direct detection methods or calculated based on the transcription reaction's performance. The direct detection method involves spotting serial dilutions of the labeled probe alongside a DIG-labeled control of known concentration on a nylon membrane, followed by chemiluminescent detection to compare signal intensities [25]. This method is straightforward and provides a functional estimate of the DIG-labeled probe concentration.
For RNA probes synthesized by in vitro transcription, yield can be calculated from the reaction itself. A standard transcription reaction using 1 µg of DNA template typically yields between 10 and 20 µg of full-length DIG-labeled RNA [25]. This represents an amplification of the template, as the DNA can be transcribed many times (up to a hundredfold) to generate a large amount of probe [16].
Table 1: Expected Probe Yields from Different DIG-Labeling Methods
| Labeling Method | Template | Typical Yield | Key Influencing Factors |
|---|---|---|---|
| In Vitro Transcription [25] | 1 µg linearized DNA | 10-20 µg RNA | DNA template purity, RNA polymerase efficiency |
| PCR-Based Labeling [24] [25] | Limited amounts of template | High, specific probe | Primer design, fidelity of polymerase |
| Random Primed DNA Labeling [25] | dsDNA template | Varies with template length | Template concentration, Klenow enzyme activity |
Assessing Label Incorporation Efficiency
Incorporation efficiency refers to the density of DIG haptens incorporated into the nucleic acid probe, which directly impacts detection sensitivity. The efficiency varies by labeling method but is generally high.
For in vitro transcription, DIG-11-UTP is added to the nucleotide mix. During the reaction, a DIG moiety is incorporated, on average, every 25 to 30 nucleotides [25]. This high density of labeling is a key advantage of the transcriptional method.
For DNA probes labeled by random primed labeling, the Klenow enzyme incorporates DIG-11-dUTP during synthesis of the complementary strand. This method also results in a high and homogeneous incorporation, with one DIG molecule inserted approximately every 20 to 25 nucleotides [25].
Table 2: Expected DIG Incorporation Efficiency and Key Metrics
| Labeling Method | DIG-Labeled Nucleotide | Average Incorporation Rate | Impact on Probe Performance |
|---|---|---|---|
| In Vitro Transcription [25] | DIG-11-UTP | Every 25-30 nucleotides | High specific activity, sensitive detection |
| Random Primed Labeling [25] | DIG-11-dUTP | Every 20-25 nucleotides | Homogeneously labeled, sensitive probe |
| PCR Labeling [24] [25] | DIG-dUTP in PCR mix | High degree of incorporation | Specific, sensitive probes from minimal template |
Methodologies for Yield and Efficiency Analysis
Direct Detection for Quantification
The direct detection procedure is a critical method for estimating the yield of DIG-labeled probes, especially for those synthesized by methods other than PCR [25].
- Preparation: Prepare serial dilutions of your labeled probe.
- Spotting: Spot these dilutions, along with a series of dilutions from a DIG-labeled control nucleic acid of known concentration, onto a nylon membrane.
- Detection: Perform chemiluminescent detection using an anti-DIG antibody conjugated to alkaline phosphatase (AP) and suitable substrates.
- Analysis: Compare the signal intensity of your probe spots to the control spots to estimate the concentration of DIG-labeled nucleic acid in your sample. This entire process can be completed in approximately 2.5 to 3 hours [25].
Gel Electrophoresis for Quality Assessment
For probes labeled by PCR, gel electrophoresis is the recommended method for evaluation [25]. It allows you to confirm:
- The size and uniformity of the synthesized probe.
- The presence of a single, distinct band of the expected size, which indicates specific synthesis.
- The absence of smearing or multiple bands, which suggests non-specific amplification or degradation.
This quality control step is essential before using a newly synthesized probe in a sensitive experiment.
Determining Functional Sensitivity
A highly recommended practice is to determine the functional sensitivity of a newly synthesized DIG-labeled antisense RNA probe [25]. This is done by:
- Using in vitro transcription to generate the corresponding unlabeled "sense" RNA transcript.
- Purifying this sense RNA and creating a dilution series to use as a target on a northern blot.
- Hybridizing the blot with your DIG-labeled antisense probe.
- The result will show the lowest amount of target RNA that can be reliably detected, providing a practical, application-specific measure of your probe's sensitivity.
Optimizing for Maximum Yield and Efficiency
Several factors are critical to achieving the high yields and incorporation efficiencies.
- Template Quality and Preparation: For in vitro transcription, the DNA template must be highly purified and linearized by a restriction enzyme that cuts downstream of the insert. Template length is also important; optimal length is around 1 kb, with a minimum of 200 bp [25].
- RNase-Free Conditions: RNA probes are highly susceptible to degradation. All steps must be performed using RNase-free reagents and consumables (e.g., DEPC-treated water, baked glassware, gloves) to prevent RNA degradation and ensure full-length probe yield [24] [16] [25].
- Enzyme Selection: The use of high-fidelity enzyme blends, such as those found in commercial PCR DIG Probe Synthesis Kits, can reduce the need for extensive optimization of parameters like MgCl₂ concentration and improve success with challenging templates [25].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for DIG-Labeled RNA Probe Synthesis and Analysis
| Reagent / Kit | Primary Function | Key Feature |
|---|---|---|
| DIG-11-UTP [25] | Labeled nucleotide for RNA probe synthesis | Alkali-labile ester bond for incorporation by RNA polymerases |
| DIG-11-dUTP [25] | Labeled nucleotide for DNA probe synthesis | Incorporated by DNA polymerases in PCR, random priming, etc. |
| SP6, T7, T3 RNA Polymerases [23] [16] [25] | In vitro transcription from specific promoters | High specificity with no cross-reactivity, high yield |
| PCR DIG Probe Synthesis Kit [25] | One-step probe synthesis and labeling | Contains optimized, high-fidelity enzyme blend; minimal optimization |
| Anti-Digoxigenin-AP (or -HRP) [25] | Immunological detection of DIG-labeled probes | High specificity and sensitivity for colorimetric/chemiluminescent readout |
| RNase Inhibitor [24] | Protection of RNA probe integrity | Prevents RNase-mediated degradation during synthesis and handling |
Step-by-Step DIG-Labeled RNA Probe Synthesis and Key Applications
Within the context of a broader thesis on digoxigenin (DIG)-labeled RNA probe protocol research, establishing an RNase-free environment is not merely a preliminary step but the foundational determinant of experimental success. Ribonucleases (RNases) are extraordinarily robust enzymes that play a critical role in nucleic acid metabolism but pose a significant threat to RNA integrity in experimental settings [26]. Their ubiquitous presence—on skin, in dust, on lab surfaces, and even in reagents—means that without rigorous pre-protocol controls, the structural integrity of RNA probes and target molecules can be compromised, leading to failed hybridizations, high background noise, and irreproducible data in sensitive techniques like in situ hybridization and Northern blotting [20] [27]. This guide provides an in-depth technical framework for researchers and drug development professionals to systematically eliminate RNase contamination before initiating critical procedures involving DIG-labeled RNA probes.
The unique vulnerability of RNA molecules demands exceptional vigilance. RNA is inherently more prone to degradation than DNA, partly due to the ubiquity and resilience of RNases [28]. Furthermore, RNA can undergo non-enzymatic strand scission when heated in the presence of divalent cations such as Mg²⁺ or Ca²⁺ at temperatures above 80°C, a process distinct from RNase-mediated degradation but equally detrimental [26]. For experiments relying on DIG-labeled riboprobes, whose quality directly impacts the sensitivity and specificity of gene expression localization in tissues or whole mounts, establishing and maintaining an RNase-free workspace is therefore the most critical pre-protocol investment [20] [29].
Effective RNase control begins with recognizing potential sources of contamination. RNases are remarkably stable enzymes, refractory to many common decontamination methods like autoclaving, and they require strong chemical treatments for reliable inactivation [26]. The primary sources of RNase contamination in a laboratory environment include:
- Human Secretions: Skin, perspiration, and mucous membranes are significant sources of RNases. Shed skin cells and contact with bare hands can directly introduce RNases to samples and reagents [26] [28].
- Microbial Contamination: Bacteria and fungi are natural reservoirs of RNases. Their spores and cells can be present in dust, on lab surfaces, and in water baths [26].
- Laboratory Surfaces and Equipment: Benchtops, pipettors, centrifuges, door handles, and tube racks are frequently contaminated through routine use and exposure to the environment [26] [30].
- Consumables and Reagents: Glassware, plasticware, water, and buffers prepared in-house can be frequent, unsuspected sources of RNases. Notably, reagents isolated from bacterial sources (e.g., some DNase preparations) can be contaminated with RNases [26] [30].
The following table systematizes the common contamination sources and their associated risks.
Table 1: Common Sources of RNase Contamination and Their Associated Risks
| Contamination Source | Specific Examples | Associated Risk |
|---|---|---|
| Human/Sample-Derived | Skin flakes, perspiration, hair, tissue samples [26] [28] | Direct introduction of RNase A family enzymes; sample RNA degradation during collection. |
| Environmental/Microbial | Dust, bacterial & fungal spores, pet dander on clothing [26] | Constant re-contamination of surfaces and solutions. |
| Lab Surfaces & Equipment | Benchtops, pipettor barrels, centrifuges, door handles, water baths [26] [31] | Cross-contamination of tubes and reagents during handling. |
| Consumables & Reagents | Non-certified water, buffers, enzymes, glassware, plasticware [26] [30] | Direct introduction of RNases into reaction mixtures and samples. |
Establishing the RNase-Free Workstation: A Practical Framework
Creating a dedicated and controlled environment is the most effective strategy to minimize cross-contamination. This involves both spatial organization and the implementation of strict personal practices.
Dedicated Workspace and Personal Practices
- Designate an "RNA Only" Zone: Establish a dedicated workstation, preferably in a low-traffic area, with its own set of equipment, including pipettors, microcentrifuges, tube racks, and reagents [30] [31]. This area should be physically separated from spaces where common laboratory activities like DNA purification, bacterial culture, or protein work are conducted.
- Employ Meticulous Gloving Technique: While wearing gloves is standard, the technique is critical. Human skin is a major RNase source, and simply wearing gloves is insufficient if they become contaminated [28]. Use only nitrile gloves, as they offer higher abrasion resistance than latex. For high-risk protocols, use individually pair-packed, sterile gloves and adopt an aseptic donning technique to ensure the outer surface remains RNase-free [28]. Longer gloves (≥30 cm) are also recommended to ensure complete overlap with lab coat sleeves and prevent exposure from the wrist [28].
- Wear a Lab Coat and Change Gloves Frequently: Always wear a clean lab coat dedicated to the RNA workspace. Change gloves anytime you touch a potential contaminant, such as a door handle, phone, computer keyboard, or your own skin [30].
Systematic Surface and Equipment Decontamination
All surfaces and equipment within the RNA workstation must be treated to inactivate RNases. A regular, documented cleaning schedule is paramount.
- Daily and Weekly Cleaning: Ambion scientists recommend thoroughly cleaning lab benchtops, pipettors, and tube racks on a weekly basis [26]. A quick wipe-down with an RNase-decontaminating solution before starting work can be part of a daily routine [30].
- Use Effective RNase Inactivating Reagents: Commercially available RNase decontamination solutions like RNaseZap are highly effective [30]. Alternatively, a laboratory-prepared regimen of 0.5% SDS followed by 3% H₂O₂ can be used [30].
- Decontaminate Non-Disposable Items: Glassware should be baked at 250°C for at least 2 hours (or overnight) to inactivate RNases [30] [31]. Plasticware that is not certified RNase-free can be rinsed with 0.1N NaOH/1mM EDTA followed by rinsing with DEPC-treated water [31].
- Use Filter Pipette Tips: Always use aerosol-barrier or filter pipette tips to prevent cross-contamination of pipettor interiors, which are difficult to decontaminate [26] [30].
Table 2: Recommended RNase Decontamination Schedule and Methods
| Frequency | Item/Surface | Recommended Method |
|---|---|---|
| Before each use | Benchtops, tube racks | Wipe with RNase decontamination solution (e.g., RNaseZap) or 0.5% SDS/3% H₂O₂ [30]. |
| Weekly | Pipettors, centrifuge rotors, door handles | Detailed cleaning with RNase decontamination solution [26]. |
| Prior to first use | Glassware | Bake at 250°C for >2 hours (up to overnight) [30] [31]. |
| Prior to first use | Plasticware (non-sterile) | Rinse with 0.1N NaOH/1mM EDTA, then with DEPC-treated water [31]. |
| Monthly / As Needed | Water sources, lab-prepared reagents | Test for RNase activity [26]. |
| As Needed | Electrophoresis equipment | Clean meticulously with an RNase decontamination solution before use [26]. |
The following workflow diagram summarizes the logical progression in establishing an RNase-free workstation.
Controlling Consumables and Reagents
The reagents and consumables that contact RNA directly are a critical control point. Trace amounts of RNase in a buffer or water can nullify all other precautions.
Water and Buffer Preparation
- DEPC Treatment: The most common method for inactivating RNases in water and salt buffers is treatment with Diethyl Pyrocarbonate (DEPC). Use 0.5 mL DEPC per liter of solution, incubate for at least 2 hours (or overnight), and then autoclave for a minimum of 45 minutes to hydrolyze and remove any residual DEPC, which is a suspected carcinogen [30] [31].
- Critical Note on Tris Buffers: DEPC cannot be used to treat Tris-based buffers because it reacts with amines. For these solutions, purchase certified RNase-free Tris or prepare them using DEPC-treated or nuclease-free water [30] [31].
- Alternative to DEPC: Ultrafiltered, molecular biology-grade water that is certified nuclease-free is a safe and convenient, though potentially more expensive, alternative [30].
Consumables and Enzymes
- Use Certified RNase-Free Consumables: Use sterile, disposable plasticware (tubes, tips) that are certified RNase-, DNase-, and pyrogen-free [31]. This eliminates the need for labor-intensive pre-treatment.
- Select RNase-Free Enzymes: Many enzymes isolated from bacterial sources (e.g., DNase I) can be contaminated with RNases. Always use molecular biology-grade enzymes certified as RNase-free [30].
- Employ RNase Inhibitors: In enzymatic reactions involving RNA, such as in vitro transcription for DIG-labeled probe synthesis or reverse transcription, include a ribonuclease inhibitor protein (e.g., RNasin) in the reaction mix. This inhibitor binds to and neutralizes common RNases of the RNase A family [26] [31]. Avoid high temperatures or denaturing conditions that could deactivate the inhibitor [30].
The Scientist's Toolkit: Essential Reagents for RNase Control
Table 3: Research Reagent Solutions for an RNase-Free Environment
| Reagent / Material | Function / Purpose | Technical Notes |
|---|---|---|
| Nitrile Gloves (Individually packed, sterile) | Creates a physical barrier against RNases from skin. | Superior abrasion resistance vs. latex; aseptic donning prevents contamination [28]. |
| RNase Decontamination Solution (e.g., RNaseZap) | Rapidly inactivates RNases on surfaces and equipment. | Effective on benchtops, pipettors, glassware; alternative: 0.5% SDS + 3% H₂O₂ [30]. |
| DEPC (Diethyl Pyrocarbonate) | Chemical inactivation of RNases in water and salt solutions. | Suspected carcinogen; requires autoclaving after incubation to remove traces [30] [31]. |
| RNase-Free Water (Certified) | Safe, pre-treated water for making solutions and reactions. | Reliable alternative to in-house DEPC treatment; ensures reagent integrity [30]. |
| Ribonuclease Inhibitor (e.g., RNasin) | Inhibits RNase A-family enzymes in enzymatic reactions. | Essential for in vitro transcription, RT-PCR; deactivated at >60°C or under denaturing conditions [26] [30]. |
| Certified RNase-Free Consumables (Tubes, tips, columns) | Prevents introduction of RNases via direct sample contact. | Individually wrapped or in sealed bags is optimal to maintain sterility [28] [31]. |
The integrity of a digoxigenin-labeled RNA probe is the cornerstone of its performance in applications ranging from whole-mount in situ hybridization to Northern blot analysis [20] [29]. A degraded probe will yield weak, non-specific, or false-negative results, rendering subsequent protocol steps futile. The rigorous pre-protocol steps outlined here—establishing a dedicated workspace, implementing systematic decontamination, and meticulously controlling reagents—are therefore not isolated tasks but an integrated system of quality assurance.
For researchers engaged in high-stakes drug development or precise gene expression mapping, adopting this holistic approach to RNase control transforms it from a reactive troubleshooting exercise into a proactive, ingrained standard of practice. By investing in this critical foundational phase, scientists ensure that the sophisticated molecular tools they create, such as DIG-labeled riboprobes, function with the sensitivity and specificity required to generate reliable and meaningful scientific data. The battle against RNases is perpetual, but with vigilance and a structured protocol, it is a battle that can be consistently won.
In the context of digoxigenin (DIG)-labeled RNA probe protocol research, the preparation of the DNA template represents the foundational step that determines the success of subsequent experimental procedures. Proper template design, linearization, and purification are prerequisite for generating high-quality, specific probes capable of detecting target RNA sequences within tissue samples via in situ hybridization (ISH). The integrity of the final RNA probe directly correlates with the precision of these initial preparative steps, ultimately influencing the sensitivity and specificity of gene expression analysis in diverse applications from developmental biology to disease pathology [4]. This technical guide outlines current best practices in template preparation, with a specific focus on methodologies supporting the synthesis of DIG-labeled RNA probes, which have become a preferred approach due to their high sensitivity and specificity for target RNA sequences [4].
Template Design Fundamentals
Vector Selection and Insert Cloning
The design phase begins with strategic vector selection to accommodate opposable promoters, such as T7, T3, and SP6 RNA polymerase binding sites, which flank the multiple cloning site. This arrangement enables transcription of both the antisense probe (experimental) and sense strand (negative control) from the same DNA template, a critical control for validating hybridization specificity [4]. The target sequence of interest is cloned into this intervening multiple cloning site, with careful consideration given to orientation relative to the promoter sequences to determine whether sense or antisense RNA will be transcribed.
For optimal results in ISH experiments, the inserted sequence should be of appropriate length. Research indicates that RNA probes should ideally be 250–1,500 bases in length, with probes of approximately 800 bases demonstrating the highest sensitivity and specificity in hybridization assays [4]. This length provides sufficient complementarity for stable hybridization while maintaining adequate diffusion properties for penetration into tissue sections.
Promoter Considerations for IVT
The in vitro transcription (IVT) reaction efficiency depends significantly on promoter strength and specificity. When designing the template, ensure that each promoter sequence is complete and optimized for the corresponding RNA polymerase. The use of pre-validated backbone tools incorporating optimized sequences can streamline this process, providing tested promoter and untranslated region combinations that enhance transcription efficiency and RNA stability [32]. While these systems are often discussed in therapeutic contexts, the same principles apply to research probe generation, particularly when consistent yield and quality are paramount.
Template Linearization Methods
Linearization Strategies
Prior to IVT, circular plasmid DNA must be linearized to prevent transcription of vector sequences and ensure defined probe length. The linearization method significantly impacts the quality and characteristics of the resulting RNA probe, making this a critical step in the preparation workflow.
Table 1: Template Linearization Methods Comparison
| Method | Procedure | Advantages | Considerations |
|---|---|---|---|
| Restriction Enzyme Digestion | Digest with enzyme cutting downstream of insert | Clean ends; defined termination; high yield | Must select enzyme that doesn't cut within insert; possible star activity |
| PCR-Generated Templates | Amplify template with incorporated promoter sequences | No vector sequences; scalable; rapid | Potential for polymerase errors; lower yield for large templates |
| Enzymatic Hydrolysis | Controlled enzymatic treatment of plasmid DNA | Applicable when suitable restriction sites are unavailable | Less precise; requires optimization to avoid template degradation |
Restriction enzyme digestion remains the most widely employed method for template linearization. The selected restriction enzyme should create either a 5' overhang or blunt end and must not cut within the insert sequence itself. Verification of complete digestion through analytical gel electrophoresis is essential, as incomplete linearization results in transcription of excessively long RNA molecules that can incorporate vector sequences, potentially increasing background noise through non-specific hybridization [4].
For templates generated via PCR amplification, the promoter sequence is incorporated directly into the PCR primer, eliminating the need for subsequent cloning steps. While this approach offers time savings, it requires stringent quality control to prevent mutations that could compromise probe specificity, particularly given that even minimal sequence mismatching (>5% non-complementary base pairs) can significantly reduce hybridization efficiency [4].
Purification Techniques for Linearized Templates
Purification Method Selection
Following linearization, effective template purification removes enzymes, salts, and other reaction components that could inhibit subsequent IVT reactions. The choice of purification method balances yield, time investment, and purity requirements.
Table 2: Purification Techniques for Linearized Templates
| Technique | Procedure | Purity Level | Recovery Efficiency | Suitability |
|---|---|---|---|---|
| Phenol-Chloroform Extraction & Ethanol Precipitation | Organic extraction followed by alcohol precipitation | Moderate to high | High (≥80%) | Standard applications; large volumes |
| Commercial Silica-Membrane Columns | Binding, wash, and elution steps | High | Moderate to high (60-80%) | Rapid processing; multiple samples |
| Magnetic Bead-Based Purification | Paramagnetic particle binding with magnetic separation | High | Consistent | Automated high-throughput applications |
| Gel Extraction | Size-selective isolation from agarose gel | Highest | Variable (40-70%) | Critical applications requiring utmost purity |
Quantitative Assessment of Purification Efficiency
Recent advances in analytical methods have enhanced our ability to quantify impurities in nucleic acid preparations. Liquid chromatography-mass spectrometry (LC-MS/MS) provides exceptional sensitivity for detecting residual contaminants, while fluorimetric quantitation using dyes like Qubit offers rapid, specific nucleic acid concentration measurements [33]. These methodologies confirm that effective purification can achieve DNA:RNA mass ratios of 1:1000 in final products, a relevant benchmark for researchers preparing templates for IVT [33]. Although this specific data comes from vaccine manufacturing, the same principles apply to research-grade template purification, particularly when high-purity templates are required for sensitive applications like single-molecule RNA FISH [34].
Quality Control Assessment
Analytical Verification Methods
Rigorous quality control ensures that linearized and purified templates meet specifications for subsequent IVT reactions. Implement a multi-parameter assessment approach:
Spectrophotometric Analysis: Determine template concentration and assess purity through A260/A280 (ideal range: 1.8-2.0) and A260/A230 (ideal range: 2.0-2.2) ratios. Significant deviations may indicate protein or organic chemical contamination, respectively.
Gel Electrophoresis: Verify template integrity, appropriate size, and complete linearization through agarose gel electrophoresis. A single, discrete band of expected size should be visible without smearing (indicating degradation) or additional bands (suggesting incomplete linearization or contamination).
Functional Testing: Perform small-scale test IVT reactions with subsequent analysis of RNA yield and integrity. This functional assessment provides the most relevant quality indicator for template performance.
The critical importance of template quality is underscored by its direct impact on molecular detection efficiency in advanced RNA imaging methods. As noted in recent optimization studies for multiplexed error robust fluorescence in situ hybridization (MERFISH), template quality directly influences signal brightness and detection efficiency, with poor template preparation contributing to increased background and reduced specificity [34].
Experimental Protocols
Detailed Linearization and Purification Protocol
This standardized protocol outlines a comprehensive procedure for template linearization and purification suitable for generating templates for DIG-labeled RNA probe synthesis.
Materials Required:
- Plasmid DNA containing insert and opposable promoters
- Appropriate restriction enzyme and corresponding buffer
- Phenol:chloroform:isoamyl alcohol (25:24:1)
- Chloroform
- 3M sodium acetate, pH 5.2
- 100% and 70% ethanol
- Nuclease-free water
Procedure:
- Restriction Digest Setup: In a 1.5 mL microcentrifuge tube, combine the following components:
- Plasmid DNA (1-10 µg)
- 10 µL appropriate 10x restriction enzyme buffer
- Restriction enzyme (10-20 units per µg DNA)
- Nuclease-free water to 100 µL final volume
Incubation: Mix thoroughly and incubate at recommended temperature for 2-4 hours. For complete digestion of larger amounts of DNA, extended incubation (overnight) with additional enzyme may be necessary.
Digestion Verification: Remove 5 µL of reaction mixture and analyze by agarose gel electrophoresis alongside undigested plasmid controls to confirm complete linearization.
Purification: a. Add equal volume phenol:chloroform:isoamyl alcohol to the remaining digest, vortex thoroughly, and centrifuge at 12,000 × g for 5 minutes. b. Transfer aqueous upper phase to a new tube and add equal volume chloroform, vortex, and centrifuge as before. c. Transfer aqueous phase to a new tube and add 0.1 volume 3M sodium acetate and 2.5 volumes 100% ethanol. d. Mix thoroughly and incubate at -20°C for at least 30 minutes. e. Centrifuge at 12,000 × g for 15 minutes at 4°C to pellet DNA. f. Carefully decant supernatant and wash pellet with 500 µL 70% ethanol. g. Centrifuge at 12,000 × g for 5 minutes, carefully remove supernatant, and air-dry pellet for 5-10 minutes. h. Resuspend DNA in 20-50 µL nuclease-free water.
Quantification: Determine DNA concentration using spectrophotometry and adjust to working concentration (typically 0.5-1.0 µg/µL) for IVT reactions.
Troubleshooting Common Issues
Incomplete Linearization: Evident by multiple bands on verification gel. Solution: Add more enzyme, extend incubation time, or ensure reaction conditions are optimal for the specific enzyme.
Low Yield After Purification: Often results from inefficient precipitation. Solution: Ensure accurate pH of sodium acetate, extend precipitation time, or increase initial DNA amount.
RNA Probe Degradation: Though occurring after IVT, this often traces back to RNase contamination during template preparation. Solution: Use RNase-free reagents and techniques throughout, including dedicated equipment and workspace.
Template Preparation Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Template Preparation and Quality Assessment
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| Restriction Enzymes | Site-specific DNA cleavage | Select enzymes that don't cut within insert; high-fidelity options available |
| Phenol-Chloroform-Isoamyl Alcohol | Organic extraction of proteins and contaminants | Phase separation removes enzymes, proteins from DNA solution |
| Silica Membrane Columns | Rapid DNA purification | Commercial kits offer standardized protocols for consistent results |
| Agarose | Gel matrix for electrophoretic separation | Verification of linearization success and DNA integrity |
| Fluorescent Nucleic Acid Stains | DNA visualization and quantification | Enables precise DNA concentration measurement beyond UV absorbance |
| LC-MS/MS Systems | High-sensitivity impurity detection | Advanced quality control for critical applications [33] |
| RNase Inhibitors | Protection of RNA products | Essential for preventing degradation in subsequent IVT steps |
Template design, linearization, and purification represent critical foundational steps in the synthesis of DIG-labeled RNA probes for in situ hybridization. By implementing these best practices—selecting appropriate vector systems, ensuring complete linearization, employing effective purification strategies, and conducting rigorous quality control—researchers can consistently generate high-quality templates that yield sensitive and specific RNA probes. These meticulous preparative procedures directly support the reliability and reproducibility of spatial gene expression analysis, enabling advancements in both basic research and drug development applications. As RNA imaging technologies continue to evolve toward increasingly multiplexed and sensitive detection methods [34], the importance of optimized template preparation protocols only grows more pronounced.
In vitro transcription (IVT) is a foundational molecular biology technique for synthesizing RNA molecules outside of a living cell, using a DNA template and a bacteriophage RNA polymerase [35]. This cell-free enzymatic reaction is critical for numerous applications, including the creation of hybridization probes, functional mRNA for vaccines and therapeutics, and RNA for structural studies [35] [36]. Within the broader scope of digoxigenin-labeled RNA probe research, IVT serves as the core production method, enabling the precise incorporation of labeled nucleotides for highly sensitive detection in techniques such as in situ hybridization and Northern blotting [37] [38]. This protocol provides a standardized, step-by-step guide for performing a robust IVT reaction, with specific considerations for the generation of non-isotopically labeled probes.
Core Components of the IVT Reaction
A successful IVT reaction requires the precise combination of several key components in an optimized buffer system. The table below summarizes these essential reagents and their functions.
Table 1: Essential Components for a Standardized IVT Reaction
| Component | Function | Standard Concentration/Final Amount |
|---|---|---|
| Linear DNA Template | Provides the sequence to be transcribed; must contain a double-stranded phage promoter (e.g., T7, T3, SP6) [35]. | 0.5–1 µg per 20 µL reaction [37]. |
| RNA Polymerase | Enzyme that synthesizes RNA complementary to the DNA template strand (e.g., T7, T3, or SP6 RNA Polymerase) [35]. | 5–20 U per µL (amount varies by supplier) [39]. |
| Ribonucleotide Triphosphates (NTPs) | The building blocks (ATP, CTP, GTP, UTP) for RNA synthesis [35]. | 0.5–1 mM of each NTP is common; up to 4 mM each for high yield [39] [40]. |
| Reaction Buffer | Provides optimal pH, ionic strength, and co-factors for polymerase activity [35]. | Typically includes Mg²⁺, DTT, and spermidine [39]. |
| Magnesium Ions (Mg²⁺) | Essential co-factor for RNA polymerase activity [35] [40]. | Concentration must be optimized; often ~20 mM, or ~6 mM above the total NTP concentration [39] [40]. |
| RNase Inhibitor | Protects synthesized RNA from degradation by RNases [41]. | 0.5–1 U per µL of reaction [41]. |
| Dithiothreitol (DTT) | Reducing agent that helps maintain enzyme stability and activity [39] [40]. | Typically 1–10 mM [39]. |
DNA Template Preparation
The quality and design of the DNA template are paramount. Templates can be prepared via plasmid linearization or PCR amplification [35] [36].
- Plasmid Linearization: Plasmid vectors containing a phage promoter should be purified and completely digested with a restriction enzyme that cuts downstream of the insert to be transcribed. Enzymes that generate a blunt end or a 5' overhang are preferred, as 3' overhangs can lead to the production of longer-than-expected transcripts [41] [36]. The linearized DNA must then be purified to remove contaminants, salts, or enzymes that could inhibit the RNA polymerase [41] [36].
- PCR Amplification: The target sequence can be amplified using a forward primer that contains the phage promoter sequence (e.g., T7 promoter) at its 5' end. This method is rapid and avoids cloning steps, making it ideal for high-throughput applications [35] [36]. Use of a high-fidelity DNA polymerase is recommended to prevent introduction of mutations [36].
Incorporation of Modified Nucleotides for Labeling
For the synthesis of digoxigenin-labeled RNA probes, a modified nucleotide (e.g., Digoxigenin-11-UTP) is incorporated during the IVT reaction [37] [38]. This is typically achieved by replacing a portion of the standard nucleotide with its labeled counterpart. A recommended starting point is a 1:3 ratio of modified UTP to standard UTP (e.g., 0.2 mM Digoxigenin-11-UTP and 0.3 mM UTP) to ensure high incorporation without significantly inhibiting the polymerase [37]. The optimal ratio should be determined empirically for each specific application and labeled nucleotide used.
Standardized IVT Protocol
Equipment and Reagent Setup
- Equipment: Microcentrifuge, dry heat block or water bath set to 37°C, vortex mixer, micropipettors, and RNase-free tips and tubes [39].
- Workspace: Dedicate a clean area and use RNase-free reagents and consumables to prevent RNA degradation. Wear gloves and routinely decontaminate surfaces with an RNase deactivating solution [42] [40].
- Reagent Preparation: Thaw all non-enzyme components at room temperature and then place them on ice. The RNA polymerase should be taken directly from storage (-20°C) and kept on ice at all times to preserve activity [42] [37].
Step-by-Step Reaction Procedure
Assemble the Reaction: In a sterile, nuclease-free microcentrifuge tube, combine the components in the order listed below at room temperature. Adding reagents at room temperature prevents precipitation of the DNA template by spermidine present in some buffers [37].
- Nuclease-free water to a final volume of 20 µL
- 2 µL of 10X Transcription Buffer
- 1 µL of 10 mM ATP
- 1 µL of 10 mM CTP
- 1 µL of 10 mM GTP
- X µL of UTP mixture (e.g., 0.6 µL of 10 mM UTP + 0.4 µL of 10 mM Digoxigenin-11-UTP for labeling) [37]
- 1 µL of linear DNA template (0.5–1 µg)
- 1 µL of RNase Inhibitor (e.g., 5 U/µL)
- 2 µL of T7 RNA Polymerase (5 U/µL)
Mix and Incubate: Gently mix the reaction by pipetting and briefly centrifuging. Incubate the tube at 37°C for 2–4 hours [37] [40]. For problematic templates with high GC-content or secondary structure, lowering the incubation temperature to 16–30°C can help increase the yield of full-length transcripts [41] [43].
DNase I Treatment (Post-Transcription): After incubation, add 1 µL of DNase I (RNase-free) to the reaction tube and mix gently. Incubate at 37°C for 15 minutes to degrade the DNA template [37] [36].
Terminate the Reaction and Purify RNA: Add 1/10 volume of 0.5 M EDTA, pH 8.0, to chelate Mg²⁺ and stop the reaction [39]. The RNA transcript can now be purified from unincorporated nucleotides and enzymes. Several methods are available:
- Lithium Chloride Precipitation: Efficient for precipitating RNA molecules >100 nucleotides but does not efficiently precipitate nucleotides, making it suitable for post-IVT purification [35].
- Spin Column Purification: A rapid and effective method based on binding RNA to a silica membrane, washing, and eluting in nuclease-free water [35] [40].
- Phenol:Chloroform Extraction and Ethanol Precipitation: A traditional method that effectively removes enzymes and other contaminants [39].
The following workflow diagram summarizes the key stages of the protocol.
Troubleshooting Common Issues
Even with a standardized protocol, challenges can arise. The table below outlines common problems, their causes, and solutions.
Table 2: IVT Troubleshooting Guide
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| No RNA Product | RNase contamination; denatured RNA polymerase; inhibitory contaminants in DNA template [41] [42]. | Use RNase-free technique and reagents; aliquot polymerase to avoid freeze-thaw cycles; ethanol-precipitate template DNA to remove contaminants [41] [42]. |
| Low Yield | Low NTP concentration; suboptimal Mg²⁺ concentration; insufficient incubation time or enzyme amount [41] [40]. | Increase NTP concentration to 2-4 mM each; optimize Mg²⁺:NTP ratio; extend incubation time to 4-6 hours; titrate enzyme concentration [41] [40] [43]. |
| Incomplete (Short) Transcripts | Premature termination due to low NTP concentration; template secondary structure; cryptic termination sites [41] [43]. | Increase concentration of the limiting nucleotide (especially for labeled NTPs); lower reaction temperature to reduce secondary structure; subclone template into a different vector [41] [43]. |
| Longer-than-Expected Transcripts | Incomplete plasmid linearization; template with 3' overhangs [41]. | Verify complete digestion of plasmid on an agarose gel; use a restriction enzyme that produces a 5' overhang or blunt end [41] [36]. |
The Scientist's Toolkit: Essential Reagent Solutions
Selecting high-quality reagents is critical for reproducibility and success. The following table lists key categories of research reagent solutions for IVT.
Table 3: Essential Research Reagent Solutions for IVT
| Reagent Solution | Function | Example Applications/Notes |
|---|---|---|
| Phage RNA Polymerase Kits | All-inclusive systems for high-yield RNA synthesis. | Often include optimized buffer, NTPs, and polymerase (e.g., T7, SP6). Ideal for standard transcript production [36]. |
| RNase Inhibitors | Protects RNA from degradation by RNases during the reaction. | Crucial for maintaining RNA integrity. Added directly to the reaction mix (e.g., RNasin Ribonuclease Inhibitor) [41]. |
| DNase I (RNase-free) | Removes the DNA template after transcription is complete. | Prevents template interference in downstream applications. Used in a post-IVT digestion step [37] [36]. |
| Labeled NTPs | Enables synthesis of non-isotopically labeled probes. | Modified nucleotides (e.g., Digoxigenin-11-UTP, Biotin-16-UTP) are mixed with standard NTPs during reaction setup [37] [38]. |
| RNA Purification Kits | Rapid cleanup of IVT reactions, removing enzymes, salts, and unincorporated NTPs. | Spin columns or magnetic bead-based systems offer quick and efficient purification [35] [40]. |
In molecular biology, the accuracy of downstream applications such as RT-PCR, RNA sequencing, and in situ hybridization is highly dependent on the purity of the RNA sample and the quality of the synthesized probes. This technical guide details two critical post-transcription processing steps: the removal of contaminating genomic DNA (gDNA) from RNA preparations and the purification of in vitro transcribed, digoxigenin (DIG)-labeled RNA probes. Within the broader context of DIG-labeled RNA probe protocol research, mastering these procedures is fundamental for generating reliable data, as impurities can lead to false-positive results in RT-PCR or high background noise in hybridization experiments [44] [20].
DNase Treatment for Genomic DNA Removal
The Necessity of DNase Treatment
The co-isolation of gDNA is a common challenge in RNA preparation. Contaminating gDNA can serve as a template during the PCR amplification step of RT-PCR, producing false-positive signals that can be misinterpreted as gene expression [44]. As shown in Table 1, no RNA isolation method consistently produces DNA-free RNA without DNase treatment, making this a critical step for any application sensitive to DNA contamination [44].
Methods for Detecting DNA Contamination
The most reliable method to detect DNA contamination is to include a "minus-RT" control (a reaction without reverse transcriptase) for each RNA sample in an RT-PCR experiment. If a PCR product is generated in this control, it was amplified from contaminating DNA [44]. While designing PCR primers to span intron-exon boundaries can help distinguish between gDNA (larger product) and cDNA, this is not foolproof due to the potential presence of intron-less pseudogenes [44].
DNase Treatment and Inactivation Protocols
DNase I treatment is the most effective method for removing gDNA contamination. However, the enzyme must be completely inactivated or removed afterward to prevent degradation of newly synthesized cDNA in subsequent steps. The following table compares common DNase inactivation methods [44]:
Table 1: Comparison of Common DNase Inactivation Methods
| Method | Procedure | Drawbacks |
|---|---|---|
| Heat Inactivation | Incubate at 75-80°C for 5 minutes. | Divalent cations in digestion buffer can cause RNA strand scission upon heating, leading to significant RNA degradation. |
| Proteinase K Treatment & Organic Extraction | Proteinase K digestion followed by phenol:chloroform extraction. | Time-consuming; risk of sample loss during extraction; use of hazardous phenol. |
| EDTA Chelation | Adding EDTA to chelate Mg²⁺ and Ca²⁺ ions required for DNase activity. | Complicates downstream enzymatic reactions (RT and PCR) which require Mg²⁺, requiring careful re-calibration of ion concentrations. |
| RNA Purification | Purifying RNA away from DNase using a filter-based column after digestion. | Adds an extra step and cost; on-column digestion may be incomplete due to suboptimal conditions. |
A robust alternative is the use of specialized kits, such as the DNA-free DNase Treatment & Removal Reagents, which include an optimized DNase I and a unique DNase Removal Reagent. This reagent binds the DNase and divalent cations after digestion, allowing their removal via a brief centrifugation without compromising RNA integrity [44]. For high-throughput workflows, magnetic bead-based RNA isolation methods like the RNAqueous-MAG technology offer advantages in consistency and ease of automation, and they can include integrated DNA removal steps [45].
The following workflow outlines the recommended steps for effective DNase treatment and inactivation:
Probe Purification for DIG-Labeled RNA
Purification in the DIG-Labeling Workflow
Following in vitro transcription to synthesize DIG-labeled RNA probes, purification is essential to remove unincorporated DIG-labeled nucleotides, salts, enzymes, and template DNA. This step is critical for reducing background signal and enhancing the sensitivity and specificity of downstream in situ hybridization assays [22] [20].
Standard Purification Methodologies
A common and effective purification strategy involves a combination of phenol-chloroform extraction and ethanol precipitation [22] [20].
- Phenol-Chloroform Extraction: This step separates the RNA probe from proteins (like the RNA polymerase) and other contaminants. The probe is recovered in the aqueous phase.
- Ethanol Precipitation: The RNA probe is precipitated from the aqueous phase using ethanol or isopropanol in the presence of a salt (e.g., ammonium acetate or lithium chloride). This also removes unincorporated nucleotides and salts.
- Column-Based Purification: As an alternative to precipitation, purified template DNA or the final RNA probe can be purified using silica-based columns, which offer convenience and speed [22].
The overall workflow for probe synthesis and purification is as follows:
The Scientist's Toolkit: Essential Reagents
Table 2: Key Research Reagent Solutions for Post-Transcription Processing
| Reagent/Kit | Primary Function | Key Features |
|---|---|---|
| DNA-free DNase Treatment & Removal Reagents [44] | Removal of genomic DNA from RNA samples. | Includes RNase-free DNase I, optimized buffer, and a proprietary removal reagent for efficient enzyme inactivation without phenol or heat. |
| RNAqueous-4PCR Kit [44] | Isolation of DNA-free RNA. | A phenol-free, glass-fiber filter-based kit that includes reagents for RNA isolation and subsequent DNA removal, yielding "RT-PCR ready" RNA. |
| RNAqueous-MAG [45] | High-throughput RNA isolation. | Magnetic bead-based technology for automated, consistent RNA purification with integrated DNA removal; ideal for processing many samples. |
| TRIzol Reagent [46] [47] | RNA isolation via guanidinium-phenol extraction. | Effective for difficult samples (e.g., snake venom); yields high RNA quantity but may require subsequent DNase treatment and cleaning [46]. |
| Phenol-Chloroform [20] | Purification of nucleic acids from enzymatic reactions. | Used to purify template DNA and synthesized RNA probes by separating nucleic acids from proteins and other contaminants. |
| RNAscope Multiplex Fluorescent Reagent Kit [48] [49] | Detection of RNA in situ. | A ready-to-use kit for sensitive detection of target RNA in fixed tissues using a proprietary signal amplification system. |
Robust and reproducible results in molecular biology hinge on meticulous post-transcription processing. The mandatory application of DNase treatment is a non-negotiable step for ensuring the integrity of RNA samples in sensitive applications like RT-PCR. Similarly, rigorous purification of DIG-labeled probes is paramount for the success of hybridization-based techniques. By understanding the principles behind these methods and leveraging the appropriate tools from the scientific toolkit, researchers can significantly enhance the reliability and quality of their data in DIG-labeled RNA probe research and beyond.
This technical guide frames the core protocols for In Situ Hybridization (ISH), Northern Blotting, and the Electrophoretic Mobility Shift Assay (EMSA) within a broader research context focused on digoxigenin (DIG)-labeled RNA probes. The choice of detection methodology is pivotal in molecular biology, influencing the sensitivity, specificity, and applicability of an assay. Non-radioactive probes, particularly those labeled with digoxigenin, have become a cornerstone of modern laboratory practice due to their safety, stability, and compatibility with sensitive detection methods [50] [51].
Digoxigenin-labeled RNA probes offer a high degree of sensitivity and specificity for the detection of nucleic acids. When integrated into the protocols for ISH, Northern blotting, and EMSA, they provide a versatile toolset for researchers and drug development professionals to visualize gene expression, quantify specific RNA molecules, and analyze protein-nucleic acid interactions. This guide details the principles, optimized methodologies, and practical applications of these techniques, emphasizing the use of DIG-labeled reagents to ensure reliable and reproducible results.
The following table summarizes the primary applications and key characteristics of the three core techniques discussed in this guide.
Table 1: Core Techniques for Nucleic Acid and Protein Analysis
| Technique | Primary Application | Key Outcome | Probe Type | Sample Type |
|---|---|---|---|---|
| In Situ Hybridization (ISH) | Localization of specific DNA/RNA sequences within cells/tissues [4] [51] | Spatial distribution of target nucleic acids within a morphological context | DIG-labeled RNA, DNA, oligonucleotides [4] [50] | Tissue sections, whole mounts (FFPE or frozen) [4] |
| Northern Blotting | Detection and quantification of specific RNA molecules [52] [53] | RNA transcript size and abundance | DIG-labeled RNA (highly sensitive), DNA, oligonucleotides [53] | Total or poly(A) RNA extracted from tissues or cells [52] |
| EMSA (Gel Shift) | Analysis of protein-nucleic acid interactions [54] [55] | Detection and characterization of DNA/RNA-binding proteins | Typically radiolabeled; non-radioactive DIG-labeled nucleic acids can be used [54] | Nuclear or whole-cell extracts, purified proteins [54] [56] |
The fundamental workflow for experiments utilizing digoxigenin-labeled probes involves a series of critical steps, from sample preparation to final detection. The following diagram illustrates the overarching logical relationship and sequence of these procedures.
In Situ Hybridization (ISH) with DIG-Labeled RNA Probes
Principle and Applications
In Situ Hybridization (ISH) enables the visualization of the spatial and temporal localization of specific nucleic acid sequences within cells, tissue sections, or entire organisms (whole mounts) [4] [51]. This technique is fundamental to understanding gene expression patterns and cellular heterogeneity in diverse fields such as developmental biology, neurobiology, and disease pathology. The use of DIG-labeled RNA probes is a preferred approach due to the high sensitivity and low background afforded by RNA-RNA hybrids, which are stable and allow for stringent washing conditions to minimize non-specific signal [20] [50].
Detailed Protocol
The following workflow details the key stages of an ISH protocol using DIG-labeled RNA probes on paraffin-embedded tissue sections.
Table 2: Key Research Reagents for In Situ Hybridization
| Reagent/Category | Specific Example | Function/Purpose |
|---|---|---|
| Tissue Fixative | 4% Paraformaldehyde [20] | Preserves tissue morphology and nucleic acid integrity |
| Permeabilization Agent | Proteinase K [4] | Digests proteins to allow probe access to target nucleic acids |
| Hybridization Buffer | Formamide, Salts, Dextran Sulfate [4] | Creates optimal conditions for specific probe-target hybridization |
| Labeled Probe | DIG-labeled Antisense RNA [4] [20] | Complementary molecule for detecting specific target mRNA |
| Blocking Agent | BSA, Milk, or Serum [4] | Reduces non-specific antibody binding to minimize background |
| Detection Antibody | Anti-DIG Antibody conjugated to Alkaline Phosphatase [51] | Binds to DIG label for subsequent colorimetric or fluorescent detection |
| Wash Buffer | SSC (Saline Sodium Citrate) [4] | Removes unbound and non-specifically bound probe; controls stringency |
Key Steps Explained:
- Sample Storage and Preparation: Tissue samples must be fixed immediately after collection to preserve RNA integrity. Common methods include flash-freezing or, more routinely for ISH, fixation in formalin followed by paraffin embedding (FFPE) [4] [51]. For FFPE tissues, sections are mounted on slides and must be deparaffinized in xylene and rehydrated through a graded ethanol series to water before hybridization [4].
- Probe Design and Synthesis: For optimal sensitivity and specificity, RNA probes should be 250–1,500 bases long, with approximately 800 bases being ideal [4]. The probe (antisense strand) is synthesized by in vitro transcription from a linearized DNA template, incorporating DIG-labeled UTP. A sense strand probe should be synthesized in parallel for use as a negative control [4] [20].
- Antigen Retrieval and Permeabilization: To make the target mRNA accessible, tissue sections are treated with proteinase K (e.g., 20 µg/mL for 10–20 min at 37°C). The concentration and time require optimization to balance signal with tissue morphology preservation [4].
- Hybridization: The DIG-labeled RNA probe is denatured and applied to the tissue in a hybridization buffer containing 50% formamide and other components to promote specificity. Hybridization is typically performed overnight at an optimized temperature (e.g., 55-65°C) [4].
- Stringency Washes: Post-hybridization, a series of washes are performed to remove unbound and non-specifically bound probe. Washes with 50% formamide in 2x SSC at 37-45°C, followed by lower concentration SSC buffers (e.g., 0.1-2x SSC) at elevated temperatures, are critical for reducing background [4].
- Immunological Detection: The hybridized DIG-labeled probe is detected by incubating with an anti-DIG antibody conjugated to an enzyme, such as alkaline phosphatase. This is followed by incubation with a colorimetric substrate (e.g., NBT/BCIP), which produces an insoluble precipitate at the site of probe hybridization [4] [51].
Northern Blotting with DIG-Labeled RNA Probes
Principle and Applications
Northern blotting is a standard technique used to detect and quantify specific RNA molecules, providing information on both transcript size and abundance [52] [53]. It remains the preferred method for identifying alternatively spliced transcripts. While historically less sensitive than nuclease protection assays or RT-PCR, the use of DIG-labeled RNA probes in combination with optimized hybridization buffers can dramatically increase sensitivity, allowing detection of as few as 100,000 molecules on a blot [53]. Research indicates that RNA probes can provide a 10-fold increase in sensitivity compared to random-primed DNA probes under standard conditions [53].
Detailed Protocol
The Northern blotting procedure involves separating RNA by size and then transferring it to a solid membrane for detection with a labeled probe.
Table 3: Key Research Reagents for Northern Blotting
| Reagent/Category | Specific Example | Function/Purpose |
|---|---|---|
| Denaturing Agent | Formaldehyde or Glyoxal [52] [53] | Prevents RNA secondary structure formation during electrophoresis |
| Separation Matrix | Denaturing Agarose Gel [52] [57] | Separates RNA molecules based on size |
| Transfer Membrane | Positively Charged Nylon Membrane [53] [57] | Immobilizes RNA for subsequent hybridization |
| Transfer Buffer | 10x SSC [52] [57] | Medium for capillary or vacuum transfer of RNA from gel to membrane |
| Hybridization Buffer | ULTRAhyb Ultrasensitive Hybridization Buffer [53] | Maximizes sensitivity and speed of hybridization |
| Labeled Probe | DIG-labeled RNA (by in vitro transcription) [53] | High-sensitivity probe for target mRNA detection |
| Wash Buffer | SSC + SDS [52] | Removes non-specifically bound probe after hybridization |
Key Steps Explained:
- RNA Isolation and Gel Electrophoresis: High-quality, intact total or poly(A) RNA is essential. RNA samples (typically 5-30 µg) are denatured using formaldehyde or glyoxal/DMSO and separated by size via electrophoresis through a denaturing agarose gel [52] [53] [57].
- Transfer and Immobilization: The separated RNA is transferred from the gel to a positively charged nylon membrane. While passive capillary transfer is common, rapid alkaline downward or vacuum transfer methods are more efficient. The RNA is then permanently immobilized onto the membrane by UV crosslinking or baking [53] [57].
- Hybridization and Washing: The membrane is pre-hybridized with a blocking solution and then hybridized with the DIG-labeled RNA probe. Using specialized hybridization buffers like ULTRAhyb can increase sensitivity up to 100-fold and reduce required hybridization times [53]. Post-hybridization, the membrane is washed with SSC buffers containing SDS to remove non-specifically bound probe. The stringency of these washes (temperature and salt concentration) is critical for specificity [52] [53].
- Detection and Analysis: The hybridized DIG-labeled probe is detected using an anti-DIG antibody conjugated to alkaline phosphatase, followed by a chemiluminescent, fluorescent, or colorimetric substrate. The signal is then captured and quantified. The distance migrated by the signal relative to an RNA ladder provides information on transcript size [53] [57].
Electrophoretic Mobility Shift Assay (EMSA)
Principle and Applications
The Electrophoretic Mobility Shift Assay (EMSA), also known as a gel shift assay, is a core technique for studying protein-nucleic acid interactions [54] [55]. Its principle is based on the observation that a protein bound to a nucleic acid (DNA or RNA) retards its electrophoretic mobility during non-denaturing gel electrophoresis. While EMSA has been traditionally performed with radiolabeled probes, non-radioactive detection methods using DIG-labeled nucleic acids are robust and sensitive alternatives [54]. EMSA is widely used to detect transcription factors, study binding kinetics, and determine binding specificity.
Detailed Protocol
The classical EMSA protocol involves incubating a protein extract with a labeled nucleic acid probe and analyzing the resulting complexes by gel electrophoresis.
Table 4: Key Research Reagents for Electrophoretic Mobility Shift Assay (EMSA)
| Reagent/Category | Specific Example | Function/Purpose |
|---|---|---|
| Nucleic Acid Probe | DIG-labeled DNA oligonucleotide or RNA transcript [54] | Target for protein binding; can be synthesized or PCR-generated |
| Protein Source | Nuclear Extract, Whole Cell Extract, Purified Protein [54] [56] | Source of DNA/RNA-binding protein(s) of interest |
| Binding Buffer | Tris-HCl, KCl, Glycerol, DTT, MgCl₂ [55] | Provides optimal ionic strength and pH for protein-nucleic acid binding |
| Non-specific Competitor | Poly(dI•dC), Sonicated Salmon Sperm DNA [54] | Binds non-specific nucleic acid-binding proteins to reduce background |
| Specific Competitor | Unlabeled probe (200-fold molar excess) [54] | Confirms binding specificity by competing for the protein of interest |
| Gel Matrix | Non-denaturing Polyacrylamide Gel [54] [55] | Resolves protein-nucleic acid complexes from free probe |
| Electrophoresis Buffer | Tris-Borate-EDTA (TBE) or Tris-Glycine [55] | Maintains native state of complexes during separation |
Key Steps Explained:
- Probe Preparation: A short, well-defined DNA oligonucleotide or RNA transcript containing the target binding site is synthesized and labeled with digoxigenin. This can be achieved during chemical synthesis for oligonucleotides or via in vitro transcription for RNA probes [54].
- Protein Extraction: The source of the DNA/RNA-binding protein is prepared. For transcription factors, this often involves preparing a nuclear extract from cultured cells or tissues [56].
- Binding Reaction: The labeled probe is incubated with the protein extract in a binding buffer. The order of addition is critical: non-specific competitor DNA (e.g., poly(dI•dC)) is added first to adsorb non-specific proteins, followed by the protein extract, and finally the labeled probe. To confirm specificity, a parallel reaction includes a 200-fold molar excess of unlabeled identical probe (specific competitor) [54].
- Electrophoresis and Transfer: The binding reactions are loaded onto a non-denaturing polyacrylamide gel. The gel matrix provides a "caging" effect that helps stabilize transient protein-nucleic acid complexes during electrophoresis [54] [55]. Free probe migrates rapidly, while protein-bound complexes are retarded. For non-radioactive detection, the resolved complexes are then transferred from the gel to a positively charged nylon membrane [54].
- Detection and Analysis: The DIG-labeled nucleic acid on the membrane is detected using an anti-DIG antibody conjugated to alkaline phosphatase and a chemiluminescent substrate. The resulting signal reveals the presence of protein-nucleic acid complexes. A "supershift" can be performed by including a specific antibody in the binding reaction; if this antibody binds to the protein in the complex, it will cause a further retardation in mobility, helping to identify the protein [54] [56].
The integration of digoxigenin-labeled RNA probes into the core protocols of ISH, Northern blotting, and EMSA provides a powerful, safe, and highly sensitive methodology for advancing research in gene expression and nucleic acid-protein interactions. This technical guide has outlined the principles, detailed protocols, and key reagents for each technique, providing a framework for their application in drug development and basic research. Mastery of these foundational methods, with a focus on optimizing probe design and detection stringency, remains essential for generating reliable and meaningful data in molecular biology.
Probe Storage Conditions and Long-Term Stability
Within the comprehensive workflow of digoxigenin (DIG)-labeled RNA probe protocols, proper storage conditions and long-term stability management represent critical pillars supporting experimental reproducibility and reagent integrity. For researchers, scientists, and drug development professionals, understanding these factors is paramount for maintaining probe functionality across extended timelines, thereby ensuring the reliability of gene expression analysis in applications ranging from basic research to pharmaceutical validation. This technical guide synthesizes current methodologies and evidence-based practices to establish optimized storage parameters that preserve the structural integrity and hybridization capacity of DIG-labeled RNA probes, ultimately supporting data consistency in longitudinal studies and multi-phase research projects.
Fundamental Storage Parameters for DIG-Labeled RNA Probes
The long-term stability of DIG-labeled RNA probes depends on meticulous control of several interconnected parameters that collectively prevent degradation and preserve functionality. RNase contamination represents the most significant threat to RNA probe integrity, as this enzyme rapidly destroys RNA molecules. Laboratory practice must therefore include rigorous RNase avoidance protocols: using sterile, RNase-free tubes and pipette tips, wearing gloves during all handling procedures, and maintaining tightly sealed containers during both storage and usage [58]. Environmental RNases present on glassware, reagents, and even operators can compromise probe stability unless these preventive measures are consistently implemented [4].
Temperature control constitutes another critical factor in probe preservation. Most protocols recommend storage at -20°C for long-term preservation of DIG-labeled RNA probes [58]. This temperature effectively slows enzymatic and chemical degradation processes that would otherwise compromise probe integrity. Additionally, avoiding repeated freeze-thaw cycles is essential, as these processes can cause RNA fragmentation through ice crystal formation and repeated stress on molecular structures. For frequently used probes, aliquoting into single-use volumes prevents quality deterioration from recurrent temperature cycling [58].
The storage buffer composition provides the chemical environment necessary for maintaining probe stability. After synthesis, DIG-labeled RNA probes are typically resuspended in pre-hybridization stock solution at standardized concentrations, often around 10 μg/mL [59]. This solution provides appropriate pH buffering and chemical conditions that stabilize the RNA molecules during storage. For purified probes, RNase-free buffers or nuclease-free water serve as appropriate suspension media, provided they maintain stable pH and lack contaminating nucleases [58].
Table 1: Key Storage Parameters for DIG-Labeled RNA Probes
| Parameter | Optimal Condition | Purpose | Implementation Considerations |
|---|---|---|---|
| Temperature | -20°C long-term storage | Slows degradation processes | Consistent maintenance; avoid temperature fluctuations |
| Freeze-Thaw Cycles | Minimize or eliminate | Prevents RNA fragmentation | Aliquot into single-use volumes |
| RNase Control | Strict RNase-free environment | Prevents RNA degradation | Use sterile tubes, wear gloves, use certified RNase-free reagents |
| Storage Buffer | Pre-hybridization solution or RNase-free buffers | Provides stabilizing chemical environment | Maintain proper pH and composition |
| Physical State | Liquid aliquots or ethanol-stored slides | Adapts to application requirements | Match storage format to intended use |
Quantitative Stability Assessment and Testing Methods
Evaluating probe stability requires both quantitative measurement and functional assessment to ensure hybridization capability remains intact. Spectrophotometric analysis at 260 nm (A260) provides a fundamental quantitative approach, where an A260 value of 1.0 corresponds to approximately 40 μg/mL of single-stranded RNA [58]. This method allows researchers to track concentration changes over time that might indicate degradation, though it does not specifically measure DIG incorporation or functional integrity.
For functional stability assessment, control hybridizations using samples with known expression patterns provide the most reliable data. As shown in validation studies, probes stored under optimal conditions consistently produce the expected signal intensity and localization patterns in situ hybridization experiments [59]. This functional testing should be conducted periodically for probes stored beyond recommended timeframes, with documentation of signal quality, background levels, and any morphological deterioration.
Electrophoretic quality control represents another valuable assessment method, enabling visualization of RNA integrity. Undegraded probes should appear as distinct bands without smearing, indicating preservation of full-length sequences. Many laboratories establish internal stability timelines based on regular quality control checks, with typical functional stability of 12-24 months reported when proper storage protocols are consistently maintained [58].
Table 2: Stability Assessment Methods for DIG-Labeled RNA Probes
| Method | Parameters Measured | Acceptance Criteria | Frequency Recommendation |
|---|---|---|---|
| Spectrophotometry | RNA concentration | Stable values over time; A260/A280 ratio ~2.0 | Pre-experiment and quarterly for long-term storage |
| Electrophoresis | RNA integrity | Sharp bands without smearing | Every 6 months |
| Control Hybridization | Functional capability | Expected signal pattern with low background | Annually or pre-critical experiments |
| Visual Inspection | Solution clarity | No precipitate or discoloration | Before each use |
Storage Conditions Across Experimental Contexts
Pre-hybridization Probe Storage
For DIG-labeled RNA probes before their application in hybridization experiments, the consolidated protocol recommends storage in pre-hybridization stock solution at concentrations of approximately 10 μg/mL at -20°C [59]. This approach maintains probes in a chemical environment similar to their working conditions, promoting stability. The Jena Biosciences HighYield T7 Digoxigenin RNA Labeling Kit protocol further emphasizes that storage at -20°C should avoid freeze-thaw cycles, with aliquoting strongly recommended for probes intended for repeated use [58].
Post-hybridization Sample Preservation
For samples that have already undergone the hybridization process, different storage considerations apply. According to Abcam's in situ hybridization protocol, slides should not be stored dry at room temperature, as this arrangement promotes degradation and increases background signal in future detection attempts. Instead, the protocol recommends storing slides in 100% ethanol at -20°C or in plastic boxes covered with saran wrap at -20°C to -80°C [4]. These conditions effectively preserve hybridized samples for several years, enabling long-term archival of experimental results and facilitating future re-analysis when required.
Commercial Kit Storage Considerations
Commercial DIG-labeling kits have specific storage requirements that users must follow to maintain component integrity. The Jena Biosciences HighYield T7 Digoxigenin RNA Labeling Kit, for instance, requires storage of all components at -20°C with protection from repeated freeze-thaw cycles [58]. The kit has a specified shelf life of 12 months when stored properly, providing a benchmark for expected stability under optimal conditions. Similar timeframes likely apply to other commercial labeling systems, though manufacturers' specifications should always take precedence.
Experimental Protocols for Stability Assessment
Protocol: Accelerated Stability Testing
Accelerated stability testing helps predict long-term storage potential under controlled stress conditions. This approach adapts pharmaceutical stability testing principles to research reagents [60].
- Aliquot Preparation: Divide the DIG-labeled RNA probe solution into multiple identical aliquots
- Stress Conditions: Expose aliquots to varying temperatures (4°C, 25°C, 37°C) for defined periods
- Periodic Sampling: Remove samples at predetermined timepoints (0, 1, 2, 4 weeks)
- Functional Assessment: Test hybridization capability using standardized control tissue
- Integrity Analysis: Evaluate RNA integrity via electrophoresis and spectrophotometry
- Data Correlation: Establish relationship between accelerated conditions and long-term stability
This protocol enables researchers to establish evidence-based expiration timeframes for their specific probe preparations and storage conditions.
Protocol: Freeze-Thaw Cycle Impact Assessment
Understanding the effect of temperature cycling is crucial for establishing proper handling procedures.
- Baseline Testing: Assess initial probe quality via electrophoresis and functional hybridization
- Cycle Simulation: Subject aliquots to defined freeze-thaw cycles (1, 3, 5, 10 cycles)
- Intercycle Storage: Maintain -20°C storage between cycles with controlled thawing at 4°C
- Post-cycle Analysis: Evaluate degradation compared to baseline using multiple methods
- Threshold Establishment: Determine the maximum acceptable cycle count before quality compromise
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for DIG-Labeled RNA Probe Workflows
| Reagent/Kit | Function | Storage Conditions | Stability Considerations |
|---|---|---|---|
| HighYield T7 Digoxigenin RNA Labeling Kit (Jena Biosciences) | Production of DIG-labeled RNA probes | -20°C for all components | 12-month shelf life; avoid freeze-thaw cycles |
| DIG-11-UTP Labeling Nucleotide | Incorporation into RNA during in vitro transcription | -20°C, protected from light | Stable for kit lifetime; sensitive to repeated thawing |
| RNA Cleanup Columns (e.g., Zymo Research, NEB) | Purification of transcribed probes | Room temperature (columns) with specific buffers | Follow manufacturer expiry dates |
| Anti-Digoxigenin Antibody (Roche) | Detection of hybridized probes | 4°C for short-term; follow manufacturer guidance | Activity decreases with improper storage; avoid bacterial contamination |
| Blocking Reagent (Roche) | Reduction of non-specific binding | Room temperature or as specified | Solutions should be prepared fresh or stored according to protocols |
Stability Optimization Workflow
The following diagram illustrates the complete workflow for optimizing DIG-labeled RNA probe stability from synthesis through storage and quality verification:
Proper storage conditions and long-term stability management of DIG-labeled RNA probes require integrated strategies addressing temperature control, RNase prevention, appropriate buffering, and rigorous quality assessment. Implementation of the protocols and practices outlined in this technical guide provides researchers with a systematic approach to maximizing probe longevity and functional integrity. Through consistent application of these evidence-based methods, scientific and drug development professionals can ensure reagent reliability across extended timelines, thereby supporting reproducible research outcomes in gene expression analysis.
Troubleshooting DIG RNA Labeling: Solving Common Problems
Within the framework of digoxigenin (DIG)-labeled RNA probe research, agarose gel electrophoresis serves as an indispensable quality control checkpoint [22]. The synthesis of RNA probes labeled with digoxigenin for techniques such as whole mount in situ hybridization (WISH) is a multi-step process, the success of which hinges on the integrity and purity of the final nucleic acid product [22]. Analyzing the resulting electrophoresis gel is therefore not merely a procedural step, but a critical diagnostic tool. It allows researchers to confirm that the in vitro transcription reaction has produced a full-length, intact probe and to identify common issues that can compromise experimental outcomes.
Interpreting these gels, however, often presents challenges. The presence of multiple bands or DNA fragments migrating at unexpected sizes frequently confounds researchers [61] [62]. Accurate interpretation is essential, as it is typically the final, low-cost verification step before committing to more expensive and time-consuming procedures like sequencing or the use of the probe in sensitive hybridization assays [61]. Misinterpretation at this stage can lead to the use of suboptimal probes, resulting in weak signals, high background noise, or failed experiments. This guide provides an in-depth technical analysis of these common electrophoretic anomalies, offering researchers a structured methodology for diagnosis and resolution.
Fundamental Principles of Gel Electrophoresis Interpretation
Gel electrophoresis separates DNA and RNA fragments based on their size and charge as they migrate through an agarose matrix under an electric field [62]. The porous network of agarose polymers acts as a molecular sieve, allowing smaller fragments to travel faster and further than larger ones [62]. The distance migrated by a nucleic acid molecule is inversely proportional to the logarithm of its molecular weight.
A key component of any gel is the DNA ladder, a mixture of DNA fragments of known sizes which serves as a reference standard [61]. To estimate the size of an unknown PCR amplicon, you can plot an imaginary line to the adjacent DNA ladder. For instance, if a band is positioned halfway between the 500 bp and 600 bp bands of a ladder, its size is approximately 550 bp [61]. For accurate analysis, it is considered best practice to load a DNA ladder in the first and last lanes of the gel. This helps identify any abnormalities in migration, such as smiling or crooked bands, and allows for more precise size estimation of amplicons across the entire gel [61].
Table: Essential Components for Gel Electrophoresis Analysis
| Component or Step | Function/Role in Analysis | Key Considerations |
|---|---|---|
| Agarose Gel | Porous matrix that separates nucleic acids by size [62]. | Gel percentage must be appropriate for the expected fragment size for optimal resolution. |
| DNA Ladder | Molecular weight standard for estimating fragment sizes [61]. | Must be run on the same gel, ideally in the first and/or last lane. |
| DNA Stain (e.g., EtBr, GelGreen) | Enables visualization of nucleic acid bands under specific light. | Some stains are more sensitive to DNA overloading, which can cause smearing [61]. |
| Digital Imaging | Creates a permanent record for annotation and analysis [61]. | Image should be captured in a dark environment with a clean filter to reduce glare and improve clarity. |
| Control Lanes | Provides benchmarks for expected results (e.g., undigested plasmid, digested plasmid) [62]. | Crucial for distinguishing between different molecular forms (e.g., supercoiled vs. linear). |
Troubleshooting Multiple Bands and Unexpected Sizes
The appearance of multiple bands or bands at unexpected sizes can stem from various issues related to the sample, reagents, or electrophoresis process itself. The following table provides a structured overview of common issues and their solutions.
Table: Troubleshooting Common Gel Electrophoresis Anomalies
| Observation | Potential Cause | Recommended Solution / Diagnostic Action |
|---|---|---|
| Multiple bands in an uncut plasmid lane [62] | Different structural forms of plasmid DNA: Supercoiled (CCC), Linear, and Open Circular (OC). | Expect multiple forms. Supercoiled (CCC) runs fastest, followed by linear, then Open Circular (OC) [62]. |
| Single band in digested plasmid, but at wrong size | Incomplete digestion by restriction enzymes. | Ensure digestion reaction is complete; check enzyme activity and incubation time. |
| Smearing across a wide range of sizes | Degraded DNA/RNA (nucleases) [61] or too much DNA loaded, overloading the gel [61]. | Use fresh, high-quality samples. Troubleshoot extraction protocol. Load less DNA. |
| Smearing | Excessive heating during electrophoresis, melting the gel [61]. | Run gel at a lower voltage; use a buffer with higher ionic strength; ensure adequate running buffer volume. |
| Bands in negative control lane | Contamination from primers (primer-dimer) [62] or foreign DNA/RNA. | Redesign primers. Use filter tips, dedicated reagents, and clean workspace to prevent contamination. |
| All bands (including ladder) are faint | Insufficient staining, degraded stain, or not enough nucleic acid loaded. | Check stain concentration and integrity. Increase loading amount. Ensure stain was well-mixed into agarose [61]. |
| Crooked or "smiling" bands | Uneven electric field or overheating in the center of the gel [61]. | Ensure the gel is run on a level surface. Run at a lower voltage to reduce heat. Check that electrodes are straight and clean [61]. |
Advanced Analysis: Plasmid DNA Forms
A classic example of expected multiple bands arises when running uncut plasmid DNA. A plasmid can exist in several physical conformations, each with distinct migration properties [62]:
- Covalently Closed Circular (CCC) Monomer: This is the intact, supercoiled form, which is compact and migrates the fastest [62].
- Linear Monomer: Results from a double-strand break and migrates slower than the CCC form [62].
- Open Circular (OC) Monomer: Results from a single-strand break (nick), is relaxed, and is the least compact, migrating the slowest [62].
- Multimeric Forms: Dimers or concatemers, which are larger and will migrate slower than their monomeric counterparts [62].
Therefore, observing two or three bands in an undigested plasmid sample is often normal. A completely digested plasmid, in contrast, should appear as a single, sharp band corresponding to the linearized form [62].
Connecting Gel Quality to DIG-Labeled RNA Probe Research
In the specific context of synthesizing DIG-labeled RNA probes, gel electrophoresis analysis takes on an added layer of importance. The protocol involves using a linearized plasmid DNA template for in vitro transcription to produce the RNA probe [22]. The quality of this starting template and the final RNA product are both assessed on a gel.
A clean, single band of the linearized plasmid DNA confirms successful and complete restriction digest. If the template DNA shows multiple bands (e.g., residual supercoiled or nicked circular DNA), it can lead to aberrant transcription products and ultimately, a heterogeneous probe population [62]. Similarly, analysis of the synthesized DIG-labeled RNA probe should reveal a single, dominant band of the expected size. Smearing or multiple lower molecular weight bands indicate RNA degradation or aborted transcription, which can significantly reduce the sensitivity and specificity of the probe in subsequent hybridization experiments like WISH [22].
The following workflow diagram outlines the key steps from experiment setup to data interpretation, highlighting where common issues arise and how to connect observations to their root causes.
The Scientist's Toolkit: Essential Reagents and Materials
Successful gel electrophoresis and accurate interpretation depend on the quality and appropriate use of key reagents. The following table details essential materials for this field.
Table: Key Research Reagent Solutions for Gel Electrophoresis
| Reagent/Material | Function | Technical Notes |
|---|---|---|
| Agarose | Forms the porous gel matrix for size-based separation of nucleic acids [62]. | Gel percentage (e.g., 1%, 2%) must be optimized for the size range of target fragments. |
| DNA/RNA Ladder | Provides molecular weight standards for estimating the size of unknown samples [61]. | A 100 bp ladder is common for analyzing PCR products and small fragments. |
| Electrophoresis Buffer (e.g., TBE, TAE) | Provides the ions necessary to conduct current and maintain stable pH during the run. | TBE provides better resolution for small fragments (< 1 kb); TAE is more common for larger fragments. |
| Nucleic Acid Stain | Intercalates with DNA/RNA to allow visualization under UV or blue light. | Includes ethidium bromide (EtBr), SYBR Safe, GelRed, GelGreen. Safety and sensitivity profiles vary. |
| Loading Dye | Contains a dense agent (e.g., glycerol) to sink samples into wells and tracking dyes to monitor migration. | Typically includes bromophenol blue and/or xylene cyanol. |
| Restriction Enzymes | Used to linearize plasmid DNA templates for RNA probe synthesis [22]. | Critical for creating a defined template; incomplete digestion leads to multiple bands and poor probes [62]. |
| RNA Polymerase (e.g., T7, SP6) | Drives in vitro transcription from a linearized template to synthesize the RNA probe [22]. | Enzyme choice is determined by the promoter sequence in the plasmid vector. |
| Digoxigenin-Labeled NTPs | Modified nucleotides incorporated during transcription to label the RNA probe [22]. | The DIG hapten allows for immunodetection in subsequent assays like in situ hybridization. |
Mastering the interpretation of gel electrophoresis results, particularly the confounding presence of multiple bands and unexpected sizes, is a non-negotiable skill for researchers engaged in precise molecular techniques like the synthesis of DIG-labeled RNA probes. By systematically evaluating gel quality, using controls effectively, and understanding the underlying molecular biology—such as the different conformations of plasmid DNA—scientists can accurately diagnose issues in their experimental workflow. This rigorous approach to analysis ensures that only high-quality, specific probes are used in downstream applications, thereby safeguarding the integrity and success of critical experiments in drug development and basic research.
In digoxigenin (DIG)-labeled RNA probe research, achieving high probe yield is critical for sensitive detection in applications such as northern blotting, in situ hybridization, and microarray analysis. Low probe yield, often resulting in faint staining and high background, can compromise experimental integrity. This technical guide synthesizes recent advances to provide a systematic framework for optimizing template quality and reaction conditions, thereby enhancing the efficiency and reliability of DIG-labeled RNA probe synthesis. The strategies outlined herein are framed within the broader objective of establishing robust, reproducible protocols for spatial transcriptomics and molecular diagnostics.
Core Principles of Probe Generation and Yield Challenges
The synthesis of DIG-labeled RNA probes typically involves in vitro transcription, where RNA polymerase synthesizes RNA strands complementary to a DNA template, incorporating DIG-labeled nucleotide analogs. The key challenges leading to low yield can be categorized into two areas:
- Template-Related Issues: Impure template DNA, short template length, or suboptimal sequence composition (e.g., high GC-content or repetitive elements) can hinder polymerase progression and reduce full-length transcript production.
- Reaction Condition Limitations: Suboptimal concentrations of nucleotides (especially DIG-UTP), magnesium ions, or RNA polymerase, as well as incorrect incubation temperature or duration, can lead to premature transcription termination, truncated probes, or low labeling efficiency.
The following sections detail targeted optimization strategies to overcome these challenges.
Template Optimization Strategies
The quality and design of the DNA template are foundational to successful high-yield probe synthesis.
Template Design and Sequence Considerations
Probe design is a critical first step. For RNA FISH methods like MERFISH and Stellaris, a primary challenge is designing a sufficient number of effective probes against a target sequence.
Increasing Probe Count: When initial designs yield fewer than the recommended 25-48 probes, several adjustments can increase the number of candidate probes [63]:
- Reduce Probe Spacing: Decreasing the minimum nucleotide spacing between probe binding sites from the default of 2 to 1 can immediately increase the number of potential binding sites, except when using certain dyes like CAL Fluor Red 635 [63].
- Vary Probe Length: For sequences with non-uniform GC content, employing a mixed-length probe set (18-22 nucleotides) can improve coverage. GC-rich regions are better targeted by shorter 18-19 mer probes, while AT-rich regions are more effectively hybridized with longer 21-22 mer probes [63].
- Expand Target Sequence: Increasing the input sequence length by including untranslated regions (UTRs) or, for intron-targeting, flanking exons provides more sequence "real estate" for probe design [63].
Managing Sequence Composition: The probe designer selects regions based on a melting temperature range, making sequences with skewed GC-content difficult [63]. Furthermore, repetitive sequences are automatically masked to prevent off-target binding.
- Relax Masking Stringency: Systematically reducing the masking level from the default (often level 5) to levels 4 or 3 can make more of the input sequence available for probe design. If necessary, further reduction to levels 2 or 1 disables species-specific repeat masking, but this must be followed by rigorous specificity checks using BLAST against the relevant transcriptome to remove probes with high complementarity to non-target RNAs [63].
Template Quality and Preparation
A pure, linearized template is essential for efficient in vitro transcription.
- Purification: Use high-purity plasmid DNA or PCR products as template. Purification via gel extraction or column-based kits is recommended to remove salts, enzymes, and impurities that inhibit polymerase activity.
- Linearization: The plasmid template must be completely linearized downstream of the insert to ensure a defined transcript length and prevent continuous transcription of the vector backbone. Verify complete digestion by agarose gel electrophoresis before proceeding to the transcription reaction.
Reaction Condition Optimization
Fine-tuning the biochemical environment of the in vitro transcription reaction is crucial for maximizing yield and label incorporation.
Biochemical Parameters
Systematic exploration of reaction components can lead to significant yield improvements. Research in multiplexed RNA imaging has shown that signal brightness—a proxy for probe assembly efficiency—depends on hybridization conditions and can be optimized [34].
- Hybridization Buffer Composition: For FISH protocols, the formamide concentration in the hybridization buffer is a key denaturant that balances specificity and efficiency. Empirical testing across a range of formamide concentrations (e.g., with a fixed temperature of 37°C) is necessary to identify the optimal concentration for a given probe set [34]. Signal brightness can remain relatively stable across a range of optimal formamide concentrations [34].
- Nucleotide Ratios: The ratio of DIG-labeled UTP to unlabeled UTP must be optimized. High concentrations of DIG-UTP can stall transcription, while low concentrations result in poor detection sensitivity. A typical molar ratio of DIG-UTP to UTP ranges from 1:3 to 1:5.
- Magnesium Ion Concentration: Mg²⁺ is a cofactor for RNA polymerase. Deviation from the optimal concentration (typically 4-8 mM) can reduce transcript yield. A titration experiment is recommended to determine the ideal concentration for a specific template and polymerase combination.
- Reaction Duration and Temperature: Standard incubation is 2 hours at 37°C. Extending the duration to 4-6 hours can increase yield, though it may also increase the rate of abortive transcripts. Maintaining a consistent temperature is critical.
Table 1: Optimization Parameters for In Vitro Transcription Reaction Components
| Parameter | Typical Range | Optimization Guidance | Impact on Yield |
|---|---|---|---|
| DIG-UTP:UTP Ratio | 1:3 to 1:5 | Titrate while holding total NTP concentration constant. | High ratio improves label density but can inhibit polymerase; low ratio reduces sensitivity. |
| MgCl₂ Concentration | 4 - 8 mM | Test in 1 mM increments. | Suboptimal levels reduce polymerase processivity; excess can promote non-specific transcription. |
| Incubation Time | 2 - 6 hours | Increase time if yield is low and template is abundant. | Longer times can increase full-length product but may also increase degraded product. |
| Polymerase Amount | 1 - 2 µL/reaction | Increase within manufacturer's guidelines for complex templates. | Insufficient enzyme limits output; excess can be wasteful without improving yield. |
| Formamide (for FISH) | Variable (e.g., 30%) [64] | Screen a range at fixed temperature [34]. | Critical for balancing hybridization efficiency and specificity in downstream applications [34]. |
Buffer Composition and Reagent Stability
The performance of reagents over the duration of multi-day experiments, such as MERFISH, can degrade, a phenomenon known as "aging." [34]
- Buffer Stability: Aliquot and store transcription and hybridization buffers at -20°C to preserve activity. Avoid repeated freeze-thaw cycles.
- Imaging Buffer Optimization: For fluorescence-based detection, the imaging buffer composition directly impacts photostability and effective signal brightness. Introducing new, optimized imaging buffers can significantly improve signal longevity and quality over long imaging sessions [34].
Experimental Protocols for Validation
Protocol: Optimized In Vitro Transcription for DIG-Labeled RNA Probes
Materials:
- Linearized, purified DNA template (1 µg)
- DIG RNA Labeling Mix (Roche)
- RNA Polymerase (SP6, T7, or T3)
- RNase-free water and tubes
- DNase I (RNase-free)
Method:
- Assemble the reaction on ice:
- Linearized template DNA: 1 µg
- DIG RNA Labeling Mix: 2 µL
- 10x Transcription Buffer: 2 µL
- RNA Polymerase: 2 µL (40 U)
- RNase-free water to 20 µL.
- Mix gently and centrifuge briefly.
- Incubate at 37°C for 2-4 hours.
- Add 2 U of DNase I (RNase-free) and incubate at 37°C for 15 minutes to remove the template DNA.
- Purify the probe using precipitation or column-based purification. Resuspend in RNase-free water with 0.1% DEPC.
- Quantify yield by spectrophotometry and assess integrity by denaturing agarose gel electrophoresis.
Protocol: Formamide Concentration Screening for FISH Hybridization
This protocol, adapted from MERFISH optimization studies [34], can be used to empirically determine the optimal stringency for your specific probe and sample.
Materials:
- Fixed and permeabilized sample
- Encoding or readout probes [34]
- Hybridization buffers with formamide concentrations ranging from 10-40%
- Wash buffer
Method:
- Divide the sample into aliquots.
- For each aliquot, hybridize with the probe set in a separate hybridization buffer, each with a different formamide concentration. Keep the hybridization temperature (e.g., 37°C) and duration constant across all conditions [34].
- Perform post-hybridization washes as per standard protocol.
- Image all samples under identical conditions.
- Quantify the signal-to-noise ratio or the brightness of single-molecule spots for each condition. The formamide concentration yielding the highest signal-to-noise is optimal for subsequent experiments [34].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for DIG-Labeled RNA Probe Workflow
| Reagent/Solution | Function | Technical Notes |
|---|---|---|
| DIG RNA Labeling Mix | Provides nucleotide substrates for polymerase, including DIG-UTP. | Pre-mixed solution ensures consistent labeling efficiency; contains ATP, CTP, GTP, UTP, and DIG-11-UTP. |
| SP6, T7, T3 RNA Polymerase | Drives in vitro transcription from specific promoters. | Select polymerase based on promoter sequence flanking the insert in the template vector. |
| RNase Inhibitor | Protects synthesized RNA probes from degradation. | Critical for maintaining probe integrity during long reactions and storage. |
| Formamide (High Purity) | Denaturant in hybridization buffers for FISH. | Enables specific probe binding by controlling hybridization stringency [34] [64]. |
| Dextran Sulphate | Component of FISH hybridization buffer. | A crowding agent that increases the effective probe concentration, enhancing hybridization kinetics [64]. |
| Anti-DIG Antibody Conjugate | Detection moiety for hybridized probes. | Conjugated to alkaline phosphatase (AP) for colorimetric detection or fluorescein (FITC) for fluorescence. |
Workflow and Pathway Visualization
The following diagram illustrates the logical workflow for diagnosing and addressing low probe yield, integrating the optimization strategies discussed in this guide.
Diagram 1: A systematic workflow for diagnosing and resolving low probe yield issues through template and reaction condition optimization.
In the broader context of digoxigenin (DIG)-labeled RNA probe protocol research, achieving optimal signal-to-noise ratio remains a fundamental challenge for researchers studying spatial gene expression. High background staining can compromise data interpretation, particularly when detecting low-abundance transcripts. This technical guide examines the interplay between stringency washes and blocking steps—two critical control points—within hybridization protocols. The principles discussed are framed within established DIG-labeled RNA probe methodologies [4] but are equally applicable to modern amplification techniques like BaseScope [65] and Hybridization Chain Reaction (HCR) [66], providing scientists with a systematic approach to background reduction across various experimental platforms.
The Science of Stringency in Hybridization
Stringency refers to the specificity of probe-target binding, which is controlled by experimental conditions that influence nucleic acid hybrid stability. In molecular hybridization, high stringency conditions ensure that only perfectly complementary nucleic acid sequences remain hybridized, while mismatched or weakly bound sequences are dissociated and removed [67].
The biochemical basis for stringency control lies in the hydrogen bonding between base pairs and the electrostatic interactions that stabilize the double-stranded complex. Salt concentration and temperature are the two primary levers for controlling stringency. Salt cations (Na⁺) neutralize the negative phosphate backbone repulsion between hybridized strands, thereby stabilizing the duplex. Lower salt concentrations reduce this shielding effect, increasing electrostatic repulsion and destabilizing imperfect hybrids. Temperature affects hydrogen bonding; higher thermal energy disrupts the hydrogen bonds in mismatched sequences more readily than in perfectly matched, stable duplexes [67].
For DIG-labeled RNA probes, typically 250-1500 bases in length with optimal sensitivity around 800 bases, these principles are paramount [4]. The goal is to establish conditions where only the specific antisense probe binding to its target mRNA is stable, while non-specific interactions with similar but not identical sequences, or interactions with cellular components, are eliminated.
Key Parameters Controlling Stringency
Table 1: Parameters for Controlling Hybridization Stringency
| Parameter | Effect on Hybridization | High Stringency Condition | Mechanism of Action |
|---|---|---|---|
| Temperature | Higher temperatures disrupt hydrogen bonds | Increase temperature (e.g., 65°C–75°C) [67] [4] | Destabilizes mismatched hybrids due to fewer hydrogen bonds |
| Salt Concentration | Salt ions neutralize negative charge repulsion | Lower salt concentration (e.g., 0.1x–0.5x SSC) [67] [4] | Reduces hybrid stability, allowing imperfect matches to dissociate |
| Formamide Concentration | Denatures nucleic acid duplexes | Include 50% formamide in hybridization buffer [4] | Lowers effective melting temperature of probe-target duplex |
| Detergent Concentration | Reduces non-specific hydrophobic interactions | Add SDS (e.g., 0.1%) to wash buffers [4] | Competes with and removes non-specifically adsorbed probe |
Experimental Protocols for Optimized Stringency Washes
Stringency is most critically applied during the post-hybridization wash steps. The following protocols detail specific methodologies for implementing controlled stringency.
Standard Stringency Wash Protocol for DIG-Labeled RNA Probes
This protocol, adapted from established DIG-labeled RNA probe methodologies [4], is fundamental for removing excess probe and non-specifically bound hybrids.
- First Wash (Removes Excess Probe & Buffer): Wash slides in a solution of 50% formamide in 2x SSC. Perform 3 times for 5 minutes each, at a temperature of 37–45°C. This initial wash removes the bulk of the unhybridized probe and hybridization buffer without being overly harsh.
- Second Stringency Wash (Removes Non-specific Hybrids): Wash slides in a low-salt SSC solution (0.1x to 2x SSC) for 3 times for 5 minutes each. The exact temperature and concentration are the primary variables for optimization [4]:
- For short or complex probes (0.5–3 kb): Use lower stringency (1–2x SSC) and lower temperature (up to 45°C).
- For single-locus or large probes: Use higher stringency (below 0.5x SSC) and higher temperature (up to 65°C).
- For repetitive probes (e.g., alpha-satellite repeats): Use the highest stringency (low SSC) and temperature (~65°C).
- Final Rinse: Wash twice in MABT (Maleic Acid Buffer with Tween 20) for 30 minutes at room temperature. MABT is gentler than PBS for nucleic acid detection and helps prepare the sample for antibody incubation [4].
Advanced Background Reduction for HCR
The in situ Hybridization Chain Reaction (HCR) is a powerful signal amplification method, but it can suffer from background caused by single probes nonspecifically binding and initiating the amplification cascade. A recent study offers a simple universal improvement [66].
- Problem: Single HCR split probes can act as bridges between hairpin DNAs and the sample via nonspecific binding, causing low but noticeable background.
- Solution: Add random oligonucleotides (e.g., random 20-mer DNA) during the pre-hybridization and hybridization steps.
- Protocol Modification:
- Pre-hybridization: Include random oligonucleotides in the pre-hybridization buffer. These random sequences bind to nonspecific sites throughout the sample, effectively blocking them.
- Hybridization: Include the same random oligonucleotides in the hybridization mixture containing the HCR probes. This continues to block nonspecific sites during the specific probe binding process.
- Outcome: This simple modification reduces background signals by approximately 3 to 90 times, significantly improving the signal-to-noise ratio for detecting low-expression mRNAs [66].
The following workflow diagram integrates both standard and advanced stringency control methods into a complete experimental process.
The Role of Blocking in Background Reduction
While stringency washes manage probe specificity, blocking agents are essential for preventing non-specific antibody binding in the detection phase, particularly in immunohistochemical detection of DIG-labeled probes. Blocking proteins occupy reactive sites on the tissue section and the slide surface that would otherwise bind the anti-DIG antibody conjugates.
For DIG-labeled RNA probe protocols, a typical blocking step involves applying a solution of MABT (Maleic Acid Buffer with Tween 20) supplemented with 2% blocking agent (e.g., Bovine Serum Albumin, milk powder, or serum) for 1–2 hours at room temperature before adding the anti-DIG antibody [4]. The detergent in MABT is gentler than PBS and is more suitable for nucleic acid detection, helping to reduce background without affecting the specific hybridized probe [4].
Integrated Troubleshooting Guide for High Background
Diagnosing the source of high background requires a systematic approach. The following table links common symptoms to their likely causes and provides targeted solutions based on the principles of stringency and blocking.
Table 2: Troubleshooting High Background in Hybridization Assays
| Symptom | Potential Cause | Corrective Action |
|---|---|---|
| General high background across entire tissue section | Inadequate blocking of non-specific antibody binding | Increase blocking agent concentration (e.g., to 2-5% BSA); extend blocking time to 2 hours; ensure slides do not dry out after rehydration [4]. |
| Punctate or speckled background, especially with HCR | Non-specific initiation of amplification by single probes | Include random oligonucleotides in pre-hybridization and hybridization steps [66]. |
| Diffuse, cloudy background with DIG probes | Insufficient post-hybridization washing | Increase volume and frequency of stringency washes; incorporate a 50% formamide wash step [4]. |
| Specific, off-target staining | Low stringency allowing probe cross-hybridization | Increase wash temperature and/or decrease salt concentration (e.g., to 0.1x SSC) of stringency washes [67]. |
| High background with poor tissue morphology | Over-digestion with protease during antigen retrieval | Titrate proteinase K concentration (e.g., test 10-20 µg/mL) and optimize incubation time [4]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Background Control in Hybridization
| Reagent | Function in Protocol | Role in Background Reduction |
|---|---|---|
| Saline-Sodium Citrate (SSC) | Buffer for dilution and washing; component of hybridization solution. | Lowering concentration (e.g., from 2x to 0.1x) in washes increases stringency, dissociating imperfect hybrids [67] [4]. |
| Formamide | Denaturing agent used in hybridization buffer and washes. | Lowers the effective melting temperature (Tm), allowing high stringency washes to be performed at lower, less damaging temperatures [4]. |
| Digoxigenin (DIG) Labeling System | Non-radioactive label for RNA, DNA, and oligonucleotide probes. | Hapten label is not naturally present in biological systems, minimizing non-specific detection compared to systems like biotin [4]. |
| Maleic Acid Buffer with Tween (MABT) | Wash and dilution buffer used before and after antibody incubation. | Gentler than PBS; its detergent action (Tween 20) helps reduce non-specific antibody binding without disrupting specific hybrids [4]. |
| Blocking Reagents (BSA, Serum, Milk) | Proteins used to saturate non-specific binding sites. | Prevents anti-DIG antibody from adhering to tissue and slide surfaces, a major source of background [4]. |
| Random Oligonucleotides | Short, nonspecific DNA sequences. | For HCR, they bind nonspecific sites, preventing single probes from initiating false-positive amplification [66]. |
| Proteinase K | Proteolytic enzyme for antigen retrieval. | Digests proteins surrounding target mRNA, improving probe access. Requires titration, as over-digestion increases background [4]. |
The following diagram illustrates the logical decision-making process for diagnosing and resolving the most common causes of high background.
Resolving high background in hybridization assays demands a methodical approach that integrates optimized stringency washes with effective blocking. The fundamental principle of increasing temperature and decreasing salt concentration during washes provides a powerful tool for enhancing specificity [67], while modern techniques like the addition of random oligonucleotides in HCR offer targeted solutions for amplification-specific noise [66]. For researchers employing DIG-labeled RNA probes, a deep understanding of these controls—combined with rigorous protocol validation using appropriate positive and negative controls as demonstrated in BaseScope probe validation [65]—is indispensable for generating reliable, publication-quality data in gene expression analysis.
In the context of digoxigenin (DIG)-labeled RNA probe synthesis, the quality of the DNA template is the foundational determinant of success. Within a broader research thesis on optimizing this protocol, incomplete linearization of plasmid DNA and the presence of contaminants represent the most significant technical hurdles, directly impacting probe specificity, yield, and experimental reliability. This guide details the core issues and provides validated solutions to ensure the production of high-quality riboprobes.
Core Challenges: Incomplete Linearization and Contaminants
The primary template-related issues can be categorized into two areas: problems arising from the linearization process itself, and those introduced by contaminants. The table below summarizes their causes and consequences.
Table 1: Core Template-Related Issues and Their Impacts
| Issue Category | Specific Problem | Consequence for DIG Probe Synthesis |
|---|---|---|
| Incomplete Linearization | Use of circular or nicked plasmid DNA in the transcription reaction [68]. | Less efficient transcription and production of non-specific, aberrant transcripts [68]. |
| Restriction enzyme digestion with 3'-overhanging or blunt ends [68]. | "Run-on" transcription, yielding unwanted transcripts of the opposite DNA strand [68]. | |
| Template Contaminants | Carryover of proteins, salts, or RNase from the template preparation [68]. | Inefficient transcription and potential degradation of the newly synthesized DIG-labeled RNA probe [68]. |
| Presence of residual template DNA in the final RNA probe mixture [69]. | Can lead to misinterpretation of gel electrophoresis results, showing multiple bands [68]. |
Detailed Experimental Protocols for Mitigation
Protocol for Template Linearization and Purification
This optimized protocol ensures complete linearization and high-purity template DNA.
Step 1: Restriction Enzyme Digestion
- Use 10 units of restriction enzyme per microgram of DNA and digest for at least 3 hours to ensure complete linearization [68].
- Select restriction enzymes that create 5'-overhanging ends. This prevents "run-on" transcription by RNA polymerase, which can occur with templates featuring 3'-overhanging or blunt ends [68].
Step 2: Template Purification
- Post-digestion gel purification is recommended to isolate the linearized DNA fragment from any undigested circular DNA or other fragments [68].
- Follow gel extraction with a spin column clean-up (e.g., using a High Pure PCR Product Purification Kit) for further purification [68].
- Alternative Method: Phenol-chloroform extraction and ethanol precipitation can also be effectively performed to remove enzymes and contaminants [68] [22].
- Final Resuspension: Resuspend the purified, linearized template DNA in RNase-free water. Check an aliquot on a gel to confirm size, purity, and concentration before use [68].
Protocol: Analysis of Template and Probe Quality
This procedure verifies the success of template preparation and the quality of the synthesized RNA probe.
- Gel Electrophoresis Analysis
- After the DIG labeling reaction, analyze the transcript by denaturing agarose gel electrophoresis (e.g., a MOPS/formaldehyde gel) or Polyacrylamide Gel Electrophoresis (PAGE) [68].
- Interpreting Multiple Bands:
- If more than one band is observed, it may be due to RNA secondary structure under non-denaturing conditions; verify with a denaturing gel [68].
- Multiple bands can also result from abortive or shortened transcripts. This can sometimes be overcome by re-cloning the template into a vector with the polylinker in the opposite orientation and transcribing with a different RNA polymerase [68].
- Critical Step: Include a DNase digestion step after the in vitro transcription reaction to remove the template DNA. This prevents residual DNA from being mistaken for a second band on the gel [68] [69].
The Scientist's Toolkit: Research Reagent Solutions
The following reagents are essential for overcoming template-related challenges in DIG-labeled RNA probe synthesis.
Table 2: Essential Reagents for Template Preparation and Quality Control
| Reagent / Kit | Function | Technical Note |
|---|---|---|
| High Pure Plasmid Isolation Kit (Roche) | For initial preparation of high-purity plasmid DNA, minimizing contaminants prior to linearization [68]. | Ensures a clean starting template. |
| Restriction Endonucleases (5' Overhang) | For complete linearization of plasmid DNA. | Prevents "run-on" transcription [68]. |
| High Pure PCR Product Purification Kit (Roche) | For post-digestion and post-gel extraction clean-up of linearized DNA template [68]. | Removes enzymes, salts, and other impurities. |
| DNase I (RNase-free) | For degradation of the template DNA after in vitro transcription is complete [68] [69]. | Prevents template DNA contamination in the final RNA probe sample. |
| DIG RNA Labeling Mix (Sigma-Aldrich) | Optimized nucleotide mixture containing DIG-11-UTP for efficient incorporation by T7, SP6, or T3 RNA polymerases [68]. | Designed for high yield; incorporates DIG-11-UTP approximately every 20-25 nucleotides [68]. |
Workflow Visualization
The following diagram illustrates the critical steps and decision points in the template preparation and verification process, integrating the protocols and solutions detailed above.
Optimization Strategies for Challenging Templates and Sensitivity
Digoxigenin (DIG)-labeled RNA probes represent a cornerstone technology in molecular biology, enabling sensitive and specific detection of nucleic acids across diverse applications from basic research to clinical diagnostics. Within the broader context of digoxigenin-labeled RNA probe protocol research, optimization strategies become paramount when working with challenging templates or when pushing the boundaries of detection sensitivity. These probes, generated through in vitro transcription with DIG-11-UTP incorporated by bacteriophage RNA polymerases (SP6, T7, T3), offer significant advantages including enhanced thermodynamic stability and superior sensitivity compared to DNA probes [70] [71]. The effectiveness of RNA-targeting approaches in drug discovery [72] further underscores the importance of robust probe technologies. However, researchers frequently encounter obstacles related to template preparation, probe integrity, and hybridization efficiency that can compromise experimental outcomes. This technical guide provides detailed methodologies and optimization strategies to overcome these challenges, ensuring reliable and sensitive detection even with the most demanding templates and applications.
Critical Challenges in Probe Synthesis and Performance
Template-Related Challenges
The quality of template DNA fundamentally determines the success of DIG-labeled RNA probe synthesis. Incomplete restriction enzyme digestion of plasmid DNA often leads to undesirable non-specific transcripts and reduced yield of full-length probes. Templates with 3'-overhanging or blunt ends frequently cause problematic "run-on" transcription, producing aberrant transcripts from the wrong DNA strand [71]. Furthermore, certain DNA sequences can induce RNA polymerases to produce abortive or shortened transcripts, while residual contaminants from template preparation—including salts, proteins, or nucleases—can severely inhibit transcription efficiency.
Sensitivity and Specificity Limitations
Achieving optimal sensitivity while minimizing background signal presents a persistent challenge in probe-based applications. The very high thermodynamic stability of RNA:RNA duplexes, while beneficial for strong hybridization, necessitates precisely optimized stringency conditions to prevent cross-hybridization, particularly to ribosomal RNAs [70]. Additionally, RNA probes are notoriously difficult to remove from membranes for re-probing due to this same stability, often requiring harsh stripping conditions that damage the immobilized target RNA [70]. Without proper optimization, these factors collectively diminish detection sensitivity and increase non-specific background, compromising data quality.
Template Optimization Strategies
Template Design and Preparation
Meticulous template preparation is the foundational step for generating high-quality DIG-labeled RNA probes. The following optimized protocol ensures template integrity and suitability for in vitro transcription:
- Vector Linearization: Use restriction enzymes that create 5'-overhanging ends whenever possible, as this significantly reduces "run-on" transcription from the opposite strand [71]. Employ 10 units of restriction enzyme per microgram of DNA and extend digestion time to at least 3 hours to ensure complete linearization.
- Template Purification: Following restriction digestion, purify linearized template DNA through gel electrophoresis or using spin column-based purification kits (e.g., High Pure PCR Product Purification Kit) [71]. Alternatively, perform phenol/chloroform extraction followed by ethanol precipitation. Resuspend the final DNA template in RNase-free water.
- Quality Control: Verify template concentration, purity, and complete linearization by running an aliquot on an agarose gel alongside uncut plasmid. Use only templates with A260/A280 ratios between 1.8-2.0 and showing single bands of expected size.
- Promoter Incorporation: For templates generated via PCR, include the appropriate bacteriophage promoter sequences (T7, T3, or SP6) directly in the PCR primers to enable direct in vitro transcription without subcloning [71].
Troubleshooting Problematic Templates
Certain template sequences present unique challenges that require specialized approaches:
- Abortive Transcription: If RNA polymerases produce truncated transcripts due to problematic sequences, reclone the insert into a vector with the opposite orientation of the polylinker, or transcribe the complementary strand using a different RNA polymerase [71].
- Secondary Structure: For templates prone to forming stable secondary structures that hinder transcription, consider adding DMSO (final concentration 2-5%) to the transcription reaction or increasing reaction temperature to 42°C if the enzyme tolerates it.
- Low Yield Templates: For difficult templates yielding insufficient probe, scale up the transcription reaction 2-5 fold while maintaining component ratios, or switch to high-yield transcription kits specifically designed for problematic templates.
Sensitivity Enhancement Approaches
Hybridization Optimization
Maximizing signal-to-noise ratio requires precise optimization of hybridization and washing conditions, particularly leveraging the high stability of RNA:RNA duplexes:
Stringency Calculation: Calculate the theoretical melting temperature (Tm) for RNA:RNA duplexes using the formula:
Tm(RNA:RNA) = 78°C + 16.6 × log10([Na+]/(1.0 + 0.7[Na+])) + 0.7 × (%GC) - 0.35 × (%formamide) - 500/(duplex length) [70]
Hybridize at 15-25°C below the calculated Tm, which typically falls between 60-65°C for standard probes—significantly higher than for DNA probes.
- Hybridization Buffer Composition: Use formamide-based buffers containing nuclease-free reagents and include 100 μg/mL total yeast RNA (superior to tRNA) as a blocking agent to reduce background and cross-hybridization [70]. Commercial buffers like ULTRAhyb have been optimized for these parameters.
- Membrane Selection: Use positively charged nylon membranes (e.g., BrightStar-Plus), as nitrocellulose cannot withstand the stringent conditions required for RNA probes and is incompatible with nonisotopic detection methods [70].
Advanced Detection and Stripping Methods
Innovative approaches to probe design and stripping can dramatically enhance sensitivity and reusability:
- StripEZE Technology: Utilize probes incorporating modified nucleotides that enable gentle removal through chemical cleavage rather than harsh thermal or chemical treatment. This approach preserves membrane integrity and allows for multiple re-probings without significant sensitivity loss [70].
- Probe Validation: Always verify probe integrity and approximate concentration by formaldehyde agarose gel electrophoresis alongside RNA standards. Note that ethidium bromide staining of RNA transcripts indicates integrity but not accurate quantification [71].
- Multiplexing Potential: While not directly discussed in DIG probe literature, principles from multiplex dPCR assays [73] suggest that using multiple probes with distinct specificities could expand application scope, though careful optimization would be required.
Table 1: Troubleshooting Guide for Common DIG RNA Probe Issues
| Problem | Possible Causes | Solutions |
|---|---|---|
| Multiple bands on gel | RNA secondary structure, template DNA contamination, abortive transcription | Use denaturing MOPS/formaldehyde gel; Include DNase digestion step; Reclone template with different polymerase [71] |
| High background | Low hybridization stringency, insufficient blocking, nuclease contamination | Increase hybridization temperature to 60-65°C; Use total yeast RNA in buffer; Use certified nuclease-free reagents [70] |
| Low signal | Probe degradation, inefficient transcription, inadequate template | Ensure RNase-free conditions; Verify template purity and complete linearization; Include positive control template [71] |
| Cross-hybridization | Overly permissive conditions, probe complementarity to non-targets | Increase stringency using calculated Tm; Verify probe specificity computationally; Shorten probe if necessary [70] |
Research Reagent Solutions
Table 2: Essential Reagents for DIG-Labeled RNA Probe Workflows
| Reagent | Function | Key Considerations |
|---|---|---|
| DIG RNA Labeling Mix | Provides DIG-11-UTP for incorporation during transcription | Optimized nucleotide mixture for SP6, T3, and T7 RNA polymerases [71] |
| RNA Polymerases (SP6, T7, T3) | Drives in vitro transcription from specific promoters | Each has specific promoter requirements; T7 generally offers highest yield |
| Positively Charged Nylon Membrane | Immobilizes target nucleic acids for hybridization | Essential for high-stringency washes; compatible with nonisotopic detection [70] |
| Anti-Digoxigenin Antibody | Binds DIG label for detection | Conjugated to alkaline phosphatase for colorimetric or chemiluminescent detection [59] |
| Total Yeast RNA | Blocking agent to reduce nonspecific binding | More effective than tRNA; must be RNase-free [70] |
| ULTRAhyb Hybridization Buffer | Optimized solution for hybridization | Pre-formulated with blocking agents; certified nuclease-free [70] |
| StripEZE Reagents | Enable gentle probe removal for reprobing | Chemical cleavage-based; preserves membrane integrity [70] |
Experimental Workflow and Applications
The following diagram illustrates the complete optimized workflow for DIG-labeled RNA probe synthesis and application, integrating the critical optimization steps discussed throughout this guide:
Optimizing digoxigenin-labeled RNA probe protocols for challenging templates and sensitivity demands a systematic approach addressing both template quality and hybridization dynamics. The strategies outlined herein—emphasizing meticulous template preparation, precise stringency control, and innovative stripping technologies—provide researchers with a comprehensive toolkit for overcoming common obstacles. As RNA-targeted therapies and diagnostics continue to advance [72], the ability to reliably generate and utilize high-quality DIG-labeled probes remains fundamental to progress in both basic research and applied clinical science. Through implementation of these optimized protocols, researchers can achieve the sensitivity and specificity required for even the most demanding applications, from single-cell RNA detection to clinical diagnostics.
Probe Validation and Comparative Analysis with Other Techniques
Methods for Determining Labeling Efficiency and Probe Concentration
In the development of digoxigenin (DIG)-labeled RNA probes, the accurate determination of labeling efficiency and probe concentration represents a critical quality control checkpoint that directly impacts experimental success across applications from in situ hybridization to advanced molecular diagnostics. These parameters dictate probe sensitivity, specificity, and ultimately, the reliability of gene expression analysis. Within the broader context of DIG-labeled RNA probe protocol research, standardized quantification methods provide essential metrics for comparing probe batches across laboratories and experimental conditions, thereby enhancing reproducibility in molecular studies. This technical guide synthesizes current methodologies to equip researchers with robust frameworks for probe characterization.
Analytical Techniques for Quantification
Spectrophotometric Analysis
Fundamental Principles: Conventional spectrophotometry provides a straightforward initial assessment of nucleic acid concentration and labeling incorporation by measuring absorbance at specific wavelengths.
Protocol:
- Dilute the DIG-labeled RNA probe in an appropriate buffer (e.g., TE buffer).
- Measure absorbance at 260 nm (A₂₆₀) for nucleic acid concentration and at 280 nm (A₂₈₀) for protein contamination assessment.
- Calculate probe concentration using the formula: Concentration (ng/μL) = A₂₆₀ × Dilution Factor × 40.
- Assess purity via the A₂₆₀/A₂₈₀ ratio, with values of 1.8-2.0 indicating minimal protein contamination.
Limitations: While spectrophotometry accurately determines RNA concentration, it cannot distinguish between labeled and unlabeled RNA molecules, providing only indirect evidence of labeling success.
Dot Blot Assay for Direct Labeling Efficiency
Principles and Applications: The dot blot assay directly semi-quantifies DIG incorporation by comparing sample signal intensity against a standardized dilution series of DIG-labeled control nucleic acids, enabling relative efficiency determination [74].
Protocol:
- Prepare a dilution series of a DIG-labeled standard with known concentration.
- Apply identical amounts of unknown probe and standard dilutions onto a positively charged nylon membrane.
- Cross-link RNA to the membrane using UV light.
- Perform immunodetection with anti-DIG-alkaline phosphatase antibodies and chromogenic substrates.
- Quantify signal intensity using imaging software and compare sample intensity to the standard curve.
Advantages: This method specifically detects DIG incorporation rather than total RNA, providing a more accurate assessment of functional labeling efficiency.
Comparative Analysis of Quantification Methods
Table 1: Technical Approaches for Assessing DIG-Labeled RNA Probes
| Method | Measured Parameter | Sample Requirement | Throughput | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Spectrophotometry | Nucleic acid concentration & purity | Low (1-2 μL) | High | Rapid, minimal sample consumption | Cannot distinguish labeled/unlabeled RNA |
| Dot Blot Assay | DIG hapten incorporation | Moderate | Medium | Direct label quantification, functional assessment | Semi-quantitative, requires standards |
| Gel Electrophoresis | RNA integrity & approximate size | Moderate | Medium | Visualizes degradation, estimates transcript size | Does not quantify DIG incorporation |
Table 2: Expected Characteristics of High-Quality DIG-Labeled RNA Probes
| Parameter | Optimal Value/Range | Significance for Experimental Quality |
|---|---|---|
| RNA Concentration | 50-100 ng/μL (post-purification) | Ensures sufficient probe for hybridization |
| A₂₆₀/A₂₈₀ Ratio | 1.8-2.0 | Indicates minimal protein contamination |
| Transcript Integrity | Discrete bands on denaturing gel | Confirms full-length probe synthesis |
| Functional Sensitivity | Detectable at ≤1 pg in dot blot | Verifies adequate DIG incorporation |
Integrated Workflow for Probe Quality Control
The following diagram illustrates the recommended sequential workflow for comprehensive quality assessment of DIG-labeled RNA probes, from synthesis to functional validation:
Research Reagent Solutions for DIG-Labeled RNA Probes
Table 3: Essential Reagents for DIG-Labeled RNA Probe Development and Quality Control
| Reagent/Category | Specific Examples | Function in Protocol |
|---|---|---|
| Labeling Nucleotides | DIG-11-UTP [74] | Hapten-labeled nucleotide incorporated during in vitro transcription |
| Enzymatic Systems | T7, T3, or SP6 RNA Polymerases [75] | DNA-dependent RNA polymerases for probe synthesis |
| Detection Antibodies | Anti-DIG-alkaline phosphatase (AP) conjugate [74] [20] | Enzyme-conjugated antibody for chromogenic or chemiluminescent detection |
| Chromogenic Substrates | NBT/BCIP [20] | Forms insoluble color precipitate for colorimetric detection |
| Membrane Materials | Positively charged nylon membrane [20] | Solid support for nucleic acid immobilization in dot blot assays |
| Hybridization Buffers | Formamide-based hybridization buffer [76] | Solution environment that promotes specific probe-target binding |
Advanced Applications and Technical Considerations
Integration with Molecular Techniques
The quantitative assessment of DIG-labeled RNA probes enables their sophisticated application in complex experimental designs. In neuroscience research, precisely quantified probes have been successfully employed for mapping neurotransmitter expression patterns in free-floating brain sections, often combined with immunofluorescence for protein co-localization studies [75]. Similarly, in developmental biology, optimized probes facilitate high-resolution mRNA localization in whole-mount specimens, with rigorous quantification preventing background staining while maximizing sensitivity [76].
Troubleshooting Common Issues
Low Labeling Efficiency: This typically results from degraded DIG-UTP, suboptimal RNA polymerase activity, or impure DNA template. Ensure fresh labeling reagents, functional enzyme lots, and purified template DNA.
High Background in Detection: Often caused by insufficient blocking or antibody overconcentration. Optimize blocking conditions with appropriate sera and titrate anti-DIG antibodies to determine optimal dilution.
RNA Degradation: Manifested as smearing on electrophoretic gels. Use RNase-free techniques throughout, including DEPC-treated water and sterile equipment [75].
Rigorous quantification of labeling efficiency and concentration of DIG-labeled RNA probes through integrated spectrophotometric, electrophoretic, and immuno-detection methods provides the foundation for reproducible, sensitive molecular detection across diverse research applications. The standardized approaches outlined in this guide enable researchers to establish critical quality control parameters that ensure experimental reliability and enhance cross-study comparisons in the continuing development of DIG-based detection methodologies.
This technical guide explores the use of digoxigenin (DIG)-labeled RNA probes in Northern blot analysis, with a specific focus on methodologies for establishing and validating assay specificity. Northern blotting remains a critical technique in molecular biology for the detection and quantification of specific RNA molecules, providing direct information about transcript size and abundance [77]. Within a broader research context involving DIG-labeled RNA probe protocols, the incorporation of competition assays is paramount for confirming the specificity of hybridization signals, thereby ensuring data reliability in gene expression studies relevant to drug development and diagnostic applications.
Northern blot analysis is a foundational technique for studying gene expression by detecting specific RNA sequences within a complex sample. Unlike newer methods like qRT-PCR, Northern blotting provides a direct relative comparison of message abundance between samples on a single membrane and is the preferred method for determining transcript size and for detecting alternatively spliced transcripts [77]. The technique involves separating RNA molecules by size via denaturing gel electrophoresis, transferring them to a solid membrane, and hybridizing them with complementary labeled probes [78].
Despite the advent of powerful techniques like RT-PCR and gene array analysis, Northern analysis remains a standard method for detection and quantitation of mRNA levels [77]. Its exceptional versatility allows the use of radiolabeled or nonisotopically labeled DNA, in vitro transcribed RNA, and oligonucleotides as hybridization probes [77]. A significant advantage of Northern blotting is the opportunity it provides to evaluate experimental progress at various points, such as assessing RNA integrity after extraction and confirming efficient transfer to the membrane [77].
The DIG system, which utilizes digoxigenin-labeled nucleic acid probes and antibody-conjugate detection, has become a cornerstone of non-radioactive detection in Northern blotting, offering high sensitivity and specificity while eliminating the safety concerns associated with radioactive isotopes.
Digoxigenin-Labeled RNA Probes: Principles and Advantages
Probe Design and Generation
RNA probes, particularly antisense RNA probes, offer significant advantages for Northern analysis due to their high sensitivity and specificity for target RNA sequences [4]. These probes are typically generated by in vitro transcription from a DNA template using bacteriophage RNA polymerases (T3, T7, or SP6) in the presence of DIG-labeled UTP [4].
For optimal results, RNA probes should be designed to be between 250–1,500 bases in length, with probes of approximately 800 bases exhibiting the highest sensitivity and specificity [4]. The in vitro transcription process allows for the incorporation of hapten-labeled nucleotides at high efficiency, producing probes with consistent labeling density essential for quantitative applications.
Detection Mechanism
The detection system for DIG-labeled probes relies on an enzyme-linked immunoassay format. After hybridization, the membrane is incubated with an anti-DIG antibody conjugated to alkaline phosphatase (AP) or horseradish peroxidase (HRP). Subsequent incubation with appropriate substrates—either colorimetric, chemiluminescent, or fluorescent—generates a detectable signal proportional to the amount of target RNA present [4].
Chemiluminescent detection, using substrates such as CDP-Star or CSPD for AP conjugates, offers the highest sensitivity, capable of detecting fewer than 100,000 molecules on a blot [77]. This sensitivity can be further enhanced through the use of optimized hybridization buffers like ULTRAhyb, which can increase sensitivity up to 100-fold compared to standard hybridization solutions [77].
Establishing Specificity Through Competition Assays
The Principle of Competition Assays
Competition assays serve as a critical control to verify that the observed hybridization signal specifically results from the interaction between the probe and its intended target sequence. The fundamental principle involves pre-incubating the labeled probe with an excess of unlabeled competitor nucleic acid before or during hybridization. The competitor molecule competes with the target RNA for binding sites on the probe, resulting in a diminished or abolished signal when specificity is confirmed.
Types of Competitor Molecules
Table 1: Types of Competitor Molecules for Specificity Validation
| Competitor Type | Description | Application | Expected Result with Specific Hybridization |
|---|---|---|---|
| Unlabeled Sense RNA | Identical sequence to target RNA | Most common competition control | Significant signal reduction |
| Unlabeled Antisense RNA | Identical to probe sequence | Direct probe binding competition | Complete signal abolition |
| Unrelated RNA | Different sequence with no homology | Non-specific binding assessment | No signal reduction |
| Mutated Target RNA | Sequence with partial homology | Epitope mapping of probe binding | Partial signal reduction depending on mismatch |
The most definitive competition control uses unlabeled antisense RNA identical to the probe sequence, which should completely abolish the signal when present in sufficient excess [4]. Alternatively, unlabeled sense RNA identical to the target RNA sequence also serves as an effective competitor.
Implementation in Northern Blot Protocol
To implement a competition assay within a standard Northern blot procedure:
Prepare competitor nucleic acid: Synthesize unlabeled RNA transcripts identical to either the probe sequence (antisense) or target sequence (sense) using the same in vitro transcription system without DIG-labeled nucleotides.
Pre-hybridization competition: Pre-incubate the labeled DIG-probe with a 10- to 100-fold molar excess of unlabeled competitor for 30 minutes at hybridization temperature before adding to the membrane.
Simultaneous competition: Add both labeled probe and excess unlabeled competitor simultaneously to the hybridization buffer.
Signal comparison: Compare the hybridization signal intensity between the competition assay and the standard hybridization without competitor.
A significant reduction (typically >80-90%) in signal intensity in the presence of specific competitor confirms the specificity of the probe-target interaction, while unchanged signal suggests non-specific hybridization.
Detailed Northern Blot Protocol with DIG-Labeled RNA Probes
RNA Isolation and Quality Control
Obtaining high-quality, intact RNA is the most critical step in Northern analysis [77]. Proper techniques must be employed to prevent RNA degradation by ubiquitous RNases:
- Use RNase-free reagents, plasticware, and glassware
- Wear gloves throughout the procedure to prevent introduction of RNases from skin
- Use dedicated RNase-free work areas
- Include RNase inhibitors in extraction buffers when appropriate
Total RNA can be isolated using organic extraction (e.g., phenol-chloroform with guanidium isothiocyanate) or silica-based column methods [79]. For low-abundance transcripts, mRNA enrichment using oligo-dT cellulose or magnetic beads can significantly enhance detection sensitivity [79]. RNA quality should be verified by electrophoresis prior to proceeding with Northern analysis.
Denaturing Gel Electrophoresis
RNA samples must be separated under denaturing conditions to prevent secondary structure formation that would affect mobility:
- Formaldehyde agarose gels: 1-1.2% agarose in MOPS buffer containing 2.2 M formaldehyde
- Glyoxal/DMSO system: Alternative denaturing system that avoids formaldehyde handling [77]
- Polyacrylamide gels with urea: For small RNAs (<200 nucleotides) [80]
The NorthernMax-Gly system provides a streamlined approach for glyoxal-based Northern analysis that eliminates safety concerns associated with formaldehyde while providing sharp RNA bands [77]. Typically, 5-30 μg of total RNA or 0.5-3 μg of poly(A)+ RNA is loaded per lane, alongside appropriate RNA molecular weight markers.
RNA Transfer and Immobilization
After electrophoresis, RNA is transferred to a positively charged nylon membrane:
- Capillary transfer: Traditional method using high-salt buffer (20X SSC) and passive capillary action overnight
- Vacuum blotting: Faster transfer (1-2 hours) with improved reproducibility [79]
- Electroblotting: Active transfer using electrical current for rapid, efficient transfer
Following transfer, RNA is immobilized on the membrane by UV cross-linking (1200 x 100 μJ/cm²) or baking at 80°C for 30-120 minutes [81]. Proper immobilization is essential to prevent RNA loss during subsequent hybridization and washing steps.
Pre-hybridization and Hybridization
The membrane is pre-hybridized for 15-60 minutes in hybridization buffer containing blocking agents (Denhardt's solution, denatured salmon sperm DNA, or commercial blocking reagents) to reduce non-specific probe binding [4]. For DIG-labeled RNA probes, hybridization is typically performed at 68°C in a buffer containing 50% formamide, which allows lower hybridization temperatures while maintaining stringency [4].
Optimized commercial hybridization buffers like ULTRAhyb can dramatically increase sensitivity, enabling detection of as few as 10,000 target molecules and reducing required hybridization times to as little as 2 hours for abundant messages [77].
Post-Hybridization Washes and Detection
Stringency washes are critical for removing non-specifically bound probe while retaining specifically hybridized probe:
- Low stringency wash: 2X SSC, 0.1% SDS at room temperature
- High stringency wash: 0.1X SSC, 0.1% SDS at 68°C (or higher temperature for probes with high GC content)
Immunological detection follows a standard protocol:
- Blocking: Incubate membrane in blocking solution (e.g., 1X Blocking Reagent) to prevent non-specific antibody binding
- Antibody incubation: Anti-DIG-alkaline phosphatase conjugate, typically diluted 1:10,000, 30 minutes
- Washing: Remove unbound antibody with multiple washes
- Detection: Incubate with chemiluminescent substrate (e.g., CSPD) and expose to X-ray film or digital imaging system
Diagram 1: Northern Blot and Competition Assay Workflow. The competition assay (diamond node) serves as a critical endpoint validation step.
Quantitative Data and Technical Specifications
Sensitivity Comparison of Probe Types
Table 2: Sensitivity Comparison of Different Northern Blot Probe Technologies
| Probe Type | Labeling Method | Relative Sensitivity | Detection Limit (Molecules) | Key Applications |
|---|---|---|---|---|
| Random-primed DNA | Random hexamer labeling | 1X | ~1,000,000 | Standard mRNA detection |
| Asymmetric PCR DNA | PCR with primer imbalance | 3-5X | ~300,000 | Low-abundance transcripts |
| DIG-Labeled RNA | In vitro transcription | 10X | ~100,000 | Highest sensitivity applications |
| LNA-modified DNA | Synthetic oligonucleotides | >10X | <100,000 | miRNA, small RNA detection [80] |
The sensitivity of DIG-labeled RNA probes can be further enhanced through the use of optimized hybridization buffers. For example, ULTRAhyb Ultrasensitive Hybridization Buffer can increase sensitivity up to 100-fold compared to standard hybridization buffers, making signals with random-primed DNA probes as intense as those seen with RNA probes [77].
Comparison of Detection Methodologies
Table 3: Comparison of RNA Detection and Localization Techniques
| Technique | Resolution | Sensitivity | Quantitation | Throughput | Key Applications |
|---|---|---|---|---|---|
| Northern Blot | Transcript size | Moderate | Good | Low | mRNA size, abundance, splice variants [77] |
| RNA Dot Blot | None | Moderate | Good | Medium | Rapid screening, multiple samples [82] |
| In Situ Hybridization | Cellular | High | Semi-quantitative | Low | Spatial localization in tissues [4] |
| RNA FISH | Subcellular | High | Semi-quantitative | Low | Subcellular localization, single-molecule detection [83] |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for DIG-Based Northern Blotting
| Reagent/Category | Specific Examples | Function/Purpose | Technical Notes |
|---|---|---|---|
| RNA Isolation | TRI Reagent, QIAGEN RNeasy, Invitrogen PureLink | High-quality RNA extraction with RNase inhibition | Column methods provide higher purity; organic extraction gives higher yield [79] |
| Membranes | BrightStar-Plus, Hybond-N+, Amersham Hybond NX | RNA immobilization for hybridization | Positively charged nylon membranes preferred for high nucleic acid binding capacity [77] |
| Labeling Systems | DIG RNA Labeling Mix, MAXIscript Kit | Probe synthesis and labeling | In vitro transcription produces high-specific-activity RNA probes [77] |
| Hybridization Buffers | ULTRAhyb, ExpressHyb | Optimized hybridization solution | Significantly enhance sensitivity and reduce hybridization time [77] |
| Detection Reagents | Anti-DIG-AP conjugate, CDP-Star, CSPD | Immunological detection and signal generation | Chemiluminescent substrates offer highest sensitivity for low-abundance targets |
| Blocking Reagents | Denhardt's solution, salmon sperm DNA, Blocking reagent | Reduce non-specific binding | Critical for minimizing background in colorimetric and chemiluminescent detection [4] |
Advanced Applications and Recent Developments
Multiplex Detection and Stripping
Northern blots can be sequentially probed for multiple targets by stripping the membrane between hybridizations. Effective stripping typically involves incubation with 0.1% SDS at 95°C or alkaline conditions (e.g., 0.2 M NaOH at 37°C) to remove bound probe without damaging the immobilized RNA [77]. Proper stripping should be verified by detecting the membrane prior to re-probing.
Small RNA Detection
Detection of small RNAs such as microRNAs presents special challenges due to their short length (19-25 nucleotides). Specialized approaches include:
- Use of high-percentage polyacrylamide gels (12-15%) for separation [80]
- Chemical modification of probes with Locked Nucleic Acid (LNA) technology, which increases thermal stability and improves sensitivity by at least 10-fold compared to DNA probes [80]
- Enhanced cross-linking methods (e.g., EDC cross-linking) to improve small RNA retention on membranes
Integration with Modern Methodologies
Recent developments have expanded the utility of Northern blotting in contemporary research:
- Single-cell Northern blotting: Adapted protocols for limited sample material
- Digital Northern analysis: Combination with microarray technology for high-throughput applications
- Multiplexed FISH techniques: Adaptation of probe design principles for spatial transcriptomics [83]
Diagram 2: Competition Assay Mechanism. The unlabeled competitor (dashed nodes) competes with the labeled probe for target binding, reducing signal output when specificity is confirmed.
Northern blot analysis with DIG-labeled RNA probes remains an essential technique for precise RNA detection and quantification, particularly when combined with competition assays to establish specificity. The methodologies outlined in this guide provide a framework for implementing these techniques with the rigor required for research and diagnostic applications. While newer technologies offer higher throughput, the unique advantages of Northern blotting—including direct size determination, alternative transcript detection, and quantitative reliability—ensure its continued relevance in molecular biology and drug development. The integration of modern enhancements such as optimized hybridization buffers and sensitive detection systems further strengthens the utility of this classic technique in contemporary research environments.
The detection of low-abundance targets is a cornerstone of modern biological research and diagnostic development. For scientists working with digoxigenin (DIG)-labeled RNA probes, achieving high sensitivity is paramount for accurately identifying rare transcripts, low-expression biomarkers, or trace pathogens. Sensitivity in molecular detection determines the minimum concentration of a target that can be reliably distinguished from its absence, fundamentally governing the efficacy of any assay [84]. While traditional immunoassays often lack the specificity or coverage needed for challenging targets, emerging technologies like mass spectrometry and CRISPR-based systems offer complementary approaches by enabling direct identification and quantification of individual molecules [85] [84]. This technical guide explores the core principles, methodologies, and advanced applications of sensitivity testing, with particular emphasis on protocols and innovations relevant to researchers utilizing DIG-labeled RNA probes.
The critical importance of sensitivity testing extends across multiple domains. In drug development, monitoring residual host cell proteins (HCPs) requires detection methods capable of identifying low-level impurities that could compromise product safety or stability [85]. In disease diagnostics, detecting minuscule quantities of nucleic acid biomarkers enables early identification of conditions like cancer or infections before clinical symptoms manifest [86] [84]. For researchers employing DIG-labeled RNA probes in situ hybridization (ISH), optimization for sensitivity directly impacts the ability to visualize spatial and temporal expression patterns of rare transcripts within tissue samples [87]. As detection technologies evolve toward increasingly sophisticated platforms, understanding the fundamental principles of sensitivity testing becomes essential for designing robust assays capable of detecting the most elusive targets.
Quantitative Landscape of Detection Technologies
The analytical performance of detection technologies varies significantly across platforms, with sensitivity ranges spanning from attomolar to nanomolar concentrations. Understanding these quantitative boundaries is essential for selecting appropriate methodologies for specific applications involving DIG-labeled RNA probes.
Table 1: Sensitivity Ranges of Detection Technologies
| Technology Platform | Detection Limit | Target Type | Time to Result | Amplification Required |
|---|---|---|---|---|
| NAPTUNE Platform [86] | Attomolar (aM) to Femtomolar (fM) | Nucleic acids, proteins | <45 minutes | Amplification-free |
| CRISPR-Cas13 (SHERLOCK) [84] | Attomolar (aM) level | RNA | Hours | Often requires pre-amplification |
| Mass Spectrometry [85] | Not explicitly quantified (sequence-specific) | Host cell proteins (HCPs) | Varies | Not applicable |
| DIG-labeled RNA Probes (ISH) [87] | Dependent on protocol optimization | RNA in tissue sections | 1-2 days | No, but signal amplification possible |
The NAPTUNE (Nucleic acids and Protein Biomarkers Testing via Ultra-sensitive Nucleases Escalation) platform represents a significant advancement with its attomolar-level sensitivity achieved through a tandem cascade of endonucleases, including apurinic/apyrimidinic endonuclease 1 (APE1) and Pyrococcus furiosus Argonaute (PfAgo) [86]. This system operates without target pre-amplification, generating DNA guides through APE1 activity that subsequently activate PfAgo-mediated cleavage of secondary probes, thereby amplifying detection signals [86]. For CRISPR-based systems, the Cas13 family exclusively targets RNA and utilizes collateral trans-cleavage activity to degrade reporter RNA molecules, generating detectable signals [84]. While these advanced platforms offer exceptional sensitivity, traditional DIG-labeled RNA probe protocols remain highly valuable for spatial context preservation in ISH applications, though they require meticulous optimization to achieve maximal sensitivity for low-abundance targets [87].
Core Principles of High-Sensitivity Detection
Achieving optimal sensitivity with DIG-labeled RNA probes requires adherence to several fundamental principles throughout the experimental workflow. Each principle addresses specific challenges associated with low-abundance target detection.
Probe Design and Labeling Efficiency
Probe characteristics directly influence hybridization efficiency and signal strength. RNA probes should ideally be 250–1,500 bases in length, with approximately 800 bases exhibiting optimal sensitivity and specificity [87]. The DIG RNA Labeling Mix is specifically formulated to incorporate digoxigenin-11-UTP at approximately every 20 to 25 nucleotides during in vitro transcription, ensuring sufficient label density for detection [88]. Using purified linearized plasmid DNA as a template is essential, as circular or nicked templates can produce non-specific transcripts and reduce efficiency. Complete restriction enzyme digestion (10 units/μg DNA for at least 3 hours) followed by gel purification ensures template quality [88]. Transcript integrity should be verified by agarose gel electrophoresis, though note that quantification by this method is not reliable due to potential secondary structures [88].
Signal Amplification Strategies
Enhancing detection signals without increasing background noise is crucial for low-abundance targets. The NAPTUNE platform employs an innovative in-situ cascade circuit where APE1 continuously generates DNA guides with 5'-phosphate ends through its cleavage activity, which then activate PfAgo-mediated cis-cleavage on secondary probes, dramatically boosting sensitivity and specificity without target amplification [86]. For ISH applications, using an anti-DIG antibody conjugated to alkaline phosphatase followed by colorimetric or fluorescent substrate development provides significant signal amplification [87]. Recent advancements in multiplexed error-robust fluorescence in situ hybridization (MERFISH) demonstrate that optimized encoding probe design and hybridization conditions can substantially enhance signal brightness and detection efficiency for individual RNA molecules [34].
Background Reduction and Specificity Enhancement
Minimizing non-specific signals is equally important as amplifying true positive signals. In ISH protocols, stringency washes using solutions with carefully controlled temperature, salt concentration, and detergent content are critical for removing imperfectly hybridized probes [87]. For short DNA/RNA probes (0.5–3 kb) or complex probes, washing temperatures should be lower (up to 45°C) with lower stringency (1–2x SSC), while single-locus or large probes benefit from higher temperatures (around 65°C) and higher stringency (below 0.5x SSC) [87]. Blocking with MABT (maleic acid buffer containing Tween 20) supplemented with 2% BSA, milk, or serum for 1–2 hours before antibody application reduces non-specific antibody binding [87]. Additionally, using sense strand RNA as a negative control helps distinguish specific from non-specific hybridization signals [87].
Experimental Protocol: DIG-Labeled RNA Probe ISH for Low-Abundance Targets
The following optimized protocol provides detailed methodologies for detecting low-abundance RNA targets using DIG-labeled probes, incorporating critical steps for sensitivity enhancement.
Tissue Preparation and Pre-Treatment
- Sample Storage and Fixation: Flash-freeze samples in liquid nitrogen or fix in formalin followed by paraffin embedding (FFPE). For long-term storage of prepared slides, store in 100% ethanol at -20°C or in a plastic box covered in saran wrap at -80°C to preserve RNA integrity for several years [87].
- Deparaffinization and Rehydration: For FFPE sections, perform sequential washes: xylene (2×3 min), xylene:100% ethanol (1:1, 3 min), 100% ethanol (2×3 min), 95% ethanol (3 min), 70% ethanol (3 min), 50% ethanol (3 min), followed by rinsing with cold tap water. Critical: Do not allow slides to dry after this point as it causes non-specific antibody binding and high background [87].
- Proteinase K Treatment: Digest with 20 µg/mL proteinase K in pre-warmed 50 mM Tris for 10–20 min at 37°C. Perform titration experiments to optimize concentration and incubation time as insufficient digestion reduces hybridization signal while over-digestion compromises tissue morphology [87].
- Acetic Acid Treatment and Dehydration: Immerse slides in ice-cold 20% (v/v) acetic acid for 20 seconds to permeabilize cells, then dehydrate through sequential 1-min washes in 70% ethanol, 95% ethanol, and 100% ethanol before air drying [87].
Hybridization and Stringency Washes
Hybridization Solution Composition:
- 50% Formamide
- 5× Salts (4 M NaCl, 100 mM EDTA, 200 mM Tris-HCl pH 7.5, 100 mM NaH₂PO₄·2H₂O)
- 5× Denhardt's solution (Ficoll, PVP, BSA)
- 10% Dextran sulfate
- 20 U/mL Heparin
- 0.1% SDS [87]
Probe Hybridization: Dilute DIG-labeled RNA probes in hybridization solution. Denature at 95°C for 2 min then immediately chill on ice. Apply 50–100 μL per section, cover with coverslip, and incubate in humidified chamber at 65°C overnight [87]. Optimal hybridization temperature depends on GC content and may require optimization between 55–62°C [87].
Stringency Washes:
Immunological Detection
- Blocking: Transfer slides to humidified chamber and add 200 μL blocking buffer (MABT + 2% BSA, milk, or serum) per section. Block for 1–2 hours at room temperature [87].
- Antibody Incubation: Drain blocking buffer and apply anti-DIG antibody at recommended dilution in blocking buffer. Incubate for 1–2 hours at room temperature [87].
- Washing: Wash slides 5×10 min with MABT at room temperature [87].
- Signal Development: Wash slides 2×10 min with pre-staining buffer (100 mM Tris pH 9.5, 100 mM NaCl, 10 mM MgCl₂) before adding colorimetric or fluorescent substrate [87].
Diagram 1: DIG-labeled RNA Probe ISH Workflow. Key sensitivity-critical steps (Deparaffinization, Stringency Washes, and Blocking) are highlighted in yellow for emphasis.
Advanced and Emerging Technologies
Beyond traditional ISH, several cutting-edge platforms offer revolutionary approaches to low-abundance target detection, providing researchers with powerful alternatives or complementary techniques.
NAPTUNE Platform Mechanism
The NAPTUNE platform employs a sophisticated enzyme cascade system for amplification-free detection. APE1 initially recognizes and cleaves at apurinic/apyrimidinic (AP) sites in the presence of target nucleic acids, generating DNA fragments with 5'-phosphate groups [86]. These fragments then serve as guide DNA for PfAgo, which activates cleavage of secondary and tertiary probes, creating a signal amplification cascade that elevates sensitivity to attomolar levels within 45 minutes [86]. This innovative approach eliminates the need for complex amplification steps while maintaining high specificity, making it particularly suitable for point-of-care testing and resource-limited environments [86].
Diagram 2: NAPTUNE Platform Cascade Mechanism. The enzyme cascade enables attomolar sensitivity without target pre-amplification in under 45 minutes.
CRISPR-Based RNA Detection
CRISPR-Cas systems have revolutionized nucleic acid detection through their programmable specificity and collateral cleavage activities. The Cas13 family exclusively targets RNA and possesses two HEPN domains that become catalytically active upon recognition of specific ssRNA targets, triggering promiscuous degradation of surrounding non-target ssRNA (trans-cleavage) [84]. This collateral activity is harnessed for diagnostics by introducing engineered reporter RNA molecules whose cleavage produces detectable fluorescence or colorimetric signals [84]. Platforms like SHERLOCK leverage this mechanism, often incorporating preamplification steps such as recombinase polymerase amplification (RPA) to enhance sensitivity to attomolar levels [84]. More recent developments focus on preamplification-free strategies using split-crRNA or split-activator systems that maintain high sensitivity while simplifying assay workflows [84].
Mass Spectrometry for Protein Detection
Advanced mass spectrometry techniques have emerged as powerful tools for monitoring low-abundance protein targets, particularly host cell proteins (HCPs) in biopharmaceutical products [85]. MS offers sequence-specific detection that complements traditional antibody-based methods, with recent innovations in data acquisition improving the ability to detect low-level impurities throughout biopharmaceutical production [85]. Different quantification strategies, including label-free, chemical labeling, and targeted detection, provide flexibility depending on application requirements. Furthermore, artificial intelligence supports more reliable analysis by improving spectral data interpretation and reducing false results [85].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for High-Sensitivity Detection with DIG-Labeled Probes
| Reagent/Category | Function/Purpose | Key Considerations |
|---|---|---|
| DIG RNA Labeling Mix [88] | Incorporates digoxigenin-11-UTP during in vitro transcription | Optimized for SP6, T7, T3 RNA polymerases; incorporates label every 20-25 nucleotides |
| Proteinase K [87] | Tissue permeabilization for probe access | Concentration (typically 20 µg/mL) and incubation time (10-20 min) require titration optimization |
| Anti-DIG Antibody [87] | Immunological detection of hybridized probes | Conjugated to alkaline phosphatase or other reporters; requires optimized dilution |
| Hybridization Buffer [87] | Optimal environment for specific probe-target binding | Contains formamide (50%), salts (5x), Denhardt's solution (5x), dextran sulfate (10%) |
| SSC Buffer [87] | Stringency washes to remove non-specifically bound probes | Concentration (0.1-2x SSC) and temperature (25-75°C) vary based on probe characteristics |
| MABT Buffer [87] | Gentle washing for nucleic acid detection | Maleic acid buffer with Tween-20; gentler than PBS for post-hybridization steps |
| Blocking Reagent [87] | Reduces non-specific antibody binding | BSA, milk, or serum (2%) in MABT; critical for minimizing background |
Sensitivity testing for low-abundance targets represents a dynamic frontier in molecular detection, with significant implications for research utilizing DIG-labeled RNA probes. While traditional ISH protocols provide robust spatial context information, emerging technologies like the NAPTUNE platform and CRISPR-based systems offer unprecedented sensitivity down to attomolar levels through innovative signal amplification mechanisms [86] [84]. The fundamental principles of optimal probe design, careful tissue processing, stringent hybridization conditions, and effective background reduction remain universally applicable across detection platforms.
For researchers working with DIG-labeled RNA probes, systematic optimization of each protocol step is essential for maximizing sensitivity. Critical parameters include probe length and labeling efficiency, proteinase K treatment conditions, hybridization temperature and stringency, and immunological detection conditions [88] [87]. By integrating these established best practices with emerging technologies and continuously evaluating assay performance through appropriate controls, scientists can push the detection boundaries for the most challenging low-abundance targets, enabling new discoveries in gene expression analysis, diagnostic development, and therapeutic monitoring.
Within molecular biology and diagnostic research, the precise detection of nucleic acids is a cornerstone technique. For decades, scientists have sought alternatives to radioactive labeling methods, with digoxigenin (DIG) and biotin emerging as the two predominant non-radioactive haptens. This whitepaper provides an in-depth technical comparison of these two systems, framing the analysis within the context of optimizing detection protocols for DIG-labeled RNA probes. Understanding the relative advantages, limitations, and appropriate applications of each system is critical for researchers and drug development professionals aiming to design robust, sensitive, and reliable assays.
Fundamental Concepts of Probe Labeling
The Digoxigenin (DIG) System
Digoxigenin is a steroid hapten derived exclusively from the Digitalis purpurea plant. Its key characteristic is that it is foreign to animal tissues, which fundamentally minimizes background interference in biological samples derived from these sources [89]. Detection of DIG-labeled probes is achieved via an antibody-based immunoassay. Typically, a high-affinity anti-DIG antibody, often conjugated to an enzyme like Alkaline Phosphatase (AP) or a fluorophore, binds to the hapten. Subsequent addition of a chromogenic, chemiluminescent, or fluorescent substrate generates the detectable signal [90] [91]. This system allows for significant signal amplification through the use of antibody sandwiches, where multiple conjugate-antibody fragments can bind a single DIG molecule [89].
The Biotin System
Biotin, also known as Vitamin B7 or Vitamin H, is an essential endogenous cofactor in all living cells. The detection of biotinylated probes relies not on an antibody, but on the high-affinity non-covalent interaction between biotin and proteins like streptavidin or avidin (Kd = 10⁻¹⁵ M) [92]. These biotin-binding proteins are then conjugated to reporters for detection. A significant consideration for this system is the ubiquitous presence of biotin in common biological samples such as liver, brain, and egg, which can lead to elevated background and false positives if not adequately controlled [92] [93].
Comparative Technical Performance
Sensitivity and Specificity
Sensitivity is a paramount metric for any detection system. A foundational 1993 study comparing non-radioactive systems for detecting amplified Herpes Simplex Virus DNA found that a DIG system with luminescent detection was equivalent in sensitivity to a radioactive (³²P) system, particularly at lower template concentrations. In contrast, biotinylated probes, whether detected colorimetrically or with photobiotin, demonstrated "clearly lower sensitivity" [90].
Specificity, or the signal-to-noise ratio, is equally crucial. Research indicates that DIG-labeled probes generally offer higher specificity with lower non-specific background compared to biotin systems [91] [89]. This is largely attributed to the absence of endogenous DIG in animal tissues, whereas endogenous biotin can cause significant background staining in certain tissues and cell types, complicating data interpretation [93] [89]. For in situ hybridization protocols, one-step detection for biotin often yields low sensitivity, whereas one-step detection for DIG can achieve high sensitivity. With multi-step detection protocols, both systems can achieve similarly high sensitivity [93].
Quantitative Analysis and Practical Workflow
A 2018 study developed a cost-effective, homemade protocol for DIG hybridization and detection, comparing it to both commercial DIG protocols and radioactive methods. The findings are summarized in the table below.
Table 1: Comparison of Nucleic Acid Detection Method Performance [91]
| Performance Metric | Radioactive Method | DIG (Commercial Protocol) | DIG (Homemade Protocol) |
|---|---|---|---|
| Sensitivity | High | Lower than homemade | High, generally better than commercial |
| Background Signal | Less background | Higher background | Reduced with optional Tween-20 |
| Quantitative Linearity | Greater linear response; more reliable for precise quantitation | Less reliable for quantitation | Less reliable for quantitation |
| Cost | High (rising costs and regulation) | Expensive | Much cheaper |
| Speed & Convenience | Time-consuming; requires protective measures | Tedious and time-consuming | Faster |
This study concluded that while radioactive methods coupled with phosphorimaging remain superior for precise quantification, the optimized DIG protocol is an excellent tool for qualitative detection and is faster, more sensitive, and much cheaper than standard commercial DIG protocols [91].
Experimental Protocols and Methodologies
Detailed Protocol: DIG-Labeled RNA Probe Synthesis
The synthesis of DIG-labeled RNA probes (riboprobes) is typically performed via in vitro transcription. The following protocol synthesizes a probe complementary to a target sequence [22] [94].
- Template Preparation: A plasmid containing the gene of interest, cloned in reverse orientation relative to the RNA polymerase promoter, is linearized using a restriction enzyme that produces a 5' overhang. Templates with 3' overhangs or blunt ends can promote "run-on" transcription of the wrong strand. The linearized template must be purified via phenol-chloroform extraction and ethanol precipitation or using a commercial purification kit to remove enzymes and salts [95].
- In Vitro Transcription: The reaction is assembled with the purified linear template, transcription buffer, RNA polymerase (SP6, T7, or T3), and a nucleotide mixture containing DIG-11-UTP. Under standard conditions, the polymerase incorporates a DIG-labeled UTP approximately every 20 to 25 nucleotides [95].
- Probe Purification and Analysis: After transcription, the DNA template is digested with DNase I. The resulting DIG-labeled RNA probe is then purified by precipitation or column-based methods. Labeling efficiency and probe integrity should be checked by running an aliquot on a MOPS-formaldehyde agarose gel to resolve any secondary structures that may form [95].
Detection Workflow for DIG-Labeled Probes
The following diagram illustrates the key steps in detecting a DIG-labeled probe after it has been hybridized to its target on a membrane.
Diagram 1: DIG Probe Detection Workflow.
The homemade detection protocol outlined in [91] recommends a blocking and antibody incubation buffer consisting of 75 mM maleic acid (pH 7.5), 200 mM NaCl, and 5% non-fat dry milk powder. Washes are performed in the same buffer with the optional addition of 0.3% Tween-20 to reduce background. The final chemiluminescent reaction is triggered by adding CSPD substrate in an alkaline buffer (100 mM Tris-HCl, 100 mM NaCl, pH 9.5) [91].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of non-radioactive detection assays requires a suite of reliable reagents. The following table catalogs key solutions for working with DIG and biotin-labeled probes.
Table 2: Key Research Reagent Solutions for Non-Radioactive Detection
| Reagent / Solution | Function / Purpose | Example & Notes |
|---|---|---|
| DIG RNA Labeling Mix | Provides nucleotides, including DIG-11-UTP, for efficient incorporation during in vitro transcription. | Roche DIG RNA Labeling Mix; optimized for SP6, T7, T3 RNA polymerases [95]. |
| Anti-DIG Antibody | The primary detection reagent that binds specifically to the digoxigenin hapten. | Often used as a Fab fragment conjugated to Alkaline Phosphatase for high sensitivity [91]. |
| Biotinylation Kits | Enable researchers to chemically label proteins or nucleic acids with biotin tags. | Thermo Fisher Pierce offers kits for cell-surface protein biotinylation and antibody labeling [92]. |
| Streptavidin/NeutrAvidin | High-affinity biotin-binding proteins conjugated to enzymes or fluorophores for detection. | NeutrAvidin (deglycosylated avidin) is recommended to reduce nonspecific binding due to its near-neutral pI [92]. |
| Homemade Hybridization Buffer | A cost-effective alternative to commercial hybridization buffers. | 250 mM sodium phosphate buffer (pH 7.4), 7% SDS, 1 mM EDTA [91]. |
| Homemade Blocking Buffer | A cost-effective alternative to commercial blocking solutions. | 75 mM maleic acid (pH 7.5), 200 mM NaCl, 5% non-fat dry milk powder [91]. |
| CSPD / CDP-Star | Ready-to-use chemiluminescent substrates for Alkaline Phosphatase. | Provides sustained light emission for high-sensitivity detection on blotting membranes [91]. |
The choice between DIG and biotin-labeled probes is not a matter of one being universally superior, but rather of selecting the right tool for the specific experimental context. The DIG system is highly recommended for applications demanding the highest specificity and sensitivity, particularly in situations involving tissues rich in endogenous biotin (e.g., liver, kidney) or when background signal is a primary concern [93] [89]. Its performance is on par with radioactive methods for qualitative detection and some quantitative applications, offering a safer and more stable alternative [90] [91].
Conversely, the biotin system remains a powerful and economical tool, especially when using modern derivatives like NeutrAvidin to mitigate nonspecific binding [92]. Its utility is greatest in applications where endogenous biotin is not an issue and when the robust avidin-biotin interaction can be leveraged for purification or pull-down assays.
For researchers focused on DIG-labeled RNA probe protocols, the evidence supports the adoption of optimized, homemade solutions for hybridization and detection to maximize performance while minimizing costs [91]. As non-radioactive techniques continue to evolve, the DIG system, with its high specificity and robust signal amplification, will undoubtedly remain an indispensable component of the molecular biologist's toolkit, particularly in sensitive diagnostic and drug development applications.
Selecting the Right Labeling Method for Your Specific Application
The ability to label RNA molecules is a cornerstone of molecular biology, enabling researchers to probe gene expression, visualize RNA localization, study RNA-protein interactions, and analyze RNA dynamics in living cells. The selection of an appropriate labeling strategy is paramount to experimental success, as the choice influences sensitivity, specificity, and the very biological questions that can be addressed. Within the broader context of digoxigenin-labeled RNA probe research, understanding the landscape of available methods—from enzymatic and chemical approaches to metabolic labeling and aptamer-based systems—provides the foundation for rigorous and reproducible science. This guide provides a comprehensive technical overview of RNA labeling methodologies, detailing their mechanisms, applications, and optimal use cases to empower researchers in making informed experimental decisions.
The fundamental principles of nucleic acid labeling involve incorporating detectable tags—such as radioactive isotopes, haptens (e.g., biotin, digoxigenin), or fluorophores—into RNA molecules without significantly altering their biochemical properties or biological function [96] [97]. These tags enable detection or purification, turning ordinary RNA into powerful probes for identifying other interacting molecules. The choice of label and incorporation method is heavily influenced by the specific application, whether it involves in situ hybridization, single-molecule tracking, pull-down assays, or studying transcriptional dynamics [96].
The table below summarizes the primary RNA labeling methods, their key characteristics, and typical applications to provide a quick reference for researchers.
Table 1: Summary of Major RNA Labeling Methodologies
| Method Category | Specific Method | Labeling Site | Key Characteristics | Recommended For |
|---|---|---|---|---|
| Enzymatic | T7 RNA Polymerase (in vitro transcription) | Uniform incorporation | Highly processive; generates long RNAs; uses [α-32P]CTP or modified NTPs [98]. | Generating large amounts of probe for Northern blotting, ribonuclease protection assays. |
| Enzymatic | T4 Polynucleotide Kinase (T4 PNK) | 5' end | Transfers phosphate from [γ-32P]ATP to 5'-OH; requires prior dephosphorylation for efficiency [96] [98]. | 5' end-labeling for gel shift assays, primer phosphorylation for cloning. |
| Enzymatic | T4 RNA Ligase | 3' end | Catalyzes attachment of 5'-phosphate (e.g., [5′-32P]pCp) to a 3'-hydroxyl group [96] [98]. | 3' end-labeling, modifying mRNA for cDNA library generation, RACE. |
| Chemical Conversion | Metabolic Labeling (e.g., SLAM-seq, TimeLapse-seq) | Newly synthesized RNA (site-specific nucleotide conversion) | Uses nucleoside analogs (4sU, 5-EU); detects RNA via T-to-C sequencing mutations [99] [100]. | High-throughput measurement of RNA synthesis/degradation dynamics in single cells. |
| Aptamer-Based | PP7/MS2 System | Specific RNA sequences | Binds fluorescently labeled coat protein to engineered RNA stem-loops; non-covalent [101]. | Live-cell RNA imaging and tracking of specific RNA molecules in vivo. |
| Proximity Labeling | OINC-seq / Halo-seq | RNA proximal to bait protein | Light-induced oxidation of RNAs near a HaloTag-fused protein; mutations read via sequencing [102]. | Mapping the RNA content of specific subcellular locations without fractionation. |
Detailed Methodologies and Protocols
Enzymatic Labeling Protocols
In Vitro Synthesis of Uniformly Radiolabeled RNA This protocol is ideal for generating probes with high specific activity, suitable for sensitive detection applications [98].
- DNA Template Preparation: The DNA template can be prepared by linearizing a plasmid downstream of a phage promoter (e.g., T7, T3, SP6), by PCR adding the promoter sequence, or by annealing oligonucleotides for short transcripts.
- Transcription Reaction: Assemble the following reaction at room temperature:
- 4 µL of 5X transcription buffer.
- 4.6 µL of nuclease-free H₂O.
- 1 µL of 3 NTP mix (10 mM each of ATP, GTP, UTP).
- 2.4 µL of 100 µM CTP.
- 5 µL of [α-32P]CTP (10 µCi/µL, 800 Ci/mmol).
- 1 µL of linear DNA template (1 µg/µL).
- 1 µL of RNase inhibitor (40 U/µL).
- 1 µL of T7 RNA Polymerase (20 U/µL).
- Incubation and Cleanup: Incubate at 37°C for 2 hours. Degrade the DNA template by adding 2 U of DNase I (RNase-free) and incubating for another 15 minutes.
- Purification: Stop the reaction with G50 buffer and purify the RNA by phenol/chloroform/isoamyl alcohol extraction followed by ethanol precipitation. For highest purity, separate the product via 8 M urea-PAGE, excise the band, and elute using a freeze-thaw method [98].
5' End-Labeling with T4 Polynucleotide Kinase (PNK) This method is critical for applications like electrophoretic mobility shift assays (EMSAs) where end-labeling minimizes steric hindrance [96] [98].
- Substrate Preparation: The RNA substrate must possess a 5'-hydroxyl group. If the RNA is transcribed in vitro or purified with a 5'-triphosphate or 5'-phosphate, it must first be dephosphorylated using calf intestinal phosphatase (CIP). Gel purification is recommended for optimal results.
- Kinase Reaction: The "forward reaction" is most efficient. Set up the reaction with the dephosphorylated RNA, T4 PNK enzyme, and [γ-32P]ATP.
- Purification: After incubation, purify the 5' end-labeled RNA using spin columns or precipitation to remove unincorporated ATP.
The workflow diagram below illustrates the two primary enzymatic labeling strategies.
Advanced and Emerging Labeling Techniques
Metabolic RNA Labeling for Transcriptional Dynamics Metabolic labeling techniques, such as SLAM-seq and TimeLapse-seq, have revolutionized the study of RNA kinetics by integrating nucleoside analogs like 4-thiouridine (4sU) with high-throughput sequencing [99] [100].
Protocol Workflow:
- Pulse-Labeling: Living cells or organisms are incubated with 4sU (typically 100 µM) for a defined pulse (e.g., 4 hours) to incorporate the analog into newly synthesized RNA.
- RNA Extraction: Total RNA is isolated, often from fixed or cryo-preserved cells to maintain cell states.
- Chemical Conversion: The key step involves a chemical reaction that specifically modifies the 4sU residue. Different methods exist:
- SLAM-seq: Uses iodoacetamide (IAA) to alkylate 4sU.
- TimeLapse-seq: Uses 2,2,2-trifluoroethylamine (TFEA) with an oxidizing agent like mCPBA.
- Library Preparation and Sequencing: The modified 4sU causes characteristic T-to-C transitions during reverse transcription and sequencing. These mutations are then computationally identified and quantified to distinguish newly synthesized (labeled) RNA from pre-existing (unlabeled) RNA [99].
Optimization and Benchmarking: Recent benchmarking studies show that the choice of chemical conversion method and sequencing platform significantly impacts data quality. "On-beads" conversion methods, particularly the mCPBA/TFEA combination, have been shown to achieve higher T-to-C substitution rates (~8%) and better RNA recovery compared to "in-situ" methods performed inside intact cells [99]. The optimal labeling time is also a critical parameter that depends on the RNA degradation rates under investigation [100].
In Vivo RNA Visualization with the PP7 Aptamer System This protocol allows for the live imaging of specific RNA molecules in cellular contexts such as tobacco pollen tubes, and is adaptable to other systems [101].
- Construct Design: Engineer the RNA of interest to include multiple repeats of the PP7 RNA stem-loop aptamer sequence.
- Reporter Protein: Use a fluorescently labeled reporter protein, such as the Pseudomonas aeruginosa PP7 bacteriophage coat protein (PCP) fused to a fluorescent protein (e.g., GFP).
- Delivery: Introduce both constructs into the target cells. For pollen tubes, biolistic gene-gun-mediated transformation is effective. For other cell types, tissue-specific promoters and appropriate transfection methods should be used.
- Imaging: The co-expressed PCP-FP reporter binds with high affinity and specificity to the PP7 stem-loops on the target RNA, allowing visualization of the resulting ribonucleoprotein particles (RNPs) using fluorescence microscopy [101].
The Scientist's Toolkit: Essential Reagents and Materials
Successful RNA labeling experiments rely on a suite of specialized reagents and materials. The following table details key components for various labeling approaches.
Table 2: Key Research Reagent Solutions for RNA Labeling
| Reagent/Material | Function/Description | Example Applications |
|---|---|---|
| Phage RNA Polymerases (T7, T3, SP6) | DNA-dependent RNA polymerases that initiate transcription with high specificity from their respective promoters. | In vitro transcription for uniform or site-specific RNA labeling [98]. |
| Modifying Enzymes (T4 PNK, T4 RNA Ligase) | T4 PNK transfers phosphate to 5'-OH; T4 RNA Ligase joins RNA ends. | 5' or 3' end-labeling with radioactive or non-radioactive tags [96] [98]. |
| Nucleoside Analogs (4-thiouridine, 5-Ethynyluridine) | Metabolically incorporated into newly synthesized RNA, serving as a chemical handle for downstream modification or detection. | Metabolic labeling experiments (SLAM-seq, TimeLapse-seq) to study RNA dynamics [99]. |
| HaloTag Fusion Protein & Halo-DBF Ligand | The HaloTag protein is targeted to a subcellular location; the DBF ligand, when activated by light, oxidizes nearby RNAs. | Proximity-dependent RNA labeling (OINC-seq/Halo-seq) [102]. |
| PP7 Coat Protein (PCP) & Aptamer | The PCP, fused to a fluorescent protein, binds with high specificity to the engineered PP7 RNA aptamer sequence. | Live-cell RNA imaging and tracking of specific RNA molecules [101]. |
| Barcoded Beads (e.g., Drop-seq) | Microbeads with oligonucleotide barcodes for capturing mRNA from single cells, enabling on-beads chemical reactions. | High-throughput single-cell RNA sequencing combined with metabolic labeling [99]. |
Strategic Selection and Concluding Remarks
Selecting the optimal RNA labeling method is a strategic decision that must align with the biological question, required resolution, and experimental constraints. The following guidelines can assist in this selection process:
- For Studying RNA-Protein Interactions: Use 5' or 3' end-labeling (e.g., with T4 PNK or T4 RNA Ligase) to avoid steric hindrance that could interfere with protein binding in assays like EMSAs or pull-downs [96].
- For High-Sensitivity Detection and Northern Blotting: Uniform labeling via in vitro transcription provides high specific activity, ideal for detecting low-abundance targets [96] [98].
- For Analyzing RNA Synthesis and Degradation Kinetics: Metabolic labeling (e.g., SLAM-seq, TimeLapse-seq) is the state-of-the-art method for transcriptome-wide, time-resolved measurement of RNA dynamics in single cells [99] [100].
- For Live-Cell RNA Imaging and Localization: Aptamer-based systems (e.g., PP7/MS2) are unparalleled for tracking the movement and localization of specific RNA molecules in real-time within living cells [101].
- For Mapping Proximal Transcriptomes without Fractionation: Proximity labeling methods (e.g., OINC-seq) are powerful for uncovering the RNA content of specific organelles or subcellular compartments [102].
In conclusion, the field of RNA labeling offers a diverse and powerful toolkit. From well-established enzymatic techniques to cutting-edge metabolic and proximity-based methods, researchers have an array of options to tackle complex biological questions. By carefully considering the trade-offs between specificity, sensitivity, and experimental throughput, scientists can select the most appropriate labeling strategy to advance their research, including the development and application of robust digoxigenin-labeled RNA probe protocols.
Conclusion
Digoxigenin-labeled RNA probes represent a powerful, versatile, and safe non-radioactive technology for sensitive nucleic acid detection. By adhering to optimized protocols for template preparation, in vitro transcription, and post-hybridization washes, researchers can achieve high-specificity results comparable to traditional radioactive methods. The robust troubleshooting and validation frameworks ensure reliability across diverse applications from basic research to drug development. Future directions include further integration with high-throughput automated platforms and expanded use in clinical diagnostics, solidifying the DIG system's role as a cornerstone technology in molecular biology.
References
- 1. Nucleic Acid Labeling [https://www.merckmillipore.com/DZ/en/products/molecular-biology-and-functional-genomics/nucleic-acid-electrophoresis-and-hybridization/nucleic-acid-labeling]
- 2. Non-Radioactive Nucleic Acid Labeling Product [https://www.archivemarketresearch.com/reports/non-radioactive-nucleic-acid-labeling-product-334158]
- 3. DIG Northern Starter Kit Protocol & Troubleshooting [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/nucleic-acid-labeling-and-detection/dig-northern-starter-kit?srsltid=AfmBOopbrMAoyr7AgR-Gxaoyl9VWNNU8gTzoeJO3VJ2DlR6XAChjopDF]
- 4. ISH: in situ hybridization protocol [https://www.abcam.com/en-us/technical-resources/protocols/in-situ-hybridization]
- 5. Digoxigenin - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/digoxigenin]
- 6. A rapid and sensitive nonradioactive method applicable for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3668450/]
- 7. Protocol for detection of glial complement expression in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11530921/]
- 8. AMPIVIEW® Ki67 (AS) Dig RNA Probes Set [https://www.enzo.com/product/ampiview-ki67-as-dig-rna-probes-set/]
- 9. Why are you still using radioactively labeled Probes? [https://www.jenabioscience.com/about-us/news-blog/3183-why-are-you-still-using-radioactively-labeled-probes]
- 10. RNA-directed RNA Polymerase - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/rna-directed-rna-polymerase]
- 11. Double in situ hybridisation protocols [https://www.ucl.ac.uk/~ucbzwdr/double%20in%20situ%20protocol.htm]
- 12. An optimized protocol for high-throughput in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4589143/]
- 13. Determination of gene expression patterns using high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2780369/]
- 14. DIG RNA Labeling Mix solution - pkg of 40 uL [https://www.sigmaaldrich.com/US/en/product/roche/11277073910?srsltid=AfmBOooqNcJQIsVXDafTGB-8ZnpBaTdh1tzAhT-ea7SsW126m-wrG2lX]
- 15. Riboprobe RNA probe templates [http://www.genedetect.com/probetemplates.htm]
- 16. RNA Probe - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/rna-probe]
- 17. DNA polymerase [https://en.wikipedia.org/wiki/DNA_polymerase]
- 18. Structure and mechanism of DNA polymerases [https://www.sciencedirect.com/science/article/pii/S0065323304710116]
- 19. Structure and Mechanism of DNA Polymerase β - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4018062/]
- 20. Production of digoxigenin-labelled RNA probes and the ... [https://www.sciencedirect.com/science/article/abs/pii/S1385299X98000105]
- 21. HighYield T7 STAR RED RNA Labeling Kit (UTP-based) [https://www.jenabioscience.com/rna-technologies/rna-labeling-modification/random-rna-labeling-in-vitro-transcription-based/rnt-101-stred-highyield-t7-star-red-rna-labeling-kit-utp-based]
- 22. Synthesis of RNA–Digoxigenin Probe [https://link.springer.com/protocol/10.1007/978-1-0716-4518-5_49]
- 23. Synthesis and purification of digoxigenin-labeled RNA ... [https://pubmed.ncbi.nlm.nih.gov/21357143/]
- 24. A PCR-Based Method for RNA Probes and Applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5942160/]
- 25. Digoxigenin (DIG) Labeling Methods [https://www.merckmillipore.com/CI/fr/technical-documents/technical-article/genomics/nucleic-acid-labeling-and-detection/dig-labeling-methods]
- 26. The Basics: RNase Control [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/nuclease-enzymes/general-articles/the-basics-rnase-control.html]
- 27. Simple and nonradioactive detection of microRNAs using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3964919/]
- 28. Tips for Maintaining an RNase-Free Environment [https://www.shieldscientific.com/glove-education/gloving-rnase-free-environment/]
- 29. Production of Digoxigenin-Labeled Riboprobes for In Situ ... [https://pubmed.ncbi.nlm.nih.gov/32436648/]
- 30. 10 Ways to Work RNase Free [https://bitesizebio.com/163/10-ways-to-work-rnase-free/]
- 31. Don't Let Ribonucleases Ruin Your Week(end) [https://www.promegaconnections.com/dont-let-ribonucleases-ruin-your-weekend-establish-a-ribonuclease-free-environment/]
- 32. Optimizing mRNA Template Design [https://www.aldevron.com/blog/optimizing-mrna-template-design]
- 33. Quantification of objective concentrations of DNA impurities ... [https://www.sciencedirect.com/science/article/pii/S0264410X25003196]
- 34. Protocol optimization improves the performance of ... [https://www.nature.com/articles/s41598-025-17477-1]
- 35. Overview of In Vitro Transcription [https://www.thermofisher.com/us/en/home/industrial/pharma-biopharma/nucleic-acid-therapeutic-development-solutions/mrna-research/overview-in-vitro-transcription.html]
- 36. Strategies for synthesizing in vitro transcribed (IVT) mRNA [https://www.neb.com/en-us/tools-and-resources/feature-articles/mind-your-caps-and-poly-a-tails-strategies-for-synthesizing-in-vitro-transcribed-ivt-mrna?srsltid=AfmBOorMtx2u3qwGjqOcM8n7Er2l9ZCh-0bJo66h9uoRbEiBH4gxGfGi]
- 37. Methods for Enzymatic Nonisotopic Labeling of RNA by in ... [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/general-articles/methods-for-enzymatic-nonisotopic-labeling-of-rna-by-in-vitro-transcription.html]
- 38. Random RNA Labeling (in vitro Transcription-based) [https://www.jenabioscience.com/rna-technologies/rna-labeling-modification/random-rna-labeling-in-vitro-transcription-based]
- 39. In Vitro Transcription from Plasmid or PCR-amplified DNA [https://www.sciencedirect.com/science/article/pii/B9780124200371000051]
- 40. In Vitro Transcription Optimization Guide [https://rna.bocsci.com/support/optimizing-in-vitro-transcription-for-high-yield-mrna-synthesis.html]
- 41. In Vitro Transcription: Common Causes of Reaction Failure [https://www.promegaconnections.com/in-vitro-transcription-common-causes-of-reaction-failure/]
- 42. Top Tips for Troubleshooting In Vitro Transcription [https://bitesizebio.com/44813/top-tips-for-troubleshooting-in-vitro-transcription/]
- 43. Practical Tips for In Vitro Transcription [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/large-scale-transcription/general-articles/practical-tips-for-in-vitro-transcription.html]
- 44. & Removal Reagents | Thermo Fisher Scientific - US DNase Treatment [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/tech-notes/a-new-method-to-remove-dna.html]
- 45. High Throughput RNA Isolation: Methods Comparison [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/posters/high-throughput-rna-isolation-methods-comparison.html]
- 46. Comparison of Four Methods of RNA Extraction and cDNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10342303/]
- 47. Comparison of RNA isolation methods on RNA-Seq [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6673-2]
- 48. Protocol for RNA fluorescence in situ hybridization in mouse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8956821/]
- 49. in situ Hybridization Kits, Hybridization Assays [https://acdbio.com/manual-assays-rnascope]
- 50. Selected In Situ Hybridization Methods: Principles and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8270300/]
- 51. In situ hybridization: Introduction to techniques ... [https://www.sciencedirect.com/science/article/abs/pii/S0740257019300620]
- 52. Northern blot - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4287216/]
- 53. The Basics: Northern Analysis [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/general-articles/the-basics-northern-analysis.html]
- 54. Gel Shift Assays (EMSA) [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/gel-shift-assays-emsa.html]
- 55. Electrophoretic Mobility Shift Assay (EMSA) for Detecting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2757439/]
- 56. Principles and problems of the electrophoretic mobility shift ... [https://www.sciencedirect.com/science/article/abs/pii/S1056871910000456]
- 57. Northern Blot- Definition, Principle, Steps, Results ... [https://microbenotes.com/northern-blot/]
- 58. HighYield T7 Digoxigenin RNA Labeling Kit (UTP-based) [https://www.jenabioscience.com/rna-technologies/rna-labeling-modification/random-rna-labeling-in-vitro-transcription-based/rnt-101-digx-highyield-t7-digoxigenin-rna-labeling-kit-utp-based]
- 59. Digoxigenin-labeled RNA probes and ISH [https://bio-protocol.org/exchange/minidetail?id=5105640&type=30]
- 60. ICH Revises Q1 Guideline Advancing Stability Testing ... [https://www.pharmaceuticalonline.com/doc/ich-revises-q-guideline-advancing-stability-testing-standards-0001]
- 61. Interpreting Electrophoresis Gels [https://bento.bio/resources/bento-lab-advice/interpreting-electrophoresis-gels-with-bento-lab/]
- 62. How to Interpret DNA Gel Electrophoresis Results [https://www.goldbio.com/blogs/articles/interpreting-gel-electrophoresis-results?srsltid=AfmBOoq-WfkfWiD-R1o25mLT9MFN2_oNUKzCcQbQlZDFSeXhzVe-ZUIe]
- 63. Designing Stellaris RNA FISH Probes Part I [https://blog.biosearchtech.com/considerations-for-optimizing-stellaris-rna-fish-probe-design]
- 64. Optimization of hybridization chain reaction for imaging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11446410/]
- 65. Protocol to validate custom-designed BaseScope probes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12615734/]
- 66. Universal Improvement of In Situ Hybridization Chain ... [https://pubmed.ncbi.nlm.nih.gov/40653625/]
- 67. Optimizing Stringency in Wash Buffers for Hybridization Assays [https://www.letstalkacademy.com/increasing-stringency-hybridization-buffers/]
- 68. DIG RNA Labeling Mix Protocol Troubleshooting [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/nucleic-acid-labeling-and-detection/dig-rna-labeling-mix?srsltid=AfmBOoqu31j6VQwhtwl-3n963l9cdB8-YVJtj1SYA1uupGbRmjSKSgyV]
- 69. Protocol to analyze tRNA and rRNA processing using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12166403/]
- 70. Increasing Sensitivity in Northern Analysis with RNA Probes [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/tech-notes/increasing-sensitivity-in-northern-analysis-with-rna-probes.html]
- 71. DIG RNA Labeling Mix Protocol Troubleshooting [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/nucleic-acid-labeling-and-detection/dig-rna-labeling-mix?srsltid=AfmBOopoeG5AnIMnVFcLMW83C8vhuR6LKL7_6WZzGHrw08Ns6xLTu3NP]
- 72. Discovery of RNA‐Targeting Small Molecules - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12375692/]
- 73. Optimization of a 6-plex Crystal Digital PCR® assay and its ... [https://www.sciencedirect.com/science/article/pii/S0048969725005741]
- 74. Digoxigenin - an overview [https://www.sciencedirect.com/topics/neuroscience/digoxigenin]
- 75. A PCR-Based Method for RNA Probes and Applications in ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2018.00266/full]
- 76. Optimized RNA ISH, RNA FISH and protein-RNA double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4126239/]
- 77. The Basics: Northern Analysis [https://www.thermofisher.com/sg/en/home/references/ambion-tech-support/northern-analysis/general-articles/the-basics-northern-analysis.html]
- 78. Southern vs Northern vs Western Blotting Techniques [https://www.labmanager.com/southern-vs-northern-vs-western-blotting-techniques-854]
- 79. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOoqPtbGhNvfJVaDw75ryLczpox_vsrtVgoLechjmGDc0VnUchR0P]
- 80. Sensitive and specific detection of microRNAs by northern blot ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC545470/]
- 81. More Than a Clever Name: Northern Blots [https://bitesizebio.com/27267/more-than-a-clever-name-northern-blots/]
- 82. RNA dot blot protocol [https://www.abcam.com/en-us/technical-resources/protocols/rna-dot-blot]
- 83. One probe fits all: a highly customizable modular RNA in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12148028/]
- 84. CRISPR–Cas based platforms for RNA detection [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc03257a]
- 85. Advanced mass spectrometry techniques for monitoring ... [https://www.sciencedirect.com/science/article/pii/S0167779925003117]
- 86. NAPTUNE: nucleic acids and protein biomarkers testing ... [https://www.nature.com/articles/s41467-025-56653-9]
- 87. In situ hybridization (ISH) protocol | Abcam [https://www.abcam.cn/technical-resources/protocols/in-situ-hybridization]
- 88. DIG RNA Mix Labeling Troubleshooting Protocol [https://www.sigmaaldrich.com/CZ/cs/technical-documents/protocol/genomics/nucleic-acid-labeling-and-detection/dig-rna-labeling-mix]
- 89. Shall you use Biotin or Digoxigenin for ISH Probe Labeling? [https://www.enzo.com/note/shall-you-use-biotin-or-digoxigenin-for-ish-probe-labeling/]
- 90. Evaluation of 3 nonradioactive DNA detection systems for identification... [https://pubmed.ncbi.nlm.nih.gov/8366171/]
- 91. A fast, sensitive and cost-effective method for nucleic acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6994052/]
- 92. Avidin-Biotin Interaction | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/avidin-biotin-interaction.html]
- 93. A comparison of digoxigenin and biotin labelled DNA and ... [https://pubmed.ncbi.nlm.nih.gov/7548436/]
- 94. 3.1 DIG labeled RNA probe synthesis by in vitro transcription [https://bio-protocol.org/exchange/minidetail?id=1848705&type=30]
- 95. DIG RNA Labeling Mix Protocol Troubleshooting [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/nucleic-acid-labeling-and-detection/dig-rna-labeling-mix?srsltid=AfmBOooJZN_Mt_80AuaVVYCRahhDvIU_KkIYHusCA6gTx3uo0wEltizP]
- 96. Methods for Labeling Nucleic Acids [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/methods-labeling-nucleic-acids.html]
- 97. Strength in numbers: quantitative single‐molecule RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5024021/]
- 98. Synthesis and Labeling of RNA In Vitro - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3736555/]
- 99. Benchmarking metabolic RNA labeling techniques for high ... [https://www.nature.com/articles/s41467-025-61375-z]
- 100. On the optimal design of metabolic RNA labeling experiments [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007252]
- 101. A protocol for in vivo RNA labeling and visualization ... [https://www.sciencedirect.com/science/article/pii/S2666166724005987]
- 102. Analyzing RNA Localization Using the RNA Proximity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12336859/]