CRISPR-Cas9 in Zebrafish: A Comprehensive Guide for Efficient Gene Editing and Disease Modeling
This article provides a comprehensive overview of CRISPR-Cas9 gene editing in zebrafish embryos, a cornerstone technique for functional genomics and drug discovery.
CRISPR-Cas9 in Zebrafish: A Comprehensive Guide for Efficient Gene Editing and Disease Modeling
Abstract
This article provides a comprehensive overview of CRISPR-Cas9 gene editing in zebrafish embryos, a cornerstone technique for functional genomics and drug discovery. It covers the foundational principles of zebrafish as a model organism and the CRISPR-Cas9 mechanism. The content details established protocols for knockout and knock-in generation, explores advanced applications like base editing for disease modeling, and addresses common troubleshooting and optimization strategies. Finally, it examines the validation of editing efficiency, comparative analysis with other models, and the direct application of zebrafish CRISPR models in high-throughput target validation and phenotypic drug screening, offering a complete resource for researchers and drug development professionals.
Why Zebrafish? Unlocking Vertebrate Biology with an Ideal Genetic Model
The tropical freshwater minnow, Danio rerio, commonly known as the zebrafish, has emerged as a powerful vertebrate model organism for biomedical research over the past few decades [1] [2]. Its unique combination of biological features provides unprecedented potential for genetic and drug screening studies, particularly when integrated with modern genome engineering technologies like the CRISPR-Cas9 system [3] [4]. The general strengths of zebrafish are well-known in the scientific community: cost-effectiveness, high fecundity, short generation time, external development, transparency during embryonic stages, and ease of genome manipulation [1]. These characteristics have positioned zebrafish as an ideal model system for addressing complex biological questions that are difficult to investigate in other vertebrate models. The relevance of zebrafish for human disease research is underscored by the high degree of genetic similarity to humans; over 80% of disease-causing human proteins have an ortholog in zebrafish, and the publishing of the zebrafish reference genome in 2013 has significantly accelerated disease modelling in this organism [1]. This application note details how these advantages, specifically external development, transparency, and high fecundity, are leveraged in CRISPR-Cas9 gene editing research, with practical protocols for implementation.
Core Advantages and Their Applications in CRISPR Research
The distinctive advantages of zebrafish are not merely convenient traits but represent fundamental characteristics that enable specific experimental approaches in genetic research, particularly in CRISPR-Cas9 based studies. The table below summarizes these key advantages and their direct research applications.
Table 1: Key Advantages of Zebrafish and Their Research Applications
| Advantage | Description | Application in CRISPR/Cas9 Research |
|---|---|---|
| External Development | Embryos develop outside the mother, enabling direct access from fertilization onward [1] [5]. | Microinjection of CRISPR components (Cas9 + gRNA) at the one-cell stage for direct genome editing [3] [6]. |
| Optical Transparency | Embryos and larvae are optically transparent during early development [1] [7]. | Real-time, high-resolution imaging of developmental processes and phenotypes in live, CRISPR-edited animals [7] [8]. |
| High Fecundity | A single pair can produce hundreds of embryos per week [1]. | High-throughput genetic screens using numerous CRISPR-injected F0 embryos [4] [8]. |
| Short Generation Time | Zebrafish reach sexual maturity in about 3-4 months [2]. | Rapid generation of stable, heritable mutant lines (F2) for analysis [3]. |
| Genetic Tractability | High degree of genetic and physiological similarity to humans [1] [7]. | Efficient modeling of human genetic diseases via targeted knockout or knock-in mutations [3] [9]. |
External Development and Direct Embryonic Access
The external fertilization and development of zebrafish embryos provide a critical technical advantage for genetic manipulation. Unlike mammalian models, researchers have direct physical access to the embryo from the moment of fertilization. This allows for the microinjection of CRISPR-Cas9 components directly into the one-cell stage zygote, ensuring that genetic modifications can be introduced at the earliest possible developmental stage [3] [6]. The procedure involves using fine needles to deliver in vitro transcribed guide RNA (gRNA) and Cas9 mRNA or protein into the cytoplasm or cell nucleus of freshly fertilized eggs [6]. This direct access is a fundamental prerequisite for efficient genome engineering, as it allows the CRISPR machinery to be present before the first cell division, increasing the likelihood of generating uniform, non-mosaic mutations in the resulting embryo [10].
Embryonic Transparency for Live Phenotyping
The optical transparency of zebrafish embryos and larvae enables direct, non-invasive observation of development in real time. This is particularly powerful when combined with transgenic reporter lines that express fluorescent proteins in specific tissues or cell types. For instance, the creation of a Tg(tg:nlsEGFP) line, which expresses nuclear-localized EGFP in thyroid follicular cells, allows researchers to monitor thyroid morphogenesis and identify developmental defects in live CRISPR-edited larvae without the need for fixation or dissection [8]. This transparency facilitates high-resolution confocal live imaging to track processes like organ formation, cell migration, and dynamic gene expression patterns in vivo. The ability to conduct such detailed phenotypic analyses in living subjects provides a direct functional readout of the effects of CRISPR-induced mutations, bridging the gap between genotype and phenotype with unprecedented clarity [7] [8].
High Fecundity and Rapid Screening
The high fecundity of zebrafish—producing hundreds of offspring per mating pair weekly—makes it uniquely suited for high-throughput genetic screens [1] [2]. This fecundity is essential for CRISPR-based functional genomics, as it allows researchers to generate and screen large numbers of F0 mosaic mutants (crispants) to rapidly assess gene function [4]. In a single experiment, dozens of genes can be targeted using multiple gRNAs, and the resulting phenotypes can be assessed at scale. This approach is exemplified by a study that systematically tested 50 different gRNAs targeting 14 genes, using pools of 20 G0 mutant embryos for each gRNA to efficiently quantify editing efficiency and functional outcomes [4]. The large number of progeny enables sufficient statistical power for these screens and also supports the subsequent breeding efforts needed to isolate and stabilize mutant alleles in the germline, generating homozygous F2 lines for definitive phenotypic analysis [3].
Advanced CRISPR-Cas9 Applications in Zebrafish
The versatility of the CRISPR-Cas9 system extends beyond simple gene knockouts. Several advanced applications have been successfully implemented in zebrafish, leveraging its unique advantages.
Programmable Base Editing
A significant advancement in zebrafish genome engineering is the adoption of "base editing" technology. This system uses a cytidine deaminase fused to a Cas9 nickase (nCas9) to directly convert one target base to another without creating double-strand breaks, enabling precise single-nucleotide changes [9]. This method has been shown to achieve site-specific single-base mutations with efficiencies of up to 28% in various gene loci [9]. For example, this technique was used to successfully model human ablepharon macrostomia syndrome (AMS) by introducing a precise p.E78K mutation in the twist2 gene, mirroring the pathogenic human mutation [9]. Base editing overcomes the key limitation of traditional homology-directed repair (HDR), which suffers from low efficiency in zebrafish, and provides a powerful tool for creating accurate models of human genetic diseases caused by point mutations.
Somatic Mutagenesis in G0 "Crispants"
The efficiency of CRISPR-Cas9 in zebrafish has enabled the widespread use of mosaic F0 generation mutants, or "crispants," for rapid phenotypic screening. In this approach, embryos injected with CRISPR components are analyzed directly for somatic mutations, bypassing the need for time-consuming generation of stable lines [4] [8]. This method is highly effective for identifying genes involved in early development and organogenesis. A prime example is a mutagenesis assay designed to identify genes crucial for thyroid morphogenesis and function. By injecting gRNAs targeting genes of interest into embryos of a transgenic thyroid reporter line, researchers could rapidly screen for thyroid-specific phenotypes like athyreosis or hypoplasia within six days, successfully validating known genes and providing a platform for testing new candidates [8]. This G0 screening strategy dramatically accelerates the functional annotation of genes.
Experimental Protocols and Workflows
Standard Workflow for CRISPR-Cas9 Gene Knockout
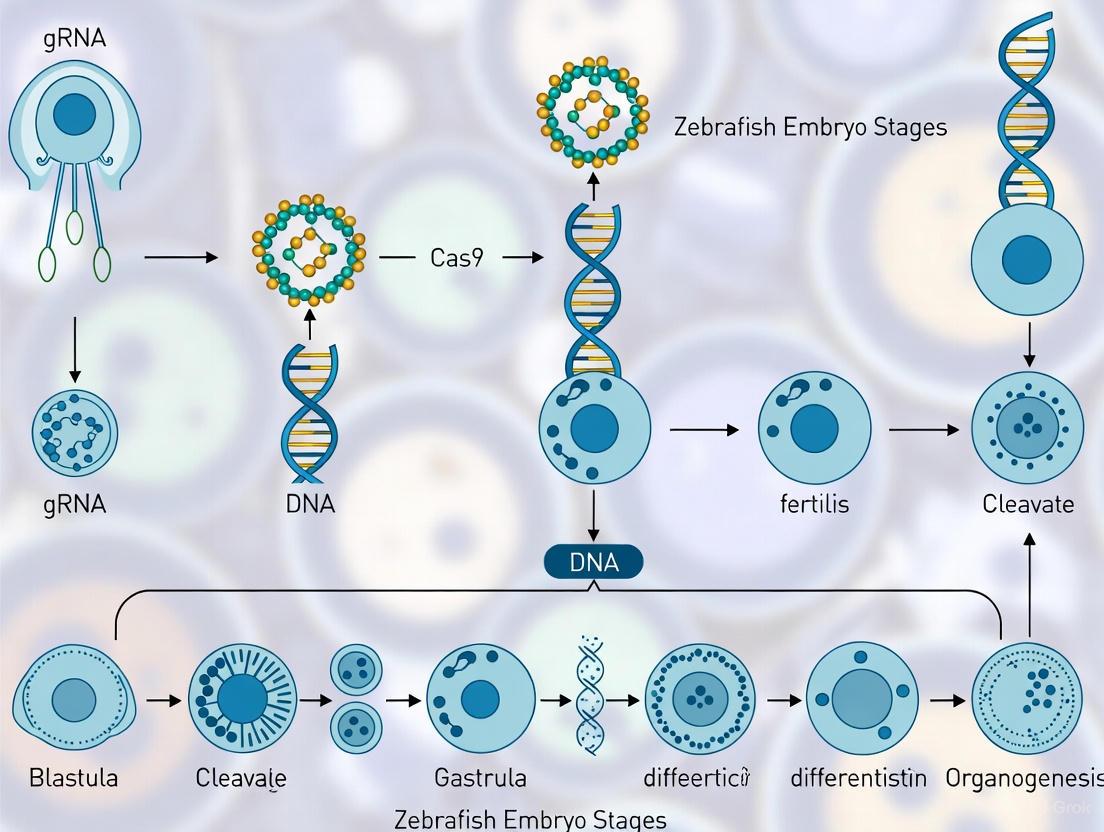
The following diagram illustrates the standard pipeline for creating and validating CRISPR-Cas9 knockout mutants in zebrafish, from gRNA design to phenotypic analysis.
Title: CRISPR-Cas9 Gene Knockout Workflow in Zebrafish
Detailed Protocol Steps:
gRNA Design and Synthesis:
- Design: Select a 20-nucleotide target sequence adjacent to a 5'-NGG Protospacer Adjacent Motif (PAM) in an early exon of the target gene. Efficiency can be predicted using tools like CRISPRScan [4] [6].
- Synthesis: The gRNA is typically synthesized by in vitro transcription using a T7 polymerase system, starting from a DNA oligonucleotide template. The Cas9 component can be delivered as mRNA or protein [3] [6].
Microinjection into One-Cell Stage Zygotes:
- Prepare an injection mix containing the sgRNA (25-50 pg) and Cas9 mRNA or protein (150-300 pg) [3] [6].
- Using a microinjection apparatus, deliver 1-2 nL of the mix directly into the cytoplasm or yolk of a freshly fertilized one-cell stage embryo. External development makes this procedure feasible and efficient [6].
Mutation Efficiency Analysis:
- At 1-5 days post-fertilization (dpf), extract genomic DNA from a pool of ~20 injected embryos or individual larvae.
- Amplify a 200-500 bp region surrounding the target site by PCR.
- Analyze the PCR products for mutagenesis efficiency using one of the following methods [4]:
- Polyacrylamide Gel Electrophoresis (PAGE): Detects heteroduplexes formed by indel mutations as smeared bands. Affordable but less quantitative.
- Tracking of Indels by Decomposition (TIDE) or Inference of CRISPR Edits (ICE): Computational tools that deconvolve Sanger sequencing traces to quantify indel frequencies.
- Illumina Sequencing: The gold standard for precise quantification of mutation efficiency and spectrum. Provides the percentage of DNA harboring indels compared to uninjected controls [4].
Protocol for Enhancing Mutagenesis Efficiency
A common challenge in zebrafish CRISPR is somatic mosaicism in F0 founders, caused by the short single-cell stage (∼40 minutes). The following protocol can be used to improve editing efficiency by extending the time window for CRISPR activity before the first cell division [10].
- Microinjection: Perform standard microinjection of CRISPR components into one-cell stage embryos as described above.
- Low-Temperature Incubation: Immediately after injection, transfer the embryos to a petri dish with embryo medium and incubate at 12°C for 30-60 minutes.
- Normal Development: After the low-temperature incubation, return the embryos to a standard incubator at 28.5°C for normal development.
- Validation: This simple temperature reduction has been shown to delay the first cell division to 70-100 minutes (compared to 40 minutes at 28°C) and is associated with a statistically significant increase in CRISPR-Cas9 mutagenesis rate without causing adverse side effects [10].
The Scientist's Toolkit: Essential Research Reagents
Successful CRISPR research in zebrafish relies on a set of core reagents and materials. The table below lists essential components and their functions.
Table 2: Essential Reagents for CRISPR/Cas9 Experiments in Zebrafish
| Reagent / Material | Function | Notes |
|---|---|---|
| Cas9 mRNA/Protein | The core endonuclease that creates double-strand breaks at the DNA target site. | Can be used as in vitro transcribed mRNA or recombinant protein. Protein may yield higher efficiency and reduce mosaicism [3] [4]. |
| Guide RNA (gRNA) | A synthetic RNA that complexes with Cas9 and directs it to a specific genomic locus via a 20-nt spacer sequence. | Can be a single-guide RNA (sgRNA) or a duplex of crRNA and tracrRNA [3]. |
| Microinjection Apparatus | For precise delivery of CRISPR components into embryos. | Includes a micropipette puller, microinjector, and micromanipulator [6]. |
| Zebrafish Transgenic Lines | Reporter lines expressing fluorescent proteins in specific tissues (e.g., Tg(tg:nlsEGFP) for thyroid). |
Enable non-invasive, live phenotyping of organ development and function in CRISPR-edited larvae [8]. |
| Rainbow Trout Ovarian Fluid (RTOF) | A specialized medium for preserving oocyte viability during in vitro manipulation. | Enables exploration of mutagenesis in oocytes prior to fertilization, though efficiency can be low [10]. |
| Polymerase Chain Reaction (PCR) | For amplifying the targeted genomic region from injected embryos. | Essential first step for genotyping and efficiency analysis via PAGE, TIDE, or sequencing [4] [6]. |
The synergistic combination of the zebrafish's inherent biological advantages—external development, transparency, and high fecundity—with the precision and power of the CRISPR-Cas9 system has created an unparalleled platform for genetic research. External development permits direct microinjection for efficient mutagenesis, transparency enables real-time visualization of phenotypic outcomes in living animals, and high fecundity supports the large-scale screens necessary for robust functional genomics. As CRISPR technologies continue to evolve, with the advent of base editing and other refined tools, the zebrafish model is poised to remain at the forefront of efforts to understand gene function, model human disease, and accelerate drug discovery.
The zebrafish (Danio rerio) has emerged as a premier model organism for biomedical research, owing to its remarkable genetic similarity to humans and experimental tractability. Approximately 70% of human genes have at least one obvious zebrafish ortholog, a figure that rises to 84% for genes known to be associated with human diseases [11] [12]. This high degree of genetic conservation, combined with the logistical advantages of zebrafish, has positioned them as an ideal platform for functional genomics and disease modeling. The advent of CRISPR-Cas9 genome editing technology has further accelerated the use of zebrafish, enabling researchers to create precise genetic models of human diseases with unprecedented efficiency [13]. This application note details the genetic similarities between zebrafish and humans and provides detailed protocols for leveraging CRISPR-Cas9 in zebrafish to study disease mechanisms and therapeutic interventions.
Genetic Similarities Between Zebrafish and Humans
The sequencing of the zebrafish genome revealed a profound level of synteny and genetic conservation with the human genome, stemming from their shared vertebrate ancestry [14] [12]. The table below summarizes the key quantitative measures of this genetic relationship.
Table 1: Quantitative Measures of Genetic Similarity Between Zebrafish and Humans
| Metric | Value | Interpretation and Significance |
|---|---|---|
| Overall Protein-Coding Gene Similarity | ~70% [11] [14] [12] | Approximately 70% of human genes have at least one zebrafish ortholog, enabling the study of a vast majority of biological pathways. |
| Disease-Associated Gene Orthologs | ~84% [11] [15] [12] | The vast majority of genes implicated in human genetic diseases have a counterpart in zebrafish, making it highly relevant for disease modeling. |
| Number of Protein-Coding Genes | ~26,000 [12] | The zebrafish has a comparable number of genes to humans, reflecting a similar level of genetic complexity. |
This genetic similarity extends beyond mere sequence conservation to functional conservation. Key biological systems, including the cardiovascular, nervous, and immune systems, rely on analogous genetic pathways in both species [12]. For instance, neurotransmitters like dopamine, which are crucial for understanding neurological disorders such as Parkinson's disease, are present and functional in zebrafish [15]. Furthermore, the external development and optical transparency of zebrafish embryos provide a unique window to observe these conserved developmental and disease processes in real time [14] [13].
CRISPR-Cas9 in Zebrafish: An Efficient Combination for Disease Modeling
The CRISPR-Cas9 system has revolutionized genetic engineering in zebrafish, enabling the efficient generation of knock-out and knock-in models to study human diseases [11] [13]. Its implementation leverages the experimental advantages of the zebrafish model.
Table 2: Key Applications of CRISPR-Cas9 in Zebrafish Disease Modeling
| Application | Description | Example in Human Disease Research |
|---|---|---|
| Knockout | Disruption of gene function to model loss-of-function disorders. | Generation of loss-of-function mutants for 17 Fanconi Anemia (FA) genes to study their role in growth and fertility [11]. |
| Knockin | Introduction of specific point mutations to replicate human genetic variants. | Creating zebrafish models of amyotrophic lateral sclerosis (ALS) and Cantú syndrome by inserting human disease-causing SNPs [11]. |
| Human Disease Validation | Functional testing of genes identified in human genomic studies. | Rapid in vivo validation of candidate genes from whole-exome sequencing of patients with developmental disorders like Miles-Carpenter syndrome [14] [13]. |
Microinjection of CRISPR-Cas9 components (Cas9 protein or mRNA along with guide RNA) into one-cell stage zebrafish embryos is the most common and efficient delivery method [11] [16]. This approach produces mosaic G0 generation fish that can be screened for desired mutations, which can then be stabilized in the germline through selective breeding to establish mutant lines [4] [17].
Experimental Protocols
Protocol 1: CRISPR-Cas9 Mediated Knockout in Zebrafish
This protocol describes the generation of knockout zebrafish models via microinjection of CRISPR-Cas9 ribonucleoprotein (RNP) complexes into one-cell stage embryos [4] [16].
Research Reagent Solutions and Essential Materials
Table 3: Key Reagents for CRISPR-Cas9 in Zebrafish
| Item | Function/Description |
|---|---|
| Cas9 Protein | The CRISPR-associated endonuclease that creates double-strand breaks in DNA. Using purified protein reduces off-target effects and shortens activity time compared to mRNA [4]. |
| Guide RNA (gRNA) | A synthetic RNA complex (crRNA:tracrRNA) or single-guide RNA (sgRNA) that directs Cas9 to the specific genomic target site [4]. |
| Microinjection Apparatus | A precision instrument including a micropipette puller, microscope, and microinjector for delivering nanoliter volumes into embryos. |
| Zebrafish One-Cell Stage Embryos | Embryos collected immediately after fertilization, which are most receptive to integration of injected genetic material [16]. |
| Agarose Injection Mold | A mold to create grooves for immobilizing embryos during the microinjection process. |
Step-by-Step Methodology
- gRNA Design and Synthesis: Design gRNAs targeting early exons of the gene of interest using predictive tools like CRISPRScan [4]. Synthesize gRNA via in vitro transcription or purchase as synthetic crRNA:tracrRNA duplexes.
- RNP Complex Formation: Complex the purified Cas9 protein with the synthesized gRNA at a molar ratio of 1:2 (e.g., 300 ng/μL Cas9 to 100 ng/μL gRNA) and incubate at 37°C for 10 minutes to form the RNP complex [4].
- Embryo Preparation: Collect freshly fertilized one-cell stage zebrafish embryos and align them in the grooves of an agarose injection plate.
- Microinjection: Load the RNP complex into a glass capillary needle and inject ~1 nL of the mixture directly into the cytoplasm of each embryo.
- Post-Injection Care and Screening: Maintain injected embryos in egg water at 28.5°C. After 24-48 hours, screen for successful mutagenesis. A common initial screening method is the heteroduplex mobility assay using polyacrylamide gel electrophoresis (PAGE), which detects the "smear" caused by indel mutations [4]. For precise quantification and identification of specific indels, genomic DNA from pooled embryos can be extracted at 5 days post-fertilization (dpf). The target locus is PCR-amplified and analyzed by Sanger sequencing followed by decomposition tools like TIDE or ICE, or more accurately, by next-generation sequencing (NGS) [4].
Protocol 2: Phenotypic Analysis of Genetic Models
Following the generation of a mutant line, robust phenotypic analysis is crucial for validating the model and understanding gene function.
Key Materials
- Mutant and wild-type (control) zebrafish larvae/adults
- RNA extraction kits and qPCR reagents for gene expression analysis
- Histochemical stains and antibodies for morphological analysis
- Behavioral tracking systems (e.g., DanioVision, ZebraBox)
Step-by-Step Methodology
- Genotypic Validation: Outcross G0 founders to wild-type fish to obtain F1 progeny. Genotype F1 to identify carriers of the mutation and establish a stable line.
- Molecular Phenotyping:
- Gene Expression: Perform RNA extraction from mutant and control larvae (e.g., at 5 dpf). Use quantitative RT-PCR or RNA-seq to analyze expression changes in the targeted gene and relevant pathway members. Note that RNA-seq of control larvae is also critical to identify potential confounders, such as differentially expressed genes resulting from the microinjection process itself [4].
- Protein Analysis: Use immunohistochemistry or western blotting with target-specific antibodies to assess protein expression and localization.
- Morphological Phenotyping: Anesthetize larvae and image them under a dissecting microscope. Screen for developmental defects in organs, brain structure, or overall body plan. For adult phenotypes, conduct similar analyses after euthanasia.
- Behavioral Phenotyping: For studies of neurological disorders, employ high-throughput behavioral assays.
- Locomotor Activity: Place individual larvae in 96-well plates and track their movement in light and dark cycles using an automated video tracking system. Models of autism spectrum disorder, for instance, have shown altered locomotor activity [11] [14].
- Social Behavior: Test adult fish in a social interaction assay to quantify shoaling and schooling behaviors, which are relevant for modeling mental disorders [14].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents and Resources for Zebrafish Research
| Resource / Reagent | Function and Utility |
|---|---|
| Zebrafish Information Network (ZFIN) | The central database for genetic, genomic, and phenotypic data. It provides expert curation of genes, mutants, phenotypes, and human disease models, and is essential for nomenclature and data retrieval [18]. |
| CRISPR Design Tools (e.g., CRISPRScan) | Online algorithms to design highly efficient gRNAs by considering factors like GC content and nucleotide position, thereby improving the success rate of mutagenesis [4]. |
| CrispRVariants Tool | A software tool for the annotation and quantification of insertion/deletion mutations from NGS data of CRISPR-edited populations, providing precise in vivo efficiency scores [4]. |
| Antisense Morpholino Oligomers (MOs) | Synthetic nucleic acids for transient gene knockdown. While useful, findings should be interpreted with caution as phenotypes may not always recapitulate those of genetic mutants [13]. |
| Tol2 Transposon System | A widely used transgenesis method in zebrafish for creating transgenic lines, facilitating tissue-specific gene expression and gene trap assays [14]. |
The combination of the zebrafish model and CRISPR-Cas9 technology represents a powerful and efficient platform for functional genomics and modeling human genetic diseases. The significant genetic similarity, with 71.4% of human genes and 84% of human disease genes having zebrafish counterparts, ensures that findings are often translatable to human physiology and pathology [11] [12]. The protocols outlined herein—from the generation of knockout models via RNP microinjection to comprehensive molecular and behavioral phenotyping—provide a robust framework for researchers. As CRISPR technology continues to evolve, its integration with the zebrafish model will undoubtedly accelerate our understanding of disease mechanisms and the development of novel therapeutic strategies, paving the way for advancements in precision medicine.
This application note provides a comprehensive breakdown of the CRISPR-Cas9 genome editing mechanism, with specific protocols for implementation in zebrafish embryo research. We detail the functional components—guide RNA (gRNA), Cas9 protein, Protospacer Adjacent Motif (PAM) sites, and DNA repair pathways—and present standardized methodologies for generating knockout lines in zebrafish. The content is structured to enable researchers to design and execute CRISPR-Cas9 experiments efficiently, with a focus on practical application in gene function studies and drug target validation.
Core Components of the CRISPR-Cas9 System
The CRISPR-Cas9 system is a revolutionary genome-editing tool derived from an adaptive immune mechanism in bacteria [19] [20]. It functions as a precise DNA-cutting system that can be programmed to target specific genomic sequences. For zebrafish research, this technology has largely replaced earlier methods like zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) due to its simplicity, efficiency, and lower cost [3] [21]. The system consists of two fundamental components:
- Cas9 Nuclease: A large (1368 amino acids) multidomain DNA endonuclease that acts as "molecular scissors" to create double-strand breaks (DSBs) in DNA [20] [22]. It contains two main lobes: a recognition lobe (REC) that binds the guide RNA, and a nuclease lobe (NUC) that houses the DNA cleavage domains [20].
- Guide RNA (gRNA): A synthetic RNA molecule that combines two natural RNA components—the CRISPR RNA (crRNA) and the trans-activating CRISPR RNA (tracrRNA) [20] [23]. The gRNA is programmable; its 5' end contains a ~20 nucleotide spacer sequence that is complementary to the target DNA site, while its 3' end forms a hairpin structure that binds the Cas9 protein [20] [21].
Table 1: Core Components of the CRISPR-Cas9 System for Zebrafish Gene Editing
| Component | Description | Function in Genome Editing |
|---|---|---|
| Cas9 Nuclease | Endonuclease from S. pyogenes; requires nuclear localization signal (NLS) for eukaryotic use [3]. | Generates a double-strand break (DSB) in the target DNA 3-4 base pairs upstream of the PAM site [21]. |
| Guide RNA (gRNA) | Single-guide RNA (sgRNA) combining crRNA and tracrRNA functionalities [3]. | Specifies target location via Watson-Crick base pairing; directs Cas9 to the precise genomic locus [20]. |
| Spacer Sequence | 17-20 nucleotide segment at the 5' end of the gRNA [21]. | Determines targeting specificity by binding to the complementary DNA protospacer sequence [19]. |
| tracrRNA Scaffold | 3' end of the sgRNA with a defined secondary structure [3]. | Serves as a binding scaffold for the Cas9 nuclease, forming the ribonucleoprotein (RNP) complex [20]. |
The Mechanism of Target Recognition and DNA Cleavage
The CRISPR-Cas9 mechanism can be systematically divided into three sequential stages: recognition, cleavage, and repair [20].
PAM-Dependent Target Recognition
The first critical step is the identification of a valid target site. Cas9 does not simply bind to any sequence complementary to the gRNA. Instead, it requires the presence of a short, conserved DNA sequence immediately downstream of the target sequence, known as the Protospacer Adjacent Motif (PAM) [19] [24]. For the most commonly used Cas9 from Streptococcus pyogenes (SpCas9), the PAM sequence is 5'-NGG-3', where "N" can be any nucleotide base (A, T, C, or G) [19] [25].
The PAM is not part of the bacterial host genome in the native CRISPR immune system, which is a key feature that prevents the Cas9 nuclease from destroying the bacterium's own DNA [19] [24]. In genome editing, the PAM requirement dictates which genomic locations can be targeted. The Cas9 protein first scans the DNA for the PAM sequence. Once it identifies a PAM, it initiates DNA melting, allowing the gRNA's spacer sequence to form base pairs with the complementary DNA strand (the "protospacer") [20] [26]. Successful hybridization activates the Cas9 nuclease domains.
DNA Cleavage by HNH and RuvC Nuclease Domains
Upon target recognition and verification, the Cas9 protein induces a blunt-ended double-strand break (DSB) [20]. This cleavage is executed by two distinct nuclease domains within Cas9, each responsible for cutting one DNA strand:
- The HNH Domain: Cleaves the DNA strand that is complementary to the gRNA (the target strand) [22] [26]. Structural studies using cryo-EM have shown that the HNH domain undergoes a large conformational shift to swing into position and cut the DNA phosphodiester bond [26].
- The RuvC Domain: Cleaves the non-complementary DNA strand (the non-target strand) [20] [22]. The catalytic residues D10, E762, H983, and D986 in the RuvC active site coordinate to hydrolyze the DNA backbone, likely via a two-metal-ion mechanism [22].
The DSB occurs 3 base pairs upstream of the PAM sequence [21]. This break then engages the cell's innate DNA repair machinery.
Diagram 1: CRISPR-Cas9 Mechanism: From Target Recognition to DNA Repair. The process begins with PAM-dependent binding, followed by coordinated cleavage by HNH and RuvC domains, and concludes with cellular repair pathways that determine the editing outcome.
Double-Strand Break Repair Pathways
The cellular response to the CRISPR-induced DSB is the cornerstone of genome editing, as the choice of repair pathway determines the final genetic outcome. Mammalian and zebrafish cells primarily utilize two distinct pathways to repair DSBs [20] [3].
Non-Homologous End Joining (NHEJ)
NHEJ is the dominant and most active repair pathway throughout the cell cycle [20]. It functions by directly ligating the two broken ends of the DNA. However, this process is error-prone and often results in small random insertions or deletions (indels) at the cleavage site [3] [23]. When these indels occur within the coding sequence of a gene, they can cause a frameshift mutation, leading to a premature stop codon and a non-functional, truncated protein. This is the basis for generating knockout alleles [23].
Homology-Directed Repair (HDR)
HDR is a precise repair mechanism that is most active in the late S and G2 phases of the cell cycle [20]. It requires a homologous DNA template—such as the sister chromatid or an exogenously supplied donor DNA—to faithfully repair the break [3]. In genome editing, researchers can harness HDR by co-injecting a designed donor DNA template along with the CRISPR-Cas9 components. This template contains the desired edit (e.g., a specific point mutation or a gene insertion) flanked by homology arms complementary to the sequences around the cut site. This allows for precise gene correction or knock-in of sequences [20] [23].
Table 2: Comparison of DNA Double-Strand Break Repair Pathways in CRISPR-Cas9 Editing
| Repair Pathway | Template Required | Mechanism | Outcome | Primary Application in Zebrafish |
|---|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | No [20] | Error-prone ligation of broken DNA ends [3]. | Small insertions or deletions (indels) [23]. | Efficient generation of gene knockouts [21]. |
| Homology-Directed Repair (HDR) | Yes (donor DNA with homology arms) [20] | Precise, templated repair using homologous sequence [3]. | Defined sequence insertion or correction [23]. | Precise nucleotide changes or gene knock-ins [23]. |
Protocol: CRISPR-Cas9 Mediated Gene Knockout in Zebrafish
This protocol outlines the steps for generating heritable knockout lines in zebrafish using CRISPR-Cas9, from gRNA design to mutant identification [21] [23].
gRNA Design and In Vitro Transcription
- Target Selection: Identify a target site of 17-20 nucleotides within the first few exons of your gene of interest, ideally in a large exon to increase the likelihood of a disruptive indel. The target must be immediately followed by a 5'-NGG-3' PAM sequence on the genomic DNA [23].
- gRNA Design:
- Use online design tools (e.g., CHOPCHOP, CRISPRscan) to select a target with high predicted efficiency and minimal off-target effects [21].
- An ideal gRNA sequence has a 5'-G in the first position for efficient T7 in vitro transcription and 40-80% GC content [23]. The sequence is: 5'-G(N)₁₈G-3', where the final G is adjacent to the PAM but is not part of it. Exclude the PAM sequence from the gRNA design [19].
- DNA Template Preparation: Synthesize the gRNA template via PCR using a two-primer system [21]:
- Gene-specific primer (crRNA segment): 5'- TTCTAATACGACTCACTATAGG(N)₁₈GGTTTTAGAGCTAGA-3'
- T7 promoter sequence is underlined.
- The gRNA target sequence is in bold.
- Common primer (tracrRNA scaffold): 5'- AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATttctagctctaaaac -3' [21]
- Gene-specific primer (crRNA segment): 5'- TTCTAATACGACTCACTATAGG(N)₁₈GGTTTTAGAGCTAGA-3'
- In Vitro Transcription (IVT): Purify the PCR product and use a T7 IVT kit to synthesize the sgRNA. Column-purify the resulting sgRNA, check its concentration on a spectrophotometer, and store in aliquots at -80°C [21] [23].
Embryo Microinjection
- Preparation of Injection Solution: Combine purified Cas9 protein (commercially available) with the synthesized sgRNA to form ribonucleoprotein (RNP) complexes. A typical 5 µL injection solution contains 2 µL of 1 mg/mL Cas9 protein and 100-200 ng of sgRNA in injection medium (200 mM KCl, 8.3 mM HEPES) [23]. Using Cas9 protein instead of mRNA reduces off-target effects and increases mutagenesis efficiency [23].
- Zebrafish Embryo Injection:
- Set up zebrafish crosses and collect one-cell stage embryos immediately after spawning.
- Using a microinjector and fine glass needles, inject ~1 nL of the RNP solution directly into the cell cytoplasm or yolk of the one-cell embryo [21] [23].
- Raise the injected embryos (F0 generation, or "crispants") in embryo medium (E3) at 28.5°C. Injected F0 fish will be genetically mosaic [21].
Identification and Validation of Mutant Alleles
- DNA Extraction: At 24-48 hours post-fertilization, pool 10-20 injected embryos and extract genomic DNA using lysis buffer (e.g., 10 µg/mL Proteinase K in Tris-EDTA buffer with Triton) [23].
- Initial Efficiency Check via Heteroduplex Mobility Assay (HMA):
- Design PCR primers that flank the target site, generating a 150-300 bp amplicon.
- Amplify the target region from the pooled embryo DNA.
- Denature and reanneal the PCR products. If indels are present, heteroduplexes (mismatched DNA strands) will form, which migrate differently on a polyacrylamide or high-percentage agarose gel than homoduplexes (perfectly matched strands). This provides a rapid, equipment-friendly assessment of mutagenesis efficiency before sequencing [23].
- Sequence Characterization:
- Clone the PCR products from individual F0 or F1 adults into a vector and perform Sanger sequencing, or use next-generation sequencing (NGS) of PCR amplicons to precisely characterize the spectrum of induced indels in a high-throughput manner [23].
- Germline Transmission:
- Raise injected F0 embryos to adulthood. Outcross individual F0 fish to wild-type partners.
- Screen the resulting F1 offspring for indels using HMA or PCR-based methods on fin-clip DNA.
- Raise F1 embryos carrying the mutant allele and establish stable homozygous lines through sibling crosses [21] [23].
Diagram 2: Workflow for Generating Zebrafish Knockout Lines. The process begins with gRNA design and culminates in the establishment of a stable mutant line, with key screening steps to confirm germline transmission.
The Scientist's Toolkit: Essential Reagents for Zebrafish CRISPR
Table 3: Key Research Reagent Solutions for CRISPR-Cas9 in Zebrafish
| Reagent / Material | Function / Description | Example / Specification |
|---|---|---|
| Cas9 Protein | Wild-type Cas9 nuclease with nuclear localization signal (NLS); used to form RNP complexes for injection [23]. | Recombinantly expressed S. pyogenes Cas9, aliquoted at 1 mg/mL in injection buffer. |
| In Vitro Transcription Kit | For synthesizing high-quality, capped sgRNA from a DNA template [21]. | T7 In Vitro Transcription Kit (e.g., Ambion). |
| Microinjection Setup | Equipment for precise delivery of CRISPR reagents into zebrafish embryos. | Micropipette puller, microinjector (e.g., Nanoliter 2000, World Precision Instruments), micromanipulator, and glass capillaries [21]. |
| gRNA Design Software | Web-based tools for selecting optimal gRNA targets with high efficiency and low off-target potential. | CHOPCHOP [21], CRISPRscan [21]. |
| Heteroduplex Mobility Assay (HMA) | A rapid, low-cost PCR-based method to detect the presence of indels in pooled or individual fish DNA before sequencing [23]. | Requires standard agarose gel electrophoresis equipment and reagents. |
| Next-Generation Sequencing (NGS) | A powerful method for the precise characterization of the spectrum and frequency of indels in mutagenized samples [23]. | Used for deep sequencing of PCR amplicons spanning the target site. |
Application Notes and Troubleshooting in Zebrafish Research
- PAM Specificity: The absolute requirement for a PAM sequence (5'-NGG-3' for SpCas9) is the primary constraint on targetable sites within the zebrafish genome. If your locus of interest lacks an NGG PAM, consider using Cas9 orthologs from other bacteria (e.g., SaCas9 with PAM NNGRRT) or engineered Cas9 variants with altered PAM specificities [25] [24].
- Minimizing Off-Target Effects: The specificity of CRISPR-Cas9 is a critical consideration. To minimize off-target cleavage:
- "Crispant" Phenotypes: Injected F0 embryos (crispants) exhibit mosaic editing, meaning different cells carry different indels. This can generate a knockdown-like phenotype useful for early functional screening, but stable lines must be established through breeding for consistent analyses [21].
- Repair Pathway Bias: The error-prone NHEJ pathway is highly efficient in zebrafish, making knockout generation straightforward. In contrast, HDR is inefficient and requires careful optimization of donor template design and concentration for successful knock-in [3] [23].
The CRISPR-Cas9 system provides a robust and adaptable framework for targeted genome engineering in zebrafish. Its programmable nature, relying on the synergy between the gRNA, Cas9 nuclease, and the PAM sequence, allows for precise genetic modifications. The resulting double-strand breaks are harnessed by cellular repair pathways to generate either knockout mutants via NHEJ or precise edits via HDR. The protocols and insights outlined in this application note empower researchers to leverage this technology effectively, accelerating functional genomics and the modeling of human diseases in a vertebrate system.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) systems originated as an intricate adaptive defense system in prokaryotic organisms, functioning as a molecular record of past viral infections that provides heritable immunity against future invasions [27]. This evolutionary adaptation has profoundly transformed modern biology and biotechnology, evolving from a bacterial immune mechanism into a versatile toolkit for precise genome manipulation in diverse organisms, including vertebrate models like zebrafish [3] [27]. The modular architecture of CRISPR-Cas systems, consisting of adaptation modules that incorporate new spacers and effector modules that execute cleavage, has enabled their repurposing for genetic engineering [27]. The classification of these systems into Class 1 (multi-subunit complexes) and Class 2 (single effector proteins) highlights the structural diversity that has been exploited for biotechnological applications, with Class 2 systems like type II Cas9 being particularly suitable for genome editing due to their simpler architecture [27].
In zebrafish (Danio rerio), a model organism with significant genomic homology to humans, CRISPR-Cas9 has emerged as a transformative technology [3]. The system's ability to bind single loci within vertebrate genomes and generate double-strand breaks (DSBs) at those sites has revolutionized genetic studies in this model organism [3]. With 70% of human genes having zebrafish orthologs and 85% of disease-associated human genes represented in the zebrafish genome, this model provides a relevant platform for studying human diseases and developing therapeutic strategies [10]. The applications of CRISPR in zebrafish have expanded from simple gene knockouts to precise base editing, knock-in strategies, and transcriptional regulation, enabling researchers to model human genetic disorders with unprecedented accuracy [28] [29] [30].
Evolution of CRISPR-Cas Systems: Mechanism and Classification
Molecular Architecture and Functional Evolution
CRISPR-Cas systems are characterized by their modular architecture, comprising two principal functional units [27]. The adaptation module, containing Cas1 and Cas2 proteins, is responsible for acquiring spacers from invading nucleic acids and integrating them into the CRISPR array, forming the genetic memory of encounters with mobile genetic elements [27]. The effector module processes CRISPR RNAs (crRNAs) and uses them to neutralize invading genetic material through sequence-specific recognition and cleavage [27]. Evolutionary studies indicate that Cas proteins evolved from transposons known as casposons, demonstrating how molecular components can be repurposed through evolution for new biological functions [27].
The transformation of CRISPR from a prokaryotic immune system to a programmable genomic scissor culminated with the discovery that the type II CRISPR-Cas9 system from Streptococcus pyogenes could be engineered as a single-guide RNA (sgRNA) system for precise genome editing in eukaryotic cells [3] [27]. This engineering feat simplified the naturally occurring dual-RNA complex (crRNA:tracrRNA) into a single chimeric guide RNA, creating a two-component system that could be programmed to target any DNA sequence adjacent to a Protospacer Adjacent Motif (PAM) [3] [27]. The Cas9 enzyme, guided by sgRNA, induces double-strand breaks in target DNA through the activation of its two nuclease domains, RuvC and HNH, which cleave the complementary and target strands, respectively [3].
Comparative Classification of CRISPR-Cas Systems
Table 1: Feature comparison between Class 1 and Class 2 CRISPR systems
| Feature | Class 1 | Class 2 |
|---|---|---|
| Structural Complexity | Multi-subunit complexes (e.g., Cascade) | Single multidomain protein (e.g., Cas9) |
| Key Proteins | Cascade and Cas3 | Cas9, Cas12, Cas13 |
| Main Function | DNA recognition and degradation through joint action | DNA/RNA recognition and cleavage in a single molecule |
| Functional Efficiency | Processive, requiring multiple proteins and steps | Direct, combining functions in a single protein |
| Applications | Less common in biotechnology due to complexity | Widely used in genome editing and biotechnology |
| Guide Structure | crRNA assembled in the complex | Dual complex of crRNA and tracrRNA or sgRNA |
| Structural Reorganization | Complex conformational changes during function | Auto-inhibited reorganization until guide RNA binding |
The diversity of CRISPR-Cas systems is organized into two main classes based on their effector module architecture [27]. Class 1 systems (types I, III, and IV) utilize multi-protein complexes for target recognition and cleavage, while Class 2 systems (types II, V, and VI) employ single effector proteins such as Cas9, Cas12, and Cas13 [27]. The structural and functional distinctions between these classes have significant implications for their biotechnological applications. Class 1 systems, with their multi-subunit complexity, have proven more challenging to adapt for genome editing applications, whereas Class 2 systems, with their single-protein effectors, have been widely adopted for their simplicity and efficiency [27].
The continuous evolution of CRISPR-Cas systems has expanded their capabilities beyond DNA targeting to include RNA manipulation with systems like Cas13 [31], precision editing with base editors [28] and prime editors [30], and epigenetic modulation with nuclease-deficient variants (dCas9) [27]. This functional diversification has transformed CRISPR from a simple genomic scissor into a versatile platform for precise genetic engineering, enabling researchers to not only cut DNA but also to rewrite genetic information and modulate gene expression with unprecedented precision.
Advanced CRISPR Applications in Zebrafish Research
Precision Genome Editing Techniques
The development of precision genome editing tools has dramatically expanded the capabilities for modeling human genetic diseases in zebrafish. Base editing technology, which uses a cytidine deaminase fused to Cas9 nickase (nCas9), enables direct, irreversible conversion of one target base to another without requiring double-strand breaks or donor templates [28]. This system has achieved site-specific single-base mutations with efficiencies up to 28% across multiple gene loci in zebrafish, with germline transmission rates of 7-37% - significantly higher than traditional homology-directed repair (HDR) methods [28]. The application of base editing has enabled the creation of precise zebrafish models of human diseases, such as the ablepharon macrostomia syndrome (AMS) model generated through E78K mutation in the twist2 gene [28].
Prime editing, a more recent advancement, has demonstrated remarkable efficiency for introducing precise genetic modifications in zebrafish. This system uses a Cas9 nickase-reverse transcriptase fusion protein coupled with a prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit [30]. Comparative studies have shown that prime editing outperforms conventional HDR, achieving up to a fourfold increase in editing efficiency for four different targets while generating fewer off-target effects [30]. This technology represents a significant advancement for creating knock-in models of human genetic diseases in zebrafish, overcoming many of the limitations associated with traditional HDR-based approaches.
RNA-Targeting CRISPR Systems
The adaptation of RNA-targeting CRISPR-Cas systems, particularly CRISPR-RfxCas13d, has expanded the genetic toolkit available for zebrafish research [31]. This technology enables efficient mRNA knockdown without altering the genome, providing a powerful approach for studying gene function during development. Optimization of delivery methods, including ribonucleoprotein (RNP) complexes and mRNA-gRNA combinations, has enhanced the efficiency and specificity of RNA targeting in zebrafish embryos [31]. Chemical modifications to guide RNAs (cm-gRNAs) have further improved the penetrance of loss-of-function phenotypes, particularly for genes expressed after 7-8 hours post-fertilization [31].
Recent work has addressed concerns about collateral RNA cleavage activity associated with Cas13 systems, demonstrating that transient delivery approaches in zebrafish embryos effectively deplete endogenous mRNAs without significant collateral effects, except when targeting extremely abundant ectopic RNAs [31]. The implementation of alternative RNA-targeting systems like CRISPR-Cas7-11 and CRISPR-DjCas13d provides additional tools for specific RNA manipulation with reduced collateral activity [31].
Homology-Directed Repair and Knock-in Strategies
Despite advancements in precision editing, homology-directed repair (HDR) remains a valuable approach for introducing larger DNA cassettes into the zebrafish genome. CRISPR-Cas9-mediated HDR has been successfully used to correct a premature stop codon at the albino (alb) locus in zebrafish, with somatic repair efficiencies up to 46% using circular donor DNA containing CRISPR target sites [29]. Germline transmission of the repaired allele was achieved in approximately 10% of adult fish [29], demonstrating the feasibility of HDR for generating stable genetic lines.
The efficiency of HDR-mediated knock-in is influenced by multiple factors, including donor template design, Cas9 concentration, and delivery method [30]. Optimization studies have shown that Alt-R HDR templates with chemical modifications improve integration efficiency, while the optimal amount of Cas9 protein ranges between 200-800 pg [30]. However, HDR using single-stranded oligodeoxynucleotides (ssODNs) can lead to complex mutational patterns, including integration of repair-template fragments at the Cas9 cut site [32]. Error-free repair typically occurs at a relatively constant rate of 1-4% across different repair templates [32], highlighting the importance of careful validation of editing outcomes through next-generation sequencing approaches.
Table 2: Comparison of precision genome editing technologies in zebrafish
| Technology | Mechanism | Efficiency | Advantages | Limitations |
|---|---|---|---|---|
| Base Editing | Chemical conversion of bases without DSBs | Up to 28% base conversion [28] | High efficiency, low indels, no donor required | Limited to specific base changes, narrow editing window |
| Prime Editing | Reverse transcription of edited sequence | Up to 4× HDR efficiency [30] | Versatile, precise, fewer off-target effects | Complex pegRNA design, variable efficiency |
| HDR | Donor template-directed repair | 1-46% (depends on template and target) [29] [32] | Flexible for various edits including insertions | Low efficiency, requires donor, error-prone |
| ssODN HDR | Short oligonucleotide template repair | 1-4% error-free repair [32] | Simple design, cost-effective | High rate of erroneous integration |
Experimental Protocols for Zebrafish Genome Editing
CRISPR-Cas9 Mutagenesis and Efficiency Optimization
The basic protocol for CRISPR-Cas9 mutagenesis in zebrafish involves microinjection of Cas9 mRNA or protein together with target-specific guide RNAs into one-cell stage embryos [3] [10]. However, standard protocols often yield mosaic founders due to the brief single-cell stage in zebrafish embryos (approximately 40 minutes) [10]. To address this limitation, researchers have developed optimization strategies that significantly improve editing efficiency:
Temperature Reduction: Lowering incubation temperature from 28°C to 12°C extends the single-cell stage from 40 minutes to 70-100 minutes, providing a longer window for CRISPR components to act before cell division. This simple modification significantly increases mutagenesis rates without causing developmental abnormalities [10].
Ribonucleoprotein (RNP) Complex Delivery: Direct injection of preassembled Cas9 protein-gRNA complexes rather than mRNA encoding Cas9 accelerates editing activity and reduces mosaicism [10] [31].
Cas9 Protein Optimization: Titration of Cas9 amounts between 200-800 pg has been shown to maximize knock-in efficiency, with excessive amounts potentially increasing off-target effects [30].
Guide RNA Modifications: Chemically modified guide RNAs with 2'-O-methyl analogs and 3'-phosphorothioate internucleotide linkages enhance stability and editing efficiency, particularly for targets transcribed after gastrulation [31].
Homology-Directed Repair and Knock-in Protocol
For precise sequence integration via HDR, the following optimized protocol has been developed:
Donor Template Design:
- For ssODN templates: Incorporate 30-40 nt homology arms on each side of the Cas9 cut site. Introduction of silent mutations in the PAM site or protospacer sequence prevents re-cleavage of edited alleles [30] [32].
- For plasmid templates: Use circular DNA containing 5'- and 3'-homology arms (≥800 bp recommended). Incorporation of CRISPR target sites flanking the insert facilitates in vivo linearization [29].
Component Preparation:
- Prepare Cas9 protein or mRNA at optimal concentrations (200-800 pg per embryo) [30].
- Synthesize target-specific sgRNAs using in vitro transcription or chemical synthesis with modifications for enhanced stability [31].
- For ssODN templates, consider chemical modifications (Alt-R HDR templates) to improve integration efficiency [30].
Microinjection Mixture:
Embryo Collection and Injection:
Screening and Validation:
- At 24-48 hours post-fertilization, extract genomic DNA from individual embryos for initial screening.
- Use allele-specific PCR, restriction fragment length polymorphism (RFLP), or T7 endonuclease I assays to identify edited founders.
- For precise editing validation, perform Sanger sequencing or next-generation sequencing of the target locus [32].
- Raise potential founders to adulthood and outcross to wild-type fish to assess germline transmission.
Table 3: Key research reagents for CRISPR-based genome editing in zebrafish
| Reagent Category | Specific Examples | Function and Application | Optimization Notes |
|---|---|---|---|
| Cas9 Variants | Wild-type Cas9, Cas9 nickase (nCas9), dCas9, Cas9-VQR | DNA cleavage, base editing, gene regulation | Cas9-VQR recognizes 5'-NGA PAM, expanding targetable sites [28] |
| Editing Platforms | Base editors (rAPOBEC1-XTEN-nCas9-UGI), Prime editors | Precision editing without DSBs | Prime editors show 4× higher efficiency than HDR for some targets [28] [30] |
| Guide RNA Formats | in vitro transcribed sgRNA, chemically modified gRNAs (cm-gRNAs) | Target recognition and Cas9 recruitment | cm-gRNAs with 2'-O-methyl and phosphorothioate modifications enhance stability [31] |
| Delivery Methods | mRNA, protein (RNP complexes), plasmid DNA | Introduction of editing components | RNP complexes reduce mosaicism; mRNA+cm-gRNA better for late zygotic genes [10] [31] |
| Donor Templates | ssODNs, plasmid vectors, long ssDNA | Homology-directed repair templates | Alt-R modified ssODNs improve HDR efficiency; circular plasmids with target sites enhance integration [29] [30] |
| Analysis Tools | T7E1 assay, BATCH-GE (NGS analysis), CRISPOR | Validation of editing efficiency and specificity | NGS essential for detecting complex repair patterns in HDR [32] |
The evolution of CRISPR from a bacterial immune system to a programmable genomic scissor has revolutionized genetic research in zebrafish and other model organisms. The continuous refinement of editing technologies - from initial CRISPR-Cas9 nucleases to base editors, prime editors, and RNA-targeting systems - has expanded the precision and scope of genetic manipulations possible in zebrafish [28] [30] [31]. These advancements have positioned zebrafish as an invaluable model for studying human genetic diseases and developing therapeutic interventions.
Future directions in CRISPR technology development for zebrafish research include the engineering of high-precision Cas9 variants with reduced off-target effects, improved delivery systems for enhanced efficiency, and the establishment of standardized validation frameworks for editing outcomes [33] [30]. The integration of computational prediction tools for guide RNA efficiency and the development of international regulatory guidelines will further advance the application of CRISPR in zebrafish research [33] [31]. As these technologies continue to evolve, they will undoubtedly uncover new insights into gene function, disease mechanisms, and therapeutic strategies, solidifying the role of zebrafish as a premier model for vertebrate functional genomics and biomedical research.
The pharmaceutical industry faces a profound productivity crisis, with the average new drug development costing $2.6 billion and taking over 10 years from discovery to regulatory approval. This application note examines how the synergistic combination of zebrafish models and CRISPR-Cas9 genome editing is accelerating therapeutic target discovery and validation. We present detailed protocols for CRISPR-mediated gene knockout in zebrafish and quantitative phenotypic screening, demonstrating how this integrated platform streamlines functional genomics and high-content drug screening to reduce costs and timelines while improving translational success.
Current drug development pipelines are hampered by high costs, extended timelines, and crowded research efforts focused on similar indications and drug targets. This results in therapeutics that often share mechanisms of action with limited efficacy improvements [34]. The discovery of novel therapeutic targets based on deeper understanding of disease biology is crucial for developing innovative medicines with potentially greater efficacy.
The convergence of zebrafish as a disease model and CRISPR-Cas9 technology presents a transformative approach to address these challenges. Zebrafish offer genetic homology of approximately 87% with humans, transparent embryos for real-time observation, and small size for high-throughput studies [35]. CRISPR-Cas9 enables precise genome editing to rapidly create disease models and systematically evaluate gene function. This combination facilitates large-scale functional genomic studies previously impractical in traditional rodent models.
The Zebrafish-CRISPR Platform: Technical Advantages
Key Features of Zebrafish for Drug Discovery
Table 1: Comparative Analysis of Animal Models in Pharmaceutical Research
| Parameter | Zebrafish | Mouse Models | In Vitro Systems |
|---|---|---|---|
| Genetic similarity to humans | ~87% conserved genes | ~95% conserved genes | Varies by cell type |
| Throughput capacity | High (100+ embryos/day) | Moderate (10-20 embryos/day) | Very high |
| Developmental timeline | 24-48 hours for organogenesis | 18-21 days gestation | Not applicable |
| Imaging capability | High (whole-organism transparency) | Limited (requires imaging techniques) | High (single-cell resolution) |
| Drug administration | Water-soluble compounds added to tank water | Oral gavage, injection | Direct to culture media |
| Cost per study | Low | High | Very low |
| Regulatory acceptance | Growing acceptance for preclinical studies | Well-established | Limited for whole-organism effects |
CRISPR-Cas9 Mechanisms for Targeted Genome Engineering
The CRISPR-Cas9 system consists of a Cas9 endonuclease and a guide RNA (gRNA) that directs the enzyme to specific genomic loci. Upon binding to the target DNA sequence adjacent to a Protospacer Adjacent Motif (PAM), Cas9 generates double-strand breaks (DSBs) that are repaired through either:
- Non-homologous end joining (NHEJ): Error-prone repair resulting in insertions or deletions (indels) that disrupt gene function
- Homology-directed repair (HDR): Precise repair using a template to introduce specific genetic modifications [3]
This system has largely superseded earlier technologies like ZFNs and TALENs due to its simpler design (requiring only guide RNA synthesis rather than custom proteins), higher efficiency, and multiplexing capabilities [3].
Application Notes: Implementing Zebrafish-CRISPR in Drug Discovery Pipelines
High-Throughput Target Validation
The zebrafish-CRISPR platform enables rapid functional assessment of candidate genes identified from human genomic studies. In a demonstration of scalability, researchers used MIC-Drop (Multiplexed Intermixed CRISPR Droplets) technology to screen 188 zebrafish genes for cardiac development roles in a single experiment [36]. This approach:
- Encapsulates guide RNAs and Cas9 enzyme in barcoded oil droplets
- Enables one researcher to inject hundreds of embryos in hours versus days
- Identified 13 genes essential for proper heart development
- Provides a framework for genome-scale functional screening
Disease Modeling: Glioma Case Study
Zebrafish glioma models illustrate the platform's versatility for studying complex diseases. Three primary modeling approaches have been developed:
- Chemical Mutagenesis: Induces random mutations for novel gene discovery
- Genetic Engineering: CRISPR-Cas9 targeting of conserved glioma genes (TP53, NF1, RB1)
- Xenotransplantation: Implantation of human glioma cells for therapeutic testing [35]
These models leverage conserved brain structures between zebrafish and humans, including telencephalon, diencephalon, and cerebellum, enabling study of tumor-brain interactions within evolutionarily conserved microenvironments [35].
High-Content Drug Screening
Zebrafish enable in vivo drug screening with cellular resolution unavailable in traditional models. The transparency of embryos and larvae permits real-time observation of:
- Tumor growth and angiogenesis
- Cell migration and metastasis
- Drug efficacy and toxicity at single-cell resolution [35]
This enables identification of compounds with inter-organ mechanisms of action that would be missed in targeted screening approaches [37].
Experimental Protocols
CRISPR-Cas9-Mediated Gene Knockout in Zebrafish
Table 2: Key Research Reagents for Zebrafish CRISPR
| Reagent/Equipment | Specification | Function | Alternative/Note |
|---|---|---|---|
| Zebrafish strain | Healthy breeding pairs (3-12 months) | Provide embryos for microinjection | Wild-type or specific mutants |
| Cas9 protein | Purified (in-house or commercial) | DNA endonuclease for target cleavage | Cas9 mRNA can be used as alternative |
| sgRNA | In vitro transcribed with T7 polymerase | Guides Cas9 to specific genomic targets | crRNA:tracrRNA duplex also effective |
| Microinjection system | FemtoJet programmable injector | Delivers CRISPR components to embryos | Manual injection systems possible |
| gRNA design tool | CRISPRScan, Benchling | Predicts optimal guide RNA sequences | Multiple tools should be compared |
| Genotyping reagents | Heteroduplex mobility assay, ICE analysis | Confirms mutagenesis efficiency | Sanger sequencing for validation |
Protocol: Targeted Gene Knockout
Before Beginning
- Secure IACUC approval and ensure compliance with animal care regulations
- Establish healthy zebrafish breeding colonies with reliable spawning
- Maintain water quality: temperature 28°C, pH 7.0-7.5, conductivity 500-1500 μS [38]
sgRNA Design and Synthesis (Timeline: 1-2 days)
- Identify target sequence using design tools (CRISPRScan recommended)
- Select 20-nucleotide target sequence adjacent to 5'-NGG-3' PAM
- Synthesize sgRNA template oligonucleotides with T7 promoter sequence
- Transcribe sgRNA using T7 High Yield RNA Synthesis Kit
- Purify sgRNA using phenol:chloroform extraction and ethanol precipitation [38]
Cas9 Protein Preparation (Timeline: 2-3 days)
- Express Cas9 protein in E. coli Rosetta (DE3) using pSHS207 plasmid
- Induce expression with 0.5 mM IPTG at 18°C for 16 hours
- Purify using Ni-NTA affinity chromatography
- Dialyze into injection buffer (150 mM KCl, 20 mM HEPES pH 7.5)
- Concentrate to 5-10 μg/μL, aliquot and store at -80°C [38]
Microinjection (Timeline: 1 day)
- Prepare injection mixture: 300 ng/μL Cas9 protein + 30-50 ng/μL sgRNA
- Add phenol red to 0.1% for visualization during injection
- Load mixture into needle using microloader tips
- Inject 1-2 nL into zebrafish embryo yolk at single-cell stage
- Transfer injected embryos to 28°C incubator for development [38]
Genotype Analysis (Timeline: 2-3 days)
- At 24-48 hours post-fertilization, collect 10-20 embryos for DNA extraction
- Amplify target region by PCR (Q5 High-Fidelity Master Mix)
- Initial screening by heteroduplex mobility assay (15% PAGE)
- Quantify indel frequency using ICE or TIDE analysis of Sanger sequences
- Confirm mutations by Illumina sequencing for precise variant characterization [4]
Quantitative Phenotypic Screening Protocol
Vascular Formation Assessment (Timeline: 3-5 days)
- Raise CRISPR-injected embryos to 2-3 days post-fertilization (dpf)
- Anesthetize with tricaine and mount in methylcellulose
- Image vasculature using confocal or fluorescence microscope
- Quantify vascular parameters: vessel length, branching points, diameter
- Compare mutants to wild-type controls for phenotypic scoring [39]
Data Analysis and Interpretation
Evaluating CRISPR Editing Efficiency
Table 3: Comparison of CRISPR Analysis Methods
| Method | Sensitivity | Throughput | Cost | Best Application |
|---|---|---|---|---|
| Heteroduplex Mobility Assay | Moderate | High | Low | Initial screening of G0 mosaics |
| TIDE Analysis | High | Moderate | Moderate | Efficiency quantification |
| ICE Analysis | High | Moderate | Moderate | Correlation with Illumina (ρ=0.88) |
| Illumina Sequencing | Very High | Low | High | Gold standard validation |
Recent evaluations of 50 different gRNAs revealed significant discrepancies between predicted and actual editing efficiencies, emphasizing the importance of empirical validation [4]. ICE analysis shows the highest correlation with Illumina sequencing results (Spearman ρ=0.88), while heteroduplex assays show weaker correlation (Spearman ρ=0.37-0.38) [4].
Addressing Technical Considerations
Off-Target Effects
- Empirical testing shows low in vivo off-target mutation rates (<1% for most loci)
- No significant inflation of de novo mutations in cross-generational studies
- Focus on top predicted off-target regions based on sequence homology [4]
Control Considerations
- "Mock" injected controls (Cas9 alone) show differential gene expression related to wound response and cytoskeleton organization
- Uninjected siblings serve as optimal controls for transcriptomic studies
- Multiple biological replicates essential for phenotypic scoring [4]
The integration of zebrafish models with CRISPR-Cas9 technology represents a transformative approach to addressing the pharmaceutical productivity crisis. This platform enables:
- Rapid functional validation of novel therapeutic targets
- High-content phenotypic screening in physiologically relevant contexts
- Accelerated timeline from target identification to preclinical validation
Future developments including advanced humanized models, CRISPR-mediated immune regulation, and high-temperature resistant strains will further enhance the translational relevance of zebrafish models in drug discovery [35]. As MIC-Drop and related technologies mature, genome-scale functional screens in zebrafish will become feasible, potentially unlocking new therapeutic strategies for complex human diseases.
From Theory to Bench: Protocols for Knockout, Knock-in, and Advanced Editing
Within the broader scope of CRISPR-Cas9 gene editing in zebrafish embryos, the microinjection of pre-assembled Cas9-gRNA ribonucleoprotein (RNP) complexes at the one-cell stage represents a foundational technique. This direct delivery method facilitates high-efficiency, heritable mutagenesis, enabling researchers to model human diseases and accelerate drug discovery pipelines [40]. The RNP approach minimizes off-target effects and mosaicism compared to mRNA injection, leading to more consistent and predictable phenotypic outcomes [41]. This protocol details a streamlined methodology for generating F0 knockout zebrafish, which is critical for functional genomics and high-throughput screening in pharmaceutical development.
Principle of the Method
The efficacy of this protocol hinges on the microinjection of a pre-formed CRISPR-Cas9 ribonucleoprotein complex directly into the cytoplasm of a single-cell zebrafish embryo. The complex comprises a guide RNA (gRNA), which confers sequence specificity, and the Cas9 nuclease, which induces a double-strand break (DSB) in the target genomic DNA [40]. The cellular repair of this break via the error-prone non-homologous end joining (NHEJ) pathway results in small insertions or deletions (indels). When these indels occur within a protein-coding exon, they can disrupt the reading frame, leading to a functional gene knockout. The use of duplex guide RNP (dgRNP) complexes, assembled from crRNA, tracrRNA, and Cas9 protein, has been demonstrated to achieve biallelic mutations in a high percentage of somatic cells in the injected F0 generation, sometimes recapitulating known genetic mutant phenotypes [41] [40].
Reagent Preparation
Research Reagent Solutions
The following table details the essential materials required for the assembly of the CRISPR-Cas9 RNP complex and the microinjection process.
Table 1: Key Research Reagents and Materials for Cas9-gRNA Microinjection
| Item | Function/Description | Example Supplier/Component |
|---|---|---|
| Alt-R Cas9 Nuclease, V3 | The engineered S. pyogenes Cas9 protein that performs the DNA cleavage. | Integrated DNA Technologies (IDT) [41] |
| Alt-R crRNA | The CRISPR RNA (crRNA) component that defines the target DNA sequence. | Integrated DNA Technologies (IDT) [41] |
| Alt-R tracrRNA | The trans-activating crRNA (tracrRNA) that facilitates the formation of the functional gRNA complex with the crRNA and Cas9. | Integrated DNA Technologies (IDT) [41] |
| Duplex Buffer | A specific buffer provided by IDT to ensure proper annealing of crRNA and tracrRNA. | Integrated DNA Technologies (IDT) [41] |
| Phenol Red (0.25%) | An injection dye added to the injection mixture to allow visual confirmation of successful delivery into the embryo. | Optional, from various chemical suppliers [41] |
| Microinjection Rig | The core setup, typically including a micromanipulator, a microinjector (air or oil pressure), a stereomicroscope, and a magnetic base. | Various manufacturers (e.g., Narishige, Eppendorf, Warner Instruments) [42] |
| Capillary Glass Needles | Fine, pulled glass capillaries used to pierce the chorion and inject nanoliter volumes into the embryo. | Various manufacturers (e.g., World Precision Instruments) [42] |
Assembling the CRISPR-Cas9 RNP Complex
The preparation of the dgRNP complex is a critical step for achieving high editing efficiency.
- Resuspend RNAs: Resuscribe the lyophilized crRNA and tracrRNA in nuclease-free duplex buffer to a stock concentration of 100 µM.
- Prepare the gRNA Duplex: In a nuclease-free microcentrifuge tube, combine 1 µL of 100 µM crRNA and 1 µL of 100 µM tracrRNA. Heat the mixture to 95°C for 5 minutes, then allow it to cool slowly to room temperature to facilitate proper annealing [41].
- Form the RNP Complex: For a single injection mixture, combine the following:
- 2 µL of the 50 µM annealed gRNA duplex (from step 2)
- 2 µL of 50 µM Cas9 protein
- 6 µL of nuclease-free water (or 5 µL water and 1 µL of 0.25% phenol red for visualization) The final concentration of the dgRNP in this 10 µL mixture is 10 µM. For co-targeting multiple genes (e.g., slc45a2 and chrna1 for pigment-free, immobilized embryos), prepare individual RNP complexes and combine them just before loading the needle [41].
- Incubate: Incubate the final mixture at 37°C for 10-15 minutes to allow the RNP complex to form completely. The complex is now ready for injection and should be used promptly.
Step-by-Step Microinjection Protocol
The diagram below illustrates the complete experimental workflow from reagent preparation to genotyping.
Detailed Injection Procedure
- Embryo Collection: Collect freshly laid zebrafish embryos within 15-20 minutes post-fertilization. Using a transfer pipette, arrange them side-by-side in the grooves of an injection mold filled with embryo medium, ensuring the cell is visible and accessible [42].
- Needle Preparation: Back-load 1-2 µL of the prepared RNP mixture into a glass capillary needle using a fine gel-loading tip. Carefully break the tip of the needle with fine forceps to an opening of approximately 1-5 µm. Calibrate the injection volume by measuring the diameter of the injection bolus against a stage micrometer; a bolus of ~50-100 µm in diameter (approximately 1-2 nL) is typically optimal.
- Microinjection: Under the stereomicroscope, orient the needle at a shallow angle (10-30 degrees). Gently pierce the chorion and enter the cytoplasm of the one-cell stage embryo. Deliver a single bolus of the RNP mixture into the cell cytoplasm using a brief pulse of air pressure. A successful injection is confirmed by the slight displacement of cytoplasm and the visible presence of the phenol red dye [42].
- Post-Injection Care: After injection, gently release the embryos from the mold into a Petri dish filled with fresh embryo medium. Incubate the injected embryos at 28.5°C, inspecting them periodically. Remove any dead or unfertilized embryos.
Quality Control and Validation
Efficiency Assessment and Genotyping
A critical step post-injection is the validation of CRISPR-induced mutations. High-Resolution Melting (HRM) analysis offers a rapid and sensitive method for this purpose.
- DNA Extraction: At 4-24 hours post-fertilization (hpf), transfer single embryos to PCR tubes. Extract genomic DNA using a rapid alkaline lysis (HotSHOT) method: add 50 µL of 50 mM NaOH, incubate at 95°C for 10-30 minutes, then neutralize with 5 µL of 1M Tris-HCl, pH 8.0 [43].
- HRM Analysis: Use the crude lysate as a template for a PCR reaction with EvaGreen dye and primers flanking the target site. Perform the HRM step on a real-time PCR machine. The presence of indels creates heteroduplex DNA that melts at a different temperature than homogenous wild-type amplicons, resulting in distinct curve profiles and allowing for genotyping within hours [43].
Quantitative Data on Mutagenesis Efficiency
The following table summarizes expected outcomes and key performance metrics from established protocols.
Table 2: Expected Efficiency Metrics for Cas9-gRNA Microinjection in Zebrafish
| Parameter | Reported Efficiency | Method of Assessment | Source/Context |
|---|---|---|---|
| Somatic Mutation Rate | >35% | SURVEYOR assay / HRM | Targeting etsrp, gata4, gata5 [40] |
| Biallelic Conversion (Phenocopy) | 24% - 38% of injected embryos | Phenotypic analysis (e.g., vessel defects, cardia bifida) | Injected embryos recapitulating etsrpy11 or fautm236a mutant phenotypes [40] |
| Earliest Indel Detection | 2-cell stage (11% of embryos) | HRM analysis | Mutagenesis kinetics for calpn1a; efficiency increases to 100% by sphere stage [43] |
| Multiplexing Efficiency | High | Phenotypic (loss of pigment, immobilization) | Co-injection of dgRNPs targeting slc45a2 and chrna1 [41] |
Application in Research
The primary application of this protocol is the rapid generation of F0 knockout embryos for functional gene analysis. A powerful extension is the use of multiplexed RNP injections to create embryos that are optimized for live imaging studies. As demonstrated, injecting dgRNPs targeting the slc45a2 (albino) gene to eliminate pigment and the chrna1 (nic1) gene to induce skeletal muscle paralysis creates "Casper"-like, immobilized embryos in any genetic background. This eliminates the need for time-consuming genetic crosses or the use of potentially toxic chemical agents like PTU, thereby facilitating high-resolution, long-term imaging of development and disease processes [41]. The genetic strategy for creating such imaging-ready embryos is outlined below.
Troubleshooting
- Low Mutation Efficiency: Ensure the RNP complex is freshly prepared and not subjected to multiple freeze-thaw cycles. Verify the quality and concentration of the guide RNA and Cas9 protein. Optimize the injection volume and pressure to ensure consistent cytoplasmic delivery.
- High Embryo Mortality: This is often caused by a needle opening that is too large, excessive injection volume, or damage to the yolk cell. Precisely calibrate the injection volume to 1-2 nL and practice needle handling to minimize physical damage.
- No Detected Mutations: Re-design and validate the gRNA target sequence using tools like CHOPCHOP or CRISPRscan [41] [43]. Confirm the genotyping assay (e.g., HRM, sequencing) is sensitive enough to detect mosaic indels in F0 embryos.
Within the broader context of CRISPR-Cas9 gene editing in zebrafish embryos, generating loss-of-function mutations remains a fundamental application for functional genomics and disease modeling. The non-homologous end joining (NHEJ) pathway serves as the dominant DNA repair mechanism in zebrafish and represents the most efficient method for creating gene knockouts [3]. Unlike homology-directed repair (HDR), which is rare and technically challenging, NHEJ actively repairs CRISPR-Cas9-induced double-strand breaks by directly ligating broken ends, often resulting in small insertions or deletions (indels) [44] [3]. When these indels occur within coding exons and disrupt the reading frame, they effectively abolish gene function, enabling researchers to study loss-of-function phenotypes directly in mosaic G0 embryos or in stable mutant lines [4] [3]. This application note details optimized protocols for leveraging NHEJ to achieve efficient gene knockouts in zebrafish, providing researchers with a robust framework for functional gene analysis.
Key Principles of NHEJ-Mediated Knockout
The theoretical foundation for NHEJ-mediated knockout is elegantly simple: CRISPR-Cas9 creates a targeted double-strand break in the genome, and the error-prone NHEJ repair pathway introduces mutations at the break site as it rejoins the DNA ends. The practical efficiency of this process, however, depends heavily on experimental parameters. The critical distinction between NHEJ and HDR pathways in zebrafish lies in their relative activities; NHEJ is highly active and constitutes the predominant repair mechanism, while HDR occurs at significantly lower frequencies [45] [3]. This biological reality makes NHEJ the preferred mechanism for generating knockouts. The key to a successful knockout experiment is a highly efficient single guide RNA (sgRNA) that creates a double-strand break at the target locus. Imperfect prediction tools mean that empirical testing of sgRNA cutting efficiency is strongly recommended before embarking on full-scale experiments [45].
The following workflow outlines the complete experimental pipeline from target design to mutant validation:
Quantitative Analysis of Knockout Efficiency Assessment Methods
Evaluating the success of a knockout experiment requires quantifying the efficiency of indel formation at the target locus. Several methods are available, each with different throughput, cost, and informational output characteristics. Next-generation sequencing provides the most accurate quantification but may be cost-prohibitive for some laboratories. Two commonly used Sanger sequencing-based tools, Inference of CRISPR Edits (ICE) and Tracking of Indels by Decomposition (TIDE), offer a reasonable balance between cost and accuracy, though they tend to underestimate efficiency compared to NGS [4]. Polyacrylamide gel electrophoresis (PAGE) provides a rapid and affordable initial assessment by visualizing heteroduplex formation, but it is only semi-quantitative and shows weaker correlation with sequencing-based methods [4].
Table 1: Comparison of Methods for Assessing CRISPR-Cas9 Knockout Efficiency in Zebrafish
| Method | Throughput | Cost | Key Advantage | Key Limitation | Correlation with NGS |
|---|---|---|---|---|---|
| Next-Generation Sequencing (NGS) | High | High | Gold standard for accuracy and indel characterization | Requires bioinformatics expertise; higher cost | Gold standard |
| Sanger Sequencing + ICE Analysis | Medium | Medium | Good balance of cost and accuracy; provides indel spectra | Underestimates efficiency compared to NGS | Spearman ρ = 0.88 [4] |
| Sanger Sequencing + TIDE Analysis | Medium | Medium | User-friendly web interface | Lower correlation with NGS than ICE | Spearman ρ = 0.59 [4] |
| PAGE Heteroduplex Assay | High | Low | Rapid and inexpensive; no sequencing required | Semi-quantitative; no sequence information | Spearman ρ = 0.37 [4] |
Step-by-Step Protocol for NHEJ-Mediated Knockout
sgRNA Design and Synthesis
Target Selection: Identify a 20-nucleotide target sequence adjacent to a 5'-NGG-3' Protospacer Adjacent Motif (PAM) in an early coding exon of your target gene. While computational prediction tools like CRISPRscan are available, their predictions show weak correlation with actual in vivo efficiency [4] [45]. Design 2-3 sgRNAs targeting different regions to maximize success probability.
sgRNA Template Preparation: Synthesize the sgRNA using the following protocol:
- Order a DNA oligo with the structure: 5'-T7 promoter - target sequence - sgRNA scaffold-3'
- Perform in vitro transcription using a commercial kit (e.g., GeneArt Precision gRNA Synthesis Kit)
- Purify the sgRNA using phenol-chloroform extraction and ethanol precipitation
- Resuspend in nuclease-free water and quantify using spectrophotometry
- Aliquot and store at -80°C
Microinjection into Zebrafish Embryos
Preparation of Injection Mixture:
- 150-300 ng/μL of sgRNA
- 300-500 ng/μL of Cas9 protein (or 150-300 ng/μL Cas9 mRNA)
- 1× injection buffer with phenol red
- Incubate the ribonucleoprotein complex for 10 minutes at 37°C prior to injection
Microinjection Procedure:
- Collect one-cell stage zebrafish embryos and align them in grooves on an injection plate
- Using a microinjector and fine glass needle, deliver 1-2 nL of the injection mixture into the cell cytoplasm
- After injection, transfer embryos to egg water and maintain at 28.5°C
- Remove unfertilized or damaged embryos after 2-4 hours
Validation of Somatic Mutagenesis
DNA Extraction (at 5 dpf):
- Pool 10-20 injected larvae or process individually in 50 μL of lysis buffer
- Incubate at 95°C for 10 minutes, then add 50 μL of neutralization buffer
- Centrifuge and use supernatant as PCR template
Efficiency Analysis:
- Amplify the target region by PCR using flanking primers
- Purify PCR products and submit for Sanger sequencing
- Analyze sequencing traces using ICE or TIDE software to determine indel percentage and spectrum
- Alternatively, run PCR products on a 10% polyacrylamide gel to visualize heteroduplex formation
Germline Transmission and Stable Line Establishment
Founder Raising: Raise injected embryos (G0) to sexual maturity. Outcross individual G0 fish to wild-type partners.
Germline Screening:
- Collect 20-30 F1 embryos from each cross at 5 dpf
- Perform fin clip biopsy on parents for genotyping
- Extract DNA and amplify the target region
- Sequence or use restriction fragment length polymorphism (RFLP) to identify heritable mutations
- Outcross confirmed founders again to establish stable lines
Table 2: Research Reagent Solutions for Zebrafish Knockout Experiments
| Reagent / Tool | Function | Specifications & Notes |
|---|---|---|
| Cas9 Protein | CRISPR nuclease; creates DSBs | Preferred over mRNA for higher efficiency and reduced off-target effects [46] |
| sgRNA | Targets Cas9 to specific genomic loci | Chemically synthesized or in vitro transcribed; design 2-3 per gene target |
| crRNA:tracrRNA Complex | Alternative to sgRNA | Two-part system; reported high efficiency in some studies [4] |
| Phenol Red | Injection tracer | Visual confirmation of successful microinjection |
| ICE Analysis Tool | Indel quantification | Free web tool for analyzing Sanger sequencing data from edited populations [4] |
| TIDE Analysis Tool | Indel quantification | Alternative web tool for decomposition of editing outcomes [4] |
| CrispRVariants | NGS data analysis | R package for characterizing mutations from multiplexed sequencing [4] |
Technical Considerations and Troubleshooting
Even with optimized protocols, several technical challenges can affect knockout efficiency. The high activity of NHEJ means that off-target effects, while generally low in zebrafish, should be considered [4]. To minimize this risk:
- Design sgRNAs with high specificity: Use BLAST against the zebrafish genome to ensure minimal off-target sites
- Consider Cas9 concentration: Higher concentrations increase on-target efficiency but may also increase off-target effects
- Outcross stable lines: To eliminate potential off-target mutations, outcross founders for at least two generations
If low knockout efficiency is observed:
- Verify sgRNA activity using CRISPR-STAT or similar methods before injection [47]
- Test different Cas9 delivery methods (protein vs. mRNA)
- Optimize injection mixture concentrations and volumes
- Screen multiple G0 founders for germline transmission
For phenotyping in G0 mosaic embryos, include appropriate controls such as uninjected siblings and Cas9-only injected embryos to account for potential confounding effects of the injection process itself, which has been shown to alter gene expression in some cases [4].
The NHEJ pathway provides a highly efficient and robust mechanism for generating gene knockouts in zebrafish, enabling both rapid assessment of gene function in G0 embryos and establishment of stable mutant lines. By following the optimized protocols outlined in this application note, researchers can reliably create loss-of-function mutations for functional genomics studies and disease modeling. The accessibility of this technology continues to drive its adoption in both academic and industrial settings, supporting drug target validation and mechanistic studies of gene function. As CRISPR technology evolves, further refinements in sgRNA design and delivery will likely enhance the precision and efficiency of NHEJ-mediated genome editing in zebrafish.
Within the broader context of CRISPR-Cas9 gene editing in zebrafish embryos, achieving precise genetic modifications through Homology-Directed Repair (HDR) represents a significant challenge and opportunity for biomedical research. Zebrafish share substantial genetic homology with humans, making them invaluable for modeling human diseases and drug development. However, the natural dominance of the error-prone non-homologous end joining (NHEJ) pathway over HDR in early embryos has historically limited efficient precise genome editing. This application note details optimized strategies and protocols to overcome these barriers, enabling researchers to reliably generate knock-in models for functional genetic studies.
The HDR Efficiency Challenge in Zebrafish
In zebrafish embryos, CRISPR-Cas9-induced double-strand breaks (DSBs) are predominantly repaired via the NHEJ pathway, which often results in insertions or deletions (indels) [48]. The HDR pathway, which can precisely incorporate an exogenous DNA donor template, occurs at significantly lower frequencies, leading to mosaicism in founder embryos (F0) and low germline transmission rates [44] [49]. This efficiency gap necessitates strategic intervention in both the cellular repair environment and the design of the donor template itself.
Quantitative in vivo reporter assays in zebrafish have demonstrated that the equilibrium between NHEJ and HDR can be chemically modulated. Inhibition of key NHEJ proteins can enhance HDR efficiency by up to 13.4-fold [48], providing a viable strategy to shift the repair balance in favor of precise editing.
Donor Template Design Strategies
The design and form of the donor template are critical determinants of HDR success. Below is a systematic comparison of the primary donor types, followed by detailed design parameters.
Table 1: Comparison of HDR Donor Templates for Zebrafish Knock-ins
| Donor Type | Typical Length | Key Features | Reported HDR Efficiency | Best Use Cases |
|---|---|---|---|---|
| Long ssDNA (zLOST) [44] | 299-512 nt | Single-stranded; requires 25+ bp homology arms | Up to 98.5% (phenotypic rescue); 31.8% germline transmission | Point mutations; small insertions; human disease modeling |
| Double-Cut dsDNA Donor [50] | ~1-2 kbp (total) | Linearized in vivo by Cas9; flanked by sgRNA target sites | 2- to 5-fold increase over circular plasmids | Insertion of larger cassettes (e.g., reporters, loxP sites) |
| ssODN [51] | Up to 200 nt | Short, single-stranded; asymmetric homology arms | Varies by locus and design | Single nucleotide changes; small tag insertions |
| Circular Plasmid [44] | >1 kbp | Double-stranded; long homology arms (0.8-2 kbp) | Low (~5%) | Large insertions requiring extensive homology |
Long Single-Stranded DNA (ssDNA) Donors
The zebrafish Long Single-Stranded DNA template (zLOST) method uses long ssDNA molecules (>200 nt) as repair templates. In a direct comparison at the tyr locus, zLOST achieved phenotypic rescue in close to 98% of injected albino embryos, a dramatic improvement over traditional donors [44] [52]. This template type is highly effective for introducing point mutations and small inserts, facilitating precise modeling of human diseases.
Double-Cut HDR Donors
For double-stranded DNA donors, a "double-cut" design significantly enhances efficiency. This strategy involves flanking the donor cassette with two sgRNA target sequences, enabling Cas9 to linearize the donor plasmid in vivo. This synchronous linearization with the genomic DSB increases HDR efficiency by twofold to fivefold in other model systems, a principle applicable to zebrafish [50]. This approach is particularly useful for inserting larger DNA fragments, such as fluorescent protein genes.
ssODN Design Parameters
When using short single-stranded oligodeoxynucleotides (ssODNs), several design parameters are critical for success [51]:
- Homology Arm Length: Asymmetric arms with 30-40 nucleotides are often optimal.
- Strand Preference: The donor strand complementary to the sgRNA (target strand) is often preferred, though this can vary by locus.
- Blocking Mutations: Incorporating silent mutations in the PAM sequence or the seed region of the sgRNA binding site is essential to prevent re-cleavage of the successfully edited allele by Cas9.
Optimized Experimental Protocols
This section consolidates the most effective methods into a single, detailed protocol for HDR-mediated knock-in in zebrafish.
Microinjection Protocol for HDR in Zebrafish
Materials:
- CRISPR-Cas9 Components: Cas9 protein (e.g., GeneArt Platinum Cas9 Nuclease) and target-specific sgRNA [53].
- HDR Donor Template: Purified long ssDNA (for zLOST) or double-cut dsDNA donor.
- Chemical Modulators: NU7441 (DNA-PK inhibitor, for NHEJ suppression) [48].
- Microinjection Setup: Standard zebrafish microinjection rig.
Procedure:
- RNP Complex Assembly: Complex the Cas9 protein and sgRNA at a final concentration of 600 ng/μL and 200 ng/μL, respectively, in injection buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5). Incubate at 37°C for 10 minutes to form the RNP complex [53] [51].
- Injection Mixture Preparation: Combine the pre-formed RNP complex with the HDR donor template. For zLOST, use a long ssDNA donor. For a double-cut donor, include a second sgRNA targeting the flanking sites on the donor plasmid [44] [50].
- Final recommended concentrations in the needle:
- Cas9 RNP: as above.
- ssDNA donor: 100-200 ng/μL.
- dsDNA donor: 50-100 ng/μL.
- Final recommended concentrations in the needle:
- Co-injection with Chemical Modulators: To shift the repair balance toward HDR, co-inject the editing components with 50 μM NU7441, a DNA-PK inhibitor proven to enhance HDR efficiency up to 13.4-fold in zebrafish [48].
- Zebrafish Embryo Injection: Inject 1-2 nL of the final mixture into the cytoplasm or cell yolk of one-cell stage zebrafish embryos.
- Post-Injection Culture: Incubate injected embryos in standard embryo medium at 28.5°C. Screen for somatic editing at 2-3 days post-fertilization (dpf), depending on the target and readout.
Screening for Germline Transmission
Screening for germline transmission requires careful design to distinguish precise HDR events from NHEJ-induced indels [49].
- Raise Injected Embryos (F0) to adulthood.
- Outcross F0 Adults to wild-type fish and collect individual clutches of F1 embryos.
- Genotype F1 Embryos: Use a combination of PCR and restriction fragment length polymorphism (RFLP) if a new site was introduced, or sequencing, to identify embryos carrying the precise HDR edit. The use of blocking mutations in the donor template is critical here to prevent false positives from persistent Cas9 activity.
The Scientist's Toolkit
Table 2: Essential Reagents and Resources for HDR in Zebrafish
| Item | Function/Description | Example Product/Source |
|---|---|---|
| Cas9 Nuclease | Creates a DSB at the target genomic locus. | GeneArt Platinum Cas9 Nuclease [53] |
| sgRNA | Guides Cas9 to the specific DNA sequence. | In vitro transcribed from a target-specific template |
| Long ssDNA Donor | Template for precise HDR repair. | Custom-synthesized (zLOST method) [44] |
| Double-Cut Donor Plasmid | dsDNA donor with sgRNA flanking sites for in vivo linearization. | Custom-cloned plasmid [50] |
| NHEJ Inhibitor | Shifts DNA repair balance from NHEJ to HDR. | NU7441 (DNA-PK inhibitor) [48] |
| Microinjection Buffer | Buffer for reagent delivery into embryos. | T10E0.1 (10 mM Tris, 0.1 mM EDTA, pH 7.5) [53] |
Visualizing the HDR Knock-in Strategy and Workflow
The following diagram illustrates the core molecular strategy for a double-cut HDR donor, which enhances knock-in efficiency by synchronizing donor linearization with the genomic DSB.
Diagram 1: Double-Cut HDR Donor Strategy. The donor plasmid and genomic locus are cleaved simultaneously by Cas9, facilitating efficient HDR.
The field of genome engineering in zebrafish has been revolutionized by the CRISPR-Cas9 system. However, a significant limitation of traditional CRISPR-Cas9 is its reliance on double-strand break (DSB) repair pathways to introduce point mutations, resulting in low efficiency and unwanted indel formations [54]. Base editing technology represents a transformative advance, enabling direct, precise conversion of single nucleotides without inducing DSBs [55] [56]. For zebrafish researchers focused on modeling human genetic diseases or performing detailed functional genomics, base editors provide an unparalleled tool for introducing single-nucleotide variants with high fidelity and efficiency.
Zebrafish are exceptionally suited for this technology due to their high genetic similarity to humans (approximately 70% of human disease-related genes have functional orthologs in zebrafish), external embryonic development, and optical transparency, which facilitates the rapid in vivo assessment of editing outcomes [55] [57]. This application note details the implementation of Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs) within the context of zebrafish embryo research, providing standardized protocols and resource guides to accelerate adoption.
Understanding Base Editor Mechanisms
Core Architecture and Function
Base editors are fusion proteins that combine a catalytically impaired Cas nuclease (such as Cas9 nickase, nCas9, or deactivated Cas9, dCas9) with a single-stranded DNA deaminase enzyme [55] [56]. They operate by chemically converting one base into another within a localized, single-stranded DNA region known as the "editing window," which is made accessible when the Cas moiety binds to the target DNA sequence guided by a sgRNA.
- Cytosine Base Editors (CBEs) typically fuse nCas9 to a cytidine deaminase enzyme (e.g., APOBEC1). Within the R-loop structure, this enzyme converts cytosine (C) to uracil (U) on the single-stranded DNA. Cellular repair mechanisms then interpret the U as a thymine (T), ultimately resulting in a C•G to T•A base pair conversion [55] [54]. The inclusion of a uracil glycosylase inhibitor (UGI) in the construct is critical, as it prevents the erroneous repair of U back to C, thereby enhancing editing efficiency [56].
- Adenine Base Editors (ABEs) fuse nCas9 to an engineered adenine deaminase (e.g., TadA). This deaminase converts adenine (A) to inosine (I), which is read by DNA polymerases as guanine (G). This process leads to an A•T to G•C base pair conversion [55] [54].
Table 1: Comparison of Base Editor Systems
| Feature | Cytosine Base Editors (CBEs) | Adenine Base Editors (ABEs) |
|---|---|---|
| Core Deaminase | APOBEC1 (or variants like PmCDA1 in Target-AID) [55] [56] | Engineered TadA (e.g., TadA-8e) [55] [56] |
| Base Conversion | C•G → T•A [56] | A•T → G•C [56] |
| Typical Editing Window | Positions ~4-8 upstream of the PAM (varies by specific editor) [55] | Positions ~4-7 upstream of the PAM (varies by specific editor) [56] |
| Key Components | nCas9, cytidine deaminase, UGI [55] | nCas9, engineered TadA adenine deaminase [55] |
| Example Editors | BE3, BE4max, AncBE4max, Target-AID, SpRY-CBE4max [55] [58] | ABE7.10, ABEmax, ABE8e, zSpRY-ABE8e [55] [58] |
Visualizing the Base Editing Mechanism
The following diagram illustrates the core mechanism of action for both CBEs and ABEs.
Diagram 1: Mechanism of CRISPR Base Editors. The process begins with sgRNA-guided binding to the target DNA. The Cas9 nickase creates a single-strand DNA bubble where the deaminase enzyme acts. CBEs convert C to U, leading to a C•G to T•A change, while ABEs convert A to I, leading to an A•T to G•C change.
Advanced Base Editing Toolkits for Zebrafish
The base editing toolbox has expanded significantly, moving beyond the canonical SpCas9 (which requires an NGG PAM) to include variants with relaxed PAM requirements. This dramatically increases the number of targetable sites in the zebrafish genome.
- SpG and SpRY Cas9 Variants: Engineered SpCas9 variants SpG and SpRY recognize alternative PAM sequences, greatly expanding the targeting scope.
- PAM-less Base Editors: Fusing deaminases to these variants creates powerful editors like SpRY-CBE4max and zSpRY-ABE8e, which have been successfully implemented in zebrafish with editing efficiencies reaching up to 96% at some loci, enabling the modeling of genetic variants previously inaccessible with traditional methods [55] [58].
Table 2: Evolution and Performance of Base Editors in Zebrafish
| Editor Name | Type | Key Features / Improvements | Reported Efficiency in Zebrafish |
|---|---|---|---|
| BE3 [55] | CBE | First nCas9-based CBE; foundational technology. | 9.25% - 28.57% [55] |
| Target-AID [55] [56] | CBE | Uses PmCDA1 deaminase; complementary editing window. | Variable, locus-dependent [55] |
| AncBE4max [55] | CBE | Codon-optimized for zebrafish; improved nuclear localization. | ~3x higher than BE3 (~90% efficiency reported in some models) [55] |
| SpRY-CBE4max [58] | CBE | Near-PAM-less targeting with high product purity. | Up to 87% - 96% [55] [58] |
| ABE7.10 [56] | ABE | First-generation efficient ABE. | ~53% average in human cells [56] |
| ABE8e / ABE8s [56] | ABE | Faster editing kinetics and wider editing window. | High efficiency demonstrated (tool used in zebrafish) [56] |
| zSpRY-ABE8e [58] | ABE | Near-PAM-less adenine base editing. | Up to 96% [58] |
Experimental Protocol: Implementing Base Editing in Zebrafish Embryos
This protocol outlines the recommended method for achieving high-efficiency base editing in zebrafish one-cell stage embryos using purified base editor protein in a ribonucleoprotein (RNP) complex, which reduces mosaicism and off-target effects [59] [58].
Reagent Preparation
- Purified Base Editor Protein: Obtain or purify the chosen base editor protein (e.g., AncBE4max, SpRY-ABE8e). Ensure the protein is tagged with a nuclear localization signal (NLS). The use of an artificial NLS (aNLS) has been shown to enhance nuclear import and editing efficiency in early zebrafish embryos [59].
- Synthetic sgRNA: Design and synthesize sgRNAs targeting the desired locus. For optimal stability and efficiency, use MS-modified sgRNAs (2'-O-methyl-3'-phosphorothioate modifications at the three terminal nucleotides at both the 5' and 3' ends) [58].
- Microinjection Buffer: Use a standard buffer such as 1x PBS or 0.5x TBE.
Microinjection Setup
- RNP Complex Assembly: Combine the purified base editor protein and MS-modified sgRNA to form the RNP complex. A typical working concentration is 5 μM for the RNP complex [58].
- Example:
- Final Injection Mix: 2.5 μl of 10 μM base editor protein + 2.5 μl of 10 μM MS-sgRNA + 5 μl injection buffer. Incubate at 37°C for 10 minutes before injection.
- Embryo Collection: Collect healthy, one-cell stage zebrafish embryos and align them in the grooves of an injection mold.
- Microinjection: Using a microinjection apparatus, inject approximately 1-2 nL of the pre-assembled RNP complex directly into the cytoplasm or cell yolk of the one-cell stage embryo [60] [16].
- Post-Injection Care: After injection, transfer embryos to egg water and maintain at 28.5°C. Monitor development and assess editing efficiency at the desired stage.
Validation and Genotyping
- Phenotypic Screening (if applicable): For edits that produce a visible phenotype (e.g., pigmentation loss in the albino gene), screen F0 mosaic larvae at 3 days post-fertilization (dpf) [59].
- Genomic DNA Extraction: At 48-72 hpf, pool or individually homogenize larvae for genomic DNA extraction.
- PCR and Sequencing: Amplify the target region by PCR and subject the product to Sanger sequencing or next-generation sequencing (NGS).
- Efficiency Analysis: Quantify editing efficiency by analyzing the Sanger sequencing chromatograms using tools like Synthego's ICE (Inference of CRISPR Edits) or by calculating the percentage of reads containing the desired edit from NGS data [58].
Table 3: Key Research Reagent Solutions for Zebrafish Base Editing
| Reagent / Resource | Function | Specific Examples & Notes |
|---|---|---|
| Base Editor Plasmids | Source of the base editor coding sequence. | BE4max, AncBE4max, ABEmax, ABE8e plasmids (Addgene) [56]. Codon-optimize for zebrafish. |
| Purified Base Editor Protein | Direct use in RNP complex for microinjection. | Commercially available or in-house purified SpRY-CBE4max, zSpRY-ABE8e. aNLS-tagged versions are preferred [59] [58]. |
| MS-modified sgRNA (EEgRNA) | Enhanced stability and efficiency; reduces degradation. | Synthesized commercially. Crucial for high efficiency with SpG and SpRY systems [58]. |
| Microinjection System | Physical delivery of RNP complexes into embryos. | Standard micromanipulators, injectors, and injection molds. |
| sgRNA Design Tools | In silico design of specific guide RNAs. | ACEofBASEs online platform for sgRNA design and off-target prediction in zebrafish [55]. Cas-OFFinder, CRISPOR [58]. |
| Genotyping Software | Analysis of sequencing data to determine editing efficiency. | Synthego ICE tool, CRISPResso2 [58]. |
Base editors have ushered in a new era of precision genetics in zebrafish research. By enabling efficient and precise single-nucleotide changes without double-strand breaks, CBEs and ABEs, particularly when combined with advanced Cas9 variants like SpRY, empower researchers to model human genetic diseases with unprecedented accuracy. The protocols and tools outlined in this application note provide a clear roadmap for the successful implementation of this transformative technology, promising to accelerate both basic functional genomics and applied drug discovery pipelines.
Within the broader thesis on CRISPR-Cas9 gene editing in zebrafish embryo research, this application note details integrated protocols for conducting high-throughput functional screens in F0 crispants. The ability to perform reverse genetics in the zebrafish model organism has been revolutionized by the CRISPR/Cas9 system, whose high efficiency in generating mutations enables F0 screens as a reality in this organism [61]. This approach combines the biological relevance of a whole vertebrate organism with the scalability required for systematic target validation in drug discovery. By circumventing the need for stable line generation, F0 screens in zebrafish provide a rapid platform for linking gene function to disease phenotypes, addressing a critical need in pharmaceutical development where erroneous target hypotheses remain a major cause of late-stage clinical failures [62]. The protocols outlined herein leverage the optical transparency and high fecundity of zebrafish embryos, enabling the screening of thousands of compounds in just days within a complex 3-dimensional context of a whole organism [63].
Key Concepts and Advantages
What are F0 Crispants?
F0 crispants ("CRISPR-ized mutants") are zebrafish embryos that exhibit mosaic mutagenesis following direct injection of CRISPR/Cas9 reagents at the single-cell stage. Unlike traditional genetic approaches that require raising mutants through generations to achieve homozygous loss-of-function, crispants enable immediate phenotypic assessment in the injected generation. This mosaic nature means that each crispant contains a spectrum of mutation types and percentages across its tissues, which can be sufficient to elicit strong, scorable phenotypes for many genetic targets.
Strategic Advantages in Drug Discovery
The application of F0 crispant screens addresses several critical challenges in modern drug development:
Rapid Target Validation: F0 screens dramatically compress the timeline for in vivo target validation from months to weeks, enabling faster progression from gene identification to functional assessment [61] [62].
Human Disease Relevance: Zebrafish share approximately 70-80% synteny with humans and possess highly conserved organ systems, providing a biologically relevant context superior to many conventional cell lines [63]. This relevance is further enhanced through the use of primary cells in some screening approaches [64].
High-Throughput Capability: The small size of zebrafish embryos permits their placement in microtiter plates, while robotic injection systems can process up to 2000 embryos per hour, enabling scalable screening approaches [65].
Phenotypic Complexity: Screens capture the integrated physiology of a complete vertebrate system, revealing effects on complex processes like angiogenesis, cardiac function, neurodevelopment, and tumor metastasis that cannot be fully modeled in monolayer cell cultures [63].
Table 1: Comparison of F0 Crispant Screening with Alternative Validation Approaches
| Parameter | F0 Crispants in Zebrafish | Traditional Zebrafish Mutants | In Vitro CRISPR Screens |
|---|---|---|---|
| Timeline to Phenotype | 1-5 days | 3-6 months | 1-4 weeks |
| Physiological Context | Whole vertebrate organism | Whole vertebrate organism | Cellular (often immortalized lines) |
| Throughput Potential | High | Low | Very High |
| Phenotypic Complexity | High (systemic, organ-level) | High (systemic, organ-level) | Limited (cell-autonomous) |
| Genetic Mosaicism | Present (enables assessment of cell-autonomous functions) | Absent (uniform genotype) | Typically absent |
| Automation Potential | High (robotic injection, automated imaging) [65] | Moderate | Very High |
The entire process for a high-throughput F0 crispant screen, from target identification to phenotypic analysis, is summarized below. This integrated workflow enables the systematic functional annotation of gene targets and modeling of human diseases.
Detailed Protocols
Protocol 1: sgRNA Design and CRISPR Reagent Preparation
Objective: To design and synthesize highly active, specific sgRNAs for target genes and prepare ribonucleoprotein (RNP) complexes for injection.
sgRNA Design and In Vitro Transcription
- Target Selection: Identify a 20-nucleotide target sequence directly upstream of a 5'-NGG-3' Protospacer Adjacent Motif (PAM) in an early exon of your target gene to maximize the likelihood of generating loss-of-function alleles.
- Specificity Check: Use tools such as those referenced by Doench et al. (2016) to minimize off-target effects while maximizing on-target activity [61].
- Oligo Template Design: Add the T7 promoter sequence (5'-TAATACGACTCACTATA-3') to the target-specific sequence.
- In Vitro Transcription: Synthesize sgRNA using a T7 in vitro transcription kit. Purify the sgRNA using standard phenol-chloroform extraction or commercial cleanup kits. Resuspend in nuclease-free water and quantify using spectrophotometry.
- Quality Control: Analyze sgRNA integrity by denaturing agarose gel electrophoresis. Store at -80°C until use.
RNP Complex Formation
- Working Solution: Dilute purified sgRNA to a concentration of 100 ng/µL in nuclease-free water.
- Complex Assembly: Combine the following in a nuclease-free microtube:
- 1 µL sgRNA (100 ng)
- 1 µL Cas9 protein (e.g., 100-200 ng)
- 1 µL 10X Injection Buffer
- Incubation: Mix gently and incubate at 37°C for 10 minutes to form RNP complexes. Keep on ice until injection (use within 2 hours).
Protocol 2: Robotic High-Throughput Embryo Injection
Objective: To achieve consistent, high-efficiency delivery of RNP complexes into single-cell zebrafish embryos for F0 crispant generation.
- Embryo Collection: Collect naturally spawned zebrafish embryos and align them along the edges of an injection mold filled with E3 embryo medium.
- Injection Plate Preparation: Load the injection needle with the prepared RNP complex mixture.
- Robotic System Setup: Configure the robotic injection system (e.g., parameters: injection pressure: 15 psi, duration: 0.1 s, injection volume: ~1 nL) [65].
- Automated Injection: Execute the injection protocol. The robotic system will position each embryo and deliver the RNP complex directly into the cell yolk or cytoplasm. Throughputs of up to 2000 embryos per hour are achievable [65].
- Post-Injection Care: Transfer injected embryos to fresh E3 medium and maintain at 28.5°C. Remove unviable embryos after a few hours.
Protocol 3: Mutation Efficiency Analysis
Objective: To quantify the mutagenesis rate induced by the CRISPR/Cas9 reagents in the injected F0 embryo population.
T7 Endonuclease I (T7E1) Assay
This method detects heteroduplex DNA formed by indel mutations.
- DNA Extraction: At 24-48 hours post-fertilization (hpf), pool 10-20 injected embryos and extract genomic DNA.
- PCR Amplification: Amplify a 300-500 bp region surrounding the target site from 50-100 ng of genomic DNA.
- Heteroduplex Formation: Denature and reanneal the PCR product by heating to 95°C for 5 minutes and then cooling slowly to room temperature.
- T7E1 Digestion: Incubate the reannealed PCR product with T7 Endonuclease I for 30 minutes at 37°C. This enzyme cleaves mismatched DNA heteroduplexes.
- Analysis: Resolve the digestion products by agarose gel electrophoresis. Cleaved bands indicate successful mutagenesis. Estimate the mutation efficiency using densitometry analysis of the gel image.
PCR and Sanger Sequencing
For a more precise assessment:
- PCR and Cloning: Amplify the target region from pooled embryo DNA and clone the products into a bacterial vector.
- Sequence Analysis: Sequence 20-50 individual clones and compare to the wild-type sequence. The percentage of clones with indels represents the mutation efficiency.
Table 2: Quantitative Standards for Mutation Efficiency and Phenotype Penetrance in F0 Screens
| Efficiency Metric | Threshold for High-Quality Screen | Measurement Method | Typical Range in Optimized Screen |
|---|---|---|---|
| Mutation Rate (Indel %) | >50% (pooled embryos) | T7E1 Assay or PCR/Sequencing | 50-90% [61] |
| Phenotype Penetrance | >70% of injected embryos | High-Content Imaging | Varies by target |
| Control Phenotype (e.g., flh) | ~100% (curved body axis) | Visual inspection at 24-48 hpf | N/A [61] |
| Embryo Viability Post-Injection | >80% at 24 hpf | Manual count | 80-95% [65] |
Protocol 4: High-Content Phenotypic Screening
Objective: To quantitatively assess complex morphological and functional phenotypes in F0 crispants using automated imaging and analysis.
Assay Setup:
- At the desired developmental stage (e.g., 1-5 days post-fertilization), anesthetize crispants and array them into multi-well plates (e.g., 96-well) containing E3 medium.
- For specific assays, add vital dyes, fluorescent probes, or chemical compounds.
Automated Image Acquisition:
- Use a high-content screening system equipped with objectives suitable for whole-embryo imaging.
- For whole-embryo views, acquire multiple Z-planes and combine them using a best-focus algorithm to create a completely in-focus image [63].
- For specific structures (e.g., heart, brain), use targeted imaging to first locate the area at low magnification and then acquire high-magnification images.
Image Analysis:
- Utilize high-content analysis software modules for specific phenotypic readouts:
- Angiogenesis: Measure intersegmental vessel length and number using tube formation application modules [63].
- Cardiac Function: Outline the heart as a region of interest in transgenic lines with fluorescent hearts. Use time-lapse imaging to measure changes in area or intensity over time to calculate heart rate and contractility [63].
- Tumor Burden: After xenotransplantation of fluorescent human cancer cells, measure total fluorescent area and intensity to quantify tumor size and metastasis [63].
- Neurotoxicity/Ototoxicity: Use spot-counting algorithms to quantify fluorescently-labeled neurons or hair cells [63].
- Utilize high-content analysis software modules for specific phenotypic readouts:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for F0 Crispant Screens
| Reagent / Solution | Function and Importance | Example Specifications / Notes |
|---|---|---|
| CRISPR Library | Provides pre-designed, arrayed or pooled sgRNAs targeting specific gene families or pathways. | Commercial libraries are available for various pathways. Essential for scalable screening [64]. |
| Recombinant Cas9 Protein | The core nuclease enzyme that creates double-strand breaks at DNA sites specified by the sgRNA. | High-purity, carrier-free protein ensures high editing efficiency and reduces toxicity in embryos. |
| Robotic Injection System | Automates the microinjection process, ensuring consistency and enabling high-throughput. | Can process up to 2000 embryos/hour; improves reproducibility vs. manual injection [65]. |
| High-Content Imaging System | Automates image acquisition and analysis of complex phenotypes in multi-well plates. | Systems like ImageXpress provide large field-of-view and z-stacking for whole embryos [63]. |
| Image Analysis Software | Quantifies complex morphological and functional readouts from embryo images. | Software like MetaXpress has application modules for angiogenesis, cell counting, etc. [63]. |
| Primary Cells | Biologically relevant cellular models for ex-vivo screening when zebrafish cell lines are insufficient. | Superior to cell lines for investigating cell signaling pathways [64]. |
Data Analysis and Presentation
Quantitative Data Analysis
Robust data analysis is critical for interpreting high-throughput screens. The quantitative data extracted from phenotypic analyses should be processed as follows:
- Normalization: Normalize raw data (e.g., vessel length, heart rate, cell counts) to internal controls within each plate. Common controls include wild-type embryos (negative control) and crispants with known strong phenotypes (positive control).
- Z-Score Calculation: For each target, calculate a Z-score based on the plate mean and standard deviation of negative controls:
(Target Value - Plate Mean) / Plate Standard Deviation. Targets with Z-scores exceeding a pre-defined threshold (e.g., |Z| > 2) are considered "hits." - Statistical Testing: Apply appropriate statistical tests (e.g., Student's t-test for comparisons between two groups, ANOVA for multiple groups) to assess the significance of observed phenotypic differences. Correct for multiple comparisons using methods like the Bonferroni correction or False Discovery Rate (FDR).
Effective Data Presentation
Presenting quantitative data clearly is essential for communication and decision-making. The principles of tabulation should be followed: tables should be numbered, have a clear title, and headings should be concise [66].
Table 4: Example Summary of Hypothetical F0 Screen Hits for Angiogenesis Phenotypes
| Target Gene | Mutation Efficiency (%) | Average Intersegmental Vessel Count | Z-Score vs. Wild-Type | p-value | Phenotype Category |
|---|---|---|---|---|---|
| flh (Control) | 92 | N/A | N/A | N/A | Axial patterning [61] |
| Gene A | 85 | 15.2 ± 1.1 | -4.6 | < 0.001 | Strong Inhibitor |
| Gene B | 78 | 28.5 ± 2.3 | 1.2 | 0.15 | No Effect |
| Gene C | 91 | 17.8 ± 1.5 | -3.1 | < 0.01 | Moderate Inhibitor |
| Wild-Type Pool | 0 | 26.3 ± 1.8 | 0.0 | N/A | Reference |
For visualizing trends over time (e.g., heart rate changes) or comparing distributions (e.g., vessel counts across multiple targets), use line graphs and bar charts (e.g., histograms) respectively [66]. These graphical presentations convey the essence of the data quickly and with striking visual impact, making them ideal for summarizing results for presentations and publications.
Troubleshooting and Optimization
Even with robust protocols, challenges can arise. The table below lists common issues and recommended solutions.
Table 5: Troubleshooting Guide for F0 Crispant Screens
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Low Mutation Efficiency | Poor sgRNA activity or stability; degraded Cas9 protein. | Redesign sgRNA using optimization tools [61]; test new aliquots of reagents; confirm RNP complex formation. |
| High Embryo Mortality | Injection volume/pressure too high; reagent toxicity. | Calibrate injection system to reduce volume; purify sgRNA to remove contaminants; try different Cas9 vendors. |
| Variable Penetrance of Phenotype | High genetic mosaicism in F0 generation. | Increase sgRNA/Cas9 concentration; inject at single-cell stage; analyze larger sample sizes; use phenotypic scoring system. |
| High Background in Assay | Autofluorescence; non-specific staining. | Include unstained controls; optimize probe concentration and wash steps; use spectral unmixing if available. |
| Poor Automated Image Segmentation | Low contrast; embryos in different orientations. | Use vital dyes for better contrast; employ orientation techniques in wells; adjust segmentation parameters in software. |
Maximizing Success: Troubleshooting Common Pitfalls and Enhancing Editing Efficiency
Within the context of a broader thesis on CRISPR-Cas9 gene editing in zebrafish embryos, the design and validation of single guide RNAs (sgRNAs) emerges as a fundamental determinant of experimental success. The simplicity of zebrafish embryology, characterized by high fecundity and externally developing embryos, combined with their genetic similarity to humans—sharing orthologs for over 70% of human protein-coding genes—makes them an indispensable vertebrate model for functional genomics and drug discovery [67] [68]. The core challenge, however, lies in the fact that sgRNAs exhibit wide variations in their mutagenic activity, and even sophisticated prediction algorithms cannot guarantee success [69]. Inconsistent sgRNA performance can lead to wasted resources, failed experiments, and inconclusive results, particularly in large-scale functional screens or when modeling human disease mutations. This application note provides a detailed, evidence-based framework for selecting high-efficiency sgRNAs and rigorously validating their cutting frequency, thereby ensuring reliable and reproducible genome editing outcomes in zebrafish research.
sgRNA Design Principles for Maximum Efficiency
The journey to a successful gene-editing experiment begins with the informed selection of a target sequence. Adherence to established design principles significantly increases the probability of obtaining a highly active sgRNA.
Sequence Selection Criteria
Optimal sgRNA design leverages both computational predictions and specific sequence features. The target site is immediately 5' of a Protospacer Adjacent Motif (PAM) sequence, which is 5'-NGG-3' for the commonly used Streptococcus pyogenes Cas9 [23] [70]. The 20-nucleotide guide sequence preceding the PAM should be selected based on the following criteria [23] [71]:
- 5' Guanines for Transcription: A 5'-G is strongly recommended for efficient transcription by the T7 RNA polymerase, which is commonly used for in vitro sgRNA synthesis. If the native target sequence does not begin with a G, one can be added to the 5' end, though this may slightly reduce cutting efficiency [23].
- Optimal GC Content: The guide sequence should have a GC content between 40% and 80%, with higher GC content generally correlating with more robust activity [23].
- Minimizing Off-Target Effects: The chosen sgRNA should be screened for potential off-target sites, especially within coding regions. Using design tools that minimize the number of predicted off-target sites is crucial for reducing confounding experimental results [23].
Utilizing Validated sgRNA Databases
Before designing a new sgRNA, researchers should consult CRISPRz (http://research.nhgri.nih.gov/CRISPRz/), a public database that catalogs experimentally validated sgRNAs for zebrafish [70]. This resource aggregates data from numerous published sources and large-scale mutagenesis projects, providing invaluable information on the relative mutagenic activity (% mutagenesis rate) of specific target sequences. Using a pre-validated sgRNA from CRISPRz can save significant time and resources, effectively de-risking the initial stages of project design [70].
Table 1: Key Criteria for Selecting an Optimal sgRNA Target Sequence
| Feature | Optimal Characteristic | Rationale & Notes |
|---|---|---|
| PAM Sequence | 5'-NGG-3' | Essential for Cas9 nuclease recognition and cleavage. |
| Guide Length | 20 nucleotides | Standard length for directing Cas9 to the target locus. |
| 5' Nucleotide | G (Guanine) | Critical for efficient T7 in vitro transcription. Can be added if not present. |
| GC Content | 40% - 80% | Higher GC content is generally associated with improved efficiency. |
| Off-Target Sites | Minimized | Use design tools to avoid guides with numerous predicted off-targets, especially in exons. |
Quantitative Assessment of sgRNA Cutting Efficiency
Once a candidate sgRNA is designed or selected, its functional activity must be quantitatively assessed. The following methods provide a robust framework for validation.
In Vitro Cleavage Assay
An efficient pre-screening method involves an in vitro cleavage test before moving to animal work. This assay uses a crude extract of Cas9 protein combined with the in vitro-transcribed sgRNA and a PCR-amplified genomic target region [69]. The reaction products are resolved on an agarose gel; cleavage of the PCR product into two smaller fragments indicates successful sgRNA activity. This method has demonstrated a perfect correlation with in vivo function, where all sgRNAs that failed the in vitro test also failed to induce mutations in zebrafish embryos, while those that passed consistently produced somatic mutations [69]. This step saves significant animal time and resources.
In Vivo Validation and Efficiency Quantification
Following in vitro confirmation, sgRNA efficiency must be measured in injected zebrafish embryos. The chosen method can influence the perceived efficiency, as demonstrated in a study targeting the cacna1c gene [72].
Table 2: Comparison of Methods for Quantifying Indel Frequency in Injected Embryos
| Method | Principle | Key Findings from Comparative Analysis [72] |
|---|---|---|
| CRISPR-STAT | Fluorescent PCR and capillary electrophoresis. | Correlated significantly with NGS (r=0.82-0.93) but tended to miss very small (1-2 bp) indels, leading to underestimation. |
| Inference of CRISPR Edits (ICE) | Computational analysis of Sanger sequencing traces. | Showed a strong correlation with NGS (r=0.90-0.92). More objective and accurate for small indels than CRISPR-STAT. |
| Next-Generation Sequencing (NGS) | Direct, high-depth sequencing of the target locus. | Considered the gold standard for precise indel estimation and profiling. Provides the most accurate and comprehensive data. |
The data show that while CRISPR-STAT and ICE are suitable for rapid sgRNA comparison, ICE provides more objective results and fewer errors in estimating small indels [72]. For critical quantifications, such as when determining the optimal conditions for knock-in experiments, NGS is the most reliable method.
Experimental Protocols
Protocol 1: In Vitro sgRNA Cleavage Assay
This protocol allows for rapid, cost-effective screening of sgRNA function before zebrafish injection [69].
- Cas9 Protein Preparation: Obtain commercially available Cas9 protein or purify it from a cell line (e.g., HEK293T). Aliquot and store at -80°C.
- Target Amplification: Design primers to amplify a 500-1000 bp genomic DNA fragment encompassing the sgRNA target site from wild-type zebrafish genomic DNA.
- Reaction Setup: In a nuclease-free tube, combine the following:
- 100-200 ng of purified PCR product.
- 100-200 ng of Cas9 protein.
- In vitro-transcribed sgRNA (molar ratio of sgRNA:Cas9 ~ 2:1).
- 1x Cas9 reaction buffer.
- Incubation: Incubate the reaction at 37°C for 30-60 minutes.
- Analysis: Resolve the reaction products on a 1.5-2% agarose gel. Compare to an uninjected control PCR product. Successful cleavage is indicated by the appearance of two smaller DNA bands corresponding to the predicted sizes of the cleaved fragments.
Protocol 2: Somatic Mutation Analysis in F0 Embryos by T7 Endonuclease I (T7EI) Assay
This protocol provides a method for assessing editing efficiency in a pool of injected embryos [23].
- DNA Extraction: At 1-2 days post-fertilization (dpf), pool 10-20 injected embryos and extract genomic DNA using a standard lysis protocol (e.g., HotShot method or commercial kit).
- PCR Amplification: Design primers flanking the target site to generate an amplicon of 200-500 bp. Perform PCR using a high-fidelity polymerase.
- Heteroduplex Formation: Purify the PCR product. Denature and reanneal it using a thermal cycler program: 95°C for 5 minutes, ramp down to 85°C at -2°C/second, then ramp down to 25°C at -0.1°C/second. This allows the formation of heteroduplexes between wild-type and mutated DNA strands.
- T7EI Digestion: Digest the reannealed PCR product with T7 Endonuclease I, which cleaves at mismatches in heteroduplex DNA.
- Visualization: Analyze the digestion products by agarose gel electrophoresis. The percentage of indel mutations can be estimated by comparing the band intensities of the cleaved and uncut PCR products.
Protocol 3: High-Accuracy Genotyping by Next-Generation Sequencing (NGS)
For precise quantification of editing efficiency and indel profiling, follow this NGS-based protocol [72] [23].
- Sample Preparation: Extract genomic DNA from individual or pooled embryos at the desired stage. For early selection, the Zebrafish Embryo Genotyper (ZEG) device can be used for minimal-invasive DNA extraction at 72 hpf [72].
- Target Amplification for NGS: Perform a two-step PCR. The first PCR uses gene-specific primers with overhangs containing Illumina adapter sequences. The second PCR adds full adapter sequences and sample-specific barcodes (indexes) to allow for multiplexing.
- Library Quantification and Sequencing: Pool the barcoded amplicons in equimolar ratios and sequence on an Illumina MiSeq or similar platform to achieve high coverage (>10,000x reads per sample).
- Data Analysis: Process the sequencing data using specialized software such as CRISPResso2 or a custom pipeline to align reads to the reference sequence and precisely quantify the spectrum and frequency of induced indels [72] [67].
Diagram 1: sgRNA Design and Validation Workflow. This flowchart outlines the stepwise process from initial guide selection to final germline transmission screening.
Advanced Applications: From Knockouts to Precise Knock-Ins
The principles of sgRNA validation are especially critical for advanced genome engineering applications beyond simple knockouts.
F0 Crispant Screening
A powerful application of highly efficient sgRNAs is the use of F0 mosaic founder zebrafish, or "crispants," for rapid phenotypic screening. Studies have shown that with high indel efficiencies (e.g., >70-88%), F0 crispants can faithfully recapitulate the phenotypes of stable germline mutants, enabling functional gene validation in a fraction of the time [67]. This approach is highly dependent on using sgRNAs with validated high activity to ensure a high proportion of mutated cells.
Improving Knock-In Efficiency
Precise knock-in via Homology-Directed Repair (HDR) has traditionally been inefficient in zebrafish. Key factors for improvement include:
- Using Cas9 Protein: Ribonucleoprotein (RNP) complexes with Cas9 protein, rather than Cas9 mRNA, significantly increase both indel and HDR efficiency [72] [23] [71].
- Optimal Repair Template: Long single-stranded DNA (lssDNA) templates, such as those used in the zLOST method, have been shown to dramatically improve HDR efficiency, with one study reporting phenotypic rescue in close to 98% of injected embryos for a visible marker [44].
- Template Conformation: For ssODN templates, a non-target asymmetric PAM-distal (NAD) conformation combined with Cas9 protein was shown to significantly outperform other configurations [72].
Diagram 2: Key Factors for Successful Zebrafish Genome Editing. The conceptual relationship between critical experimental choices that determine the outcome of CRISPR experiments.
Table 3: Key Research Reagent Solutions for Zebrafish CRISPR/Cas9 Work
| Reagent / Resource | Function / Description | Examples & Notes |
|---|---|---|
| Cas9 Nuclease | Engineered Cas9 protein for RNP complex formation. | Commercial suppliers provide high-purity, ready-to-use Cas9 (e.g., GenCRISPR NLS-wtSpCas9). Using protein over mRNA increases efficiency and reduces mosaicism [23] [71]. |
| Synthetic sgRNA | Chemically synthesized, high-purity guide RNA. | Companies like GenScript offer modified sgRNAs (EasyEdit, SafeEdit) with improved stability and reduced off-target effects compared to in vitro transcribed (IVT) sgRNAs [73]. |
| Validated sgRNA Database | Public repository of tested sgRNA sequences. | CRISPRz provides a curated list of sgRNAs with known mutagenic efficiency, saving design and validation time [70]. |
| Homology-Directed Repair (HDR) Templates | Donor DNA for precise knock-in of sequences. | Long ssDNA (lssDNA) templates (e.g., zLOST method) show superior HDR efficiency compared to short ssODNs or dsDNA [44]. |
| Genotyping Tools | Software and assays for quantifying editing. | CRISPResso2 for NGS data analysis; ICE (Synthego) for Sanger sequencing analysis; T7 Endonuclease I assay for quick confirmation [72] [67]. |
The application of CRISPR-Cas9 technology in zebrafish research has revolutionized our ability to model human diseases and understand gene function. A critical component of this system is homology-directed repair (HDR), which enables precise genome modifications when coupled with an exogenous donor template. The design of this donor template, particularly the length of homology arms flanking the desired insertion, is a primary determinant of knock-in efficiency. This application note synthesizes current research to provide optimized protocols for designing both single-stranded oligodeoxynucleotide (ssODN) and plasmid-based donor templates, specifically within the context of zebrafish embryo research.
The fundamental challenge in zebrafish genome engineering lies in the relatively low efficiency of HDR compared to the dominant non-homologous end joining (NHEJ) pathway [32] [44]. While double-strand breaks induced by CRISPR-Cas9 are highly efficient, the precise incorporation of exogenous DNA sequences via HDR requires optimization of multiple parameters, with homology arm design being paramount. Research indicates that HDR efficiency is strongly determined by repair-template composition, with homology-arm length being one of the most influential factors [32].
Homology Arm Length Guidelines for Different Donor Types
The optimal length for homology arms is intrinsically linked to the type of donor template used. The choice between ssODNs and plasmid-based donors is typically governed by the size of the intended insertion.
Table 1: Recommended Homology Arm Lengths by Donor Type
| Donor Template Type | Insert Size | Recommended Homology Arm Length | Key References & Evidence |
|---|---|---|---|
| ssODN | Short inserts (< 50 bp), point mutations | 30–60 nt [74], with 40 nt being a common starting point [51]. | IDT recommendations; empirical testing in Jurkat and HAP1 cells showing functional HDR [51] [74]. |
| Long ssDNA (lssDNA) | ~200 bp to > 500 bp | 50–300 nt; can be asymmetric. A shorter 3' arm (50 nt) outperformed a longer one (300 nt) at some loci [75]. | Zebrafish studies at sox3 and pax6a loci; zLOST method showed high efficiency [44] [75]. |
| Plasmid (dsDNA) | Large fragments (> 1 kb), e.g., fluorescent reporters | At least 500 bp [74]; often 800 bp to 1 kb per arm for traditional gene targeting vectors [76]. | Historical gene targeting data; design considerations for large inserts where efficiency drops significantly [74] [76]. |
Special Considerations for ssODN Design
Single-stranded oligodeoxynucleotides are the donor of choice for introducing point mutations and short tags. Beyond arm length, several factors are critical for success:
- Strand Preference: The choice of which strand to use as the donor (the "target" strand bound by the gRNA or the "non-target" strand containing the PAM) can impact efficiency. While one study in mammalian cells found strand preference to be locus-dependent [51], another in zebrafish found that anti-sense asymmetric oligo design achieved around 2% HDR efficiency [44]. Testing both orientations is recommended if initial efficiency is low.
- Blocking Mutations: To prevent re-cleavage of the successfully modified locus by Cas9, incorporate silent "blocking" mutations in the PAM sequence or the seed region of the protospacer within the donor template [51]. This ensures the modified sequence is no longer recognized by the gRNA, enriching for correctly edited cells.
- Placement of the Edit: The intended modification should be placed as close as possible to the Cas9 cut site (within 10 bp is ideal), as HDR efficiency drops significantly for edits distal to the break [76].
The Rise of Long ssDNA Donors
Recent advances have demonstrated that long single-stranded DNA (lssDNA) donors offer a superior alternative for knocking in sequences of a few hundred base pairs, such as composite tags or small genes. The zLOST (zebrafish long single-stranded DNA template) method reported a dramatic increase in HDR efficiency, with phenotypic rescue rates jumping from approximately 5% with other donors to 98.5% at the tyr locus, albeit with varying numbers of rescued cells [44]. A key finding with lssDNA donors is the potential for asymmetric arm design. For example, when knocking a composite tag into the sox3 and pax6a genes, a shorter 50 nt 3' homology arm resulted in a higher knock-in efficiency than a longer 300 nt arm [75]. This suggests that the optimal configuration may be locus-specific and should be empirically tested.
Table 2: Comparative Efficiency of Different Donor Templates in Zebrafish
| Donor Template | Model System | Key Finding | Reported Efficiency |
|---|---|---|---|
| Asymmetric ssODN | Zebrafish embryos | Precise HDR knock-in assessed by high-throughput sequencing. | ~2% [44] |
| Plasmid dsDNA (with dual gRNAs) | Zebrafish embryos | HDR-mediated integration of a 5.5-kb CAG-GFP vector into the Rosa26 locus. | Achieved via ssODN-mediated end joining [77] |
| lssDNA (zLOST) | Zebrafish tyr mutant | Phenotypic rescue via HDR-mediated correction of a mutation. | Up to 98.5% (somatic); Germline transmission up to 31.8% [44] |
| lssDNA (Composite Tag) | Zebrafish sox3, sox11a, pax6a | Precise knock-in of ~200 bp composite tag with optimized lssDNA design. | Germline transmission rates as high as 21% [75] |
Detailed Experimental Protocols
Protocol 1: Knocking-in Point Mutations with ssODNs
This protocol is adapted from methods used to introduce point mutations in zebrafish and other model organisms [51] [78].
Reagents and Materials:
- CRISPR-Cas9 Components: Cas9 protein or mRNA; target-specific gRNA.
- Donor Template: ssODN with 30-60 nt homology arms, designed with silent blocking mutations.
- Injection Buffer: 0.5X Tango buffer or Danieau buffer.
- Zebrafish: Wild-type or Casper zebrafish embryos at the one-cell stage.
Procedure:
- Design and Obtain Donor ssODN:
- Design an ssODN with your desired mutation (e.g., a point mutation) flanked by homology arms of 40 nt.
- Modify the PAM sequence in the donor (e.g.,
NGGtoNGC) to create a blocking mutation. - Order the ssODN from a commercial supplier with phosphorothioate modifications at the terminal 2-3 bases of each end to enhance stability.
Prepare the Injection Mix:
- Prepare a working injection mix containing:
- 150-300 ng/μL of Cas9 protein complexed with gRNA (as an RNP) OR 100-200 ng/μL of Cas9 mRNA + 20-50 ng/μL of gRNA.
- 100-200 ng/μL of the purified ssODN donor template.
- Centrifuge the mixture briefly and keep it on ice until injection.
- Prepare a working injection mix containing:
Microinjection into Zebrafish Embryos:
- Load the injection mix into a needle and backfill with immersion oil.
- Inject approximately 1 nL of the mixture directly into the cytoplasm or cell yolk of one-cell stage zebrafish embryos.
- Raise the injected embryos (F0) to adulthood. These are potential founders.
Screening and Validation:
- At 1-3 days post-fertilization (dpf), screen a subset of F0 embryos for the desired edit using a restriction fragment length polymorphism (RFLP) assay or PCR followed by Sanger sequencing.
- Raise mosaic F0 founder fish to adulthood and outcross them to wild-type fish.
- Screen the resulting F1 progeny for germline transmission of the point mutation by sequencing tail-clip genomic DNA.
Protocol 2: Knocking-in Large Inserts with lssDNA Donors
This protocol is based on the highly efficient zLOST and related methods [44] [75].
Reagents and Materials:
- CRISPR-Cas9 Components: Cas9 protein; two target-specific gRNAs (one for the genomic locus, one for the donor, if using a universal gRNA strategy).
- Donor Template: lssDNA (200-2000 nt) containing the insert and homology arms (e.g., 50-300 nt).
- Injection Buffer: As in Protocol 1.
Procedure:
- Design and Synthesis of lssDNA Donor:
- Design a double-stranded DNA fragment containing your insert (e.g., a GFP cassette) flanked by homology arms of 48-300 nt. Incorporate a universal gRNA (UgRNA) target site flanking the 5' and 3' homology arms to liberate the homology arms in vivo [79].
- Generate the lssDNA donor using PCR-based methods followed by bead-based purification or order it from a commercial supplier.
Prepare the Injection Mix:
- Assemble a ribonucleoprotein (RNP) complex by pre-incubating Cas9 protein with both the genomic-targeting gRNA and the UgRNA.
- Combine the RNP complex with the lssDNA donor (final concentration 50-100 ng/μL) in injection buffer.
Microinjection and Screening:
- Inject the mixture into one-cell stage zebrafish embryos as described in Protocol 1.
- For reporters like GFP, screen injected F0 embryos for somatic expression at the appropriate developmental stage.
- Raise reporter-positive F0 embryos to adulthood. The high efficiency of lssDNA methods increases the probability of germline transmission.
- Outcross F0 founders and screen F1 progeny for stable integration of the knock-in.
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagent Solutions for CRISPR Knock-ins
| Reagent / Solution | Function | Example & Notes |
|---|---|---|
| CRISPR-Cas9 Nuclease | Induces a targeted double-strand break in the genome. | S.p. Cas9 is most common; Cas12a is an alternative for AT-rich regions [51]. |
| Guide RNA (gRNA) | Directs the Cas nuclease to the specific genomic target. | Can be a single guide RNA (sgRNA) or a complex of crRNA and tracrRNA [51]. |
| HDR Donor Template | Serves as the repair template for precise integration. | ssODN: For point mutations/short inserts. lssDNA: For tags/small genes. Plasmid dsDNA: For large inserts [44] [76]. |
| Universal Guide RNA (UgRNA) | A standardized, highly efficient gRNA sequence used to linearize the donor template in vivo, exposing homology arms. | Simplifies donor design and improves knock-in efficiency via the HMEJ pathway [79]. |
| Microinjection Apparatus | For the precise delivery of editing components into zebrafish embryos. | Includes a micromanipulator, injector, and needles. |
| Nucleofection System | For the delivery of editing components into mammalian cells (for validation studies). | Used in studies optimizing HDR in cell lines like Jurkat and HAP1 [51]. |
Workflow and Decision Diagram
The following diagram illustrates the logical workflow for selecting the appropriate donor template and designing homology arms based on the intended genetic modification.
Optimizing the length of homology arms in donor templates is a critical step in designing efficient CRISPR-Cas9 knock-in experiments in zebrafish. The prevailing data indicates a clear distinction between donor types: short homology arms (30-60 nt) are optimal for ssODNs, while longer arms (≥ 500 bp) are necessary for plasmid-based donors. The emergence of lssDNA donors with homology arms of 48-300 nt presents a highly efficient option for a wide range of applications, from inserting composite tags to correcting disease-associated mutations. By following the guidelines and detailed protocols outlined in this document, researchers can systematically enhance the precision and efficiency of their genome engineering efforts in zebrafish, thereby accelerating functional genomics and disease modeling studies.
Early-Stage Embryo Injection: The Critical Importance of the One-Cell Stage for Germline Transmission
The generation of stable, genetically modified zebrafish lines is a cornerstone of developmental biology and biomedical research. Achieving high rates of germline transmission is the critical step that separates transient somatic editing from heritable genetic models. This application note delineates the foundational protocol and underlying principles for microinjecting CRISPR-Cas9 components into the one-cell stage zebrafish embryo, a method demonstrated to be paramount for maximizing the efficiency of germline transmission. We present quantitative evidence that this approach, when optimized with ribonucleoprotein (RNP) complexes and long single-stranded DNA (lssDNA) donors, can yield germline transmission rates as high as 21-28% [75] [80]. Furthermore, we detail the supporting methodologies for genotyping and validation, providing a complete workflow for researchers aiming to accelerate the creation of precise knock-in and knockout models.
The zebrafish (Danio rerio) has emerged as a preeminent model organism for validating candidate human disease genes due to its high genetic conservation, external development, and fecundity [80]. The advent of CRISPR-Cas9 technology has revolutionized genetic engineering in this model, shifting the paradigm from random mutagenesis to precise, targeted genome modifications.
A fundamental challenge in this process is mosaicism, a condition where the founder generation (F0) embryo contains a mixture of cells with different genetic alterations. This occurs when the CRISPR-Cas9 system acts after the zygote has already undergone one or more rounds of cell division. Mosaicism severely complicates the recovery of mutant alleles in the next generation (F1), as the edited genetic material may not be incorporated into the germline cells [81].
Injection at the one-cell stage is the most effective strategy to circumvent this problem. By introducing the CRISPR machinery at the earliest possible opportunity, the likelihood that the editing event will be replicated in all subsequent cells, including the primordial germ cells, is vastly increased. This principle is supported by studies across species; for instance, research in human and non-human primate embryos has shown that the timing of CRISPR-Cas9 delivery is critical, with co-injection with sperm into M-phase oocytes resulting in significantly reduced mosaicism [81]. In zebrafish, streamlined protocols for microinjection at the one-cell stage have been established as the gold standard for generating heritable mutations [42] [80]. The subsequent sections of this note will dissect the experimental protocol, present quantitative data on efficiency, and provide a toolkit for successful implementation.
Experimental Protocol: One-Cell Stage Microinjection and Knock-In Strategy
Critical Reagents and Equipment
The following table catalogues the essential materials required for the preparation of injection samples and the microinjection procedure itself.
Table 1: Research Reagent Solutions and Essential Materials
| Item | Function/Description | Key Considerations |
|---|---|---|
| Cas9 Protein | Recombinant Cas9 nuclease that forms the core of the RNP complex. | Using protein, rather than mRNA, leads to faster activation and degradation, reducing off-target effects and mosaicism [81]. |
| Target-Specific gRNA | Guide RNA that directs Cas9 to the specific genomic locus. | Can be synthesized via a cloning-free, high-throughput method using overlapping oligonucleotides [80]. |
| Long ssDNA (lssDNA) Donor | Single-stranded DNA template for HDR-mediated precise knock-in. | Superior to double-stranded DNA (dsDNA) templates due to lower cytotoxicity and higher integration specificity [75]. |
| Microinjection Apparatus | System including a micropipette puller, microscope, and microinjector. | Essential for the precise delivery of nanoliter volumes into the one-cell embryo [42]. |
| Zebrafish Embryos | Wild-type or genetically defined strain embryos. | Must be collected within 20-30 minutes post-fertilization to ensure injection at the one-cell stage [42]. |
Step-by-Step Workflow for Injection and Screening
The workflow for generating germline-transmitting zebrafish involves a sequence of optimized steps, from embryo preparation to the identification of positive founders. The following diagram outlines this comprehensive process.
Detailed Protocol:
Embryo Collection and Preparation: Collect freshly fertilized zebrafish embryos and align them on an agarose plate submerged in embryo medium using a transfer pipette. The one-cell stage is identifiable by a single, large cell. Injection should be performed before the first cleavage division [42].
CRISPR-Cas9 RNP Complex Formation:
- For Knock-Outs: Synthesize sgRNA as described in [80]. Mix the sgRNA with recombinant Cas9 protein to form the RNP complex. Incubate at 37°C for 10 minutes to allow complex formation. A typical injection mixture might contain 300 ng/μL Cas9 protein and 30-100 ng/μL sgRNA [42].
- For Precise Knock-Ins: Include a long ssDNA (lssDNA) donor template in the injection mix. Key design parameters for the lssDNA donor include:
- Homology Arm Length: A 50-nucleotide (nt) 3' homology arm can yield higher knock-in efficiency than a 300-nt arm for certain loci (e.g., sox3, pax6a) [75].
- Strand Selection: The choice of the "target" or "non-target" lssDNA strand can significantly impact efficiency and is locus-dependent [75].
- Insertion Site: The distance between the Cas9 cleavage site and the tag insertion site should be minimized to ensure precise editing [75].
Microinjection: Load the injection mixture into a fine glass needle. Using a micromanipulator, carefully puncture the chorion and inject the mixture directly into the cytoplasm of the one-cell embryo. The volume delivered is typically 1-2 nL, containing the RNP complex with or without the donor template [42].
Post-Injection Culture and Screening: After injection, remove damaged embryos and incubate the survivors at 28.5°C. Raise the injected embryos (F0 founders) to sexual maturity. Outcross each F0 adult to wild-type fish. To screen for germline transmission, genitely screen the resulting F1 progeny. Efficient methods include:
- Fluorescence PCR: A rapid method for identifying insertions or deletions (indels) [80].
- High-Throughput Sequencing: Allows for the verification of a large number of unique alleles and precise knock-in events [75] [80].
- Junction PCR: For knock-ins, allele-specific qPCR for both the 5' and 3' junctions can confirm precise integration [75].
Quantitative Data and Efficiency Analysis
The optimization of injection parameters and donor design directly translates to quantifiable improvements in editing efficiency. The data below summarize key findings from the literature.
Table 2: Quantitative Data on Knock-In and Germline Transmission Efficiency
| Target Gene | Donor Template & Strategy | Key Optimized Parameter | Reported Efficiency | Source |
|---|---|---|---|---|
| sox3, sox11a, pax6a | lssDNA with composite tag | lssDNA strand selection; 50-nt 3' homology arm | Precise knock-in with germline transmission rates as high as 21% | [75] |
| 83 genes (162 loci) | CRISPR-Cas9 RNP for KO | High-throughput sgRNA synthesis; one-cell injection | Average germline transmission rate of 28% (99% mutation success) | [80] |
| Ybx1 | HDR-mediated point mutation | Use of Cas9 protein + HDR stimulation | Germline transmission efficiency up to 25% | [82] |
The underlying molecular mechanisms that make one-cell injection so critical are tied to the DNA repair processes in the early embryo. The following diagram illustrates the competitive repair pathways activated after a CRISPR-Cas9-induced double-strand break (DSB) and how the presence of a donor template at the one-cell stage favors precise editing.
The Scientist's Toolkit
Beyond the core protocol, several reagents and strategies are pivotal for success.
Table 3: Advanced Reagent Solutions for Precision Editing
| Tool | Application | Mechanism and Benefit |
|---|---|---|
| lssDNA Donor | Precise insertion of epitope tags, point mutations. | Serves as a repair template for HDR. Demonstrates higher precision and lower toxicity compared to dsDNA donors [75]. |
| Composite Tags (e.g., FLAG-Bio-HiBiT) | Protein detection, purification, and functional analysis. | Combines multiple tags to increase functionality and circumvent antibody cross-reactivity issues, especially in gene families like Sox and Pax [75]. |
| Tissue-Specific Cas9 Vectors | Somatic cell-specific gene disruption. | Allows for spatial control of gene knockout by driving Cas9 expression with a tissue-specific promoter, broadening loss-of-function studies [83]. |
| Chicken U6 (cU6.3) Promoter | Enhanced gRNA expression in chick models. | An example of species-specific optimization; provides 4-fold higher gRNA expression than a human U6 promoter, improving knockout efficiency [84]. |
The data and protocols presented herein unequivocally establish microinjection at the one-cell stage as a non-negotiable best practice for maximizing germline transmission in zebrafish CRISPR-Cas9 experiments. The combination of this temporal precision with biochemical optimizations—such as the use of RNP complexes and lssDNA donor templates—creates a robust framework for efficient genome engineering.
The quantitative evidence demonstrates that this optimized approach consistently achieves germline transmission efficiencies exceeding 20%, a critical threshold for making high-throughput mutagenesis and phenotyping projects feasible [75] [80]. Furthermore, the ability to phenotype in the F1 generation by inbreeding founder fish can reduce the timeline for mutant characterization by an entire generation (approximately 3-4 months), offering a significant acceleration for research pipelines [80].
In conclusion, the meticulous application of the principles outlined in this application note—embryo timing, reagent quality, and donor design—empowers researchers to reliably generate stable zebrafish lines. This capability is fundamental for advancing our understanding of gene function and for modeling human diseases with high fidelity.
The generation of precise knock-in zebrafish models using CRISPR-Cas9 represents a powerful approach for functional genomics and human disease modeling. However, the low efficiency of homology-directed repair (HDR) in zebrafish poses significant challenges for identifying founder fish carrying precise edits. This application note details robust screening methodologies that combine fluorescent PCR-based techniques with ICE (Inference of CRISPR Edits) analysis and allele-specific principles to efficiently detect precise integration of epitope tags and point mutations. Our optimized pipeline enables researchers to overcome the bottleneck of traditional screening methods, facilitating the reliable establishment of stable zebrafish lines with customized genetic modifications. The protocols described herein have been successfully implemented for inserting epitope tags at defined loci and recapitulating human disease-associated point mutations, with germline transmission rates of 1-5% achievable through systematic screening of fewer than 12 founder fish per gene.
CRISPR-Cas9 has revolutionized genome editing in zebrafish, enabling the generation of targeted knockout models through error-prone non-homologous end joining (NHEJ). However, the creation of precise knock-in models via homology-directed repair remains challenging due to the inherent inefficiency of HDR in zebrafish embryos [85]. A significant bottleneck in this process is identifying the rare founder fish that transmit precisely integrated sequences amid a background of random indels and complex repair outcomes [85] [86].
Traditional screening methods, including cloning and sequencing of numerous clones or next-generation sequencing of pooled embryos, are often cost-prohibitive and labor-intensive for many laboratories [85]. While computational tools like ICE analysis can infer editing efficiency from Sanger sequencing data, they require high-quality sequence reads and may miss low-abundance edits in mosaic founders [85]. Similarly, conventional allele-specific PCR is difficult to scale and standardize across multiple targets [85].
This application note addresses these challenges by presenting optimized screening strategies that combine the sensitivity of fluorescent detection with the analytical power of capillary electrophoresis and ICE analysis. Built upon established principles of allele-specific recognition, these methods provide a robust framework for identifying precise edits in both somatic and germline tissues of zebrafish founders [85] [86]. The protocols are specifically tailored for the context of a broader thesis on CRISPR-Cas9 gene editing in zebrafish embryos, with emphasis on practical implementation for research and drug development applications.
Key Concepts and Definitions
Foundational Techniques
Table 1: Core Methodologies for Precise Edit Detection
| Technique | Principle | Application in Knock-in Screening | Key Advantage |
|---|---|---|---|
| Allele-Specific PCR | Selective amplification based on perfect primer-template matching at 3' end | Distinguishes precise knock-in alleles from wildtype and NHEJ-induced indels [85] | High specificity for single-nucleotide changes |
| ICE (Inference of CRISPR Edits) Analysis | Computational decomposition of Sanger sequencing chromatograms | Quantifies editing efficiency and characterizes mutation spectra [85] | Works with standard Sanger sequencing data |
| Fluorescent PCR & Capillary Electrophoresis | Fluorescence-labeled primers with size-based separation via capillary electrophoresis | Detects size variations in PCR products; enables multiplexing [85] [86] | High sensitivity and quantitative capabilities |
| CRISPR-STAT (Somatic Tissue Activity Test) | Fluorescent PCR analysis of somatic edits in embryo tissues | Rapid validation of sgRNA and repair template efficiency [85] [86] | Early-stage assessment before germline screening |
The following diagram illustrates the comprehensive workflow for founder screening, integrating both somatic and germline analysis phases:
Research Reagent Solutions
Table 2: Essential Reagents and Materials for Founder Screening
| Category | Specific Reagents/Items | Function/Application | Implementation Notes |
|---|---|---|---|
| CRISPR Components | sgRNA synthesis reagents (HiScribe T7 Kit) [86], Cas9 protein/mRNA, pT3TS-nls-zCas9-nls plasmid [86] | Generation of editing machinery | Use highly active sgRNAs validated by CRISPR-STAT |
| Repair Templates | Single-stranded oligodeoxynucleotides (ssODNs) with asymmetric homology arms [85] [86] | HDR template for precise edits | 40-60 nt homology arms; include blocking mutations in PAM |
| PCR Reagents | Fluorescently labeled primers (FAM, HEX), M13F-FAM primer-/56-FAM/TGTAAAACGACGGCCAGT [86], high-fidelity DNA polymerase | Amplification of target loci | Use M13-tailed forward and pig-tailed reverse primers |
| Electrophoresis & Analysis | Capillary sequencer (3730xl DNA Analyzer), GeneScan 400HD ROX size standard [86], Hi-Di formamide [86] | Fragment size separation and detection | Enables single-base pair resolution |
| DNA Processing | Restriction enzymes (NEB) with appropriate buffers [86], DNA extraction solutions (Extraction, Tissue Preparation, Neutralization) [86] | Digestion and preparation of genomic DNA | Critical for RFLP-based screening of point mutations |
| Software & Analysis Tools | ICE CRISPR Analysis Tool (Synthego), TIDER, Primer3 [86] | Computational assessment of editing efficiency | Free online tools for design and analysis |
Comprehensive Protocols
Phase 1: Somatic Screening in Injected Embryos
This initial phase allows rapid assessment of editing efficiency before investing in raising fish to adulthood.
sgRNA Design and Validation
- Design: Select target sites within 20 bp of the desired edit location using tools like Benchling or Synthego CRISPR Design Tool [87]. For epitope tag insertion, target regions immediately before the stop codon; for point mutations, position the cut site as close as possible to the target nucleotide.
- Validation: Assess sgRNA activity by CRISPR-STAT:
- Inject sgRNA/Cas9 complex into 20-30 embryos
- Extract genomic DNA from pooled 1 dpf embryos
- Perform fluorescent PCR with M13-tailed primers
- Analyze by capillary electrophoresis
- Select sgRNAs inducing >50% indels in somatic tissue [85]
ssODN Repair Template Design
- Homology Arms: Incorporate asymmetric homology arms (40-60 nucleotides total) with the longer arm on the side with more sequence complexity [86].
- Modification Elements:
- For epitope tags: Insert tag sequence in-frame with surrounding coding sequence
- For point mutations: Include desired nucleotide change + silent mutation to destroy PAM sequence (e.g., NGG → NCG) [85]
- Optional: Introduce novel restriction site for secondary screening validation
- Synthesis: Order as ultramers (100 nt+) with phosphorothioate modifications at ends to enhance stability [86].
Microinjection and Embryo Analysis
- Injection Mix Preparation:
- Microinjection: Inject 1-2 nL into cell cytoplasm of 1-cell stage zebrafish embryos.
- Somatic Screening:
- At 1 dpf, collect 8-10 injected embryos for DNA extraction
- Perform fluorescent PCR with knock-in screening primers
- Analyze by capillary electrophoresis for expected size shifts
- Compare with sgRNA-only controls to confirm HDR-specific enrichment [85]
Phase 2: Germline Transmission Screening
This critical phase identifies founders that transmit precise edits to the next generation.
Founder Selection and Fin Biopsy
- Raise injected embryos (G0) to adulthood (approximately 3 months).
- Perform fin clip biopsies on anesthetized fish, preserving each sample separately in 95% ethanol or DNA extraction buffer.
- Extract genomic DNA using commercial kits or established protocols [86].
Screening by Fluorescent PCR and Capillary Electrophoresis
The screening approach varies based on the type of edit being detected, as illustrated in the following genotyping strategy diagram:
For Epitope Tag Insertion:
- Perform fluorescent PCR using primers flanking the insertion site
- Prepare samples for capillary electrophoresis:
- 1 μL PCR product + 9 μL Hi-Di formamide + 0.5 μL GeneScan 400HD ROX size standard [86]
- Run on capillary sequencer and analyze for expected size increase corresponding to tag insertion
- Expected outcomes: Wildtype (single peak), precise knock-in (peak at WT size + insertion length), indels (multiple variant peaks) [85]
For Point Mutations:
- Perform fluorescent PCR with primers introducing a restriction site difference
- Digest PCR products with appropriate restriction enzyme (2-4 hours, manufacturer's recommended conditions)
- Analyze digests by capillary electrophoresis as above
- Expected outcomes: Wildtype (specific fragment pattern), precise knock-in (altered fragment pattern due to gained/lost restriction site) [85] [86]
ICE Analysis for Mutation Characterization
- PCR and Sequencing: Amplify target region with standard primers and submit for Sanger sequencing.
- ICE Analysis:
- Upload sequencing chromatograms (.ab1 files) to ICE analysis tool (e.g., Synthego ICE)
- Compare with reference sequence and uninjected controls
- Determine precise editing efficiency and identify specific mutations
- Threshold for positive founder: >5% precise knock-in sequence in background of indels [85]
Quantitative Data and Expected Outcomes
Table 3: Expected Efficiency Metrics for Knock-in Screening
| Parameter | Epitope Tag Insertion | Point Mutation | Measurement Method |
|---|---|---|---|
| Somatic Editing Rate | 15-25% of injected embryos (n=40) show expected size peak [85] | 10-20% of injected embryos (n=40) show expected size peak [85] | CRISPR-STAT capillary electrophoresis |
| Germline Transmission Rate | 1-5% of raised founders [85] | 1-5% of raised founders [85] | Germline screening of F1 progeny |
| Precise Integration Rate | ~30% of edited alleles (5/15 clones sequenced) [85] | ~25% of edited alleles [85] | Clone sequencing validation |
| Founders to Screen | ≤12 founders per gene to achieve positive line [85] | ≤12 founders per gene to achieve positive line [85] | Statistical estimation |
| False Positive Rate | <5% (1/26 embryos in sgRNA-only control) [85] | <5% with restriction digest confirmation [85] | Control comparisons |
Troubleshooting and Optimization
Common Challenges and Solutions
- Low HDR Efficiency: Increase ssODN concentration (up to 300 ng/μL); optimize homology arm length; use high-fidelity Cas9 variants.
- High Background Indels: Lower sgRNA concentration to reduce NHEJ competition; use Cas9 ribonucleoprotein complexes for shorter activity windows.
- Poor Primer Specificity: Redesign with stricter parameters; optimize annealing temperature gradients; include positive and negative controls.
- Weak Capillary Electrophoresis Signals: Increase PCR cycles (up to 35); check fluorescent primer labeling efficiency; concentrate DNA samples.
Validation Strategies
- Sequential Confirmation: Combine size-based screening with restriction digest and ICE analysis for multi-layered validation.
- Clone Sequencing: Gel-purify PCR products and clone into sequencing vectors for analysis of 10-20 clones per positive founder.
- Functional Validation: For epitope-tagged lines, confirm protein expression and localization via immunohistochemistry or Western blot.
The integration of allele-specific principles with fluorescent PCR detection and ICE analysis provides a robust framework for identifying precise CRISPR-Cas9 edits in zebrafish founders. This comprehensive pipeline addresses the critical bottleneck in knock-in generation by enabling efficient screening of both somatic and germline events. The methodologies outlined herein have been successfully implemented for the establishment of stable zebrafish lines with customized genetic modifications, supporting advanced research in functional genomics and human disease modeling. Through systematic application of these protocols, researchers can significantly enhance their capability to generate precise genetic models in zebrafish, accelerating discoveries in basic biology and therapeutic development.
The CRISPR/Cas9 system has revolutionized functional genomics in zebrafish (Danio rerio), enabling rapid generation of knockout models for studying human diseases and developmental processes [88]. This model organism offers exceptional advantages for high-throughput genetic screens, including external fertilization, high fecundity, embryonic transparency, and rapid development [88] [4]. However, a significant challenge persists beyond intended gene knockouts: unintended on-target effects comprising complex rearrangements and large genomic deletions. These alterations can confound phenotypic interpretations and compromise experimental validity, necessitating robust detection and mitigation strategies.
While off-target effects occurring at genomic sites with sequence similarity to the guide RNA have received considerable attention, on-target effects at the intended cleavage site present a more insidious challenge. The repair of CRISPR/Cas9-induced double-strand breaks via error-prone non-homologous end joining (NHEJ) can generate not only simple indels but also complex chromosomal rearrangements, large deletions, and inversions. In zebrafish models, where G0 mosaic mutants are increasingly used for rapid functional assessment, these unintended on-target events can create misleading phenotypes if not properly characterized [4]. This application note details comprehensive assays and strategic approaches to detect, quantify, and avoid these complex rearrangement events in zebrafish embryo research.
Understanding and Detecting Complex On-Target Rearrangements
Experimental Approaches for Detection
Effective detection of complex rearrangements requires moving beyond standard genotyping techniques. While simple indels can be detected using methods like polyacrylamide gel electrophoresis (PAGE) or T7 endonuclease I assays, these approaches often miss larger structural variants [4]. Advanced methodologies provide more comprehensive detection capabilities:
High-Resolution Melting (HRM) Analysis: This technique offers a rapid, sensitive approach for identifying CRISPR-induced mutations, including complex mosaic patterns. The process involves extracting genomic DNA via HotSHOT method, performing EvaGreen-based PCR amplification of the target locus, and analyzing melting curves with high temperature resolution (0.02°C per second) [43]. HRM can detect heteroduplex formation resulting from multiple mutant alleles in mosaic embryos, with irregular melting profiles indicating mutation diversity. This method successfully identifies mutations as early as the 2-cell stage in zebrafish embryos, enabling rapid assessment of editing efficiency and mutation complexity [43].
Next-Generation Sequencing (NGS): For comprehensive characterization of on-target effects, Illumina sequencing of amplified target regions provides quantitative assessment of indel spectra and frequency. This approach enables detection of complex mutation patterns that might be missed by Sanger sequencing or decomposition tools [4]. The CrispRVariants software package facilitates analysis of these sequencing data, quantifying the proportion of reads carrying different indel alleles and providing a more accurate efficiency score than Sanger-based methods [4].
Long-Range PCR and Electrophoretic Separation: Large deletions spanning hundreds to thousands of base pairs can be detected through long-range PCR amplification followed by agarose gel electrophoresis. Significantly larger or smaller amplicons than the wild-type product indicate substantial deletions or rearrangements. This approach is particularly valuable for identifying promoter deletion strategies used to generate RNA-less mutants that avoid transcriptional adaptation [89].
Quantitative Assessment of Detection Methods
The table below compares the capabilities of different detection methods for identifying various types of on-target effects:
Table 1: Comparison of Detection Methods for CRISPR On-Target Effects
| Method | Detection Capability | Time Requirement | Cost Factor | Sensitivity Limit | Best Use Case |
|---|---|---|---|---|---|
| HRM Analysis | Simple indels, mutation complexity | ~2 hours (including DNA extraction) | Low | Single nucleotide changes | Rapid screening of G0 embryos, early efficiency checks |
| PAGE Heteroduplex | Simple indels, mutation presence | 4-6 hours | Low | ~5% mosaic detection | Quick assessment of editing efficiency |
| Sanger + Decomposition (TIDE/ICE) | Simple indels, efficiency scores | 1-2 days | Medium | ~5-10% allele frequency | Efficiency quantification when NGS unavailable |
| Illumina Amplicon Sequencing | Complete indel spectrum, complex rearrangements | 3-5 days | High | <1% allele frequency | Comprehensive characterization, publication data |
| Long-Range PCR | Large deletions, structural rearrangements | 1-2 days | Medium | ~100bp+ deletions | Detecting promoter deletions, major rearrangements |
Recent evaluations of 50 different gRNAs in zebrafish revealed that standard Sanger-based decomposition tools (TIDE and ICE) significantly underestimate editing efficiencies compared to Illumina sequencing, with Illumina estimates averaging 19.4% higher than ICE scores [4]. This discrepancy highlights the importance of selecting appropriate detection methods based on the required sensitivity and comprehensiveness.
Workflow for Comprehensive On-Target Assessment
The following diagram illustrates a recommended workflow for systematic detection and mitigation of complex on-target effects:
Strategic Approaches to Avoid Unintended On-Target Effects
gRNA Design and Validation
Careful gRNA design represents the first line of defense against unintended on-target effects. Empirical data from zebrafish studies reveal significant discrepancies between gRNA efficiency predictions from different computational tools and actual in vivo performance [4]. When designing gRNAs:
- Utilize multiple prediction tools including CRISPRScan, which was specifically developed using zebrafish experimental data and considers factors like nucleotide GC content and nucleosome positioning [4]
- Select gRNAs with moderate efficiency scores rather than maximum scores, as extremely high-efficiency gRNAs may increase the likelihood of complex rearrangements
- Avoid repetitive genomic regions where homologous recombination may promote larger deletions
- Validate multiple gRNAs per gene target to identify candidates with clean editing profiles
Experimental validation of gRNA efficiency should employ quantitative methods such as Illumina amplicon sequencing rather than relying solely on predictive algorithms or qualitative assessments [4].
Alternative Targeting Strategies
Promoter Deletion Approaches: Traditional frameshift mutations generated by CRISPR/Cas9 can trigger transcriptional adaptation, where degradation of mutant transcripts leads to upregulation of homologous genes, potentially masking phenotypes [89]. Generating RNA-less mutants by deleting gene promoters with dual gRNAs avoids this compensation effect. This strategy involves:
- Designing two gRNAs flanking the promoter region of the target gene
- Co-injecting with Cas9 protein to delete the entire promoter sequence
- Verifying complete absence of zygotic transcription via RT-PCR
- Outcrossing potential off-target mutations before phenotypic analysis [89]
Early Embryo Screening: Implementing HRM analysis at the blastula stage (4 hours post-fertilization) enables early assessment of editing efficiency and mutation complexity [43]. This approach allows researchers to identify gRNAs that produce complex mutation patterns before investing in raising embryos to adulthood.
The Researcher's Toolkit: Essential Reagents and Materials
Table 2: Essential Research Reagents for On-Target Effect Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Cas9 Protein or mRNA | CRISPR endonuclease component | Protein allows more rapid clearance, reducing off-target potential |
| Gene-specific guide RNAs | Targets Cas9 to genomic loci | Designed using zebrafish-optimized tools like CRISPRScan |
| HotSHOT Reagents (NaOH, Tris-HCl) | Rapid genomic DNA extraction | Enables genotyping of embryos or fin clips in <2 hours [43] |
| EvaGreen PCR Master Mix | Fluorescent DNA intercalation | Enables HRM analysis post-amplification without specialized probes |
| HRM-Compatible Real-Time PCR System | High-resolution melting curve analysis | Requires temperature resolution of 0.02°C/s for optimal detection |
| Illumina Sequencing Reagents | Comprehensive mutation profiling | Provides quantitative assessment of full indel spectrum |
| Long-Range PCR Kit | Amplification of large genomic regions | Detects deletions >100bp that might be missed by standard PCR |
Mitigating complex on-target effects in zebrafish CRISPR research requires a multifaceted approach combining careful experimental design, appropriate detection methodologies, and strategic editing approaches. Based on current research, the following best practices are recommended:
- Employ tiered detection strategies beginning with rapid HRM screening followed by comprehensive NGS characterization for clones showing complex melting profiles
- Validate gRNA efficiency empirically using quantitative methods rather than relying solely on predictive algorithms
- Consider promoter deletion approaches when studying genes with known homologs to avoid transcriptional adaptation effects
- Screen for large deletions using long-range PCR, particularly when phenotypic discrepancies occur
- Implement early embryo screening at 4 hours post-fertilization to identify optimal gRNAs before investing in raising adult fish
As CRISPR applications in zebrafish models continue to expand toward higher-throughput functional genomics, robust strategies for detecting and avoiding unintended on-target effects will be essential for generating reliable, interpretable data in both basic research and drug development contexts.
Ensuring Rigor: Validating Models, Comparing Tools, and Translating Data
Within zebrafish (Danio rerio) research, the need to rapidly and accurately quantify the efficiency of CRISPR-Cas9 genome editing is paramount. The accuracy of this initial efficiency assessment directly impacts downstream decisions, from screening candidate genes in F0 mosaic crispants to establishing stable mutant lines [4] [67]. While several methods have been developed to analyze the insertions and deletions (indels) resulting from non-homologous end joining (NHEJ), researchers are often faced with a choice between cost, throughput, and analytical depth. This application note provides a structured comparison of three prominent methods—ICE (Inference of CRISPR Edits), TIDE (Tracking of Indels by Decomposition), and Next-Generation Sequencing (NGS)—framed within the context of a streamlined workflow for zebrafish embryos. We summarize quantitative performance data and provide detailed protocols to guide researchers in selecting and implementing the optimal quantification strategy for their experimental goals.
The following table summarizes the core features, performance, and practical considerations of ICE, TIDE, and NGS for CRISPR analysis in zebrafish.
Table 1: Core Features and Performance of CRISPR Quantification Methods
| Feature | ICE (Synthego) | TIDE | Next-Generation Sequencing (NGS) |
|---|---|---|---|
| Underlying Data | Sanger Sequencing | Sanger Sequencing | High-throughput sequencing of amplicons |
| Typical Workflow Speed | Medium | Medium | Slow |
| Relative Cost | Low | Low | High |
| Key Performance Metric | High correlation with NGS (R² = 0.96) [90] | Good correlation with NGS, but can underestimate efficiency [4] [46] | Gold standard for accuracy and sensitivity [91] |
| Detection of Complex Indels | Good; can detect large insertions/deletions [90] | Limited; best for +1 bp insertions, struggles with complex indels [90] [92] | Excellent; provides complete spectrum of edits [93] [91] |
| Best Use Case | High-throughput screening of F0 crispants where NGS is not feasible | Preliminary, low-cost assessment of editing success | Unbiased, comprehensive analysis of editing outcomes and off-target effects |
Experimental Comparison and Workflow
Quantitative Performance in Zebrafish
Direct experimental comparisons in zebrafish models highlight critical performance differences between these tools. A 2022 study in BMC Genomics systematically evaluated CRISPR tools and found that while both ICE and TIDE scores were correlated with Illumina-based editing scores, ICE showed a higher correlation (Spearman ρ = 0.88) than TIDE (Spearman ρ = 0.59) [4]. The study also noted that both Sanger-based methods significantly underestimated editing efficiencies compared to NGS, with Illumina estimates being 19.4% higher on average than ICE scores [4].
A 2024 systematic comparison of computational tools using artificial sequencing templates confirmed that while all tools are reasonably accurate for simple indels, their performance varies with complexity. DECODR (a tool similar to ICE and TIDE) was found to provide the most accurate estimations for the majority of samples, while TIDE-based TIDER was better suited for analyzing knock-in efficiency [93]. This underscores the importance of tool selection based on the expected editing profile.
Analysis Workflow Diagram
The following diagram illustrates the decision pathway for selecting and applying these quantification methods within a typical zebrafish CRISPR experiment.
Detailed Protocols for Key Experiments
Protocol 1: ICE Analysis for High-Throughput Screening
This protocol is optimized for the rapid genotyping of pooled G0 crispant zebrafish embryos [4] [46].
Sample Preparation:
- Microinjection: Co-inject one-cell stage zebrafish embryos with Cas9 protein (recommended for higher efficiency) or mRNA and sgRNA complexes [46].
- DNA Extraction: At 5 days post-fertilization (dpf), pool approximately 20 injected embryos. Extract genomic DNA using a standard protocol (e.g., proteinase K digestion followed by purification) [4].
- PCR Amplification: Amplify a ~500-700 bp region surrounding the CRISPR target site using high-fidelity DNA polymerase.
- Sanger Sequencing: Purify the PCR product and submit for Sanger sequencing in the forward and reverse directions.
Data Analysis with ICE (Synthego):
- Access the ICE web tool.
- Upload the
.ab1chromatogram files from both the edited sample and an uninjected control sibling. - Input the exact sgRNA target sequence used for the experiment.
- Run the analysis. The tool will output an ICE Score (indel frequency percentage), a Knockout Score (proportion of frameshift indels), and a detailed breakdown of the specific indel sequences and their relative abundances [90].
Protocol 2: NGS Amplicon Sequencing for Comprehensive Analysis
This protocol provides a definitive analysis of on-target editing and can be adapted for off-target assessment [46] [91].
Library Preparation (rhAmpSeq-based):
- DNA Extraction: Extract genomic DNA from individual or pooled embryos. For early screening, the Zebrafish Embryo Genotyper (ZEG) device can be used for minimal-invasive biopsy at 72 hpf [46].
- Multiplex PCR Amplification: Using a system like the IDT rhAmpSeq CRISPR Analysis System, perform a multiplex PCR to simultaneously amplify the on-target locus and a panel of nominated off-target sites. This step incorporates Illumina sequencing adapters and sample barcodes.
- Library Pooling and Purification: Combine the uniquely barcoded PCR products from multiple samples, then purify the pooled library.
Sequencing and Data Analysis:
- Sequencing: Load the pooled library onto an Illumina sequencer (e.g., MiSeq) for high-depth, targeted sequencing.
- Bioinformatic Analysis: Process the raw sequencing data through a dedicated pipeline (e.g., the one provided with the rhAmpSeq system or CRISPRESSO2 [67]).
- Quantification: The pipeline aligns reads to a reference sequence, identifies indels, and calculates the precise frequency of each editing event, providing a complete picture of editing efficiency and specificity [91].
Zebrafish Embryo Editing and Genotyping Workflow
The diagram below details the core steps for generating and genotyping CRISPR-edited zebrafish, incorporating the quantification methods discussed.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these protocols relies on key laboratory reagents and tools. The following table lists essential components for a CRISPR quantification workflow in zebrafish.
Table 2: Essential Reagents and Tools for Zebrafish CRISPR Analysis
| Category | Item | Function & Application Notes |
|---|---|---|
| CRISPR Components | Alt-R S.p. Cas9 Nuclease V3 (IDT) | High-activity Cas9 protein for efficient editing when complexed with sgRNA [93] [46]. |
| crRNA & tracrRNA (IDT) | Components for forming guide RNA; can be annealed to form sgRNA [4] [93]. | |
| Sample Prep & Analysis | rhAmpSeq CRISPR Analysis System (IDT) | An end-to-end solution for designing and sequencing multiplexed amplicons for on- and off-target analysis by NGS [91]. |
| Proteinase K | For digestion of zebrafish embryos and tissue to release genomic DNA [93]. | |
| High-Fidelity DNA Polymerase | For accurate PCR amplification of the target locus prior to sequencing. | |
| Software & Design Tools | ICE Analysis Tool (Synthego) | Web-based tool for deconvoluting Sanger sequencing data to quantify indel efficiency [90]. |
| CRISPRscan | Algorithm for predicting gRNA efficiency, trained on zebrafish data [4]. | |
| CRISPRESSO2 | Bioinformatics tool for analyzing and visualizing NGS data from CRISPR experiments [67]. |
Within zebrafish-based functional genomics and drug target validation, CRISPR-Cas9 technology enables rapid generation of knockout models, often directly in F0 mosaic mutants (crispants) to accelerate research [4] [94]. A critical determinant in the reliability of these experiments is the comprehensive assessment of off-target effects—unintended edits at genomic sites with sequence similarity to the target. Accurate off-target evaluation is essential for attributing observed phenotypes to the intended genetic modification, a cornerstone for valid scientific conclusions and robust drug development pipelines.
The scientific community primarily employs two complementary approaches for off-target assessment: in silico prediction, which uses computational algorithms to forecast potential off-target sites, and in vivo empirical analysis, which experimentally identifies edits that actually occur in the living organism. While in silico tools offer speed and cost-efficiency, their predictions often vary and may not fully capture the complexity of a living system [4]. Conversely, empirical data derived from zebrafish provide a ground-truth validation but can be limited by the sensitivity of detection methods. This Application Note delineates the strengths and limitations of each approach, provides detailed protocols for their application in zebrafish, and synthesizes a framework for their integrated use to enhance the fidelity of CRISPR-Cas9 gene editing.
Comparative Analysis: In Silico Prediction vs. In Vivo Empirical Data
A systematic evaluation of 50 guide RNAs (gRNAs) in zebrafish embryos revealed a significant discrepancy between the editing efficiencies predicted by eight common in silico design tools and the empirical efficiencies observed in vivo [4]. This underscores the inherent challenge of relying solely on computational forecasts.
However, the same study, along with other foundational work, found that the frequency of off-target mutations at loci predicted in silico or identified in vitro was generally low (<1-3%) in zebrafish G0 embryos [4] [95]. This suggests that while prediction tools may not be perfectly calibrated for on-target efficiency in zebrafish, they can still flag sites where off-target activity is possible.
A critical advancement in this field is the discovery that CRISPR-Cas9 can induce large structural variants (SVs), such as deletions and insertions ≥50 bp, at both on-target and off-target sites. One study demonstrated that 6% of editing outcomes in founder larvae were SVs, and these mutations could be transmitted to the next generation, with 9% of F1 offspring carrying an SV and 26% carrying a traditional off-target mutation [96]. Many of these larger aberrations escape detection by standard Sanger sequencing or short-read next-generation sequencing methods, highlighting a potential blind spot in conventional off-target assessment and the need for more sophisticated empirical validation [96].
The table below summarizes the core characteristics of each approach.
Table 1: Core Characteristics of In Silico and In Vivo Off-Target Assessment Methods
| Feature | In Silico Prediction | In Vivo Empirical Validation |
|---|---|---|
| Basis | Computational algorithms based on sequence homology, mismatch tolerance, and genomic context [97]. | Direct sequencing of DNA from edited zebrafish embryos or tissues. |
| Key Tools | CHOPCHOP, CRISPRScan, CCTop, E-CRISP [4] [94] [97]. | Long-read sequencing (PacBio, Nanopore), Amplicon Sequencing, CIRCLE-Seq, GUIDE-seq [96] [97] [98]. |
| Typical Reported Off-Target Rate in Zebrafish | Varies widely based on tool and parameters. | Generally low (often <1-3% for small indels) but can include SVs [4] [96] [95]. |
| Advantages | Rapid, inexpensive, guides initial gRNA design and prioritization. | Provides direct, factual evidence of editing events; can detect unexpected mutations and SVs [96]. |
| Limitations | Discrepancies with in vivo efficiency; may miss off-targets with low sequence homology [4]. | More resource-intensive; sensitivity depends on method depth (e.g., detection limit of ~0.5% for standard NGS) [98]. |
Integrated Experimental Protocol for Off-Target Assessment
This section outlines a multi-tiered protocol for a rigorous off-target assessment in zebrafish CRISPR experiments, integrating both predictive and empirical elements.
Stage 1: gRNA Design and In Silico Pre-screening
- gRNA Design: Design gRNAs targeting early coding exons of your gene of interest. Tools like CHOPCHOP or CRISPRScan can be used for initial selection [4] [94].
- Specificity Check: Run the selected gRNA sequence through multiple in silico prediction tools (e.g., CHOPCHOP, CCTop) to generate a list of potential off-target sites.
- Filtering Criteria: Exclude candidate gRNAs that have potential off-target sites with fewer than 2 mismatches within the genome. For sites with 3 mismatches, confirm they are not located in exons or untranslated regions (UTRs) of other protein-coding genes [94]. A final BLAST check is recommended to rule out off-target interactions.
Stage 2: Zebrafish Embryo Injection and DNA Extraction
- Microinjection: Prepare a CRISPR-Cas9 ribonucleoprotein (RNP) complex by combining purified Cas9 protein with the synthesized gRNA. Microinject this complex into the yolk of one-cell stage wild-type (e.g., AB strain) zebrafish embryos [96].
- Rearing: Raise the injected embryos and uninjected control siblings under standard conditions to 5 days post-fertilization (dpf) [4].
- DNA Extraction: At 5 dpf, euthanize and pool approximately 20 G0 larvae. Extract high-quality genomic DNA using a standard phenol-chloroform protocol or commercial kit. The uninjected control DNA is crucial for downstream bioinformatic analysis.
Stage 3: Empirical On- and Off-Target Analysis
The choice of empirical method depends on the required sensitivity and the need to detect structural variants.
Table 2: Selection Guide for Empirical Off-Target Detection Methods
| Method | Best Suited For | Key Procedural Step | Sensitivity Limit |
|---|---|---|---|
| Targeted Amplicon Sequencing (Short-Read) | High-throughput screening of in silico-predicted off-target sites. | Amplify ~200 bp regions surrounding the on-target and predicted off-target sites from the pooled DNA. Sequence on an Illumina platform and analyze with tools like CrispRVariants [4]. | ~0.5% [98] |
| Long-Range Amplicon Sequencing (PacBio) | Detecting large structural variants (SVs) and complex rearrangements at known sites. | Construct large amplicons (2.6–7.7 kb) spanning the Cas9 cleavage sites. Sequence using the PacBio Sequel system for long, high-quality reads and analyze with specialized software (e.g., SIQ) [96]. | Not specified, but higher for SVs than short-read. |
| CRISPR Amplification Method | Ultrasensitive validation of very low-frequency off-target mutations (<0.5%). | For each candidate site, perform multiple rounds of PCR amplification where the Cas9-gRNA complex is used to cleave and remove wild-type DNA, thereby enriching for mutant DNA fragments before final NGS [98]. | As low as 0.00001% [98] |
The following workflow diagram illustrates the integrated protocol for comprehensive off-target assessment.
Table 3: Key Research Reagents and Resources for Zebrafish CRISPR Off-Target Analysis
| Item | Function/Application | Example/Note |
|---|---|---|
| CHOPCHOP Web Tool | Online resource for gRNA design and initial in silico off-target prediction [94]. | Freely available at https://chopchop.cbu.uib.no. |
| Purified Cas9 Protein | Formation of RNP complexes for highly efficient and specific microinjection [96]. | Commercially available from multiple suppliers (e.g., IDT, Thermo Fisher). |
| CrispRVariants R Package | Bioinformatics tool for quantifying and visualizing indel mutations from targeted amplicon sequencing data [4]. | Available through Bioconductor. |
| PacBio Sequel System | Long-read sequencing platform for identifying large structural variants and complex edits at on- and off-target sites [96]. | Provides continuous long reads (CLR) or high-fidelity (HiFi) reads. |
| SIQ Analysis Software | Computational tool for detecting and quantifying genome editing outcomes from PacBio long-read sequencing data [96]. | Specifically designed for analyzing editing outcomes from long-read sequencers. |
A rigorous approach to off-target assessment is non-negotiable for high-quality CRISPR research in zebrafish. While in silico predictions are an invaluable first step, they are not a substitute for empirical validation. The emerging evidence of heritable large structural variants at off-target sites [96] mandates the use of sensitive, long-read sequencing technologies in critical applications, especially those with therapeutic implications. By adopting the integrated framework outlined in this protocol—combining computational pre-screening with empirical validation tailored to the required sensitivity—researchers can significantly de-risk their zebrafish CRISPR models, leading to more reliable genotype-phenotype correlations and more confident decision-making in drug development.
Within the burgeoning field of zebrafish functional genomics, the CRISPR-Cas9 system has become an indispensable tool for generating targeted mutations. A critical factor influencing the success of these gene-editing experiments is the efficient delivery of CRISPR components into the embryo. While microinjection is the most common delivery method, alternative strategies are being actively developed to potentially improve efficiency, reduce mosaicism, and enable higher-throughput workflows. This application note provides a comparative analysis of three such delivery methods—electroporation, lipid nanoparticles (LNPs), and superparamagnetic iron oxide nanoparticles (SPIONs)—within the context of CRISPR-Cas9 gene editing in zebrafish embryos. We summarize key quantitative data, provide detailed protocols for electroporation, and outline essential reagents to equip researchers with the knowledge to select and optimize delivery strategies for their specific experimental needs.
Performance Data Comparison
A recent comparative study evaluated these three delivery strategies for CRISPR-Cas9-mediated editing of the ifi27l2a gene in two marine teleost cell lines, DLB-1 and SaB-1 [99]. The performance metrics are summarized in the table below.
Table 1: Comparative Performance of CRISPR-Cas9 Delivery Methods in Marine Teleost Cell Lines
| Delivery Method | Cell Line | Editing Efficiency | Key Observations |
|---|---|---|---|
| Electroporation | SaB-1 | Up to 95% | High efficiency under optimized conditions [99]. |
| DLB-1 | ~30% | Locus-specific genomic rearrangements observed [99]. | |
| Lipid Nanoparticles (LNPs) | DLB-1 | ~25% | Enabled intracellular delivery and moderate editing [99]. |
| SaB-1 | Minimal | Very low editing efficiency in this cell line [99]. | |
| SPION-based Magnetofection | DLB-1 & SaB-1 | Not Detected | Efficient cellular uptake, but no detectable editing, suggesting post-entry barriers [99]. |
This data highlights that delivery efficiency is highly dependent on both the method and the specific cell type, with electroporation currently showing the highest potential efficacy. Intracellular trafficking, nuclear localization, and Cas9 aggregation were identified as key factors influencing the final editing outcome [99].
Detailed Experimental Protocols
Electroporation of Zebrafish Embryos
Electroporation presents a viable alternative to microinjection for delivering plasmid DNA, recombinant Cas9 nuclease, and synthetic guide RNAs into zebrafish embryos [100]. The following protocol is optimized for high efficiency and low toxicity.
Table 2: Key Reagents for Embryo Electroporation
| Component | Specification/Note |
|---|---|
| Electroporation System | Square wave electroporator [100]. |
| Plasmid DNA | Dissolved in calcium-free Ringer's solution or 0.9% NaCl [101]. |
| CRISPR RNP Complex | Recombinant Cas9 protein pre-complexed with sgRNA [100]. |
| Electroporation Buffer | Calcium-free Ringer's solution (119 mM NaCl, 2.9 mM KCl, 5 mM HEPES; pH 7.2) [101]. |
Step-by-Step Workflow:
- Preparation of Electroporation Mixture: Dilute the plasmid DNA or pre-assembled CRISPR RNP complex in calcium-free Ringer's solution to a final concentration of approximately 1 µg/µL. A small amount (e.g., ≤3%) of a tracking dye like Fast Green can be added to visualize the solution during handling [102].
- Embryo Collection and Preparation: Collect freshly laid fertilized zebrafish eggs. Dechorionate the embryos if necessary. Efficient gene delivery can be achieved during the cleavage stage [100].
- Electroporation Setup: Place the embryos in the electroporation chamber. Ensure they are surrounded by the DNA/RNP solution.
- Pulse Application: Apply a series of square wave electrical pulses. The exact parameters (voltage, pulse duration, number of pulses) must be optimized for the specific electroporator and chamber setup. The goal is to achieve efficient delivery while maintaining high embryo survival [100].
- Post-Electroporation Care: Immediately transfer the electroporated embryos to standard embryo medium (e.g., E3 water). Incubate them under standard conditions (28.5°C) and monitor for development.
Microinjection of CRISPR-Cas9 Components
Microinjection remains the gold standard for generating mutant alleles in zebrafish. The protocol below uses a Cas9 RNP complex for high efficiency.
Table 3: Microinjection Reagent Setup
| Component | Stock Concentration | Final Concentration | Volume (for 5 µL total) |
|---|---|---|---|
| sgRNA | 1500 ng/µL | 200 ng/µL | 0.7 µL |
| Cas9 Protein | 3000 ng/µL | 600 ng/µL | 1.0 µL |
| Injection Buffer (T10E0.1) | - | - | 3.3 µL |
| Total Volume | - | - | 5.0 µL |
Step-by-Step Workflow:
- RNP Complex Assembly: Mix the sgRNA and Cas9 protein to the final concentrations listed in Table 3. Incubate at 37°C for 10-15 minutes to allow RNP complex formation.
- Needle Preparation: Prepare glass capillary needles using a micropipette puller.
- Embryo Preparation: Collect freshly laid fertilized eggs and align them on an injection agar plate.
- Microinjection: Load the RNP mixture into the needle and inject ~1 nL directly into the blastomere of one-cell stage embryos.
- Post-Injection Care: Transfer injected embryos to embryo medium and incubate at 28.5°C. Phenotypes, such as loss of pigmentation when targeting the tyrosinase gene, can be observed in mosaic G0 embryos within 2-3 days [103].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these protocols relies on a set of key reagents and tools. The following table details these essential components.
Table 4: Key Research Reagent Solutions for CRISPR Delivery in Zebrafish
| Reagent / Tool | Function / Application | Examples / Notes |
|---|---|---|
| CRISPR RNP Complex | Direct delivery of active nuclease; reduces off-target effects and mosaicism. | Recombinant Cas9 protein + in vitro transcribed sgRNA [103]. |
| sgRNA Design Tools | Predict on-target efficiency and potential off-target sites. | CRISPRScan, a tool built from zebrafish data, accounts for GC content and nucleosome positioning [4]. |
| Indel Detection Kits | Analyze mutagenesis efficiency in injected embryos. | TIDE (Tracking of Indels by Decomposition) and ICE (Inference of CRISPR Edits) deconvolve Sanger sequencing data [4]. Heteroduplex mobility assays by PAGE are a cost-effective alternative [4]. |
| Electroporation Apparatus | Enables bulk delivery of molecules into cells via electrical pulses. | Square wave electroporator; custom chambers for embryos [100]. |
| Ionizable Lipids | Core component of LNPs for encapsulating and delivering nucleic acids or RNPs. | Used in formulations for in vivo delivery to tissues like liver and lungs [104]. |
| Square Wave Electroporator | Applies controlled electrical pulses for efficient macromolecule delivery into embryos. | Optimized parameters are key for high efficiency and low embryo toxicity [100]. |
The choice of delivery method for CRISPR-Cas9 in zebrafish research is a critical determinant of experimental success. This analysis demonstrates that while electroporation can achieve high editing efficiencies comparable to microinjection in certain contexts, its performance, like that of LNP and SPION methods, is subject to cell-type-specific and locus-specific variables. Electroporation offers a promising alternative for bulk delivery of CRISPR components into embryos, whereas LNP-mediated delivery shows potential but requires further optimization for robust application in zebrafish. SPIONs, while facilitating cellular uptake, face significant post-entry barriers that currently preclude efficient gene editing. Researchers are thus advised to select a delivery strategy based on their target cells, desired efficiency, and technical capabilities, and to conduct pilot experiments to optimize conditions for their specific application.
The integration of CRISPR/Cas9 genome editing has positioned the zebrafish (Danio rerio) as a powerful vertebrate model that effectively bridges the gap between high-throughput capability and physiological relevance in biomedical research. This application note details how zebrafish, particularly within the first 5 days post-fertilization, offer a unique combination of scalability, genetic tractability, and systemic in vivo data that aligns with the 3Rs principles (Replacement, Reduction, and Refinement). We provide a comparative analysis against traditional mammalian models and present detailed protocols for implementing a CRISPR/Cas9-based functional genomics workflow, enabling researchers to streamline target validation and phenotypic screening within a more ethical and efficient research pipeline.
The pharmaceutical industry faces a significant productivity crisis, with an exceedingly high rate of drug attrition in clinical trials due largely to lack of efficacy and safety liabilities [57]. A critical factor in this challenge is the limited predictive value of current preclinical models. Zebrafish have emerged as a transformative model organism that addresses this need, combining the systemic complexity of a vertebrate with the scalability and ethical advantages of lower organisms [105] [57]. When combined with the precision of CRISPR/Cas9 gene editing, zebrafish present a robust platform for high-throughput functional genomics and phenotypic drug screening, serving as a strategic intermediary between simplistic in vitro systems and costly, low-throughput mammalian studies [106] [57]. This document outlines the specific advantages of this model and provides practical protocols for its implementation in a research setting focused on the 3Rs.
Comparative Analysis: Zebrafish vs. Mammalian Models
The choice of an animal model involves balancing throughput, physiological relevance, and ethical considerations. The following tables provide a quantitative and qualitative comparison to guide this decision.
Table 1: Quantitative Comparison of Key Model Organisms
| Feature | Zebrafish | Mouse | C. elegans | D. melanogaster |
|---|---|---|---|---|
| Genetic Similarity to Humans | ~84% of disease-related genes have orthologs [107] | >80% genetic similarity [107] | 65% of disease genes are homologous [108] | ~75% of disease-related genes [107] |
| Generation Time | ~3 months [106] | ~3 months | ~3 days [108] | ~12 days [107] |
| Offspring per Mating | 200-300 embryos/week [107] | ~6-8 pups/litter | ~140 eggs/day [108] | ~50-100 eggs/day |
| Embryo Development | External, ex utero | Internal, in utero | External | External |
| Maintenance Cost | Low | High | Very Low | Very Low |
| Regulatory Status (Substitute) | Larvae ≤5 dpf not considered protected animals in EU [105] | Regulated as protected animals | Largely unregulated | Largely unregulated |
Table 2: Qualitative Analysis of Physiological Relevance and 3Rs Impact
| Aspect | Zebrafish | Mouse | C. elegans / D. melanogaster |
|---|---|---|---|
| Systemic Physiology | Complex organ systems (functional liver, kidney, heart, CNS) [57] | High physiological similarity to humans | Lack key mammalian systems (e.g., circulatory, complex CNS) [107] |
| Drug Metabolism (ADME) | Recapitulates mammalian ADME features; "body-on-chip" setup [57] | Gold standard for pharmacokinetics | Not applicable |
| Throughput for Genetic/Phenotypic Screens | High-throughput capable [106] [57] | Low-throughput | Very high-throughput [107] |
| 3Rs - Replacement | Replaces mammals for early-stage discovery and toxicity screening [105] [109] | N/A | Can replace vertebrates for basic genetic studies |
| 3Rs - Reduction | One zebrafish can replace multiple mammals; reduces numbers in later stages [105] | Higher numbers typically required | Drastically reduces animal use |
| 3Rs - Refinement | Transparency enables non-invasive imaging; minimal stress [105] | Procedures can cause more stress and discomfort | Minimal suffering due to lower sentience |
CRISPR/Cas9 Workflow in Zebrafish: A High-Throughput Protocol
The following protocol, adapted from a established high-throughput functional genomics pipeline [106], allows for the efficient generation and analysis of mutant zebrafish lines. Realistically, two researchers can target tens to hundreds of genes per year using this methodology.
Materials and Reagents
Table 3: Research Reagent Solutions for CRISPR/Cas9 Workflow
| Reagent / Material | Function | Brief Description / Note |
|---|---|---|
| CRISPR/Cas9 Plasmid | Expression of Cas9 protein and sgRNA | For stable expression; can be replaced with recombinant Cas9 protein for increased efficiency. |
| sgRNA Synthesis Kit | Production of single-guide RNA | Cloning-free in vitro transcription is recommended for scalability [106]. |
| Microinjection Apparatus | Delivery of CRISPR components | Standard equipment for zebrafish embryo microinjection. |
| Phenol Red Solution | Injection tracer | Allows for visual confirmation of successful microinjection. |
| Fluorescence PCR Reagents | High-sensitivity mutant identification | Used for genotyping and screening of founder fish (F0) [106]. |
| Next-Generation Sequencing (NGS) Platform | Determination of exact genetic lesion | For precise characterization of induced mutations. |
Step-by-Step Protocol
Goal: Generate a stable zebrafish mutant line within a 6-month timeframe.
Workflow Overview:
Procedure:
Target Selection and sgRNA Synthesis (3 Days)
- Design sgRNAs targeting your gene of interest using specialized software.
- Synthesize sgRNAs using a cloning-free in vitro transcription method to enhance throughput and scalability [106].
Microinjection and Embryo Rearing
- Co-inject synthesized sgRNA and Cas9 mRNA/protein into the yolk or cell of one-cell stage zebrafish embryos.
- Raise the injected embryos (F0 generation) to sexual maturity (~3 months). These are the potential founder fish.
Founder (F0) Screening and Identification (Within 3 months of injection)
- Outcross the adult F0 fish to wild-type partners.
- Collect a small number of embryos from each clutch and pool them for DNA extraction at 24-48 hours post-fertilization.
- Perform fluorescence PCR on the pooled DNA to identify clutches carrying mutations. This high-sensitivity method allows for efficient screening of germline-transmitting events [106].
Establishment of a Stable Mutant Line (Within 6 months)
- For F0 fish identified as positive, outcross them again to obtain individual F1 progeny.
- Genotype the individual F1 offspring using Sanger sequencing or next-generation sequencing to determine the exact nature of the induced mutation.
- Raise heterozygous F1 fish to adulthood.
- Incross the heterozygous F1 adults to generate the first generation (F2) of homozygous mutants for phenotypic analysis.
The 3Rs Framework: Zebrafish as an Ethical and Efficient Solution
The use of zebrafish, particularly in CRISPR-based research, directly aligns with and advances the 3Rs principles.
Replacement: The EU Directive 2010/63/EU classifies zebrafish embryos and larvae within the first 5 days post-fertilization (dpf) as non-protected, in vitro models. This is because they are not capable of independent feeding. At this stage, they already possess fully developed organ systems, including a beating heart and functional nervous system, making them a robust and predictive alternative non-animal model for toxicity screening, disease modeling, and early drug discovery [105] [57].
Reduction: Zebrafish contribute to reduction in two key ways. First, their high fecundity (hundreds of embryos per week) and small size enable high-throughput experimental designs, where one experiment can yield data that would require many more mammalian subjects [105]. Second, by using zebrafish for early-stage target validation and compound screening, researchers can "fail fast and fail early," narrowing down the selection of compounds and targets before committing to more costly and regulated mammalian tests, thereby reducing the overall number of mammals required [105] [57].
Refinement: The biological characteristics of zebrafish lead to significant refinement of experimental techniques. The optical transparency of larvae allows for non-invasive, real-time in vivo imaging of internal processes such as organ function, blood flow, and disease progression without causing stress or harm to the animal. This transparency reduces the need for invasive terminal procedures, thereby minimizing pain, suffering, and distress [105].
The following diagram illustrates how zebrafish integrate into a drug discovery pipeline optimized for the 3Rs:
The synergistic combination of zebrafish biology and CRISPR/Cas9 technology presents a paradigm shift in preclinical research. This model offers an unparalleled balance of throughput, physiological relevance, and adherence to the 3Rs. By serving as a strategic bridge between in vitro systems and mammalian models, zebrafish enhance the predictive validity of early-stage research, reduce overall costs and timelines, and promote more humane research practices. The provided protocols and analyses offer a roadmap for research teams to integrate this powerful approach into their drug discovery and functional genomics pipelines.
Application Note: Precision Disease Modeling with CRISPR/Cas9
The zebrafish (Danio rerio) has emerged as a powerful vertebrate model for biomedical research, occupying a strategic position between in vitro high-throughput screenings and mammalian preclinical models. With approximately 84% of human disease-associated genes having functional counterparts in zebrafish, this model offers exceptional translational potential for understanding disease mechanisms and accelerating drug discovery [11] [57]. The optical transparency of embryos, rapid external development, small size, and high fecundity enable large-scale genetic and pharmacological studies that would be prohibitively expensive or time-consuming in mammalian systems [11] [110]. The combination of these inherent advantages with CRISPR/Cas9 genome editing technology has established zebrafish as a premier system for generating precise human disease models that faithfully recapitulate patient pathology.
CRISPR/Cas9 Revolution in Zebrafish Genetics
The advent of CRISPR/Cas9 has fundamentally transformed zebrafish genetic engineering, enabling researchers to move beyond traditional loss-of-function approaches to precise nucleotide editing. This technological advancement allows for the introduction of patient-specific point mutations into the zebrafish genome, creating models that more accurately mimic human genetic disorders than conventional knockout approaches [111] [112]. The ability to perform homology-directed repair (HDR) using single-stranded DNA oligonucleotide templates has been particularly impactful for modeling diseases caused by single nucleotide changes, which represent the majority of human genetically inherited conditions [111]. This precision editing capability has opened new avenues for studying complex genetic disorders while maintaining endogenous gene expression levels and cell-type specificity that are often lost in overexpression models.
Protocol: Cantú Syndrome Knock-in Model Generation
Experimental Design and Workflow
The generation of zebrafish Cantú syndrome models demonstrates an optimized CRISPR/Cas9-mediated knock-in approach for introducing patient-specific cardiovascular-disorder-causing mutations. This protocol outlines a streamlined 22-week workflow from sgRNA design to establishment of stable heterozygous lines carrying missense mutations in zebrafish orthologs of KCNJ8 and ABCC9 genes [111] [112].
Reagent Preparation and Microinjection
sgRNA Design and Synthesis: Design sgRNAs following established guidelines [112] using online tools such as ChopChop. Select target sites as close as possible to the desired mutation site (within 4 bp of PAM sequence when possible) to maximize editing efficiency. Transcribe sgRNAs in vitro using appropriate RNA polymerase systems.
Single-Stranded Oligodeoxynucleotide (ssODN) Template Design: Design 50-bp ssODN templates containing the patient-specific mutation flanked by homologous arms. Incorporate silent mutations that disrupt the PAM recognition sequence to prevent re-cutting by Cas9 after successful incorporation. Include restriction enzyme sites when possible to facilitate genotyping [112].
Microinjection Mixture Preparation: Prepare injection mixture containing:
- Cas9 protein or mRNA: 100-200 ng/μL
- sgRNA: 50-100 ng/μL
- ssODN template: 100-200 ng/μL
- Phenol red tracer: 0.1%
Inject 1-2 nL of the mixture into the cell of one-cell stage zebrafish embryos. Culture injected embryos at 28.5°C in E3 embryo medium [112].
Screening and Validation
F0 Mosaic Screening: At 24 hours post-fertilization (hpf), assess a subset of injected embryos for editing efficiency using PCR amplification of the target region followed by restriction fragment length polymorphism (RFLP) analysis or Sanger sequencing. For Cantú syndrome models, reported indel introduction efficiencies typically range from 71.4% to 100% [112].
Germline Transmission: Raise microinjected embryos to adulthood (F0 founders). Outcross F0 fish to wild-type partners and screen F1 progeny for precise incorporation of the desired mutation. Efficiency of precise knock-in in germline transmission ranges from 3.8% to 21.4% in published Cantú syndrome models [112].
Establishment of Stable Lines: Outcross confirmed F1 heterozygous carriers to establish stable lines. Validate mutations through Sanger sequencing and maintain through standard zebrafish husbandry practices.
Table 1: Cantú Syndrome Knock-in Line Generation Efficiency
| Target Gene | Mutation | Indel Efficiency (%) | Precise KI Efficiency (%) | Reference |
|---|---|---|---|---|
| KCNJ8 | V65M | 100 | 21.4 | [112] |
| ABCC9 | G989E | 75 | 3.8 | [112] |
| ABCC9 | C1043Y | 75 | 12.5 | [112] |
Application Note: Phenotypic Characterization of Cantú Syndrome Models
Cardiovascular Phenotype Analysis
Zebrafish knock-in models of Cantú syndrome faithfully recapitulate the cardiovascular abnormalities observed in human patients. Heterozygous kcnj8+/V65M larvae demonstrate significantly elevated mean end-diastolic (mEDV) and end-systolic (mESV) volumes with strikingly enhanced cardiac output (53% increase) due to equally increased stroke volume at 5 days post-fertilization (dpf) [112]. Additionally, contractile function is elevated and an increased amount of pericardial edema is observed, mirroring the cardiomegaly characteristic of Cantú syndrome patients.
Vascular analysis reveals significantly reduced cardinal vein and dorsal aorta blood flow velocity in heterozygous kcnj8+/V65M larvae, corresponding to the low blood pressure and diminished vascular tone reported in CS patients [112]. Adult heterozygous fish exhibit significantly enlarged ventricular chamber volume, while atrial area remains similar to wild-type size, demonstrating the specific cardiovascular effects of these KATP channel mutations.
Signaling Pathway Alterations in Cantú Syndrome
Cantú syndrome is caused by gain-of-function mutations in genes encoding the pore-forming (Kir6.1, KCNJ8) and regulatory (SUR2, ABCC9) subunits of the predominantly cardiovascular isoforms of ATP-sensitive potassium channels (KATP) [111] [112]. These mutations lead to constitutive channel activation, resulting in the pathological vasodilation and cardiovascular remodeling observed in patients and zebrafish models.
Protocol: High-Throughput Epilepsy Modeling and Drug Screening Platform
F0 Crispant-Based Screening Approach
Traditional establishment of isogenic zebrafish lines requires extensive time and resources. The F0 crispant platform enables rapid functional assessment of gene-disease relationships within days rather than months, making it ideal for high-throughput drug screening applications [113] [114]. This approach is particularly valuable for epilepsy research, where numerous genetic candidates have been identified through patient sequencing studies.
Tyrosinase Co-targeting Selection System: To reduce variability and maximize utility of F0 knock-out approaches, simultaneously target the gene of interest with the tyrosinase (tyr) reporter gene. The tyr gene encodes a protein essential for melanin production, and its disruption results in easily detectable absence of pigmentation [113] [114]. This pigmentation phenotype serves as a visual indicator of efficient CRISPR/Cas9 activity, allowing enrichment for larvae with high mutation rates in the co-targeted gene of interest.
Multiparametric Behavioral Analysis: Combine highly efficient gene inactivation with automated analysis of morphological developmental defects and complex multiparametric behavioral analysis to describe seizure-like events. Use kinematic parameters such as locomotor activity and characteristic circular swimming patterns to identify seizure-like events in response to epileptogenic stimuli [113] [114].
Experimental Workflow for Epilepsy Drug Screening
Crispant Generation: Co-inject Cas9 protein with sgRNAs targeting both tyrosinase and epilepsy-associated genes into one-cell stage embryos. Culture embryos at 28.5°C until phenotypic screening.
Phenotypic Selection: At 48-120 hours post-fertilization, select larvae with complete loss of pigmentation indicating efficient tyr gene disruption. These crispants likely carry high rates of mutations in the co-targeted epilepsy genes as well [113] [114].
Pentylenetetrazole (PTZ) Seizure Induction: Expose 6-7 dpf larvae to PTZ, a GABAA receptor antagonist that induces seizure-like activity. Use concentration ranges of 5-15 mM PTZ dissolved in embryo medium with exposure times of 10-60 minutes [113] [115].
High-Throughput Behavioral Analysis: Use automated video tracking systems to quantify seizure-associated behaviors including:
- Increased locomotor activity
- Circular swimming patterns
- Burst swimming events
- Loss of posture
Compound Screening: Array selected crispants into multi-well plates and expose to test compounds dissolved in embryo medium. Include positive controls (established anti-seizure medications) and vehicle controls. Assess both efficacy in suppressing seizure-like behaviors and potential toxicity through morphological assessment.
Table 2: Epilepsy Gene Targets for F0 Crispant Screening
| Gene | Human Disorder | Crispant Phenotype | Validation Method |
|---|---|---|---|
| scn1lab | Dravet Syndrome | Spontaneous seizures, behavioral defects | Electrophysiology, locomotor analysis [115] |
| Multiple targets (6 genes) | Childhood Epilepsy | PTZ-induced seizure susceptibility | Multiparametric behavioral analysis [113] |
| Various NDD genes | Neurodevelopmental Disorders | Altered seizure threshold | High-throughput video tracking [114] |
Application Note: Translational Success Stories
From Zebrafish to Clinical Candidates
The zebrafish platform has demonstrated remarkable success in identifying and validating novel therapeutic compounds for epilepsy. The most prominent example comes from Dravet syndrome modeling, where scn1lab mutant zebrafish have led to multiple clinical candidates [115].
In a landmark study, a phenotype-based screen of 320 compounds in scn1lab mutant zebrafish identified clemizole, an FDA-approved compound with anti-histaminic properties, as an effective inhibitor of spontaneous convulsive behaviors and electrographic seizures [115]. Clemizole (EPX-100) has subsequently progressed through Phase I clinical trials and is under investigation as an 'add-on treatment' in a pivotal Phase II clinical trial for Dravet syndrome.
Further screening in the scn1lab model identified fenfluramine (now FDA-approved as Fintepla) and synthetic cannabinoids (similar to the FDA-approved cannabidiol Epidiolex) as effective anti-seizure medications [115]. Additionally, repurposed drugs including trazodone (Desyrel) and lorcaserin (Belviq) showed efficacy in zebrafish models, with lorcaserin demonstrating significant seizure reduction in a compassionate-use trial involving Dravet syndrome patients [115].
Advantages of Zebrafish in Antiepileptic Drug Discovery
The zebrafish model offers several distinct advantages for antiepileptic drug discovery:
Conservation of Drug Response: Zebrafish show high conservation of convulsive behavioral responses and drug metabolism pathways with mammals, providing superior predictive value for human efficacy and toxicity [115] [57].
Blood-Brain Barrier Assessment: Unlike in vitro systems, zebrafish larvae possess a functional blood-brain barrier, enabling evaluation of compound penetration into the central nervous system [115].
Integrated Physiology: Zebrafish provide a complete in vivo system with functional ADME (Absorption, Distribution, Metabolism, Excretion) properties, allowing simultaneous assessment of efficacy and toxicity [57].
High-Throughput Capacity: The small size and external development of zebrafish larvae enable screening of hundreds of compounds in multi-well plates at a fraction of the cost of mammalian studies [113] [114].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Zebrafish CRISPR-based Disease Modeling
| Reagent/Category | Specific Examples | Function and Application | Technical Notes |
|---|---|---|---|
| Genome Editing Tools | Cas9 protein/mRNA, sgRNAs | Introduction of targeted DNA double-strand breaks | Protein more efficient for F0 crispants [113] [112] |
| Reporter Systems | tyrosinase (tyr) sgRNA | Visual selection of high-efficiency crispants | Non-pigmented larvae indicate efficient editing [113] [114] |
| Knock-in Templates | Single-stranded oligodeoxynucleotides (ssODNs) | Homology-directed repair for point mutations | Include PAM-disrupting silent mutations [111] [112] |
| Seizure Induction | Pentylenetetrazole (PTZ) | Chemical induction of seizure-like events | GABAA receptor antagonist [113] [115] |
| Phenotypic Assessment | High-speed video microscopy, behavioral tracking systems | Quantitative analysis of cardiac and neurological phenotypes | Automated systems enable high-throughput screening [113] [112] |
| Validation Methods | Sanger sequencing, restriction enzyme digestion | Confirmation of precise genome editing | RFLP analysis for efficient genotyping [112] |
Concluding Remarks
The integration of CRISPR/Cas9 genome editing with zebrafish disease modeling has created a powerful platform for translational research, enabling rapid generation of precise genetic models that faithfully recapitulate human disorders. The Cantú syndrome knock-in models demonstrate how patient-specific point mutations can be introduced to study complex cardiovascular pathologies, while the F0 crispant platform for epilepsy research showcases the potential for high-throughput functional genomics and drug screening.
These approaches significantly reduce the time and resources required for preclinical validation, bridging the gap between target identification and therapeutic development. As CRISPR technologies continue to advance, zebrafish models will play an increasingly important role in personalized medicine, allowing researchers to rapidly validate newly identified disease variants and screen for targeted therapies in a physiologically relevant in vivo context.
The success stories in antiepileptic drug discovery, particularly for Dravet syndrome, highlight the translational impact of zebrafish-based approaches and provide a roadmap for applying these strategies to other genetic disorders. By combining genetic precision with high-throughput capability, zebrafish models are poised to accelerate the development of novel therapeutics for a wide range of human diseases.
Conclusion
The synergy between CRISPR-Cas9 technology and the zebrafish model has fundamentally accelerated the pace of functional genomics and preclinical research. This powerful combination enables rapid gene function annotation, highly efficient disease modeling, and direct phenotypic drug screening, often within months. As the field progresses, future directions will focus on enhancing precision with next-generation editors like prime editors, improving the efficiency of 'hard-to-edit' loci, and standardizing high-throughput platforms for personalized medicine. The continued refinement of these tools solidifies the zebrafish's role as an indispensable vertebrate bridge between in vitro assays and clinical application, poised to deliver novel therapeutic targets and candidates to the pipeline.
References
- 1. Models for Human Disease Studies | Frontiers Zebrafish ... Research [https://www.frontiersin.org/research-topics/13286/zebrafish-models-for-human-disease-studies/impact]
- 2. Practical approaches for implementing forward genetic strategies in... [https://pubmed.ncbi.nlm.nih.gov/21805265/]
- 3. Zebrafish genome engineering using the CRISPR-Cas9 system - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5127170/]
- 4. Evaluation of CRISPR gene-editing tools in zebrafish | BMC Genomics | Full Text [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-08238-1]
- 5. Duke School of Medicine Z-Core Zebrafish Programs [https://sites.duke.edu/zebrafishfacility/zebrafish-research/]
- 6. Generation of zebrafish models by CRISPR /Cas9 genome editing - PubMed [https://pubmed.ncbi.nlm.nih.gov/25431076/]
- 7. : an efficient vertebrate model for understanding role of gut... Zebrafish [https://molmed.biomedcentral.com/articles/10.1186/s10020-022-00579-1]
- 8. A Rapid CRISPR/Cas-based Mutagenesis Assay in Zebrafish for Identification of Genes Involved in Thyroid Morphogenesis and Function | Scientific Reports [https://www.nature.com/articles/s41598-018-24036-4]
- 9. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5524635/]
- 10. Improving CRISPR/Cas9 mutagenesis efficiency by delaying the early development of zebrafish embryos | Scientific Reports [https://www.nature.com/articles/s41598-020-77677-9]
- 11. Editing Using CRISPR: Gene as a Model System for... Zebrafish [https://www.synthego.com/blog/crispr-zebrafish/]
- 12. Between Genetic and Similarity | ZeClinics CRO Zebrafish Humans [https://www.zeclinics.com/blog/genetic-similarity-between-zebrafish-and-humans/]
- 13. CRISPR/Cas9 in zebrafish: an efficient combination for human genetic diseases modeling - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5214880/]
- 14. as an animal model for biomedical research | Experimental... Zebrafish [https://www.nature.com/articles/s12276-021-00571-5?error=cookies_not_supported&code=46b9d8b2-4baf-40b4-b348-82d3ba9d286c]
- 15. -Based Parkinson's Disease Models: Unveiling Zebrafish ... Genetic [https://pubmed.ncbi.nlm.nih.gov/40511807/]
- 16. Generation of Functional Genetic Study Models in Zebrafish Using... [https://pubmed.ncbi.nlm.nih.gov/32813255/]
- 17. / CRISPR -Mediated Cas in 9 | SpringerLink Genome Editing Zebrafish [https://link.springer.com/protocol/10.1007/978-1-0716-2990-1_17]
- 18. ZFIN - Database Commons [https://ngdc.cncb.ac.cn/databasecommons/database/id/5072]
- 19. and considerations for PAM | IDT sequences CRISPR [https://eu.idtdna.com/page/support-and-education/decoded-plus/importance-of-the-pam-sequences-in-crispr-cas9-gene-editing/]
- 20. Mechanism and Applications of CRISPR / Cas - 9 -Mediated Genome... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388126/]
- 21. CRISPR/Cas9-Mediated Genome Editing in Zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11753827/]
- 22. Frontiers | Active - Site Models of Streptococcus pyogenes Cas in... 9 [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.653262/full]
- 23. Efficient Production and Identification of CRISPR/Cas9-generated Gene Knockouts in the Model System Danio rerio - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6231919/]
- 24. Protospacer adjacent motif - Wikipedia [https://en.wikipedia.org/wiki/Protospacer_adjacent_motif]
- 25. What is the PAM in Sequence Experiments, and Why is it... CRISPR [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 26. insights into DNA cleavage... | Nature Communications Structural [https://www.nature.com/articles/s41467-017-01496-2?error=cookies_not_supported&code=13228fd1-239f-4c65-acca-945dff36d44f]
- 27. - CRISPR Systems: Origins and Their Impact on Cas ... Functional [https://premierscience.com/pjs-25-732/]
- 28. Programmable base editing of zebrafish using a modified... genome [https://www.nature.com/articles/s41467-017-00175-6?error=cookies_not_supported&code=40c85def-4380-473a-b0ae-584ce2942aa8]
- 29. Precise and efficient genome in editing using the... zebrafish [https://pmc.ncbi.nlm.nih.gov/articles/PMC4299274/]
- 30. Prime editing outperforms homology-directed repair as a tool for CRISPR-mediated variant knock-in in zebrafish | Lab Animal [https://www.nature.com/articles/s41684-025-01560-1]
- 31. Enhanced RNA-targeting CRISPR-Cas technology in zebrafish | Nature Communications [https://www.nature.com/articles/s41467-025-57792-9]
- 32. CRISPR/Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments - PubMed [https://pubmed.ncbi.nlm.nih.gov/30355591/]
- 33. Frontiers | Mapping the therapeutic landscape of CRISPR - Cas for... 9 [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1558432/full]
- 34. Meets CRISPR : Accelerating the Discovery of New... Zebrafish [https://pubmed.ncbi.nlm.nih.gov/32462967/]
- 35. Frontiers | Zebrafish in glioma models : advances in... research [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1601656/full]
- 36. -Based Technology To Speed Identification of CRISPR Involved... Genes [https://healthcare.utah.edu/press-releases/2021/08/crispr-based-technology-speed-identification-of-genes-involved-health]
- 37. disease Zebrafish in models ... | Nature Reviews drug Drug Discovery [https://www.nature.com/articles/s41573-021-00210-8?error=cookies_not_supported&code=0da6eb4c-e29d-4832-a69a-05946e5256f6]
- 38. CRISPR-Cas9-induced gene knockout in zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9617198/]
- 39. Protocol for generating mutant zebrafish using CRISPR-Cas9 followed by quantitative evaluation of vascular formation - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2666166723007207]
- 40. Genome editing with RNA-guided Cas nuclease in 9 embryos Zebrafish [https://www.nature.com/articles/cr201345?error=cookies_not_supported]
- 41. Rapid Generation of Pigment Free, Immobile Zebrafish and... Embryos [https://pmc.ncbi.nlm.nih.gov/articles/PMC8392167/]
- 42. of Antisense Morpholinos, CRISPR/ Microinjection RNP, and... Cas 9 [https://link.springer.com/protocol/10.1007/978-1-4939-7665-2_18]
- 43. A simplified method for identifying early CRISPR -induced indels in... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-2881-1]
- 44. / CRISPR -mediated precise genome modification by a long ssDNA... Cas 9 [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6493-4]
- 45. The State of Knock -ins in Zebrafish - InVivo Biosystems [https://invivobiosystems.com/crispr/the-state-of-knock-ins-in-zebrafish/]
- 46. Improved selection of zebrafish CRISPR editing by early next-generation sequencing based genotyping | Scientific Reports [https://www.nature.com/articles/s41598-023-27503-9]
- 47. A robust pipeline for efficient -in of point mutations and epitope... knock [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08971-1]
- 48. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos | Communications Biology [https://www.nature.com/articles/s42003-019-0444-0]
- 49. An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediated gene editing - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S002192582039757X]
- 50. Efficient precise knockin with a double cut HDR donor after... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1164-8]
- 51. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair | Scientific Reports [https://www.nature.com/articles/s41598-021-98965-y]
- 52. / CRISPR - Cas precise 9 modification by a long... mediated genome [https://pubmed.ncbi.nlm.nih.gov/31964350/]
- 53. Microinjection With Embryo - CRISPR in Mice and Cas 9 Zebrafish [https://www.thermofisher.com/ht/en/home/life-science/genome-editing/genome-editing-learning-center/genome-editing-resource-library/genome-editing-application-notes/protocol-embryo-microinjection-crispr-cas9-mice-zebrafish.html]
- 54. CRISPR-Cas9 DNA Base-Editing and Prime-Editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7503568/]
- 55. Frontiers | Base in editors : a new era for functional... zebrafish [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1598887/full]
- 56. 101: Cytosine and Adenine CRISPR Base Editors [https://blog.addgene.org/single-base-editing-with-crispr]
- 57. Frontiers | Combining Zebrafish and CRISPR/Cas9: Toward a More Efficient Drug Discovery Pipeline [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2018.00703/full]
- 58. SpG and SpRY variants expand the CRISPR ... | Nature Communications [https://www.nature.com/articles/s41467-022-31034-8?error=cookies_not_supported&code=d99cc31b-81a2-48c7-9dfd-c22864ecc96e]
- 59. Efficient genome editing using modified Cas9 proteins in zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10997048/]
- 60. Request your copy: New Efficient Mouse and Zebrafish ... embryos [https://www.thermofisher.com/us/en/home/global/forms/crispr-cas9-embryos-microinjection-protocol.html]
- 61. / CRISPR -Directed Gene Editing for the Generation of... Cas 9 [https://pubmed.ncbi.nlm.nih.gov/28678442/]
- 62. Applications of CRISPR genome editing technology in drug ... target [https://pubmed.ncbi.nlm.nih.gov/28388235/]
- 63. - High imaging assays using throughput , a zebrafish organism... model [https://www.moleculardevices.com/en/assets/app-note/dd/img/high-throughput-imaging-assays-using-zebrafish-a-model-organism-for-human-disease]
- 64. and its Applications in CRISPR Discovery | Lonza Drug [https://bioscience.lonza.com/lonza_bs/CH/en/crispr-screening-and-it-s-applications-in-drug-discovery]
- 65. Robotic injection of zebrafish embryos for high - throughput ... [https://pubmed.ncbi.nlm.nih.gov/23769806/]
- 66. Presentation of Quantitative Data | PSM Made Easy [https://ihatepsm.com/blog/presentation-quantitative-data]
- 67. Crispant analysis in zebrafish as a tool for rapid functional screening of... [https://elifesciences.org/articles/100060]
- 68. Progress in Gene-Editing Technology of Zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8468279/]
- 69. CRISPR Guide RNA Validation In Vitro - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5549792/]
- 70. CRISPRz: a database of zebrafish validated sgRNAs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4702947/]
- 71. -Cas9-Based Methods for Generating... Highly Efficient CRISPR [https://pubmed.ncbi.nlm.nih.gov/31708433/]
- 72. Improved selection of zebrafish editing by early... CRISPR [https://www.nature.com/articles/s41598-023-27503-9?error=cookies_not_supported&code=bd2a76c8-8dac-40e5-93f8-cc51c72d74f8]
- 73. Synthetic Cas9 CRISPR Solutions | GenScript sgRNA [https://www.genscript.com/crispr-synthetic-sgrna.html]
- 74. Optimal Length for Left & Right Homology Arms for CRISPR | IDT [https://www.idtdna.com/pages/support/faqs/when-designing-donor-dna-for-use-in-homology-directed-repair-(hdr)-what-are-the-optimal-lengths-of-the-left-and-right-homology-arms-and-what-is-the-maximum-size-of-sequence-that-can-be-efficiently-inserted-in-mammalian-cells]
- 75. Frontiers | Efficient CRISPR-Cas9-Mediated Knock-In of Composite... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.598634/full]
- 76. Homology arm design [https://groups.google.com/g/crispr/c/7wjdosrnuYY]
- 77. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes | Nature Communications [https://www.nature.com/articles/ncomms10431]
- 78. Efficient In Vivo Introduction of Point Mutations Using ssODN and a Co-CRISPR Approach | Biological Procedures Online | Full Text [https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/s12575-020-00123-7]
- 79. Efficient targeted integration directed by short homology in zebrafish ... [https://elifesciences.org/articles/53968]
- 80. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9 - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4484386/]
- 81. CRISPR: Established Editor of Human Embryos? - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5819596//]
- 82. An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediated gene editing - PubMed [https://pubmed.ncbi.nlm.nih.gov/29500194/]
- 83. A CRISPR / Cas vector system for tissue-specific gene disruption in... 9 [https://pubmed.ncbi.nlm.nih.gov/25752963/]
- 84. Optimization of CRISPR-Cas9 genome editing for loss-of-function in the early chick embryo - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5728388/]
- 85. A robust pipeline for efficient knock-in of point mutations and epitope... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9730659/]
- 86. Fluorescent PCR–based Screening Methods for Precise Knock-in of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10415208/]
- 87. Efficient Cas9‐based Genome Editing Using CRISPR Analysis Webtools in Severe Early‐onset‐obesity Patient‐derived iPSCs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9377717/]
- 88. / CRISPR in Cas : an efficient combination for human genetic... 9 zebrafish [https://pmc.ncbi.nlm.nih.gov/articles/PMC5214880/]
- 89. Generating Zebrafish RNA-Less Mutant Alleles by Deleting Gene... [https://pubmed.ncbi.nlm.nih.gov/34913119/]
- 90. How to Pick the Best CRISPR Data Analysis Method for Your Experiment [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 91. off-target detection with CRISPR | IDT NGS [https://sg.idtdna.com/pages/technology/crispr/crispr-genome-editing/crispr-detection/next-generation-sequencing]
- 92. Discrepancies in indel software resolution with somatic CRISPR/Cas9 tumorigenesis models | Scientific Reports [https://www.nature.com/articles/s41598-023-41109-1]
- 93. Systematic Comparison of Computational Tools for Sanger Sequencing-Based Genome Editing Analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10854981/]
- 94. A Combined Human in and Silico / CRISPR -Mediated Cas ... 9 in Vivo [https://pmc.ncbi.nlm.nih.gov/articles/PMC9082939/]
- 95. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish - PubMed [https://pubmed.ncbi.nlm.nih.gov/24257628/]
- 96. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations | Nature Communications [https://www.nature.com/articles/s41467-022-28244-5]
- 97. Using traditional machine learning and deep learning methods for on- and off-target prediction in CRISPR/Cas9: a review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10199778/]
- 98. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment | Nature Communications [https://www.nature.com/articles/s41467-020-17418-8]
- 99. Analysis of Comparative / CRISPR Methods in Marine... Cas 9 Delivery [https://pubmed.ncbi.nlm.nih.gov/41226739/]
- 100. Transgenic Expression and Genome Editing by Electroporation of... [https://pubmed.ncbi.nlm.nih.gov/32748174/]
- 101. Frontiers | Fast gene transfer into the adult zebrafish brain by herpes... [https://www.frontiersin.org/journals/neural-circuits/articles/10.3389/fncir.2014.00041/full]
- 102. Method for In Vivo Electroporation of Plasmid DNA in the... Delivery [https://www.jove.com/pl/t/60066/electroporation-method-for-vivo-delivery-plasmid-dna-adult-zebrafish]
- 103. Embryo Microinjection With CRISPR-Cas9 in Mice and Zebrafish | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/genome-editing/genome-editing-learning-center/genome-editing-resource-library/genome-editing-application-notes/protocol-embryo-microinjection-crispr-cas9-mice-zebrafish.html]
- 104. Lung and liver editing by lipid nanoparticle delivery of a stable... [https://www.nature.com/articles/s41587-024-02437-3?error=cookies_not_supported&code=6c7d99a5-2a3d-48d9-9619-ddcd880f4d48]
- 105. and the Zebrafish : Ethical and Efficient Research Solutions 3 Rs [https://www.zeclinics.com/blog/zebrafish-and-the-3rs/]
- 106. A high- throughput functional genomics workflow based on... [https://pubmed.ncbi.nlm.nih.gov/27809318/]
- 107. Comparing Common Model Organisms | ZeClinics CRO [https://www.zeclinics.com/blog/comparing-common-model-organisms/]
- 108. An Updated Comparison of Common Model Organisms - InVivo Biosystems [https://invivobiosystems.com/disease-modeling/an-updated-comparison-of-common-model-organisms/]
- 109. Comparison of toxicity values across zebrafish early life stages and mammalian studies: Implications for chemical testing - PubMed [https://pubmed.ncbi.nlm.nih.gov/25261610/]
- 110. Spotlight on zebrafish: the next wave of translational research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6451428/]
- 111. Effective CRISPR/Cas9-based nucleotide editing in zebrafish to... [https://pubmed.ncbi.nlm.nih.gov/30355756/]
- 112. Effective CRISPR/Cas9-based nucleotide editing in zebrafish to ... [https://ncbi.nlm.nih.gov/pmc/articles/PMC6215435]
- 113. A Zebrafish -Based Platform for High-Throughput Epilepsy Modeling... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10931767/]
- 114. A Zebrafish-Based Platform for High-Throughput Epilepsy Modeling and Drug Screening in F0 [https://www.mdpi.com/1422-0067/25/5/2991]
- 115. Innovative Epilepsy Modeling with Zebrafish | ZeClinics CRO [https://www.zeclinics.com/blog/innovative-alternatives-for-modeling-epilepsy/]