Cry2/CIB vs. LOV Domains: A Comprehensive Guide for Choosing Optogenetic Tools in Biomedical Research
This article provides a systematic comparison of two dominant optogenetic systems, Cry2/CIB and light-oxygen-voltage (LOV) domains, for controlling protein-protein interactions in mammalian cells.
Cry2/CIB vs. LOV Domains: A Comprehensive Guide for Choosing Optogenetic Tools in Biomedical Research
Abstract
This article provides a systematic comparison of two dominant optogenetic systems, Cry2/CIB and light-oxygen-voltage (LOV) domains, for controlling protein-protein interactions in mammalian cells. Tailored for researchers and drug development professionals, it explores the foundational biology, mechanisms, and distinct kinetic profiles of these tools. We detail their methodological applications in activating intracellular signaling and gene regulation, offer practical strategies for troubleshooting and performance optimization, and present a direct, evidence-based comparison of their spatial resolution, efficiency, and suitability for specific experimental goals. The synthesis empowers scientists to select and implement the optimal optogenetic system for precise spatiotemporal control in basic research and therapeutic development.
Unpacking the Core Biology: From Natural Photoreceptors to Engineered Actuators
Origins and Natural Functions of Cry2 and LOV Photoreceptors
Photoreceptors are fundamental molecular switches that allow organisms to perceive and respond to light. In the field of optogenetics, scientists have co-opted these natural light-sensing proteins to precisely control biological processes in living cells. Among the most valuable tools are Cryptochrome 2 (Cry2) from Arabidopsis thaliana and various Light-Oxygen-Voltage (LOV) domains derived from plants, bacteria, and other organisms. These photoreceptors enable researchers to manipulate protein-protein interactions, intracellular signaling pathways, and cellular behaviors with unprecedented spatial and temporal precision using light. This guide provides a comprehensive comparison of the Cry2 and LOV domain systems, examining their natural origins, molecular mechanisms, and experimental applications to inform selection for optogenetic research and therapeutic development.
Natural Origins and Biological Functions
Cry2 Photoreceptors
- Origin: Cryptochrome 2 (Cry2) is a blue-light photoreceptor originally identified in the model plant Arabidopsis thaliana [1] [2]. It regulates various aspects of plant growth and development.
- Natural Functions in Plants: Cry2 mediates numerous blue-light-dependent processes in Arabidopsis, including photoperiodic flowering, inhibition of hypocotyl elongation, shade avoidance responses, and seed germination [1] [2]. Recent research has surprisingly revealed that Cry2 also maintains functionality in darkness, where it inhibits root growth by suppressing cell division in the root apical meristem [2].
- Structural Organization: Cry2 consists of two primary domains: an N-terminal photolyase homology region (PHR) that binds the flavin adenine dinucleotide (FAD) chromophore, and a C-terminal extension (CCE) important for signaling [1]. The PHR domain (approximately residues 1-498) is sufficient for light sensing and oligomerization [3].
LOV Domain Photoreceptors
- Origin: LOV domains are widespread throughout the tree of life, found in plants, bacteria, fungi, and archaea [4] [5]. They represent a subfamily of the Per-Arnt-Sim (PAS) domain superfamily.
- Natural Functions: LOV domains serve as blue-light sensors in various proteins that regulate diverse processes including phototropism, gene expression, and circadian rhythms [4]. Notable examples include the LOV2 domain from Avena sativa (oat) phototropin 1 and the LOV domain from Vaucheria frigida aureochrome1 (VfAU1-LOV) [4].
- Structural Organization: LOV domains are compact modules of approximately 120 residues that non-covalently bind a flavin chromophore (FMN or FAD). Photoactivation induces reversible conformational changes, such as the undocking of the C-terminal Jα helix in AsLOV2 [4].
Table 1: Natural Origins and Physiological Roles
| Feature | Cry2 Photoreceptor | LOV Domains |
|---|---|---|
| Organism of Origin | Arabidopsis thaliana (plant) | Various plants, bacteria, fungi, archaea |
| Chromophore | Flavin adenine dinucleotide (FAD) | Flavin mononucleotide (FMN) or FAD |
| Primary Natural Functions | Regulation of flowering time, hypocotyl elongation, root growth, shade avoidance | Phototropism, gene regulation, circadian rhythms, environmental adaptation |
| Dark State | Monomeric [1] | Jα helix associated with core (AsLOV2) [4] |
| Light-Activated State | Tetramerizes and interacts with partner proteins (e.g., CIB1) [1] | Conformational change (e.g., Jα helix undocking), dimerization (VfAU1-LOV) [4] |
Molecular Mechanisms of Photoactivation
Cry2 Photoactivation Mechanism
The Cry2 photoactivation cycle begins with the absorption of a blue light photon by the FAD chromophore. This triggers a well-orchestrated molecular rearrangement:
- Electron Transfer and Proton Donation: Light absorption initiates electron transfer from a conserved tryptophan residue, followed by proton transfer from a critical aspartate residue (D393 in Arabidopsis Cry2) to the FAD, generating the signaling-active FAD neutral radical (FADH•) state [1].
- Oligomerization: In the dark, Cry2 exists as a monomer. Upon blue light illumination, it undergoes homo-tetramerization, forming the active oligomeric state [1] [3].
- Partner Interaction: The photoexcited and oligomerized Cry2 exposes interaction surfaces that allow binding to various signaling partners, most notably the transcription factor CIB1 (CRY2-INTERACTING bHLH1), in a blue-light-dependent manner [1].
- Post-Translational Regulation: Cry2 activity is modulated by phosphorylation via Photoregulatory Protein Kinases (PPKs) and by ubiquitination through distinct E3 ubiquitin ligases (COP1/SPAs and LRBs), which target the receptor for degradation [6].
Figure 1: Cry2 Photoactivation and Signaling Mechanism. Blue light triggers electron/proton transfer to FAD, inducing oligomerization and CIB1 interaction to initiate signaling.
LOV Domain Photoactivation Mechanism
LOV domains undergo a distinct photocycle characterized by a reversible covalent bond formation:
- Covalent Adduct Formation: Upon blue light excitation, the conserved cysteine residue within the LOV domain forms a covalent bond with the C4a carbon of the flavin chromophore, resulting in a metastable adduct state [4].
- Conformational Change: This adduct formation triggers structural rearrangements within the LOV domain. In the well-characterized AsLOV2 domain, this causes the undocking of the C-terminal Jα helix from the core PAS domain [4].
- Dimerization or Allostery: Depending on the specific LOV protein, the light-induced conformational change can lead to dimerization (as in VfAU1-LOV) or can regulate the activity of an fused effector domain [4].
- Dark Recovery: The adduct state is thermally unstable, and the LOV domain spontaneously reverts to the dark state, breaking the covalent bond and restoring the original conformation [4].
Figure 2: LOV Domain Photoactivation Mechanism. Blue light induces flavin-cysteine adduct formation, leading to conformational changes that drive dimerization or effector regulation.
Experimental Applications in Optogenetics
Cry2-Based Optogenetic Systems
The Cry2/CIB1 and Cry2 oligomerization systems have been extensively engineered for optogenetic applications due to Cry2's dual interaction capabilities:
- CRY2-CIB1 Heterodimerization: This naturally occurring pair provides a specific light-induced heterodimerization system. It has been widely used to recruit proteins to specific cellular locations, control transcription, and modulate signaling pathways by bringing two different proteins into proximity [4] [3].
- CRY2 Homo-oligomerization: The light-induced self-association of Cry2 enables clustering of target proteins, which is useful for activating signaling pathways that respond to oligomerization (e.g., receptor tyrosine kinases, Raf pathway) and for sequestering proteins into inactive condensates [4] [3].
- Engineered Variants: Researchers have developed Cry2 mutants with enhanced properties:
- CRY2olig (E490G): Exhibits enhanced oligomerization capability for applications requiring robust clustering [3].
- CRY2high: Contains mutations that further promote homo-oligomerization via manipulation of C-terminal electrostatic charges [3].
- CRY2low: Engineered with reduced oligomerization tendency to minimize unintended clustering in CRY2-CIB1 applications [3].
LOV-Based Optogenetic Systems
LOV domains have been adapted into various optogenetic tools that primarily exploit their light-induced conformational changes:
- LOV2-Based Effector Regulation: The AsLOV2 domain is commonly used as a photoswitchable steric blocker. In the dark, the Jα helix binds to the LOV core, potentially occluding a fused effector domain. Light illumination releases the Jα, unmasking the effector activity [4].
- iLID/SspB Heterodimerization System: This engineered system consists of a modified AsLOV2 domain (iLID) that binds tightly to its partner SspB upon blue light illumination, providing a high-affinity heterodimerization tool [4].
- LOV Dimerization Systems: Natural LOV domains like VfAU1-LOV undergo light-induced dimerization, which can be used to induce homo-interaction of fused target proteins [4].
- Extremophile LOV Domains: Recently characterized LOV domains from extreme environments, such as the archaeal ALovD-1 from Lake Diamante, show remarkable stability under high salt concentrations (up to 3M), expanding the range of experimental conditions for optogenetics [5].
Table 2: Key Optogenetic Applications and Properties
| Property | Cry2 Systems | LOV Domain Systems |
|---|---|---|
| Primary Optogenetic Applications | Heterodimerization (CRY2-CIB1), Homo-oligomerization, Cluster formation | Steric occlusion (LOV2), Heterodimerization (iLID/SspB), Homo-dimerization (VfAU1) |
| Key Engineered Variants | CRY2olig, CRY2high, CRY2low [3] | iLID, Magnets, LOV2 circular permutants [4] |
| Activation Kinetics | Rapid activation (seconds), slow dark recovery (minutes) [3] | Fast activation (seconds), tunable recovery (seconds to minutes) [4] |
| Spectral Sensitivity | Blue light (∼450 nm) [1] | Blue light (∼450 nm) [4] |
| Exogenous Cofactor | No (binds FAD endogenously) [4] | No (binds FMN/FAD endogenously) [4] |
| Environmental Robustness | Standard physiological conditions | Archaeal LOV domains function in extreme conditions (e.g., high salinity) [5] |
Key Experimental Protocols
Assessing Cry2-CIB1 Interaction via Yeast Two-Hybrid
The yeast two-hybrid (Y2H) system provides a powerful genetic method to validate and characterize Cry2-CIB1 interactions [1].
Protocol:
- Strain and Plasmids: Use MaV203 yeast strain or similar. Clone CRY2 (residues 1-535) into pDBTrp plasmid as fusion with Gal4 DNA-binding domain (GalBD). Clone CIB1 into pGADT7rec plasmid as fusion with Gal4 activation domain (GalAD).
- Transformation: Co-transform both plasmids into yeast and select on SC -Trp/-Leu medium.
- Interaction Selection: Plate transformed yeast on SC -Trp/-Leu/-Ura medium. Interaction between CRY2 and CIB1 reconstitutes Gal4 function and activates URA3 expression, allowing growth on -Ura plates.
- Light Control: Incubate plates under continuous blue light for interaction or in complete darkness for negative control. Wild-type CRY2 should only interact with CIB1 under blue light [1].
Testing CRY2 Oligomerization via Size Exclusion Chromatography
Size exclusion chromatography (SEC) can directly monitor the light-dependent oligomerization of Cry2 [1].
Protocol:
- Protein Preparation: Express and purify recombinant CRY2PHR domain (residues 1-498) with an appropriate tag (e.g., His-tag, GFP-tag).
- Chromatography Setup: Equilibrate SEC column (e.g., Superdex 200) with suitable buffer. Maintain temperature at 4°C or use cold room.
- Light Stimulation: Divide protein sample into two aliquots. Keep one in darkness and illuminate the other with blue light (e.g., 450 nm LED, 10-100 μmol m⁻² s⁻¹) for 5-15 minutes before injection.
- Analysis: Inject samples and monitor elution profile. Compare elution volumes between dark and light samples. CRY2 tetramers elute earlier than monomers due to larger hydrodynamic radius [1].
Characterizing LOV Domain Photocycling via UV-Vis Spectroscopy
UV-visible spectroscopy monitors the spectral changes associated with LOV domain photocycling, particularly the formation and decay of the cysteinyl-flavin adduct [5].
Protocol:
- Sample Preparation: Express and purify LOV domain protein (e.g., ALovD-1). Desalt into appropriate buffer to remove contaminants.
- Dark Adaptation: Incubate sample in complete darkness for >1 hour to ensure complete transition to dark state.
- Spectral Acquisition: Record absorption spectrum from 300-600 nm. Characteristic dark state spectrum shows peaks at ∼450 nm (flavin) and ∼370 nm.
- Light Illumination: Expose sample to blue light (450 nm) and immediately record spectrum. Adduct formation decreases 450 nm peak and increases ∼390 nm peak.
- Dark Recovery: Monitor recovery by taking sequential spectra in darkness. Fit decay to exponential function to determine recovery half-time [5].
Research Reagent Solutions
Table 3: Essential Research Reagents and Their Applications
| Reagent / Tool | Function in Research | Example Applications |
|---|---|---|
| CRY2PHR (1-498) | Core light-sensing domain for optogenetic constructs | Base for CRY2-CIB1 and CRY2 oligomerization systems [3] |
| CIB1 | Native CRY2 interaction partner | CRY2-CIB1 heterodimerization systems [1] |
| AsLOV2 | Photoswitchable steric block module | LOV2-based optogenetic tools (e.g., LOVTRAP) [4] |
| iLID/SspB | Engineered high-affinity LOV heterodimerization pair | Precise protein recruitment and complex formation [4] |
| VfAU1-LOV | Natural light-induced homodimerizing LOV | Inducing protein dimerization with light [4] |
| pDBTrp & pGADT7rec | Yeast two-hybrid vectors | Testing protein-protein interactions (e.g., CRY2-CIB1) [1] |
| Anti-CRY2 Antibodies | Immunodetection of CRY2 | Western blot, immunoprecipitation to study CRY2 expression and degradation [6] |
| ALovD-1 | Halophilic archaeal LOV domain | Optogenetics under extreme salt conditions [5] |
Cry2 and LOV domains provide complementary tools for the optogenetic toolbox, each with distinct advantages rooted in their natural functions and molecular mechanisms. Cry2 systems offer the unique capability for both heterodimerization and homo-oligomerization, making them versatile for applications ranging from transcriptional control to signaling pathway activation. The extensive engineering of Cry2 variants (CRY2high, CRY2low) allows researchers to fine-tune oligomerization properties for specific experimental needs. LOV domain systems excel in applications requiring reversible conformational changes, with tools available for steric occlusion, heterodimerization, and homodimerization. The discovery and characterization of extremophile LOV domains further expands their utility under challenging experimental conditions.
Selection between these systems should be guided by the specific biological question: Cry2 is ideal when leveraging its natural interaction partners or when induced clustering is desired, while LOV domains offer precision in controlling protein conformation and activity with minimal perturbation. As optogenetic applications continue to evolve in complexity, particularly in drug development and synthetic biology, both systems will play crucial roles in enabling precise spatial and temporal control of cellular processes.
Optogenetics has revolutionized the biological sciences by enabling precise, light-controlled manipulation of cellular processes with high spatiotemporal resolution. Unlike chemical inducers that diffuse rapidly and lack spatial precision, optogenetic systems offer reversible, non-invasive control over biological functions simply by applying light [7]. Two principal photochemical mechanisms—light-induced homo-oligomerization and hetero-dimerization—form the cornerstone of many optogenetic tools. These systems allow researchers to control diverse processes, including gene expression, signal transduction, and organelle distribution, by bringing specific proteins together in response to light [8] [7]. This guide provides a detailed comparison of these two mechanisms, focusing on the well-characterized CRY2/CIB system and representative LOV-domain systems, to aid researchers in selecting the appropriate optogenetic strategy for their experimental needs.
Core Mechanisms and Molecular Components
The CRY2/CIB System: A Dual-Function Tool
The photoreceptor Cryptochrome 2 (CRY2) from Arabidopsis thaliana is a flavin-binding protein that exhibits complex behavior under blue light (430-490 nm). Upon photoexcitation, CRY2 can undergo two distinct types of interactions:
- Hetero-dimerization: CRY2 binds to its natural partner protein, CIB1 (CRY2-Interacting bHLH1) [8] [9].
- Homo-oligomerization: CRY2 molecules self-associate to form higher-order clusters or "photobodies" [8].
This dual functionality makes CRY2 a versatile but complex optogenetic tool. The heterodimerization with CIB1 occurs rapidly within subseconds after light illumination and dissociates with a half-life of approximately 5.5 minutes in darkness, allowing for repeated induction over many cycles [8]. Concurrently, the homo-oligomerization property enables the formation of protein clusters that can be harnessed to modulate various cellular functions [8].
LOV-Domain Systems: Simplicity and Single-Component Design
Light-Oxygen-Voltage (LOV) domains are another class of blue-light-sensitive photoreceptors that utilize flavin nucleotides as chromophores. A prominent example is the EL222 protein from Erythrobacter litoralis, which consists of a light-sensitive LOV domain and a helix-turn-helix (HTH) DNA-binding domain [10]. Unlike the dual-functionality of CRY2, EL222 operates primarily through a light-induced homodimerization mechanism. In the dark state, the HTH domain is sterically inhibited by the LOV domain. Blue light absorption triggers the formation of a covalent adduct between a cysteine residue in the LOV domain and the flavin mononucleotide, causing a conformational change that releases the HTH domain, enabling receptor dimerization and subsequent DNA binding [10]. A key advantage of certain LOV-based systems like EL222 is their single-component nature, reducing genetic complexity and variability associated with multi-component systems [10].
Table 1: Fundamental Properties of CRY2/CIB and LOV-Domain Systems
| Property | CRY2/CIB System | LOV-Domain System (EL222) |
|---|---|---|
| Photoreceptor Origin | Arabidopsis thaliana (plant) | Erythrobacter litoralis (bacterium) |
| Chromophore | Flavin | Flavin Mononucleotide (FMN) |
| Activation Light | Blue light (430-490 nm) | Blue light |
| Core Mechanism | Dual: Hetero-dimerization with CIB1 & Homo-oligomerization | Light-induced homodimerization & DNA binding |
| Number of Components | Two (CRY2 & CIB1) for heterodimerization | One (single polypeptide) |
| Dark State | Monomeric/cytosolic (CRY2) | HTH domain inhibited |
| Light State | Hetero-dimer with CIB1 or CRY2 clusters | Homodimer bound to DNA |
| Key Structural Domains | Photolyase Homology Region (PHR, aa 1-498) | LOV domain + HTH DNA-binding domain |
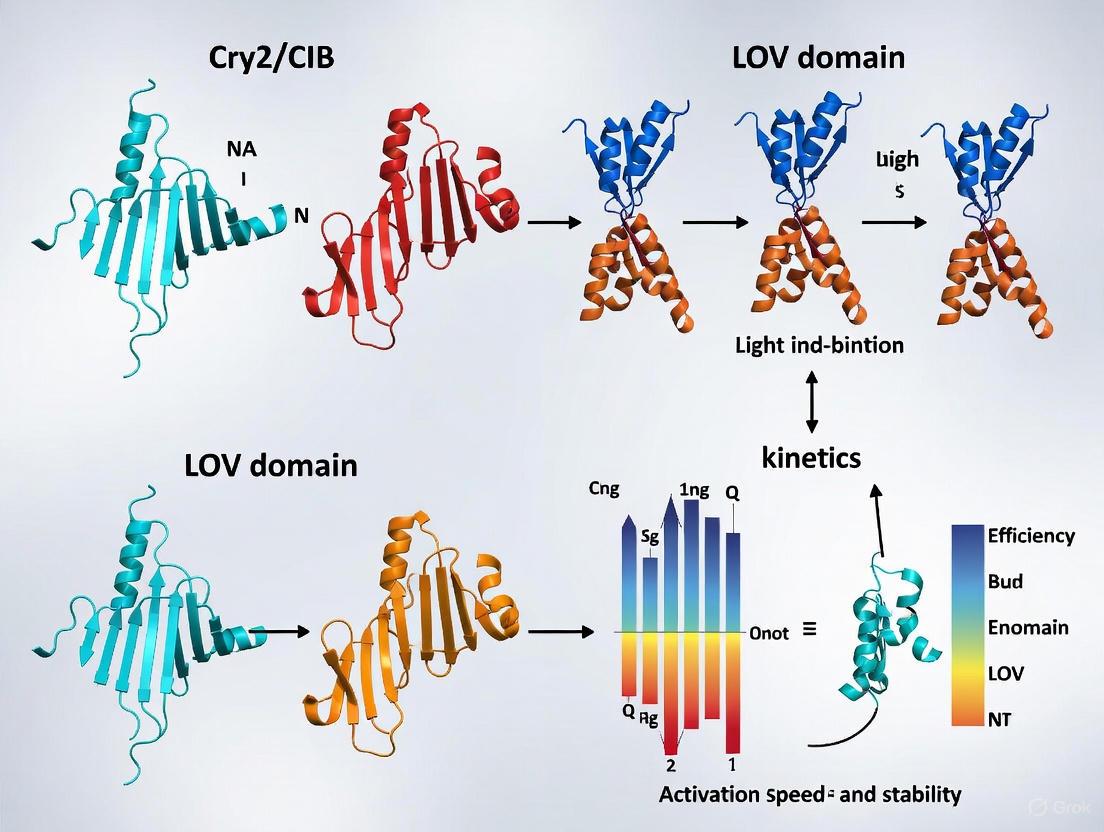
Figure 1: Core signaling pathways of CRY2/CIB and LOV-domain optogenetic systems. The CRY2/CIB system exhibits dual functionality upon blue light activation, enabling both hetero-dimerization with CIB1 and homo-oligomerization. In contrast, the LOV-domain system EL222 operates through a single-component mechanism where light-induced conformational change enables homodimerization and DNA binding.
Quantitative Performance Comparison
Efficiency and Kinetics
The operational characteristics of CRY2 and LOV-domain systems differ significantly in their kinetics, reversibility, and induction efficiency, which determines their suitability for various experimental applications.
CRY2/CIB System Performance:
- Hetero-dimerization Kinetics: CRY2-CIB1 heterodimerization occurs rapidly within subseconds after blue light illumination and dissociates with a half-life of approximately 5.5 minutes upon light withdrawal [8].
- Oligomerization Efficiency: The oligomerization efficiency of CRY2 is highly dependent on its subcellular localization. Membrane-bound CRY2 (targeted to plasma membrane, ER, or mitochondria) exhibits drastically enhanced oligomerization compared to its cytoplasmic form. While cytoplasmic CRY2 formed clusters in only about 20% of cells with an average of 6.4 small clusters per cell, membrane-tethered CRY2 consistently formed hundreds to thousands of bright clusters within seconds of blue light exposure in every transfected cell [8].
- Dual Mechanism Interference: The presence of CIB1 can influence CRY2 oligomerization. Bulky CIB1 fusion proteins can suppress CRY2 cluster formation, while cytoplasmic CRY2 recruitment to the membrane via membrane-bound CIB1 can intensify its oligomerization [8].
LOV-Domain System Performance:
- Kinetics: The EL222 LOV system features fast activation kinetics (seconds) with spontaneous reversion in the dark (approximately 50 seconds at ambient temperature) [10].
- Transcriptional Activation: When fused to the potent VPR transactivation domain (VP64-p65-Rta), the resulting DEL-VPR system achieved up to 570-fold induction of target gene expression in mammalian cells under blue light, reaching expression levels comparable to strong constitutive promoters like CMV [10].
- Basal Activity: EL222-based systems typically exhibit minimal basal activity in the dark state, making them suitable for applications requiring tight control [10].
Table 2: Quantitative Performance Metrics of CRY2/CIB vs. LOV-Domain Systems
| Performance Metric | CRY2/CIB System | LOV-Domain System (EL222) |
|---|---|---|
| Activation Kinetics | Subseconds (heterodimerization) [8] | Seconds [10] |
| Deactivation Half-life | ~5.5 minutes (heterodimerization) [8] | ~50 seconds [10] |
| Maximum Induction Fold | Varies by application; typically high for membrane-recruitment | Up to 570-fold (DEL-VPR for gene expression) [10] |
| Basal Activity (Dark State) | Low, but context-dependent | Very low [10] |
| Spatial Precision | High (subcellular clustering possible) | High (nuclear gene control) |
| Reversibility | High (fully reversible) | High (rapid spontaneous reversion) |
| Key Limitation | Dual mechanisms can interfere | Single-component but limited to DNA-binding applications |
Experimental Factors Affecting Performance
Several experimental factors significantly impact the performance of these optogenetic systems:
Cellular Context Dependence:
- CRY2 oligomerization is markedly enhanced when the protein is localized to cellular membranes (plasma membrane, ER, mitochondrial membrane) compared to its cytoplasmic form [8].
- Fusion partner size can affect CRY2 behavior—bulky CIB1 fusion proteins can suppress CRY2 homo-oligomerization [8].
Light Delivery Parameters:
- Both systems require precise blue light delivery, but specific parameters (intensity, duration, pulse frequency) must be optimized for each application.
- For transcriptional activation with DEL-VPR, specific illumination regimes are necessary to achieve maximal induction [10].
Experimental Protocols and Methodologies
Characterizing CRY2 Oligomerization in Different Cellular Compartments
Objective: To systematically analyze the oligomerization behavior of CRY2 in cytoplasmic versus membrane-bound contexts.
Key Reagents:
- CRY2-PHR (amino acids 1-498) fused to fluorescent protein (e.g., CRY2-mCh or CRY2-GFP)
- Membrane-targeting sequences: CaaX motif (plasma membrane), Sec61TM (ER membrane), Miro1TM (mitochondrial outer membrane)
- Control: Light-insensitive CRY2 mutant (CRY2(D387A)) [8]
Methodology:
- Cell Culture and Transfection: Culture appropriate mammalian cells (e.g., COS-7, HEK293T, 3T3) and transfect with CRY2 constructs targeted to different cellular compartments.
- Light Stimulation: Expose cells to intermittent blue light pulses (460-480 nm, 200 ms exposure every 5 s at 9.7 × 10³ mW/cm²) for durations ranging from 1 to 10 minutes.
- Image Acquisition and Analysis: Capture time-lapse fluorescence images to monitor cluster formation. Quantify the percentage of cells showing clusters and the number/size of clusters per cell.
- Experimental Controls: Include cells expressing CRY2(D387A) mutant to confirm light-specific effects, and use green light illumination (~550 nm) to verify wavelength specificity [8].
Expected Outcomes: Membrane-bound CRY2 (CRY2-mCh-CaaX, CRY2-mCh-Sec61, CRY2-mCh-Miro1) will show rapid and dramatic oligomerization within seconds of blue light exposure, while cytoplasmic CRY2 will exhibit limited and inconsistent cluster formation [8].
Assessing CRY2-CIB1 Heterodimerization and Interference with Oligomerization
Objective: To examine how CRY2 homo-oligomerization and CRY2-CIB1 heterodimerization activities mutually affect each other.
Key Reagents:
- CRY2 fused to fluorescent reporter (e.g., CRY2-mCh)
- CIB1 (amino acids 1-170) fused to various protein domains of different sizes
- Membrane-targeted CIB1 constructs [8]
Methodology:
- Co-expression Studies: Co-transfect cells with CRY2-mCh and various CIB1 fusion constructs.
- Light Activation and Imaging: Apply blue light illumination and monitor both CRY2 cluster formation and recruitment to CIB1 localization sites.
- Quantitative Analysis: Measure the extent of CRY2 oligomerization in the presence versus absence of different CIB1 fusions, and quantify recruitment efficiency of cytoplasmic CRY2 to membrane compartments via membrane-bound CIB1 [8].
Expected Outcomes: Certain bulky CIB1 fusion proteins will suppress CRY2 homo-oligomerization, while membrane-bound CIB1 will recruit cytoplasmic CRY2 to membranes and enhance its local oligomerization [8].
Measuring Transcriptional Activation with LOV-Domain Systems
Objective: To quantify light-induced gene expression using the EL222-based DEL-VPR system.
Key Reagents:
- DEL-VPR construct (EL222 fused to VPR transactivation domain)
- Reporter plasmid with firefly luciferase (fLuc) or mCherry under C120 promoter with TATA box
- Control constructs (e.g., VP-EL222, VEL) for comparison [10]
Methodology:
- Cell Line Preparation: Culture HEK293T or CHO-K1 cells and co-transfect with DEL-VPR and reporter constructs.
- Light Stimulation Regime: Illuminate cells with blue light using optimized protocols. For high induction, continuous or specific pulsed illumination may be required.
- Reporter Quantification:
- For luciferase: Measure luminescence at specified time points after light induction.
- For fluorescent proteins: Analyze fluorescence intensity by flow cytometry or microscopy.
- Data Analysis: Calculate fold induction by comparing light-induced expression to dark controls. Normalize for transfection efficiency using internal controls [10].
Expected Outcomes: The DEL-VPR system should show strong light-induced reporter expression (up to 570-fold induction) with minimal basal activity in dark conditions [10].
Figure 2: Experimental workflow for optogenetic manipulation. The generalized protocol begins with system selection and proceeds through construct design, cell preparation, light stimulation, and quantitative analysis. The CRY2 system is particularly suited for membrane recruitment and subcellular clustering applications, while LOV-domain systems excel in transcriptional control.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of optogenetic experiments requires careful selection of molecular tools and reagents. The following table summarizes key components for working with CRY2/CIB and LOV-domain systems.
Table 3: Essential Research Reagents for Optogenetic Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| CRY2 Constructs | CRY2-PHR (aa 1-498), CRY2-mCh, CRY2-GFP, CRY2(D387A) mutant | Core light-sensing component for oligomerization and heterodimerization [8] |
| CIB1 Constructs | CIB1 (aa 1-170), various CIB1 fusion partners | CRY2 binding partner for heterodimerization applications [8] |
| Membrane Tags | CaaX motif, Sec61TM, Miro1TM | Target proteins to plasma membrane, ER, or mitochondrial membranes [8] |
| LOV-Domain Tools | EL222, VP-EL222, DEL-VPR (EL222-VPR) | Single-component optogenetic systems for transcriptional control [10] |
| Reporter Systems | C120-minP-FLuc, C120-minP-mCherry | Quantify optogenetic system performance and induction efficiency [10] |
| Cell Lines | HEK293T, CHO-K1, COS-7, 3T3 | Standard mammalian cell lines for optogenetic validation [8] [10] |
| Fluorescent Proteins | mEGFP, mCherry, mCherry2 (optimal for oligomerization studies) | Protein tagging and quantification; mCherry2 shows superior properties for quantifying oligomerization [11] |
Application Scenarios and System Selection Guide
Comparative Advantages and Limitations
CRY2/CIB System Advantages:
- Versatility: The dual functionality enables both recruitment-based applications (via heterodimerization) and local concentration enhancement (via oligomerization) [8].
- Established Protocols: Widely adopted with extensive validation in diverse cellular processes.
- Membrane Applications: Particularly effective for membrane-associated signaling manipulation due to enhanced oligomerization at membranes [8].
CRY2/CIB System Limitations:
- Complex Behavior: The interplay between oligomerization and heterodimerization requires careful experimental design and controls [8].
- Context Dependence: Performance varies significantly based on subcellular localization and fusion partners [8].
LOV-Domain System Advantages:
- Simplicity: Single-component design reduces experimental variables and ensures consistent expression ratios [10].
- Precise Transcriptional Control: Excellent for gene expression applications with high induction folds and low background [10].
- Fast Kinetics: Rapid activation and deactivation suitable for dynamic control [10].
LOV-Domain System Limitations:
- Application Scope: Primarily optimized for transcriptional control rather than general protein-protein interaction studies.
- Limited Versatility: Less adaptable to diverse cellular processes compared to CRY2.
Selection Guidelines for Specific Research Applications
Choose CRY2/CIB when:
- Studying processes involving membrane signaling or compartmentalization
- Designing systems that benefit from local protein concentration (clustering)
- Working with established CRY2-based optogenetic tools from the literature
- Applications require the specific biological context where CRY2's dual functionality is beneficial
Choose LOV-Domain systems when:
- Prioritizing simple, single-component design
- Needing precise control over gene expression with minimal basal activity
- Fast activation/deactivation kinetics are critical
- Conducting all-optical experiments requiring minimal genetic footprint
Both light-induced homo-oligomerization (exemplified by CRY2) and hetero-dimerization (exemplified by CRY2-CIB1 and LOV-domain systems) provide powerful, genetically encoded strategies for controlling cellular processes with high spatiotemporal precision. The CRY2 system offers unique versatility through its dual functionality but requires careful consideration of cellular context and potential interference between its oligomerization and heterodimerization activities. In contrast, LOV-domain systems like EL222 provide simplified, single-component operation optimized particularly for transcriptional control applications. The choice between these mechanisms should be guided by the specific biological question, desired spatial control, kinetic requirements, and the cellular context of the intended application. As optogenetic technology continues to evolve, both approaches will remain essential tools in the molecular biology toolkit, enabling increasingly precise interrogation and manipulation of cellular function.
Chromophore Requirements and Practical Considerations for Mammalian Systems
Optogenetic control of cellular processes has revolutionized biological research and therapeutic development by enabling precise, light-dependent manipulation of protein interactions, gene expression, and signaling pathways. For researchers working with mammalian systems, the choice of optogenetic tool involves critical considerations of chromophore requirements, activation kinetics, and practical implementation. Two predominant blue-light-responsive systems—cryptochrome 2/CIB1 (Cry2/CIB1) and light-oxygen-voltage (LOV) domain-based tools—offer distinct advantages and limitations for mammalian applications [10] [12] [13]. This guide provides an objective comparison of their performance, supported by experimental data, to inform selection for specific research needs.
Fundamental System Architectures and Chromophore Requirements
Cry2/CIB1 System Architecture
The Cry2/CIB1 system derives from Arabidopsis thaliana and consists of two primary components: the photolyase homology region of Cry2 (amino acids 1-498) and the N-terminal domain of CIB1 (amino acids 1-170) [13] [14]. Upon blue light exposure (peak activation ~450 nm), Cry2 undergoes conformational changes enabling interaction with CIB1, facilitating recruitment of proteins of interest to specific cellular locations.
Chromophore Requirement: Cry2 non-covalently binds flavin adenine dinucleotide (FAD) as its endogenous chromophore [15]. While FAD is ubiquitously present in mammalian cells as a redox cofactor, its availability and incorporation into Cry2 can vary across cell types and affect system performance.
LOV Domain System Architecture
LOV domains, found in phototropins from various species, form covalent adducts with their chromophore upon blue light exposure. The EL222 protein from Erythrobacter litoralis represents a well-characterized LOV-based optogenetic tool comprising a light-sensitive LOV domain and a helix-turn-helix DNA-binding domain [10]. In darkness, the LOV domain sterically occludes the functional domain; blue light illumination triggers conformational changes that release this inhibition.
Chromophore Requirement: LOV domains covalently bind flavin mononucleotide (FMN) through a conserved cysteine residue [10]. Similar to FAD, FMN is endogenously available in mammalian cells, eliminating the need for exogenous chromophore supplementation.
Table 1: Fundamental Characteristics of Cry2/CIB1 and LOV Domain Systems
| Characteristic | Cry2/CIB1 System | LOV Domain Systems |
|---|---|---|
| Origin | Arabidopsis thaliana (plant) | Various (e.g., Erythrobacter litoralis, Avena sativa) |
| Core Components | Cry2 (1-498 aa) + CIBN (1-170 aa) | Single-component (e.g., EL222) or two-component (e.g., iLID) |
| Chromophore | Flavin Adenine Dinucleotide (FAD) | Flavin Mononucleotide (FMN) |
| Chromophore Binding | Non-covalent | Covalent (cysteine-FMN adduct) |
| Endogenous Chromophore in Mammalian Cells | Yes | Yes |
| Peak Activation Wavelength | ~450 nm (blue light) | ~450 nm (blue light) |
Diagram: Chromophore Binding and Activation Mechanisms
Performance Comparison in Mammalian Systems
Activation Kinetics and Spatial Precision
The temporal and spatial resolution achievable with optogenetic tools directly impacts their utility for controlling fast cellular processes and targeting specific subcellular compartments.
Cry2/CIB1 Kinetics: The Cry2/CIB1 system exhibits relatively slow switch-off kinetics (τ₁/₂OFF = 290 ± 30 s at 37°C), which limits its temporal resolution [13]. This prolonged active state allows activated Cry2 to diffuse widely within the cell, resulting in poor spatial confinement when targeting small subcellular volumes.
LOV Domain Kinetics: LOV-based systems like iLID and Magnets demonstrate significantly faster switch-off kinetics (seconds to minutes) [13]. This enables superior spatial precision, confining dimerization to the illuminated volume. However, this enhanced spatial resolution often comes at the expense of total dimer yield compared to Cry2/CIB1 [13].
DEL-VPR Performance: A recently developed LOV-based system called DEL-VPR, which fuses EL222 to the potent VPR transactivation domain (VP64-p65-Rta), achieves up to 570-fold induction of target gene expression in HEK293T and CHO-K1 cells [10]. This system combines minimal basal activity in darkness with expression levels comparable to strong constitutive promoters like CMV upon blue light illumination.
Table 2: Performance Characteristics in Mammalian Cells
| Performance Metric | Cry2/CIB1 | LOV Domain Systems | Experimental Context |
|---|---|---|---|
| Switch-On Half-Time (τ₁/₂ON) | 3.7 ± 0.9 s | Seconds (varies by construct) | In vivo at 37°C [13] |
| Switch-Off Half-Time (τ₁/₂OFF) | 290 ± 30 s | Seconds to minutes | In vivo at 37°C [13] |
| Spatial Confinement | Low | High | Illumination of 3μm × 3μm ROI [13] |
| Gene Induction Fold-Change | Not specified | Up to 570-fold (DEL-VPR) | HEK293T/CHO-K1, light vs. dark [10] |
| Basal Activity (Dark) | Variable | Low (DEL-VPR) | Reporter gene expression [10] |
| Cytotoxicity | Cluster formation observed [13] | Minor basal cytotoxicity [10] | Long-term expression studies |
Experimental Workflow for Mammalian Gene Expression Control
The following diagram illustrates a generalized protocol for implementing optogenetic gene expression control in mammalian cells, adaptable for both Cry2/CIB1 and LOV-based systems.
Key Experimental Protocols
Light-Induced Gene Expression with DEL-VPR
Objective: Achieve high-level, light-inducible expression of target proteins (e.g., monoclonal antibodies) in mammalian cell lines [10].
Materials:
- Plasmids:
- pcDNA3.1_CMV-DEL-VPR (optogenetic actuator)
- Reporter/target gene under C120-minP promoter (5xC120 repeats with minimal promoter)
- Cell Lines: HEK293T or CHO-K1
- Light Source: Blue LED array (470 nm)
Methodology:
- Cell Culture and Transfection: Maintain cells in DMEM (HEK293T) or appropriate medium. Co-transfect with DEL-VPR and reporter plasmids using standard transfection methods.
- Light Stimulation: Illuminate cells with pulsed blue light (470 nm). Optimal parameters include 200-ms pulses every 2 seconds at 7.07 W/cm² for 5 minutes to several hours, depending on application [10] [13].
- Expression Analysis: Quantify target gene expression via fluorescence (for reporters like mCherry), luciferase activity, or specific immunoassays (e.g., ELISA for antibodies). Compare light-stimulated samples to dark controls.
Key Considerations: DEL-VPR exhibits minimal basal activity in darkness and achieves maximal induction approximately 24 hours post-stimulation [10]. For protein production applications, extended illumination (24-48 hours) may be beneficial.
Subcellular Protein Recruitment with Cry2/CIB1
Objective: Recruit cytosolic proteins to specific organelles with light activation [13].
Materials:
- Plasmids:
- Organelle-targeted CIBN (e.g., ER-CIBN, CIBN-Mito)
- mCherry-Cry2 fusion (cytosolic prey)
- Cell Line: Appropriate mammalian cell line (e.g., HeLa, HEK293)
- Microscopy System: Confocal microscope with 488 nm laser
Methodology:
- Cell Preparation: Co-transfect cells with bait (organelle-targeted CIBN) and prey (mCherry-Cry2) constructs.
- Localized Illumination: Define a small region of interest (e.g., 3×3 μm) on the target organelle. Illuminate with 200-ms blue light pulses every 2 seconds (488 nm, 7.07 W/cm²) for 5 minutes [13].
- Image Acquisition: Monitor mCherry fluorescence redistribution during and after illumination.
- Data Analysis: Quantify prey recruitment efficiency and spatial spread outside the illuminated area.
Key Considerations: Cry2/CIB1 recruitment spreads beyond the illumination area due to slow dissociation kinetics [13]. Cry2 also exhibits light-dependent clustering, which may confound interpretation of recruitment experiments.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Mammalian Optogenetics
| Reagent Category | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| Optogenetic Actuators | DEL-VPR (EL222-VPR fusion) [10]CRY2PHR (1-498 aa) [13]CIBN (1-170 aa) [13]iLID [13] | Light-sensitive protein modules for controlling cellular processes | DEL-VPR offers high induction; CRY2/CIBN enables two-component control |
| Reporter Plasmids | 5xC120-minP-FLuc [10]5xC120-minP-mCherry [10] | Quantify system performance and induction levels | C120 promoter optimized for EL222; minimal promoter reduces background |
| Mammalian Cell Lines | HEK293T [10]CHO-K1 [10]HEKblue IFN reporter cells [10] | Expression hosts with high transfection efficiency | Industry-relevant for bioproduction; specialized reporter lines available |
| Light Delivery Systems | Blue LED arrays (470 nm)Confocal microscope lasers (488 nm) | Precise light application with controlled parameters | LED arrays suitable for culture dishes; microscopes enable spatial patterning |
| Chromophore Sources | Standard cell culture media | Provides FAD/FMN cofactors | No supplementation typically needed in mammalian systems [10] [15] |
The selection between Cry2/CIB1 and LOV domain optogenetic systems for mammalian applications involves balancing multiple factors including temporal resolution, spatial precision, induction strength, and experimental simplicity. LOV-based systems like DEL-VPR offer superior induction levels, fast kinetics, and minimal basal activity, making them ideal for applications requiring high-precision temporal control and strong gene expression, such as biopharmaceutical production [10]. The Cry2/CIB1 system provides robust interaction strength but suffers from slower off-kinetics and lower spatial resolution, potentially limiting its utility for subcellular manipulation despite its effectiveness for whole-cell activation [13]. Both systems leverage endogenous flavin-based chromophores in mammalian cells, eliminating the need for exogenous supplementation that plagues phytochrome-based systems. Researchers should select tools based on their specific needs for kinetics, spatial control, and expression levels, while considering the continuous emergence of engineered variants with enhanced properties.
In the field of optogenetics, blue-light-sensing photoreceptors provide powerful tools for controlling cellular processes with high spatiotemporal precision. Among these, cryptochrome 2 (Cry2) and various LOV (Light-Oxygen-Voltage) domains represent two principal classes of photoreceptors that have been extensively engineered for optogenetic applications [16]. While both systems respond to blue light, they differ fundamentally in their structural organization, photochemical mechanisms, and functional outputs. This guide provides an objective comparison between the Cry2/CIB system and LOV-based systems, focusing on their core structural components: the PHR (Photolyase Homology Region) domain of Cry2 and the Jα helix of LOV domains. Understanding these key structural elements is essential for selecting the appropriate optogenetic tool for specific research applications, particularly in neuroscience and drug development where precision control of biological processes is critical.
Structural Architecture and Photoactivation Mechanisms
The PHR Domain of Cryptochrome 2 (Cry2)
The Cry2 photolyase homology region (PHR) is an approximately 500-residue domain that serves as the photosensory core of the cryptochrome 2 photoreceptor [17] [18]. This domain contains two structural subdomains: an N-terminal α/β subdomain and a C-terminal α subdomain, connected by a flexible loop [18]. The PHR domain noncovalently binds the flavin adenine dinucleotide (FAD) chromophore within its core, which is essential for blue light absorption [17]. A defining feature of Cry2 activation is its propensity for light-induced homo-oligomerization, a process critical for its physiological function [17].
Upon blue light absorption, the FAD chromophore undergoes photoreduction through an electron transfer chain involving conserved tryptophan residues known as the "Trp-triad" [17]. This photochemical event triggers significant conformational changes that enable the formation of CRY-CRY homo-oligomers. Structural studies of Arabidopsis CRY2 have revealed that photoactivated PHR domains can form tetrameric complexes arranged in ring-like structures with two distinct interfaces: head-to-tail (H-T) and head-to-head (H-H) interactions [18]. The transition from monomeric to oligomeric states represents the fundamental activation mechanism of Cry2, creating interaction surfaces for signaling partners such as CIB1 (CRY2-interacting bHLH1) [17]. The Cry2 system is notably regulated by inhibitory proteins called BICs (Blue-light Inhibitors of CRYs), which bind to the PHR domain and physically block both photoreduction and homo-oligomerization [17].
The Jα Helix of LOV Domains
LOV domains represent a subgroup of the PAS (Per-Arnt-Sim) domain superfamily that sense blue light using a noncovalently bound flavin mononucleotide (FMN) chromophore [19] [20]. A defining structural feature of many LOV domains, particularly the well-characterized Avena sativa LOV2 (AsLOV2) domain, is the C-terminal Jα helix, which plays a critical role in signal transduction [19]. In the dark state, the Jα helix is folded against the core LOV domain, maintaining the system in an inactive conformation.
Blue light absorption triggers the formation of a covalent adduct between a conserved cysteine residue and the C4a atom of the FMN isoalloxazine ring [19] [21]. This photochemical event initiates a signal transduction pathway that leads to the unfolding and dissociation of the Jα helix from the LOV core, thereby activating fused effector domains [19]. Key residues mediating this process include a conserved glutamine (Q513 in AsLOV2) that rotates out of the flavin binding pocket upon photoactivation, and asparagine (N414) that facilitates the allosteric coupling between adduct formation and Jα helix unfolding through hydrogen bond rearrangements [19]. Unlike Cry2, some LOV domains such as Aureochrome 1a from Phaeodactylum tricornutum undergo light-induced dimerization as part of their activation mechanism [20].
Table 1: Comparison of Structural Components and Photoactivation Mechanisms
| Feature | Cry2 PHR Domain | LOV Jα Helix |
|---|---|---|
| Chromophore | Flavin Adenine Dinucleotide (FAD) [17] | Flavin Mononucleotide (FMN) [19] |
| Dark State | Monomeric form [17] | Jα helix folded against LOV core [19] |
| Activation Trigger | Photoreduction of FAD via Trp-triad [17] | Cysteine-FMN adduct formation [19] |
| Light-Induced Structural Change | Homo-oligomerization (dimers/tetramers) [17] [18] | Jα helix unfolding and dissociation [19] |
| Key Conserved Residues | Trp-triad (W374, W397 in Arabidopsis CRY2) [17] | Cysteine, Gln, Asn (C450, Q513, N414 in AsLOV2) [19] |
| Regulatory Proteins | BICs inhibit oligomerization [17] | Typically none (intrinsic regulation) |
Quantitative Performance Comparison
Kinetic Parameters and Operational Characteristics
The Cry2 and LOV systems exhibit markedly different kinetic properties that determine their suitability for various experimental applications. The Cry2/CIB1 system demonstrates relatively slow activation and deactivation kinetics, with in vivo studies reporting switch-on half-times (τ₁/₂ON) of approximately 3.7 seconds and switch-off half-times (τ₁/₂OFF) of about 290 seconds at 37°C [13]. This prolonged activated state contributes to limited spatial resolution due to diffusion of activated Cry2 from illumination sites [13].
In contrast, LOV-based systems such as iLID typically display significantly faster kinetics, with some engineered variants achieving deactivation half-times on the order of seconds [13]. The wild-type AsLOV2 domain exhibits dark state recovery with a half-life of approximately 80 seconds, though mutagenesis has yielded variants with activated-state half-lives ranging from 6 seconds to 20 minutes [16]. The Aureochrome 1a LOV domain from Phaeodactylum tricornutum shows particularly slow dark recovery kinetics with time constants of 826-1500 seconds depending on construct length [20].
Spatial Resolution and Practical Performance
Spatial confinement of dimer formation represents a critical performance metric for optogenetic tools. Studies comparing three blue-light-dependent dimerization systems (Cry2/CIB1, iLID, and Magnets) have demonstrated that systems with faster switch-off kinetics achieve superior spatial resolution [13]. The slow off-kinetics of Cry2 result in significant diffusion of the activated photoreceptor away from the illumination site, leading to dimer formation in non-illuminated cellular regions [13]. This limitation persists even when Cry2 is used as the membrane-tethered component rather than the soluble prey.
LOV-based systems like iLID achieve markedly improved spatial confinement due to their faster deactivation kinetics [13]. However, this enhanced spatial resolution often comes at the expense of total dimer yield, creating a trade-off that researchers must consider based on their experimental needs [13]. The propensity of activated Cry2 to form clusters and aggregates represents an additional consideration that may complicate experimental interpretation [13].
Table 2: Quantitative Performance Comparison of Optogenetic Systems
| Parameter | Cry2/CIB1 System | LOV-Based Systems (e.g., iLID) |
|---|---|---|
| Switch-On Half-Time (τ₁/₂ON) | ~3.7 seconds (in vivo) [13] | Faster than Cry2 (specific values vary by variant) [13] |
| Switch-Off Half-Time (τ₁/₂OFF) | ~290 seconds (in vivo) [13] | Seconds to minutes (engineerable) [13] [16] |
| Spatial Resolution | Limited due to slow off-kinetics and diffusion [13] | Superior confinement with fast-cycling variants [13] |
| Dimerization Efficiency | High recruitment level [13] | Moderate (trade-off with spatial resolution) [13] |
| Tendency for Clustering | Yes (can form aggregates) [13] | Typically minimal |
| Experimental Flexibility | Primarily heterodimerization with CIB1 [17] | Various outputs: dimerization, dissociation, conformational change [19] [16] |
Experimental Protocols and Methodologies
Assessing Cry2 PHR Domain Oligomerization
Protocol 1: Analyzing Light-Induced Cry2 Oligomerization
- Purpose: To demonstrate and quantify blue light-induced oligomerization of Cry2 PHR domains.
- Materials: Purified Cry2 PHR domain protein (e.g., residues 1-489 of Arabidopsis CRY2), size exclusion chromatography (SEC) column, multi-angle light scattering (MALS) detector, blue light source (450 nm, ~5-10 μmol m⁻² s⁻¹), UV-Vis spectrophotometer.
- Method:
- Divide the purified Cry2 PHR protein into two aliquots. Keep one aliquot in complete darkness and expose the other to continuous blue light for 5-15 minutes.
- Analyze both samples using SEC-MALS under respective light conditions (requires light-safe SEC setup for light-treated sample).
- Monitor elution profiles at 280 nm (protein) and 450 nm (FAD chromophore).
- Compare molar masses calculated from light scattering data between dark and light conditions.
- Confirm protein oligomerization by a shift to earlier elution volumes and increased molar mass in light-treated samples.
- Validation: Include constitutively oligomerizing mutants (e.g., CRY2W374A) as positive controls and oligomerization-deficient mutants (e.g., CRY2W374A/W349A) as negative controls [17].
- Related Assays: Co-immunoprecipitation with CIB1 to demonstrate functional consequences of oligomerization [17].
Measuring Jα Helix Unfolding in LOV Domains
Protocol 2: Monitoring Jα Helix Dynamics in LOV Domains
- Purpose: To characterize light-induced conformational changes in the Jα helix of LOV domains.
- Materials: Purified LOV domain protein (e.g., AsLOV2), circular dichroism (CD) spectrometer, time-resolved infrared (TRIR) spectroscopy setup, blue light source.
- Method:
- Record far-UV CD spectra (190-250 nm) of the LOV protein in dark-adapted and blue light-illuminated states.
- Quantify changes in α-helical content by monitoring the signal at 222 nm.
- For kinetic analysis, use TRIR spectroscopy to track changes in the amide I region (1600-1700 cm⁻¹) after laser flash photolysis.
- Identify the specific spectral signature of Jα helix unfolding (~1620-1640 cm⁻¹) [19].
- Fit kinetic data to determine rates of helix unfolding and refolding.
- Validation: Utilize Jα helix deletion mutants or point mutants (e.g., N414A in AsLOV2) that alter unfolding kinetics [19].
- Related Assays: Hydrogen/deuterium exchange coupled to mass spectrometry (HDX-MS) to probe solvent accessibility changes [20].
Cellular Recruitment Assays
Protocol 3: Comparing Spatial Confinement in Live Cells
- Purpose: To evaluate the spatial precision of Cry2 and LOV systems in a cellular environment.
- Materials:
- Method:
- Co-transfect cells with appropriate bait-prey pairs for each system.
- Identify cells with moderate expression levels of both components.
- Irradiate a small (3 μm × 3 μm) region of interest (ROI) with 200-ms pulses of 488 nm light every 2 seconds for 5 minutes.
- Acquire time-lapse images of the entire cell to monitor prey redistribution.
- Quantify the fraction of total cellular prey recruited to the target organelle outside the illuminated ROI over time.
- Analysis: Systems with faster off-kinetics (e.g., iLID) will show minimal recruitment outside the ROI, while Cry2 will display substantial diffuse recruitment due to its slow dissociation [13].
Signaling Pathways and Molecular Mechanisms
The diagrams below illustrate the distinct photoactivation pathways and signal transduction mechanisms for Cry2 and LOV domain systems.
Research Reagent Solutions
Table 3: Essential Research Reagents for Cry2 and LOV Studies
| Reagent Category | Specific Examples | Function/Application | System |
|---|---|---|---|
| Photoreceptor Constructs | Arabidopsis CRY2 PHR (aa 1-498) [13] | Core photoreceptor component for Cry2 experiments | Cry2 |
| AsLOV2 (Avena sativa LOV2) domains [19] | Benchmark LOV domain for structural/functional studies | LOV | |
| Interaction Partners | CIBN (N-terminal 170 aa of CIB1) [13] | Cry2 binding partner for heterodimerization systems | Cry2 |
| SspB/SsrA peptide pairs [13] | Binding partners for iLID dimerization system | LOV | |
| Key Mutants | CRY2W374A (constitutively active) [17] | Control for oligomerization-dependent effects | Cry2 |
| CRY2 interface mutants (W349A, R439L) [17] | Negative controls for oligomerization studies | Cry2 | |
| AsLOV2 N414A/Q mutants [19] | Probing allosteric pathways in Jα helix unfolding | LOV | |
| Expression Systems | Mammalian expression vectors (e.g., pcDNA3.1) | Cellular recruitment and functional assays | Both |
| E. coli protein expression systems | Large-scale protein production for biophysics | Both | |
| Detection Tools | Anti-GFP/FLAG antibodies | Immunoprecipitation and Western blot analysis | Both |
| Fluorescent protein fusions (EGFP, mCherry) [13] | Live-cell imaging and recruitment assays | Both |
Application Considerations and System Selection
Cry2/CIB1 System Advantages:
- Strong signaling output with high recruitment levels in cellular assays [13]
- Established protocol with extensive validation in literature [17] [13]
- Compatibility with various fusion partners and subcellular localizations [13]
Cry2/CIB1 System Limitations:
- Slow off-kinetics limit temporal and spatial resolution [13]
- Tendency to form clusters or aggregates upon activation [13]
- Endogenous activity in some mammalian cell types may cause background effects
LOV-Based System Advantages:
- Engineerable kinetics with variants spanning wide temporal ranges [13] [16]
- Superior spatial confinement with fast-cycling variants [13]
- Minimal cluster formation providing more homogeneous cellular distribution
- Versatile output modalities including dimerization, dissociation, and conformational changes [16]
LOV-Based System Limitations:
- Reduced total dimer yield in fast-cycling variants [13]
- Potential dark activity in some engineered systems
- Shorter signaling duration requiring sustained illumination for prolonged effects
Selection Guidelines for Specific Applications
- For high spatial precision applications (subcellular perturbation, patterned stimulation): Choose fast-cycling LOV variants (e.g., iLID) with superior spatial confinement [13].
- For maximal signal amplification (transcriptional activation, strong pathway activation): Select Cry2/CIB1 for its high recruitment efficiency, despite lower spatial resolution [13].
- For rapid, reversible control (kinetic studies, oscillatory signaling): Prefer LOV systems with tuned off-kinetics matching experimental timescales [19] [13].
- For prolonged signaling events (sustained transcription, developmental processes): Cry2/CIB1 may be preferable due to its persistent activated state [13].
- For minimal system cross-talk: Consider LOV systems to avoid potential interference with endogenous cryptochromes in mammalian systems.
The continuing development of both Cry2 and LOV systems promises further expansion of the optogenetic toolkit, with computational design and directed evolution approaches yielding variants with enhanced properties for specialized applications in basic research and therapeutic development [22].
Practical Implementation: Activating Signaling Pathways and Controlling Cellular Processes
Optogenetics has evolved beyond the control of neuronal excitability to enable precise, light-dependent manipulation of fundamental cellular processes, including protein-protein interactions, signal transduction, and organelle positioning. Among the diverse photosensory proteins available, the CRY2/CIB1 system from Arabidopsis thaliana and various light-oxygen-voltage (LOV) domains have emerged as particularly versatile platforms for controlling protein localization and function. These systems enable three primary strategic approaches for optical control: membrane recruitment, cytosolic assembly, and sequestration.
This guide provides a detailed comparative analysis of the CRY2/CIB and LOV domain systems, focusing on their operational principles, kinetic properties, and experimental performance in diverse cellular contexts. We present quantitative data from key studies, detailed methodological protocols, and visual representations of signaling pathways to assist researchers in selecting and implementing the optimal system for their specific applications in basic research and drug development.
System Fundamentals and Mechanisms of Action
The CRY2/CIB System
The CRY2/CIB system is based on the blue light-dependent interaction between the plant cryptochrome 2 (CRY2) and its binding partner CIB1 (CRYPTOCHROME-INTERACTING BASIC HELIX-LOOP-HELIX 1). Upon illumination with blue light (peak activation ~450 nm), CRY2 undergoes a conformational change that enables rapid binding to CIB1 [23] [24]. This interaction is reversible in the dark, with dissociation half-lives ranging from minutes to tens of minutes depending on the specific CRY2 variant used [25]. The system requires flavin adenine dinucleotide (FAD) as an endogenous chromophore, which is ubiquitously present in mammalian cells, eliminating the need for exogenous cofactor addition [26].
Key structural insights have led to optimized system performance. The minimal photolyase homology region (PHR) of CRY2 (residues 1-498) is sufficient for light-dependent interaction, while further truncations (e.g., CRY2(535)) show improved dynamic range with reduced dark activity [25]. Similarly, the N-terminal fragment of CIB1 (CIBN, residues 1-170) or an even smaller domain (CIB81, residues 1-81) maintains robust light-dependent binding while minimizing potential non-specific interactions [25].
LOV Domain-Based Systems
LOV domains are blue light-sensitive photoreceptor domains found in various plants, bacteria, and fungi. Unlike CRY2/CIB, LOV domains typically utilize the light-induced conformational change to control protein function through intramolecular folding or homodimerization [23]. Upon blue light illumination (440-473 nm), a conserved cysteine residue in the LOV domain forms a covalent adduct with the flavin mononucleotide (FMN) chromophore, leading to structural rearrangements that can relieve autoinhibition or promote dimerization [23].
The specific mechanisms of LOV-based optogenetic tools vary considerably depending on their origin and engineering. Some LOV systems directly fuse the domain to an effector protein, using the light-induced conformational change to control its activity. Others exploit LOV domain heterodimerization with natural or engineered binding partners to recruit signaling domains [23]. Like CRY2/CIB, LOV domains utilize endogenous flavin chromophores, making them readily applicable across cell types and model organisms.
Figure 1: Mechanism of Action for CRY2/CIB and LOV Domain Systems. The CRY2/CIB system operates through blue light-induced heterodimerization, enabling recruitment of cytosolic proteins to specific cellular locations. LOV domains typically function through intramolecular conformational changes that release autoinhibition of fused effector domains upon blue light illumination.
Quantitative Performance Comparison
Kinetic Properties and Spectral Characteristics
Table 1: Fundamental Properties of CRY2/CIB and LOV Domain Systems
| Parameter | CRY2/CIB System | LOV Domain Systems | Experimental Context |
|---|---|---|---|
| Activation Wavelength | 450 nm [26] | 440-473 nm [23] | Mammalian cells |
| Reversion Mechanism | Dark reversion [26] | Dark reversion or photoreversion [23] | Varies by specific LOV variant |
| Activation Time | Seconds [26] | Seconds to minutes [23] | Mammalian cells |
| Dark Reversion Half-Life | ~5.5 min (wild-type) [25]; 2.5-24 min (mutants) [25] | Seconds to hours [23] | Mammalian cells, 34°C [25] |
| Chromophore | FAD (endogenous) [26] | FMN (endogenous) [23] | No exogenous addition needed |
| Dynamic Range (Fold Induction) | Up to 158-fold [24] | Variable by application | Split Cre recombination [24] |
| Spatial Resolution | Subcellular [24] | Subcellular [23] | Limited by diffraction |
| Multiphoton Activation | 820-980 nm [24] | Typically single-photon | Demonstrated in brain slices [24] |
Experimental Performance Metrics
Table 2: Performance in Key Experimental Applications
| Application | CRY2/CIB Performance | LOV Domain Performance | References |
|---|---|---|---|
| Membrane Recruitment | Translocation in <300 ms; >95% cells responsive [24] | Generally slower; application-dependent | HEK293 cells [24] |
| Transcriptional Activation | Strong dose-dependence to light pulses [24] | Successful but kinetics vary | Yeast system [24] |
| Cre Recombinase Activation | 158-fold induction over dark control [24] | Limited demonstrations | HEK293T cells [24] |
| Cytosolic Assembly | Efficient clustering [23] | Application-dependent efficiency | Various cell types |
| Sequestration | Effective for inactivation [23] | Effective for inactivation [23] | Various cell types |
| Bacterial Applications | Functional in E. coli, B. subtilis, C. crescentus, S. pneumoniae [27] | Limited reports in bacteria | Multiple bacterial species [27] |
Experimental Protocols and Implementation
Membrane Recruitment Assay Using CRY2/CIB
Principle: This assay demonstrates light-induced recruitment of cytosolic CRY2-fused proteins to membrane-tethered CIBN, enabling quantitative analysis of translocation kinetics and efficiency [25] [24].
Key Reagents and Constructs:
- CIBN-pmGFP: N-terminal fragment of CIB1 (residues 1-170) fused to a prenylated EGFP for plasma membrane localization [24]
- CRY2-mCherry: Full-length CRY2 or CRY2PHR (residues 1-498) fused to mCherry [24]
- Cell Line: HEK293T cells or other easily transfectable mammalian cells
- Imaging System: Confocal microscope with 488 nm and 561 nm laser lines, temperature control (34°C), and precise light stimulation capability
Methodology:
- Cell Preparation and Transfection:
- Plate HEK293T cells on glass-bottom dishes at 50-70% confluence
- Co-transfect with CIBN-pmGFP (0.5-1 μg) and CRY2-mCherry (1-2 μg) using standard transfection reagents
- Incubate for 24-48 hours to allow protein expression
- Maintain cells in darkness or dim red light to prevent pre-activation
Image Acquisition:
- Use a confocal microscope with temperature maintained at 34°C
- Acquire baseline images of both GFP and mCherry channels in the dark
- Apply blue light stimulation (488 nm laser, 1-5% power) with continuous or pulsed illumination
- Capture time-lapse images every 1-5 seconds for 10-15 minutes
- For dissociation kinetics, cease blue light and continue imaging in the dark for 30-60 minutes
Data Analysis:
- Quantify mCherry fluorescence intensity at the plasma membrane versus cytosol over time
- Calculate translocation half-time (t1/2) from exponential fits to association curves
- Determine dissociation half-life from decay curves following light removal
- Compare CRY2 variants (e.g., wild-type vs. L348F vs. W349R) for altered kinetics [25]
Expected Results: Wild-type CRY2 typically shows translocation within seconds of illumination and dissociation with a half-life of approximately 5.5 minutes at 34°C. The L348F mutant exhibits prolonged membrane association (half-life ~24 minutes), while W349R shows faster dissociation (half-life ~2.5 minutes) [25].
Bacterial Protein Recruitment Using CRY2/CIB
Principle: This protocol adapts the CRY2/CIB system for subcellular protein targeting in bacterial cells, enabling spatial control of cellular processes in E. coli and other model bacteria [27].
Key Reagents and Constructs:
- TetR-CIBN: Tetracycline repressor fused to CIBN for chromosomal DNA targeting [27]
- CRY2-mCherry: CRY2 photolyase homology region fused to mCherry [27]
- Bacterial Strain: E. coli with 240x tetO array integrated near the origin of replication [27]
- Expression System: Single plasmid with coupled expression or two-plasmid system with independent promoters [27]
Methodology:
- Strain Preparation:
- Use E. coli strain harboring 240x tetO array sequence inserted near oriC [27]
- Transform with plasmid expressing TetR-CIBN and CRY2-mCherry
- Grow cultures in appropriate antibiotics at 37°C with protection from light
Microscopy and Light Activation:
- Mount bacterial cells on agarose pads for microscopy
- Image using widefield or confocal microscopy with 488 nm and 561 nm excitation
- Apply blue light activation (488 nm, 30 ms pulses at 84.6 W/cm² every 5 seconds) [27]
- Monitor CRY2-mCherry localization over time
Quantification:
Expected Results: In >95% of cells, CRY2-mCherry forms distinct foci at tetO array locations within minutes of blue light illumination. The system shows complete reversibility, with foci dispersing within approximately 40 minutes after light removal, and can be reactivated multiple times with consistent efficiency [27].
Comparative Analysis and Strategic Implementation
Application-Specific System Selection
Figure 2: Decision Framework for Selecting Between CRY2/CIB and LOV Domain Systems. The optimal choice depends on specific experimental requirements, with CRY2/CIB excelling in reversible heterodimerization applications and LOV domains offering advantages for single-component conformational control.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Implementing CRY2/CIB and LOV Domain Systems
| Reagent Category | Specific Examples | Function and Utility | Source/Reference |
|---|---|---|---|
| CRY2 Constructs | CRY2PHR (1-498), CRY2(535), CRY2(L348F), CRY2(W349R) | Light-sensing components with varying kinetics and dark activity | [25] |
| CIB1 Constructs | CIBN (1-170), CIB81 (1-81) | Binding partners with minimal size and optimized performance | [25] |
| LOV Domain Tools | AsLOV2, LOV2-based conformational switches | Intramolecular control of fused effector domains | [23] |
| Expression Systems | Single plasmid (coupled), Two-plasmid (independent) | Flexible control of component expression levels | [27] |
| Membrane Tags | CIBN-pmGFP (prenylated) | Plasma membrane targeting for recruitment assays | [24] |
| Localization Tags | TetR-CIBN, histone fusions, organelle markers | Subcellular targeting to specific compartments | [27] |
| Reporters | Split Cre, Gal4-based transcription, fluorescent proteins | Readout systems for dimerization efficiency | [24] |
| Bacterial Strains | E. coli with tetO arrays, B. subtilis, C. crescentus | Model systems for bacterial optogenetics | [27] |
The strategic implementation of optogenetic control through membrane recruitment, cytosolic assembly, and sequestration has revolutionized our ability to manipulate cellular signaling with unprecedented spatial and temporal precision. Both CRY2/CIB and LOV domain systems offer distinct advantages that make them suitable for different experimental scenarios.
The CRY2/CIB system excels in applications requiring rapid, reversible heterodimerization with tunable kinetics, particularly for membrane recruitment and split protein reconstitution. Its validation across diverse model systems, including mammalian cells, neurons, and bacteria, combined with the availability of engineered variants with optimized properties, makes it an exceptionally versatile platform. The recent development of photocycle mutants with extended or shortened signaling state lifetimes further enhances its experimental utility [25].
LOV domain systems offer complementary strengths, particularly for applications requiring intramolecular control of protein function with minimal component complexity. Their well-characterized photocycle and the availability of established conformational switches make them ideal for all-or-none activation of specific effector domains.
Future developments in both systems will likely focus on expanding the spectral range for multiplexed control, enhancing dynamic range further, and improving tissue penetration capabilities. The continued optimization of these optogenetic tools will undoubtedly yield even more powerful approaches for dissecting complex biological processes and developing light-controlled therapeutic strategies.
Activating Receptor Tyrosine Kinase Signaling (e.g., Trk, FGFR, Ephrin) with Optogenetic Tools
Receptor Tyrosine Kinases (RTKs) are crucial regulators of cellular processes, including proliferation, differentiation, and survival. Traditional methods for studying RTK signaling rely on ligand stimulation, which lacks spatiotemporal precision and often activates multiple receptor subtypes simultaneously. Optogenetic tools have emerged as powerful alternatives that enable unprecedented control over RTK activation using light. These tools primarily utilize light-sensitive proteins from plants and microbes, such as cryptochrome 2 (CRY2) and light-oxygen-voltage-sensing (LOV) domains, which undergo conformational changes or oligomerization upon illumination [4] [28]. By fusing these photoreceptors to RTK signaling domains, researchers can achieve precise spatial and temporal control over intracellular signaling pathways, bypassing the need for natural ligands and enabling sophisticated experiments dissecting signaling mechanisms in living cells [29] [28].
This review compares two principal optogenetic systems—CRY2/CIB and LOV domains—for activating RTK signaling, focusing on their mechanistic bases, experimental performance, and practical applications. We provide structured comparisons of their quantitative parameters, detailed experimental protocols, and visualization of signaling pathways to guide researchers in selecting appropriate tools for specific biological questions.
Comparative Analysis of Optogenetic Systems
CRY2/CIB vs. LOV Domain Systems: Mechanisms and Benchmarking
The CRY2/CIB system derives from Arabidopsis thaliana cryptochrome 2. Blue light exposure (typically 440-488 nm) induces both CRY2-CIB1 hetero-dimerization and CRY2-CRY2 homo-oligomerization [3] [4]. This dual capability has been exploited to activate various RTKs, including EphB2, RET, and Trk receptors [30] [31]. A key advantage is its robustness without requiring exogenous cofactors, though its inherent homo-oligomerization can complicate applications designed purely for hetero-dimerization [3]. Protein engineering has produced optimized CRY2 variants: CRY2olig (E490G) enhances clustering for applications requiring higher-order oligomerization [31], while CRY2high and CRY2low mutants feature tuned oligomerization capacities through C-terminal charge modifications [3].
The LOV domain system, sourced from phototropins such as Avena sativa LOV2 (AsLOV2), undergoes conformational changes in its C-terminal Jα helix upon blue light exposure, undocking from its core domain [4]. This mechanism has been harnessed in tools like the improved light-inducible dimer (iLID), which hetero-dimerizes with its binding partner SspB [4]. Unlike CRY2, LOV-based systems typically exhibit less background homo-oligomerization, making them preferable for applications requiring precise hetero-dimerization without unintended clustering.
A direct benchmarking study comparing optical dimerizer systems revealed significant differences in their performance characteristics [32]. While CRY2/CIB and TULIPs (a LOV-based system) showed similar light sensitivity and activation profiles in yeast transcriptional assays, CRY2/CIB demonstrated slightly lower background activity in the dark when regulating a yeast MAPK pathway [32]. Red-light regulated systems (phyB/PIF3 and phyB/PIF6) showed varied light sensitivity and fold-activation levels, but their requirement for exogenous chromophores can limit experimental ease in some biological systems.
Table 1: Performance Benchmarking of Optogenetic Dimerizer Systems
| Optogenetic System | Light Wavelength | Key Interactions | Dark Activity | Fold-Activation | Cofactor Requirement |
|---|---|---|---|---|---|
| CRY2/CIB | Blue (440-488 nm) | Hetero-dimerization + Homo-oligomerization | Low to moderate | High | No (endogenous FAD) |
| TULIPs (LOV-based) | Blue (~450 nm) | Primarily hetero-dimerization | Moderate | Comparable to CRY2/CIB | No |
| PhyB/PIF3 | Red/Far-red | Hetero-dimerization | Variable | Variable | Yes (phycocyanobilin) |
| PhyB/PIF6 | Red/Far-red | Hetero-dimerization | Variable | Variable | Yes (phycocyanobilin) |
Table 2: Engineered CRY2 Variants for Enhanced Optogenetic Control
| CRY2 Variant | Key Mutation(s) | Oligomerization Behavior | Primary Applications |
|---|---|---|---|
| CRY2wt | None | Moderate homo-oligomerization | General use |
| CRY2olig | E490G | Enhanced clustering | RTKs requiring higher-order oligomerization (e.g., EphB2) |
| CRY2high | C-terminal charge modifications | Elevated oligomerization | Applications demanding robust clustering |
| CRY2low | C-terminal charge modifications + fluorescent protein fusion | Suppressed oligomerization | CRY2-CIB1 applications requiring minimal homo-oligomerization |
Quantitative Comparison of Optogenetic RTK Tools
Various optogenetic RTK tools have been developed and characterized across different cellular and model systems. The following table summarizes key operational parameters for selected tools, demonstrating the range of light conditions and experimental contexts in which they have been successfully applied.
Table 3: Performance Characteristics of Selected Optogenetic RTK Tools
| Optogenetic Tool | Targeted RTK | Light Parameters | Activation Kinetics | Key Readouts | Reference |
|---|---|---|---|---|---|
| OptoEphB2 | EphB2 | 440 nm, ~10 mW/cm², pulsed | Phosphorylation: τ ~50 s; Clusters: τ ~15 s | Receptor phosphorylation, cell rounding, dendritic filopodia growth | [31] |
| optoRET | RET | Blue light, various intensities | ERK/AKT activation within minutes | Grb2 recruitment, ERK/AKT phosphorylation, axon filopodia formation | [30] |
| Opto-hRET (LOV-based) | RET | Blue light, optimized parameters | Not specified | Neuronal survival, potential therapeutic applications | [30] |
| OptoTrk | TrkA/B/C | 470 nm, ~1 mW/cm² | Not specified | Neuronal differentiation, survival | [4] |
| OptoFGFR | FGFR | 488 nm, 1.30-64.94 mW/cm² | Not specified | Cell proliferation, differentiation | [4] |
Experimental Protocols for Key Applications
General Workflow for Optogenetic RTK Activation
The experimental process for implementing and validating optogenetic RTK tools typically follows a standardized workflow, from molecular construct design to functional validation. The diagram below illustrates this generalized protocol.
Protocol 1: Activation and Validation of OptoEphB2 Signaling
The OptoEphB2 tool exemplifies the CRY2olig approach for activating EphB2 forward signaling without ephrin ligand stimulation [31].
Molecular Design:
- Replace EphB2 extracellular and transmembrane domains with N-terminal myristoylation signal from c-Src for plasma membrane targeting
- Fuse CRY2olig (E490G mutant) to EphB2 intracellular domain
- Include mCherry or other fluorescent protein for visualization
- Consider kinase-dead mutant (K99M) as negative control
Transfection and Culture:
- Transfect HEK293T cells or mouse embryonic fibroblasts (MEFs) using standard transfection reagents (e.g., Calfectin)
- Culture in Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS and 1% Penicillin/Streptomycin
- Maintain at 37°C with 5% CO₂
- For neuronal studies: transfect hippocampal neurons and culture in appropriate neuronal medium
Light Stimulation Parameters:
- Wavelength: 440 nm blue light
- Intensity: 10 mW/cm²
- Pulse regimen: Three 250-ms pulses delivered 4.5 s apart, or 0.1 Hz pulsed illumination
- For spatial control: use digital light patterning to illuminate specific subcellular regions
Validation assays:
- Western blotting: Assess tyrosine phosphorylation 3-10 minutes after illumination
- Immunoprecipitation: Verify OptoEphB2 phosphorylation specifically
- Live-cell imaging: Monitor cluster formation kinetics (time constant ~15 s)
- Phenotypic analysis: Quantify cell rounding in fibroblasts or filopodia formation in neurons
Expected Results:
- Robust tyrosine phosphorylation visible within 1-3 minutes of illumination
- Light-induced cluster formation at plasma membrane
- Kinase-dependent cell rounding in fibroblasts (≈30% reduction in cell area)
- Dendritic filopodia growth in hippocampal neurons via Abl2/Arg pathway
Protocol 2: Light Control of RET Signaling with optoRET
The optoRET system enables optical control of RET signaling, important in neuronal development and maintenance [30].
Construct Design:
- Fuse human RET common cytoplasmic region (aa 658-1062) to CRY2PHR
- Include Lyn N-terminal myristoylation sequence for membrane targeting
- Incorporate mCitrine or similar fluorescent tag
- Use CRY2 E281A mutation to minimize basal activity
Neuronal Culture and Transfection:
- Culture HEK293T cells or primary neurons in appropriate media
- For neuronal differentiation: treat Neuro2a cells with 20 µM retinoic acid in DMEM with 1% FBS for 3 days
- Transfert using calcium phosphate or lipofection methods
- For in vivo applications: express in dopaminergic neurons of substantia nigra
Light Activation:
- Blue light illumination (parameters optimized for specific experimental setup)
- Vary illumination duration to dynamically modulate signaling strength
- For localized activation: illuminate distal axons to study retrograde signaling
Downstream Signaling Assessment:
- ERK-KTR reporter: Monitor ERK activity via nuclear-cytoplasmic shuttling
- Immunoblotting: Detect phospho-ERK and phospho-AKT at various time points
- Grb2 recruitment: Assess via co-immunoprecipitation or proximity assays
- Morphological changes: Quantify filopodia-like protrusions in stimulated regions
Key Findings:
- Robust, sustained ERK activation compared to other optoRTKs
- Efficient AKT pathway stimulation
- Retrograde signaling from distal axons to soma
- Cdc42-dependent filopodia formation in stimulated regions
Signaling Pathways and Molecular Mechanisms
RTK Activation Mechanisms via Optogenetic Tools
Optogenetic RTK tools emulate natural activation mechanisms by inducing light-controlled receptor oligomerization, which triggers autophosphorylation and downstream signaling cascade initiation. The following diagram illustrates the key signaling pathways activated by optogenetic RTK tools and their biological outcomes.
CRY2 Molecular Interactions and Engineering Strategies
Understanding CRY2 interaction mechanisms has enabled engineering of optimized variants for specific applications. Research has revealed that CRY2-CIB1 hetero-dimerization and CRY2-CRY2 homo-oligomerization are governed by distinct molecular interfaces [3].
Molecular Interfaces:
- CRY2-CIB1 interaction: Dependent on positively charged residues at the N-terminus of CRY2 (Lys-2, Lys-5, Lys-6)
- CRY2-CRY2 interaction: Governed by electrostatic charges at C-terminal residues 489 and 490
- Engineering principle: Positive charges facilitate oligomerization, while negative charges inhibit it
Structural Insights:
- No crystal structure available for monomeric or oligomeric CRY2
- Homology modeling based on Arabidopsis CRY1 structure informs engineering approaches
- Membrane localization enhances CRY2 oligomerization capacity
- Tagging with bulky proteins (e.g., tdTomato) sterically hinders oligomer formation
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Optogenetic RTK Studies
| Reagent/Category | Specific Examples | Function/Application | Source/Reference |
|---|---|---|---|
| CRY2 Constructs | CRY2wt, CRY2olig (E490G), CRY2high, CRY2low | Light-sensitive actuators with tuned oligomerization properties | [3] [31] |
| LOV Constructs | iLID, TULIPs, VfAu1-LOV | Alternative blue-light systems with different oligomerization characteristics | [32] [4] |
| OptoRTK Tools | OptoEphB2, optoRET, OptoTrk, OptoFGFR | Light-activatable RTK signaling domains | [33] [30] [31] |
| Cell Lines | HEK293T, MEFs, Neuro2a | Model systems for validation and signaling studies | [3] [30] [31] |
| Signaling Reporters | ERK-KTR, AKT biosensors, Ca²⁺ indicators | Live-cell monitoring of pathway activation | [33] [30] |
| Light Equipment | Blue LED arrays (440-488 nm), digital light patterning systems | Controlled illumination with spatial and temporal precision | [4] [31] |
Optogenetic tools for activating RTK signaling, particularly those based on CRY2/CIB and LOV domain systems, provide powerful methods for dissecting complex signaling pathways with high spatiotemporal precision. The CRY2 system offers robustness and engineering flexibility through variants with tuned oligomerization properties, making it particularly suitable for applications requiring higher-order clustering or specific subcellular localization. LOV-based systems provide alternative approaches with potentially lower background homo-oligomerization. Selection between these systems should be guided by specific experimental needs, including desired oligomerization state, kinetic requirements, and compatibility with biological models. As these tools continue to evolve through protein engineering and better understanding of molecular mechanisms, they will further enhance our ability to precisely manipulate and understand cellular signaling networks in health and disease.
Opto-CRISPR systems represent a groundbreaking fusion of optogenetics and CRISPR-based genome editing, enabling unprecedented spatial and temporal control over gene regulation and editing processes. By integrating light-sensitive proteins with CRISPR-Cas machinery, researchers can now manipulate cellular functions with exceptional precision, overcoming limitations of traditional chemically inducible systems that lack rapid reversibility and spatial resolution [10] [34]. These innovative tools have revolutionized our ability to study complex biological processes, from fundamental gene regulation to therapeutic applications in disease models.
The core principle underlying Opto-CRISPR technology involves the coupling of photosensory domains with CRISPR components, creating systems that remain inactive in darkness but become functional upon light illumination. This paradigm shift allows researchers to precisely dictate when and where gene editing or transcriptional regulation occurs, thereby minimizing off-target effects and enabling the study of dynamic cellular processes with minute-scale precision [12]. As these technologies continue to evolve, they hold tremendous promise for both basic research and clinical applications, particularly in the realm of precision medicine and synthetic biology.
This review comprehensively compares the two dominant optogenetic systems used in Opto-CRISPR applications: the Cry2/CIB system derived from Arabidopsis thaliana cryptochrome 2, and various LOV domain-based systems originating from light-oxygen-voltage sensing domains. We examine their respective mechanisms, performance characteristics, and optimal applications within gene regulation and editing contexts, providing researchers with the necessary framework to select appropriate tools for their specific experimental needs.
Comparative Analysis of Cry2/CIB vs. LOV Domain Systems
Core Characteristics and Mechanisms
Table 1: Fundamental Properties of Cry2/CIB and LOV Domain Optogenetic Systems
| Property | Cry2/CIB System | LOV Domain Systems |
|---|---|---|
| Origin | Arabidopsis thaliana cryptochrome 2 | Various organisms (e.g., Erythrobacter litoralis, Avena sativa) |
| Chromophore | Flavin adenine dinucleotide (FAD) | Flavin mononucleotide (FMN) |
| Activation Wavelength | Blue light (~450 nm) | Blue light (~450 nm) |
| Key Components | CRY2 (photosensor), CIB1 (interactor) | LOV domain (photosensor), various interaction partners |
| Typical Response Time | Seconds to minutes | Milliseconds to seconds |
| Reversion Kinetics | Slow (t½ ~5.5 minutes) [25] | Fast (t½ ~50 seconds) [10] |
| Multimerization Tendency | CRY2 clusters upon activation [13] | Minimal clustering |
| Spatial Resolution | Moderate [13] | High [13] |
The Cry2/CIB system utilizes the light-sensitive CRY2 protein that undergoes a conformational change upon blue light exposure, enabling interaction with its binding partner CIB1. This interaction can be harnessed to bring together split CRISPR-Cas components or recruit transcriptional effectors to specific genomic loci. A notable characteristic of CRY2 is its tendency to form light-dependent clusters, which can be advantageous for applications requiring strong signal amplification but may reduce spatial precision [13] [25].
In contrast, LOV domain-based systems such as EL222 and iLID employ a different mechanism where blue light triggers the formation of a covalent adduct between a conserved cysteine residue in the LOV domain and the FMN chromophore. This results in conformational changes that either expose hidden interaction surfaces or disrupt intramolecular interactions. LOV domains typically exhibit faster reversion kinetics compared to Cry2/CIB, allowing for more rapid on/off switching and finer temporal control [10] [13].
Performance Metrics in Gene Regulation Applications
Table 2: Performance Comparison in Gene Regulation Applications
| Parameter | Cry2/CIB System | LOV Domain Systems | Experimental Context |
|---|---|---|---|
| Fold Induction | Up to 260-fold [25] | Up to 570-fold [10] | Transcriptional activation |
| Dark State Activity | Low to moderate background [25] | Minimal basal activity [10] | Mammalian cells |
| Activation Kinetics | t½ ON ~5.5 min [25] | t½ ON: seconds [10] | Recruitment to DNA |
| Deactivation Kinetics | t½ OFF ~5.5-24 min [25] | t½ OFF: ~50 sec [10] | Dark reversion |
| Dynamic Range | 26-fold improvement with CRY2(535) [25] | 570-fold vs. dark state [10] | Light vs. dark conditions |
| Spatial Precision | Moderate due to clustering [13] | High with minimal diffusion [13] | Subcellular recruitment |
When deployed for gene regulation applications, the DEL-VPR system (a LOV domain-based tool utilizing the EL222 protein from Erythrobacter litoralis fused to the VPR transcriptional activator) demonstrates exceptional induction capabilities, achieving up to 570-fold increase in target gene expression under blue light illumination [10]. This performance surpasses many conventional CRISPR activation systems and even rivals strong constitutive promoters like CMV. The system exhibits minimal basal activity in darkness, making it particularly suitable for applications requiring tight regulation of toxic or influential genes.
The Cry2/CIB system also shows robust induction capabilities, with optimized CRY2 truncations (CRY2(535)) demonstrating a 26-fold reduction in dark activity compared to earlier variants while maintaining strong light-induced activation [25]. The development of CRY2 photocycle mutants with altered kinetics further enhances its versatility, with the L348F variant exhibiting an extended signaling state half-life of approximately 24 minutes compared to the wild-type ~5.5 minutes, and the W349R variant showing a shorter half-life of about 2.5 minutes [25]. These engineered variants enable researchers to match the tool's kinetic properties to their specific experimental timeframe.
Experimental Protocols for Key Opto-CRISPR Applications
Protocol for Light-Activated Transcriptional Control Using DEL-VPR
The DEL-VPR system represents one of the most potent LOV domain-based tools for optogenetic gene regulation. Below is a standardized protocol for implementing this system for transcriptional control in mammalian cells:
Reagents and Equipment:
- DEL-VPR construct (EL222-VPR fusion) [10]
- Reporter plasmid with 5xC120-minP promoter driving target gene [10]
- HEK293T or CHO-K1 cells [10]
- Blue LED light source (450-470 nm, 1-10 μW/mm²) [10]
- Standard cell culture materials and transfection reagents
Procedure:
- Cell Seeding and Transfection:
- Seed HEK293T or CHO-K1 cells in appropriate multi-well plates 24 hours prior to transfection
- Co-transfect cells with DEL-VPR expression plasmid and reporter plasmid using preferred transfection method (e.g., lipofection, electroporation)
- Maintain cells in darkness or under red safelight conditions to prevent premature activation
Light Stimulation:
- 24-48 hours post-transfection, expose experimental groups to blue light (450-470 nm)
- Optimize light intensity (typically 1-10 μW/mm²) and duration based on experimental requirements
- For temporal control applications, utilize pulsed illumination (e.g., 200-ms pulses every 2 seconds) [10]
- Maintain control groups in complete darkness
Analysis and Validation:
- Quantify gene expression changes via RT-qPCR, fluorescence measurement, or Western blotting
- Assess induction fold-change by comparing light-stimulated vs. dark controls
- Evaluate potential phototoxicity through cell viability assays
Troubleshooting Notes:
- High basal activity may indicate excessive DEL-VPR expression; titrate plasmid amounts
- Low induction may require optimization of light parameters or promoter elements
- The 5xC120 promoter with minimal promoter (minP) shows optimal performance [10]
Protocol for CRISPRi Regulation Using PhoBIT1 System
The PhoBIT1 system exemplifies how LOV domains can be engineered for optogenetic control of CRISPR interference (CRISPRi) applications:
Reagents and Equipment:
- dCas9-ssrA fusion construct [12]
- sspB(LOV2)-BFP-KRAB construct (PhoBIT1) [12]
- sgRNA expression vector targeting gene of interest
- Blue light source (470 nm)
- Flow cytometer or fluorescence microscope for readout
Procedure:
- System Assembly:
Cell Transfection and Light Application:
- Co-transfect all components into target cells
- For repression, maintain cells in darkness to allow PhoBIT1-mediated KRAB recruitment to dCas9
- For de-repression, expose to 470-nm blue light to induce dissociation of KRAB domain
- For sustained control, utilize light-dark cycling
Assessment of Gene Repression:
- Monitor target gene expression via reporter (eGFP) fluorescence or mRNA quantification
- Measure repression efficiency by comparing to non-targeting sgRNA controls
- Kinetics analysis: PhoBIT1 shows dissociation t½ ~8.5 sec and reassociation t½ ~28.1 sec [12]
Applications:
- This system enables reversible gene silencing with ~60% repression efficiency in the dark state [12]
- Particularly useful for studying essential genes or achieving graded transcriptional control
- Compatible with multiplexed gene regulation through use of multiple sgRNAs
Signaling Pathways and Molecular Mechanisms
The following diagrams illustrate the core molecular mechanisms and experimental workflows for Cry2/CIB and LOV domain-based Opto-CRISPR systems.
Cry2/CIB Opto-CRISPR Activation Mechanism
LOV Domain Opto-CRISPR Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Opto-CRISPR Experiments
| Reagent/Solution | Function | Examples/Specifications |
|---|---|---|
| DEL-VPR System | High-potency light-activated transcription | EL222-VPR fusion; 570-fold induction [10] |
| CRY2/CIB Variants | Light-dependent dimerization | CRY2(535) with reduced dark activity [25] |
| C120 Promoter System | Light-responsive promoter | 5xC120-minP for optimal induction [10] |
| PhoBIT Systems | Reversible protein interaction control | PhoBIT1 (light-OFF), PhoBIT2 (light-ON) [12] |
| Engineered CRY2 Mutants | Tunable kinetics | L348F (t½ ~24 min), W349R (t½ ~2.5 min) [25] |
| Light Delivery Systems | Precise illumination | Blue LED arrays (450-470 nm), confocal microscopy |
| Cell Lines | Implementation platform | HEK293T, CHO-K1, HEKblue IFN reporter cells [10] |
Successful implementation of Opto-CRISPR systems requires careful selection of appropriate reagents and tools. The DEL-VPR system stands out for applications demanding high-level gene expression, such as production of difficult-to-express proteins like monoclonal antibodies and bispecific antibodies [10]. For kinetic studies requiring different timescales, engineered CRY2 mutants with altered photocycle properties provide valuable flexibility, enabling researchers to match the tool's dissociation kinetics to their experimental timeframe [25].
The emerging PhoBIT systems offer unique advantages for applications requiring minimal basal activity and reversible control. PhoBIT1 functions as a light-OFF switch where blue light induces dissociation of bound components, while PhoBIT2 operates as a light-ON switch with CRY2-mediated control [12]. These systems are particularly valuable for controlling CRISPRi activity and signaling pathways where background activity must be minimized. When implementing these systems, the C120 promoter provides optimal performance with LOV domain-based transcription factors, while proper light delivery systems must be calibrated to ensure sufficient penetration without causing cellular photodamage.
The systematic comparison of Cry2/CIB and LOV domain-based Opto-CRISPR systems reveals distinct advantages for different research applications. LOV domain systems, particularly the DEL-VPR platform, excel in scenarios requiring high induction levels, fast kinetics, and minimal background activity. These characteristics make them ideal for bioproduction applications and studies demanding precise temporal control. In contrast, Cry2/CIB systems offer advantages through their tunable kinetics and strong dimerization capabilities, making them suitable for processes that benefit from sustained activation.
Future developments in Opto-CRISPR technology will likely focus on expanding the color palette beyond blue light-responsive systems to enable multi-channel control of different cellular processes. Additionally, continued engineering of both Cry2/CIB and LOV domains will further improve their dynamic range, kinetics, and orthogonality. The integration of these systems with emerging CRISPR modalities, such as base editing and prime editing, will open new frontiers for precision genome manipulation with spatiotemporal control. As these tools mature, they hold tremendous potential for therapeutic applications where precise control over gene editing activity is paramount for safety and efficacy.
The expanding toolkit of Opto-CRISPR systems empowers researchers to address biological questions with unprecedented precision, bridging the gap between observational studies and functional manipulation. By carefully matching system properties to experimental requirements, scientists can now explore gene function and regulation with temporal and spatial resolution that was previously unattainable.
Liquid-liquid phase separation (LLPS) has emerged as a crucial biological process underlying the formation of membraneless organelles and protein condensates, with its dysregulation increasingly implicated in neurodegenerative pathologies such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. Optogenetic tools provide unprecedented spatiotemporal control over protein clustering, enabling precise investigation of phase separation dynamics in living cells. This guide compares two principal optogenetic dimerization systems—CRY2/CIB and LOV-based platforms—for probing phase separation in neurodegenerative disease models. We present quantitative benchmarking data, detailed experimental protocols, and reagent specifications to inform tool selection for specific research applications in neuroscience and drug development.
Biomolecular condensates formed through phase separation concentrate specific proteins and nucleic acids, facilitating diverse cellular functions from stress response to synaptic regulation. In neurodegenerative disease, proteins such as TDP-43, FUS, and hnRNPA1 undergo pathogenic phase transitions into solid-like aggregates that disrupt cellular homeostasis. Optogenetic systems allow researchers to mimic and manipulate these processes by using light to induce controlled protein-protein interactions that nucleate condensate formation.
The ideal optogenetic tool for studying phase separation exhibits minimal dark activity, rapid activation kinetics, high dynamic range, and tunable dissociation kinetics. Two of the most widely employed systems are based on the Arabidopsis thaliana cryptochrome 2 (CRY2) and various Light-Oxygen-Voltage (LOV) domains, each with distinct photophysical properties and implementation considerations [32] [35]. This guide provides a systematic comparison of these systems, emphasizing their application in neurodegenerative disease research.
System Comparison: CRY2/CIB vs. LOV Domains
Core Photophysical Properties
Table 1: Fundamental Characteristics of Optogenetic Dimerization Systems
| Property | CRY2/CIB System | LOV Domain Systems |
|---|---|---|
| Native Source | Arabidopsis thaliana | Various plants, bacteria, fungi |
| Chromophore | Flavin Adenine Dinucleotide (FAD) | Flavin Mononucleotide (FMN) |
| Activation Wavelength | Blue light (~450 nm) | Blue light (~450 nm) |
| Primary Mechanism | Light-induced heterodimerization | Light-induced conformational change |
| Dark State Recovery | Thermal relaxation (~5.5 min at 34°C) [25] | Varies by variant: AsLOV2 (~80 s), VVD (~5 h) [35] |
| Key Engineering Targets | Reduced dark interaction, altered photocycle kinetics [25] | Improved dynamic range, fusion compatibility [35] |
Experimental Performance Metrics
Table 2: Quantitative Performance Benchmarks in Cellular Systems
| Performance Metric | CRY2/CIB | LOV-Based Systems | Experimental Context |
|---|---|---|---|
| Fold-Activation | Up to 26-fold improvement with CRY2(535) [25] | Varies by application and LOV variant | Transcriptional activation in yeast [32] |
| Dark State Activity | Significantly reduced with CRY2(535) [25] | Application-dependent | Membrane recruitment assays |
| Dissociation Half-life | Wild-type: ~5.5 min; L348F: ~24 min; W349R: ~2.5 min [25] | AsLOV2: ~80 s; EL222: ~30 s; YtvA: ~100 min; VVD: ~5 h [35] | Following pulse illumination |
| Spatial Precision | Subcellular compartment targeting demonstrated [14] | High precision in engineered fusions | Bacterial and mammalian cells |
Applications in Phase Separation Research
Table 3: Implementation for Biomolecular Condensate Studies
| Application | CRY2/CIB Approach | LOV-Based Approach |
|---|---|---|
| Condensate Nucleation | CRY2-fused intrinsically disordered regions (IDRs) cluster with light | LOV2-fused self-associating domains |
| Reversibility Studies | Tunable dissociation via photocycle mutants [25] | Rapid vs. slow cycling LOV variants [35] |
| Signaling Modulation | Recruitment to pathway components [36] | Allosteric control of enzymatic activity |
| Endogenous Protein Control | Limited without tagging | Opto-nanobodies for native targets [37] |
Experimental Protocols for Phase Separation Studies
CRY2-Based Condensate Formation Assay
Principle: Blue light-induced CRY2 clustering nucleates phase separation of fused protein domains.
Materials:
- Plasmid encoding CRY2 fusion protein (e.g., CRY2-IDR)
- Appropriate cell line (HEK293T or neuronal cell culture)
- Blue light illumination system (LED array or laser)
- Live-cell imaging setup with environmental control
Procedure:
- Cell Preparation: Plate cells on imaging-appropriate dishes and transfert with CRY2 fusion construct.
- Expression Optimization: Titrate DNA concentration to achieve moderate expression (high levels may cause saturation).
- Image Acquisition: Maintain cells at 37°C with 5% CO₂ during imaging.
- Light Activation: Illuminate with 450 nm light at 0.1-1 mW/mm² intensity with 100-500 ms pulses every 5-30 seconds.
- Data Collection: Capture images of fluorescent protein fusion every 30-60 seconds.
- Quantification: Analyze condensate formation kinetics using particle analysis and thresholding.
Key Considerations:
- Include dark-state controls without illumination
- Test multiple expression levels to avoid artifactual condensation
- For reversibility studies, monitor recovery after illumination cessation
LOV-Based Allosteric Control of Phase Separation
Principle: Light-induced conformational change in LOV domain modulates binding interface accessibility.
Materials:
- LOV fusion construct (e.g., AsLOV2-zip or opto-nanobody)
- Target protein or co-expression system
- Blue light illumination capable of precise spatial patterning
Procedure:
- System Validation: Confirm light-dependent binding in control assays (e.g., membrane translocation).
- Fusion Design: Integrate LOV domain at positions determined to maximize dynamic range.
- Illumination Pattern: Apply spatially restricted light to subcellular regions.
- Kinetic Analysis: Monitor binding and dissociation with high temporal resolution.
- Specificity Controls: Verify target specificity through competition or knockout.
Applications in Neurodegeneration:
- Light-controlled inhibition of pathogenic protein aggregation
- Spatially restricted condensate dissolution
- Allosteric regulation of condensate composition
Optogenetic System Selection Guide
Decision Framework for Optogenetic Tool Selection in Neurodegeneration Research
Signaling Pathways in Optogenetic Phase Separation
Molecular Pathways of Optogenetic Condensate Control
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Optogenetic Phase Separation Studies
| Reagent Category | Specific Examples | Function | Implementation Notes |
|---|---|---|---|
| CRY2/CIB Constructs | CRY2(535), CIBN (1-170), CIB81 (1-81) [25] | Light-induced heterodimerization | CRY2(535) shows reduced dark activity vs CRY2PHR |
| LOV Domain Variants | AsLOV2, VVD, FKF1, EL222, sLOV [35] [37] | Conformational switching | sLOV eliminates nuclear export sequences |
| Expression Systems | Single plasmid (CRY2/CIBN) [14] | Controlled co-expression | Balanced expression minimizes background |
| Fluorescent Reporters | mCherry, iRFP, RCaMP [14] [36] | Live-cell visualization | Red-shifted reporters minimize spectral overlap |
| Illumination Systems | LED arrays, laser scanning | Precise light delivery | Intensity control critical for kinetic studies |
| Neuronal Cell Models | iPSC-derived neurons, cell lines | Disease-relevant contexts | Enable study of endogenous protein dynamics |
Both CRY2/CIB and LOV domain systems offer powerful, complementary approaches for investigating phase separation in neurodegenerative disease models. The CRY2/CIB system excels in direct clustering applications with its tunable kinetics and well-characterized optimization variants, making it ideal for studying condensation dynamics and nucleation thresholds. In contrast, LOV-based systems provide sophisticated allosteric control and the unique capability to modulate endogenous proteins through opto-nanobody technology, enabling targeted manipulation of specific condensate components.
Selection between these systems should be guided by specific experimental goals: CRY2 for direct assembly studies and LOV systems for precise perturbation of endogenous condensation processes. As optogenetic tools continue to evolve through protein engineering and computational design, their integration with neurodegenerative disease models promises to unravel the pathological significance of phase transitions and accelerate therapeutic discovery.
Solving Common Problems and Enhancing System Performance
A pivotal challenge in the application of Arabidopsis thaliana Cryptochrome 2 (CRY2) within optogenetic systems is its inherent capacity to undergo two distinct, simultaneous light-dependent interactions: CRY2-CIB1 hetero-dimerization and CRY2-CRY2 homo-oligomerization [3]. While both phenomena are useful, a significant complication arises because the CRY2-CIB1 hetero-dimerization system, widely used for optical control of processes like gene expression and organelle transport, is often confounded by unintended CRY2 homo-oligomerization [3]. This unintended clustering can lead to experimental noise, reduced specificity, and difficulty in interpreting results. For years, selecting the desired type of interaction was not possible because the molecular mechanisms governing these interactions were unknown [3]. This review, set within a broader thesis comparing CRY2/CIB and LOV domain systems, details how mechanistic insights powered the rational engineering of CRY2high and CRY2low mutants, providing researchers with a refined toolkit for precise optogenetic control.
Mechanistic Insights: The Electrostatic Basis for Controlling CRY2 Interactions
The breakthrough in controlling CRY2 clustering came from the discovery that CRY2–CIB1 and CRY2–CRY2 interactions are governed by well-separated protein interfaces [3]. Research revealed that charged residues at the N-terminus of CRY2 are critical for its interaction with CIB1, while electrostatic charges at the C-terminus, specifically at residues 489 and 490, drastically affect light-induced CRY2 homo-oligomerization [3].
The fundamental principle uncovered was that positive charges at the C-terminus facilitate oligomerization, whereas negative charges inhibit it [3]. This understanding of the role of electrostatic forces provided a clear path for engineering CRY2 variants with tailored oligomerization tendencies, moving beyond random mutagenesis to a rational design strategy.
Diagram: Mechanism of CRY2 Interactions and Engineering Strategy
Comparative Performance: CRY2high vs. CRY2low vs. CRY2wt
The engineering of C-terminal charges led to the development of two specialized CRY2 variants with opposing functions, quantitatively compared in the table below.
Table 1: Quantitative Comparison of Key CRY2 Variants
| Feature | CRY2wt (Wild-type) | CRY2high (Enhanced) | CRY2low (Suppressed) |
|---|---|---|---|
| Oligomerization Propensity | Baseline (Constitutive) | Drastically Enhanced [3] [4] | Significantly Reduced [3] [4] |
| Primary Engineering | N/A | Introduction of positive charges at C-terminus to facilitate oligomerization [3] | Introduction of negative charges at C-terminus to inhibit oligomerization [3] |
| Typical Fusion Strategy | Standard fusion to protein of interest (POI) | Fusion of POI to engineered CRY2high | Fusion of POI to CRY2low-tdTomato; the bulky fluorescent protein sterically hinders oligomer formation [3] |
| Ideal Application | General use where some oligomerization is acceptable | Strategies requiring robust clustering (e.g., activating Raf, Rho GTPases) [3] [4] | CRY2-CIB1 hetero-dimerization applications requiring minimal unintended homo-oligomerization [3] |
| Impact on Signaling | Can activate pathways but with potential for unintended clustering | Enables strong activation of membrane-bound and cytosolic signaling pathways via clustering [4] | Improves specificity of membrane recruitment and downstream activation [3] |
Experimental Workflow for Characterizing CRY2 Variants
The characterization of these optogenetic tools relies on well-established cellular assays that visually and quantitatively report on protein interaction dynamics.
Table 2: Key Experimental Protocols for Characterizing CRY2 Mutants
| Protocol Name | Experimental Workflow | Key Readout / Data Analysis |
|---|---|---|
| Membrane Recruitment Assay (for CIB1 binding) | Co-transfect cells with CRY2 variant fused to a fluorescent protein (e.g., mCherry) and CIB1-GFP fused to a transmembrane domain (e.g., Sec61β for ER targeting) [3]. Image cells before and after delivery of intermittent blue light pulses (e.g., 200-ms pulses). | Quantify the fraction of CRY2 signal recruited from the cytosol to the membrane after each light pulse. CRY2low shows reduced recruitment efficiency, indicating weaker CIB1 binding, while CRY2wt is rapidly and completely recruited [3]. |
| Cytosolic Clustering Assay (for homo-oligomerization) | Transfert cells with the CRY2 variant (e.g., CRY2-iRFP) alone. Image cells over time during continuous or pulsed blue light illumination using fluorescence microscopy [3] [4]. | Measure the formation, size, and number of distinct CRY2 puncta in the cytosol over time. CRY2high shows rapid and large cluster formation, while CRY2low shows significantly suppressed clustering [3]. |
| Signaling Pathway Activation Assay | Fuse a CRY2 variant to a signaling domain (e.g., the cytoplasmic tail of a receptor or an intracellular kinase like Raf). Stimulate with blue light and measure downstream signaling events (e.g., ERK phosphorylation for Raf) via immunoblotting [3] [4]. | Quantify the level of phosphorylated downstream effector relative to a total protein control. CRY2high enables tuning of signaling levels based on its enhanced clustering ability [3]. |
Diagram: Experimental Workflow for Membrane Recruitment Assay
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for CRY2 Engineering and Application
| Reagent / Tool | Function and Description | Relevance to CRY2 Studies |
|---|---|---|
| CRY2 PHR Domain (aa 1-498) | The core, light-sensitive region of CRY2 used as the actuator in most optogenetic tools; binds the FAD chromophore endogenously [3] [4]. | The foundational scaffold for engineering CRY2high and CRY2low mutants. |
| CIB1 / CIBN | CRY2's natural binding partner. CIBN is a truncated version (aa 1-100) that minimizes constitutive activity [38]. | Used in membrane recruitment assays to test hetero-dimerization specificity and efficiency of CRY2 variants [3] [38]. |
| Membrane Targeting Sequences | Short peptide sequences (e.g., CAAX box for plasma membrane, Sec61β for ER membrane) that anchor proteins to specific cellular membranes [3] [37]. | Critical for recruiting CIBN to membranes to test light-induced translocation of CRY2 variants [3] [37]. |
| tdTomato Fluorescent Protein | A large, bright, tandem dimeric fluorescent protein [3]. | When fused to CRY2low, it provides steric hindrance to further suppress residual oligomerization, creating the CRY2low-tdTom tool [3]. |
| Opto-Raf / Opto-RTK Systems | CRY2 fused to cytoplasmic signaling domains (e.g., Raf kinase, receptor tyrosine kinase domains) [4] [38]. | Used as a cellular readout to demonstrate how CRY2high's enhanced clustering can tune the level of MAPK/ERK pathway activation [3] [4]. |
The rational engineering of CRY2high and CRY2low mutants, guided by a deep understanding of electrostatic mechanisms, has directly addressed the long-standing challenge of unintended clustering in CRY2-based optogenetics. These tools provide researchers with an unprecedented level of control, enabling them to selectively potentiate or minimize CRY2-CRY2 homo-interactions based on their experimental needs. This specialization enhances the precision of optogenetic perturbations across biology, from decoding signal transduction networks to modeling disease-associated protein aggregation. Within the competitive landscape of optogenetic systems, the CRY2/CIB system, now refined with these specialized variants, remains a premier choice for blue-light-controlled dimerization and clustering, distinguished by its robust function without the need for exogenous cofactors.
The physiological function of proteins within cells is critically dependent on their subcellular distribution and local regulation. Optogenetic tools, which use light to control protein-protein interactions, offer a powerful method for manipulating these processes with high precision [13]. The ability to confine protein activity to a specific subcellular volume enables researchers to dissect complex signaling pathways and understand protein function with unprecedented detail [39] [13]. However, not all optogenetic systems achieve equivalent spatial confinement, and a key determinant of this capability lies in their switch-off kinetics—the rate at which the photoreceptor returns to its inactive state once illumination ceases [13].
Spatial control at the subcellular level represents a significant advantage of light-dependent systems over chemically inducible dimerization tools, which do not allow for spatial control of dimer formation [13]. The optimal system for regulating biological processes at the subcellular level would allow redistribution of the desired amount of protein with the highest spatial precision and fastest on and off kinetics [13]. This comparison guide objectively evaluates the performance of two major classes of blue-light-dependent optogenetic systems—Cry2/CIB and LOV domain-based tools—focusing specifically on how their switch-off kinetics determine their effectiveness for spatially-confined experiments.
System Architectures and Molecular Mechanisms
Cry2/CIB System Fundamentals
The Cry2-CIB1 system was one of the first blue-light-dependent dimerization systems developed and remains widely used in cell biology [13]. This system utilizes the photolyase homology region of the Arabidopsis thaliana photoreceptor cryptochrome 2 (amino acids 1-498) and the 170 N-terminal amino acids of its interacting partner, the transcription factor CIB1 [13]. In the most common configuration, the photoreceptor (CRY2PHR) serves as the cytosolic prey, while CIBN (a truncated version of CIB1) is anchored as the bait to specific subcellular locations [13].
Upon blue light exposure, CRY2 undergoes a conformational change that enables binding to CIBN, leading to rapid recruitment of the prey to the bait-anchored compartment. A significant limitation of this system, however, is its slow relaxation kinetics. The Cry2-CIBN dimer displays relatively fast switch-on kinetics (τ₁/₂ON = 3.7 ± 0.9 seconds) but notably slow switch-off kinetics (τ₁/₂OFF = 290 ± 30 seconds in vivo at 37°C) [13]. This slow disassociation rate substantially impacts its ability to maintain spatial confinement, as activated prey molecules can diffuse away from the illumination area and continue to interact with bait proteins outside the targeted region.
LOV Domain-Based Systems and Variants
Light-oxygen-voltage (LOV) domains constitute another major class of blue-light-responsive optogenetic tools, with several engineered variants offering distinct performance characteristics. The fundamental LOV domain originates from phototropin photoreceptors and undergoes a conformational change upon blue light absorption through flavin mononucleotide (FMN) binding [39] [12].
iLID (Improved Light-Induced Dimer) represents a key engineered LOV-based system that utilizes the interaction between a modified LOV2 domain from Avena sativa phototropin 1 and its binding partner SspB [13] [12]. In the dark state, the C-terminal Jα helix of the LOV2 domain sterically hinders access to the SspB binding site. Blue light illumination causes unfolding of the Jα helix, exposing the binding motif and enabling interaction with SspB [12].
Magnets, another LOV-based system, were developed through multidirectional engineering of the Neurospora crassa photoreceptor Vivid (VVD) [13]. A defining feature of Magnets is that both components of the dimer are photoreceptors, and both must be activated by light to achieve dimerization [13]. This system typically demonstrates faster switch-off kinetics compared to Cry2/CIB.
LightR (Light-regulated allosteric switch) represents an innovative application of LOV domains where two VVD domains are connected in tandem to create a light-sensitive clamp [39]. This system operates through a distinct mechanism—in the dark state, the clamp remains open, distorting the catalytic domain of a fused enzyme and inactivating it. Blue light illumination causes clamp closure, restoring the enzyme's native structure and activity [39]. Engineered variants like FastLightR incorporate specific mutations (I85V in both VVD domains) to accelerate activation-inactivation dynamics [39].
PhoBITs (Photo-inducible binary interaction tools) constitute recently developed LOV-based systems that employ innovative engineering approaches. PhoBIT1 functions as a "light-OFF" switch where LOV2 is inserted into SspB, and blue light illumination allosterically modulates the ssrA binding pocket, triggering dissociation [12]. This system demonstrates a dissociation half-life of 8.5 seconds and a re-association half-life of 28.1 seconds [12].
Table 1: Molecular Characteristics of Major Optogenetic Systems
| System | Photoreceptor Origin | Binding Partners | Chromophore | Primary Mechanism |
|---|---|---|---|---|
| Cry2/CIB | Arabidopsis thaliana cryptochrome 2 | CIBN (N-terminal fragment of CIB1) | FAD | Light-induced heterodimerization |
| iLID | Avena sativa phototropin 1 LOV2 | SspB | FMN | Steric occlusion release |
| Magnets | Neurospora crassa VVD | Engineered VVD variants | FMN | Bidirectional photoactivation |
| LightR | Neurospora crassa VVD | Intramolecular clamping | FMN | Allosteric control of enzyme activity |
| PhoBIT1 | Avena sativa phototropin 1 LOV2 | ssrA peptide | FMN | Allosteric modulation of binding pocket |
Direct Performance Comparison: Quantitative Kinetics and Spatial Confinement
Systematic investigation of these optogenetic systems under identical experimental conditions has revealed critical differences in their spatial confinement capabilities [13]. In studies where a 3μm × 3μm region of interest was illuminated for 5 minutes with 200-ms-long blue-light pulses every 2 seconds, the Cry2/CIB system showed significant leakage outside the illumination area [13]. The mCh-Cry2 prey populated large portions of the endoplasmic reticulum or mitochondrial network despite restricted illumination, a direct consequence of the slow switch-off kinetics (τ₁/₂OFF = 290 ± 30 seconds) that allow activated prey to diffuse extensively and bind to bait outside the irradiated region [13].
In contrast, LOV domain-based systems, particularly Magnets and fast iLID variants, demonstrated superior spatial confinement under the same experimental conditions [13]. The highest confinement was achieved by Magnets and by iLID variants with very fast switch-off kinetics, although this enhanced spatial resolution came at the expense of the total amount of dimers formed [13]. This trade-off between spatial precision and recruitment efficiency represents an important consideration for experimental design.
The engineered FastLightR system, incorporating I85V mutations in both VVD domains, exhibits accelerated activation-inactivation dynamics that enable subcellular regulation [39]. This fast-cycling variant requires more frequent illumination for sustained activation but shows rapid inactivation when illumination ceases, making it particularly suitable for applications requiring precise temporal control [39].
Table 2: Quantitative Comparison of Kinetic Parameters and Spatial Confinement
| System | Switch-On Half-Time (τ₁/₂ON) | Switch-Off Half-Time (τ₁/₂OFF) | Spatial Confinement Rating | Dimer Formation Efficiency |
|---|---|---|---|---|
| Cry2/CIB | 3.7 ± 0.9 seconds | 290 ± 30 seconds | Low | High |
| iLID (standard) | 1-5 seconds | 20-60 seconds | Medium | Medium-High |
| iLID (fast variants) | <1 second | 5-15 seconds | High | Medium |
| Magnets | <1 second | 10-30 seconds | High | Medium |
| PhoBIT1 | Not specified | 8.5 seconds (dissociation) | High | Medium |
Beyond these direct comparisons, the Cry2/CIB system exhibits an additional limitation: activated Cry2 has a propensity to oligomerize, both in solution and on target membranes, leading to the formation of clusters that further compromise spatial precision [13]. These aggregates result in a delayed redistribution of the dimerized components and can introduce experimental artifacts that complicate data interpretation.
Experimental Protocols for Assessing Spatial Confinement
Subcellular Recruitment Assay Protocol
The following methodology has been validated for direct comparison of spatial confinement across different optogenetic systems [13]:
Cell Preparation and Transfection:
- Culture fibroblastic cells (e.g., HEK-293, CHO-K1) on glass-bottom dishes suitable for high-resolution microscopy.
- Co-transfect with plasmids encoding: (a) a cytosolic prey fusion (e.g., mCherry-Cry2 for Cry2/CIB; miRFP670-SspB for iLID) and (b) an organelle-targeted bait fusion (e.g., ER-CIBN-EGFP for Cry2/CIB; mitochondrial-OMP25-ssrA for iLID systems).
- Incubate for 24-48 hours to allow protein expression.
Microscopy and Illumination:
- Use a confocal microscope equipped with a 488nm laser for photoactivation and appropriate lasers for fluorescent protein excitation.
- Select cells with moderate expression levels of both bait and prey constructs.
- Define a small (3μm × 3μm) region of interest (ROI) within the cytoplasm, avoiding the perinuclear region.
- Illuminate the ROI for 5 minutes with 200-ms-long blue-light pulses every 2 seconds (488nm; 7.07 W/cm² intensity).
- Continuously acquire images of the entire cell at 5-10 second intervals throughout the illumination period and for at least 15 minutes post-illumination.
Data Analysis:
- Quantify prey fluorescence intensity within the illuminated ROI, a distal control region of equivalent size, and the entire target organelle (ER or mitochondria).
- Calculate the confinement ratio as: (ROI intensity - distal intensity) / (total organelle intensity).
- Determine the temporal dynamics of prey accumulation and dissipation by fitting exponential curves to the fluorescence intensity data.
Figure 1: Experimental workflow for quantifying spatial confinement of optogenetic tools
FastLightR Kinetics Characterization Protocol
For characterizing systems with engineered kinetics such as FastLightR, the following protocol adaptations are recommended [39]:
Sample Preparation:
- Generate LightR construct by inserting the LightR domain (two VVD domains connected with flexible linkers) into a flexible loop of the target protein.
- For FastLightR, introduce I85V mutations into both VVD domains using site-directed mutagenesis.
- Transfect mammalian cells and allow 24-48 hours for protein expression.
Kinetic Measurements:
- Use time-lapse microscopy to monitor cellular responses (e.g., cell spreading for Src kinase, phosphorylation signals).
- Apply localized blue light illumination (465nm) using a digital micromirror device or similar patterning system.
- Vary illumination frequency and duration to assess activation and inactivation kinetics.
- For fast-cycling variants, utilize shorter, more frequent light pulses (e.g., 30s ON/30s OFF) compared to standard variants (e.g., 2min ON/5min OFF).
Application-Based System Selection Guidelines
Signaling Pathway Dissection
For investigating fast signaling processes with subcellular resolution, LOV domain-based systems with rapid switch-off kinetics are strongly preferred. The ability to confine activation to specific cellular compartments enables researchers to establish causal relationships in signaling cascades that would be impossible with slower systems [39] [12]. FastLightR variants have successfully demonstrated subcellular regulation of Src kinase activity, inducing local protrusions and cell polarization that align with known physiological roles [39]. Similarly, PhoBIT systems have enabled optogenetic control of GPCR signaling, calcium influx, and necroptotic cell death with high spatial precision [12].
Transcriptional Control
For applications involving gene expression control where rapid spatial confinement is less critical, Cry2/CIB remains a viable option despite its slower kinetics [40] [41]. The CRY2/CIB system has been successfully implemented for light-activated protein expression of BDNF in human cells, showing a 64.7-fold increase in expression upon light induction [41]. The system's sustained activation state can be beneficial for processes requiring prolonged transcriptional activity, though spatial control remains limited.
Therapeutic Development
In drug discovery and therapeutic applications, the choice of optogenetic system depends on the specific requirements. Integrated Biosciences has developed an optogenetic screening platform that enables precise control of biological targets, leading to the discovery of compounds with unprecedented mechanisms of action [42] [43]. For these applications, LOV domain-based systems may provide advantages in high-content screening where subcellular resolution is valuable, while Cry2/CIB might be suitable for whole-cell responses.
Table 3: Application-Based System Selection Guidelines
| Application | Recommended System | Rationale | Key Considerations |
|---|---|---|---|
| Subcellular signaling | Fast iLID, Magnets, FastLightR | Fast off-kinetics enable high spatial precision | May require higher expression or illumination intensity |
| Transcriptional control | Cry2/CIB, LOV-based gene switches | Sustained activation beneficial for gene expression | Spatial control limited with Cry2/CIB |
| 3D tissue engineering | Red/far-red systems, LOV switches | Better tissue penetration; genomic stability needed | Requires exogenous chromophore for PhyB/PIF |
| High-throughput screening | iLID, Cry2/CIB | Balance between efficiency and precision | Cry2 oligomerization may complicate readouts |
| Therapeutic protein control | LightR, PhoBIT systems | Allosteric control maintains natural regulation | Requires extensive protein-specific optimization |
Table 4: Key Research Reagent Solutions for Optogenetic Studies
| Reagent Category | Specific Examples | Function/Application | Implementation Notes |
|---|---|---|---|
| Photoreceptor Plasmids | mCh-Cry2 (Addgene #78771), iLID (Addgene #124319), LightR variants | Core optogenetic actuators | Select codon-optimized versions for mammalian expression |
| Targeting Sequences | ER-CIBN, CIBN-Mito, Lyn-FP | Subcellular localization of bait components | Verify localization with fluorescent tags |
| Expression Systems | Sleeping Beauty transposase, lentiviral vectors | Stable genomic integration | Enables uniform expression in cell populations |
| Illumination Systems | Digital micromirror devices (DMD), LED arrays, confocal microscopy | Precise spatial-temporal illumination | DMD enables complex patterning; LEDs suitable for uniform stimulation |
| Kinetic Analysis Tools | Fluorescence recovery after photobleaching (FRAP), confinement ratio calculations | Quantitative assessment of spatial precision | Custom scripts available for automated analysis |
The direct comparison between Cry2/CIB and LOV domain-based optogenetic systems reveals a fundamental trade-off: the slow switch-off kinetics of Cry2/CIB (τ₁/₂OFF = 290 ± 30 seconds) enable robust dimer formation but severely limit spatial confinement, while LOV systems with faster off-kinetics (e.g., PhoBIT1 dissociation τ₁/₂ = 8.5 seconds) offer superior spatial precision at the potential expense of total recruitment efficiency [13] [12].
For researchers requiring high spatial resolution at subcellular scales, LOV domain-based systems—particularly engineered variants with accelerated kinetics—provide clearly superior performance. The development of tools like FastLightR [39] and PhoBITs [12] with optimized switching dynamics represents a significant advancement for applications demanding precise spatial control. These systems enable mechanistic dissection of signaling pathways that was previously impossible with slower optogenetic tools.
Future directions in the field include further engineering of switch-off kinetics to balance spatial precision with functional efficacy, development of orthogonal systems for simultaneous control of multiple processes, and adaptation of these tools for in vivo therapeutic applications [42] [43] [12]. As optogenetic methodologies continue to evolve, the fundamental principle established by these comparisons remains: switch-off kinetics are not merely a secondary characteristic but a primary determinant of spatial confinement that must be carefully matched to experimental requirements.
In optogenetics, the dynamic range of a tool—defined as the ratio of its response in the "on" state (light) versus the "off" state (dark)—is paramount for achieving precise experimental control. A high dynamic range, characterized by minimal activity in the dark (low baseline leakiness) and strong activation upon illumination, is essential for cleanly manipulating biological processes. The challenge of baseline leakiness is a common hurdle, particularly when working with light-inducible dimerization systems. This guide objectively compares two principal classes of optogenetic dimerizers—the Cry2/CIB system from plants and various LOV domain-based systems (including iLID and Magnets)—focusing on strategies to engineer improved dynamic range by suppressing unwanted dark activity, supported by direct experimental evidence.
The core function of optical dimerizers is to control protein-protein interactions with light. Despite this shared goal, the underlying mechanisms and performance characteristics of Cry2/CIB and LOV-based systems differ significantly.
Cry2/CIB System
This system is based on the Arabidopsis thaliana photoreceptor cryptochrome 2 (Cry2) and its binding partner CIB1. Upon blue light illumination, Cry2 undergoes a conformational change, enabling its interaction with CIB1 [25] [13]. A widely used construct consists of the photolyase homology region of Cry2 (Cry2PHR, amino acids 1-498) and a truncated CIB1 (CIBN, amino acids 1-170) [13].
LOV Domain-Based Systems
These systems are derived from Light-Oxygen-Voltage (LOV) sensing domains. Two prominent examples are:
- iLID: Developed from the Avena sativa phototrophin 1 LOV2 domain, which, upon light activation, exposes a peptide (SsrA) that binds to its partner SspB [13].
- Magnets: Engineered from the Neurospora crassa photoreceptor Vivid. A key feature is that both components (VvdN and VvdC) are photosensitive and must be activated by light to dimerize [13].
Table 1: Head-to-Head Comparison of Key Performance Metrics
| Feature | Cry2/CIB (CRY2PHR/CIBN) | iLID (LOV-based) | Magnets (LOV-based) |
|---|---|---|---|
| Dark State Leakiness | Moderate to High [25] | Low [13] | Low [13] |
| Dissociation Half-life (τ₁/₂OFF) | ~5.5 minutes (at 34°C) [25], ~290 seconds (at 37°C) [13] | Fast (seconds; specific variants can be ~10x faster than Cry2) [13] | Fast (seconds) [13] |
| Spatial Confinement | Lower (due to slow off-kinetics) [13] | Higher [13] | Highest [13] |
| Tendency to Cluster/Oligomerize | Yes (Cry2 forms clusters in light) [25] [13] | No [13] | No [13] |
Engineering Strategies to Minimize Leakiness
The performance data reveals a direct connection between a system's off-kinetics and its baseline leakiness. Slower off-kinetics allow activated proteins to diffuse and interact outside the illuminated area, increasing background noise. The primary strategy for improving dynamic range, therefore, centers on optimizing these kinetics and minimizing premature interactions.
Strategy 1: Protein Truncation
A direct approach to improving a tool's dynamic range is to identify the minimal functional domains required for light-dependent interaction, thereby removing regions that may promote non-specific binding.
Experimental Protocol for CRY2/CIB Truncation Screening [25]:
- Construct Design: Generate truncated versions of Cry2 (e.g., CRY2(1-515), CRY2(1-535)) and CIB1 (e.g., CIBN(1-170), CIB81(1-81)).
- Functional Assay: Test constructs in a standardized assay. A common method is a transcriptional activation assay in mammalian cells where one protein is fused to a DNA-binding domain (e.g., LexABD) and the other to a transcription activation domain (e.g., VP16).
- Quantification: Measure the activity of a reporter gene (e.g., via luminescence) under both dark and light conditions. The ratio of light-to-dark activity defines the dynamic range.
- Validation: Confirm findings using an orthogonal method, such as visualizing the recruitment of a cytosolic protein to a membrane-tethered partner.
Key Findings:
- The CRY2(535) truncation demonstrated a 26-fold reduction in dark activity compared to the commonly used Cry2PHR (1-498) in a split transcription factor assay, while maintaining strong light-induced interaction [25].
- A CIB81 truncation (first 81 amino acids of CIB1) was found to be sufficient for light-dependent binding to CRY2, offering a smaller alternative to CIBN [25].
Strategy 2: Mutagenesis for Altered Photocycle Kinetics
The spontaneous return of the photoreceptor to its ground state after light pulses is a key determinant of leakiness. Engineering mutations that accelerate this process directly reduces the window for unwanted interactions.
Experimental Protocol for Isolating Photocycle Mutants [25]:
- Library Creation: Generate a mutagenesis library targeting the photosensory domain of the photoreceptor (e.g., residues 290-498 of Cry2).
- Selection/Screening: Use a yeast-two-hybrid system where interaction between Cry2 and CIB1 activates a survival reporter (e.g., URA3).
- For long-lived mutants, select for growth on plates lacking uracil after a light pulse, followed by a counter-selection (e.g., 5-FOA) in the dark to eliminate constitutively active or short-lived mutants.
- For short-lived mutants, the selection strategy is reversed.
- Kinetic Characterization: Validate hits in mammalian cells using a membrane recruitment assay. A pulse of blue light is delivered, and the dissociation rate of the prey from the bait is quantified over time to determine the new half-life (τ₁/₂OFF).
Key Findings:
- A Cry2 L348F mutant was identified as a long-cycling variant, with a dissociation half-life of ~24 minutes (vs. ~5.5 min for wild-type) [25].
- A Cry2 W349R mutant was identified as a short-cycling variant, with a dissociation half-life of ~2.5 minutes [25].
- While not explicitly shortening the off-kinetics, the fast inherent kinetics of iLID and Magnets are a fundamental reason for their lower baseline leakiness and superior spatial confinement compared to first-generation Cry2/CIB [13].
Strategy 3: System Selection and Configuration
The choice of dimerization system and how its components are deployed (which partner is the "bait" and which is the "prey") significantly impacts performance.
Experimental Protocol for Testing Spatial Confinement [13]:
- Construct Configuration: Fuse the "bait" protein (e.g., CIBN for Cry2/CIB, SspB for iLID) to a marker protein (e.g., EGFP) and target it to a specific organelle membrane (e.g., ER, mitochondria).
- Localized Illumination: Express the cytosolic "prey" protein (e.g., Cry2-mCherry, iLID-mCherry) and illuminate a very small (e.g., 3x3 μm) region of interest (ROI) with brief pulses of blue light.
- Imaging and Analysis: Monitor the redistribution of the prey. High spatial confinement is indicated by the prey accumulating only within the illuminated ROI, with little to no spread to other parts of the organelle.
Key Findings:
- The Magnets system and fast iLID variants achieved the highest spatial confinement, with dimerization effectively restricted to the illuminated volume [13].
- In the Cry2/CIB system, the slow off-kinetics and propensity for Cry2 to form clusters led to the prey protein populating large areas of the organelle network outside the initial illumination site, contributing to higher effective baseline leakiness in applications requiring spatial precision [13].
Visualizing the Experimental Workflow
The following diagram illustrates the core methodology for quantifying the dynamic range and dissociation kinetics of optogenetic dimerizers.
Experimental Workflow for Dynamic Range
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of the protocols above requires a suite of reliable reagents. The table below lists key materials for working with Cry2/CIB and LOV-based systems.
Table 2: Essential Research Reagents for Optogenetic Dimerization Studies
| Reagent / Resource | Function / Description | Example Systems |
|---|---|---|
| CRY2(535) truncation | A Cry2 truncation with reduced dark-state self-interaction and improved dynamic range [25]. | Cry2/CIB |
| CIB81 truncation | A minimal CIB1 domain (first 81 aa) for light-dependent binding to CRY2 [25]. | Cry2/CIB |
| Cry2 Photocycle Mutants (L348F, W349R) | Engineered Cry2 variants with extended or shortened signaling state lifetimes, respectively [25]. | Cry2/CIB |
| Fast iLID Variants | iLID proteins engineered for extremely rapid off-kinetics, enabling high spatial precision [13]. | iLID |
| Magnets (VvdN, VvdC) | A dimerization system where both partners are photoreceptors, requiring dual illumination and offering high confinement [13]. | Magnets |
| Membrane Targeting Sequences | Peptide sequences (e.g., for ER, mitochondria) to anchor the "bait" protein to specific organelles [13]. | Universal |
| Split Transcription Factor Assays | Assays (e.g., Gal4, LexA) to quantitatively measure interaction strength and dynamic range in cells [25]. | Universal |
Reducing baseline leakiness is a multi-faceted challenge in optogenetics, directly addressed by strategic protein engineering and system selection. For the Cry2/CIB system, leveraging second-generation components like CRY2(535) and photocycle mutants such as W349R can significantly enhance dynamic range. However, for applications demanding the highest spatial precision and lowest possible background, LOV domain-based systems like iLID and Magnets present a superior alternative due to their intrinsically faster off-kinetics and lack of dark-state clustering. The choice between systems ultimately depends on the specific experimental needs, including the required temporal control, spatial precision, and tolerance for baseline activity.
In the precise world of cell biology, optogenetic dimerization tools provide unparalleled control over protein interactions. A central challenge in employing these tools lies in navigating the inherent trade-off between two critical parameters: the efficiency of protein recruitment and the spatial resolution of the control. Systems that excel at robustly recruiting large amounts of a protein of interest often do so at the cost of confining the activity to a small, well-defined cellular volume, and vice versa.
This guide objectively compares the performance of two major classes of blue-light-responsive optogenetic systems—CRY2/CIB and LOV domain-based systems (specifically iLID)—in the context of this trade-off. We summarize direct comparative experimental data, detail the methodologies used to generate it, and provide a toolkit to help researchers select the optimal system for their specific application.
Performance Comparison at a Glance
The following table synthesizes quantitative data from direct, side-by-side comparisons of CRY2/CIB and the LOV-based iLID system, highlighting the core trade-offs.
Table 1: Direct Performance Comparison of CRY2/CIB and iLID
| Performance Metric | CRY2/CIB System | iLID System |
|---|---|---|
| Switch-On Kinetics (τ₁/₂ON) | ~3.7 ± 0.9 seconds [13] | Faster than CRY2/CIB [13] |
| Switch-Off Kinetics (τ₁/₂OFF) | ~290 ± 30 seconds (slow) [13] | ~24-30 seconds (fast) [13] |
| Spatial Confinement | Low: Activation spreads far beyond the illuminated region [13] | High: Dimerization is efficiently confined to the target area [13] |
| Recruitment Efficiency | High: Can recruit a large amount of protein [13] | Lower than CRY2/CIB, but sufficient for many applications [13] |
| Key Compromise | High recruitment at the expense of spatial resolution [13] | Excellent spatial resolution at the expense of total recruited protein [13] |
| Notable Artifacts | Prone to light-induced clustering and oligomerization [13] [3] | More defined binary interaction [44] |
Experimental Insights and Methodologies
The data presented in Table 1 is derived from standardized experimental protocols designed to quantify spatial control and efficiency.
Core Experimental Workflow
A typical experiment to assess spatial confinement involves a structured workflow, as outlined below.
Figure 1: Generalized experimental workflow for comparing optogenetic tools.
Detailed Experimental Protocol
The following steps detail the methodology used to generate the comparative data [13]:
Construct Design:
- Bait Protein: The binding partner (CIBN for the CRY2/CIB system, SspB for the iLID system) is fused to a fluorescent protein (e.g., EGFP) and targeted to specific organelle membranes, such as the Endoplasmic Reticulum (ER) using the N-terminal sequence of cytochrome b5, or to mitochondria using the C-terminal sequence of OMP25.
- Prey Protein: The photosensitive partner (CRY2 for CRY2/CIB, the LOV2-SsrA fusion for iLID) is fused to a cytosolic fluorescent reporter (e.g., mCherry).
Cell Transfection & Expression: The bait and prey constructs are co-transfected into mammalian cells (e.g., COS-7 or HeLa cells). Cells are incubated for 24-48 hours to allow for protein expression.
Targeted Illumination & Imaging:
- A confocal microscope is used to define a small (e.g., 3 μm x 3 μm) Region of Interest (ROI) on a part of the ER or mitochondrial network.
- This ROI is irradiated with a series of short blue-light pulses (e.g., 200 ms pulses every 2 seconds at 488 nm, 7.07 W/cm²) for a set duration (e.g., 5 minutes).
- The redistribution of the cytosolic prey (mCherry signal) is imaged in real-time during and after the local illumination.
Quantitative Analysis:
- Spatial Confinement: The extent to which the prey protein accumulates outside the illuminated ROI is measured. Efficient systems show sharp boundaries at the ROI edge.
- Recruitment Efficiency: The total fluorescence intensity of the prey protein recruited to the target organelle within the illuminated ROI is quantified.
- Kinetics: The rate of prey accumulation after light onset (switch-on) and its dissipation after light cessation (switch-off) are calculated.
Mechanistic Basis of the Observed Trade-offs
The fundamental difference in performance between CRY2/CIB and iLID stems from their distinct molecular mechanisms, particularly their switch-off kinetics.
Molecular Mechanisms and Spatial Spread
The slow relaxation of CRY2 leads to the spread of activity, while the fast off-kinetics of iLID enables superior spatial confinement.
Figure 2: Mechanism of spatial confinement. CRY2's slow off-kinetics lead to poor confinement, while iLID's fast off-kinetics enable high spatial precision.
The CRY2/CIB system's slow off-kinetics (~5 minutes) mean that once a CRY2 molecule is activated by light, it remains in its active state long enough to diffuse throughout the cell and bind CIB1 on membranes far from the original illumination site [13]. Furthermore, activated CRY2 has a propensity to form large, stable clusters, which exacerbates the loss of spatial control and can lead to unintended experimental artifacts [13] [3].
In contrast, the iLID system's fast off-kinetics (~30 seconds) ensure that the LOV2 domain quickly returns to its dark state, re-caging the SsrA peptide. Any prey molecule that diffuses away from the illuminated volume rapidly deactivates and dissociates, effectively limiting the dimerization to the targeted area [13]. This fast off-rate is the key to iLID's superior spatial resolution.
The Scientist's Toolkit: Key Research Reagents
Selecting the appropriate tools is critical for a successful optogenetic experiment. The table below lists essential reagents and their functions based on the cited studies.
Table 2: Key Research Reagent Solutions for Optogenetic Dimerization Studies
| Reagent / Tool Name | Type/Function | Key Feature(s) & Application |
|---|---|---|
| CRY2PHR (1-498)-mCherry | Photosensory Prey | Standard CRY2 photolyase homology region; used for light-induced heterodimerization with CIBN [13] [3]. |
| CIBN-GFP-Sec61β | Membrane-Tethered Bait | N-terminal fragment of CIB1; targets to ER membrane via Sec61β trans-membrane domain for recruiting cytosolic CRY2 [13] [3]. |
| iLID (LOV2-SsrA) | Photosensory Prey | Light-induced unfolding of LOV2 exposes SsrA peptide for binding SspB [13]. |
| SspB-GFP-Mito | Membrane-Tethered Bait | Binds exposed SsrA tag; targeted to mitochondria via OMP25 targeting sequence for high-resolution recruitment studies [13]. |
| CRY2low-tdTom | Engineered CRY2 Variant | CRY2 mutant with suppressed oligomerization; improves specificity for heterodimerization applications by reducing unintended clustering [3]. |
| Magnets | Engineered Vivid System | Fast, reversible homodimerizer; both components are photosensory, offering an alternative design paradigm [13]. |
| PhoBITs | Engineered ssrA-sspB System | Utilizes a minimal 7-residue ssrA tag; designed for low basal activity and versatile control of diverse protein functions [44]. |
The choice between the CRY2/CIB and iLID optogenetic systems is a direct reflection of the experimental priorities. There is no universally superior tool, only the most appropriate one for a given biological question.
- Choose CRY2/CIB when the primary goal is to achieve strong, robust recruitment of a target protein, and high spatial precision is not the critical factor. Be mindful of its tendency to form clusters.
- Choose iLID (or related LOV-based tools like PhoBITs or Magnets) when high spatial resolution is paramount, and a potentially lower total recruitment yield is acceptable. Its fast kinetics are essential for mimicking rapid, native biological processes in confined subcellular domains.
Understanding the mechanistic underpinnings of this performance trade-off empowers researchers to make informed decisions, optimizing their experimental design for precise and biologically relevant discovery.
Head-to-Head Comparison: Quantifying Performance Metrics for Informed Tool Selection
This guide provides a direct performance comparison between two primary blue-light-induced dimerization systems, Cry2/CIB and iLID, with a focused analysis on their ability to confine protein interactions to small subcellular volumes. The capacity for high spatial resolution is critical for optogenetic experiments that aim to mimic natural signaling patterns or manipulate specific cellular compartments. Based on current experimental data, systems with faster off-kinetics, particularly iLID and its variants, achieve superior spatial confinement. In contrast, the slower reversion kinetics of the Cry2/CIB system limit its spatial precision, though it may offer advantages in the total amount of protein recruited [13].
The table below summarizes the core characteristics of these two systems.
| Feature | Cry2/CIB System | iLID System |
|---|---|---|
| Origin | Arabidopsis thaliana cryptochrome 2 (CRY2) and its binding partner CIB1 [25] | Engineered from Avena sativa LOV2 domain and bacterial SsrA-SspB pair [13] |
| Core Components | CRY2 (photosensory) and CIB1 (binding partner); common truncations include CRY2PHR (1-498) and CIBN (1-170) [25] [14] | iLID (photosensory, contains LOV2 and SsrA) and SspB (binding partner) [13] |
| Light Response | Light-induced heterodimerization (ON switch) [25] | Light-induced heterodimerization (ON switch) [13] |
| Typical Activation | Blue light (~450 nm) [25] [14] | Blue light (~450 nm) [13] |
| Key Limitation | Slower off-kinetics and propensity to cluster limit spatial resolution [13] | Faster off-kinetics can limit the total amount of dimer formed [13] |
Quantitative Benchmarking of System Kinetics and Performance
Direct comparative studies have quantified the kinetic parameters and spatial performance of the Cry2/CIB and iLID systems under identical experimental conditions. The dissociation kinetics (switch-off half-life) are the primary determinant of spatial confinement.
Table 2: Direct Kinetic and Spatial Performance Comparison [13]
| Parameter | Cry2/CIB System | iLID System | Experimental Context |
|---|---|---|---|
| Switch-On Half-Life (τ₁/₂ON) | 3.7 ± 0.9 s | Information missing | Measured in vivo at 37°C in fibroblastic cells. |
| Switch-Off Half-Life (τ₁/₂OFF) | 290 ± 30 s (∼5 min) | Ranges from seconds to minutes (fastest variants achieve sub-minute off-kinetics) | Measured in vivo at 37°C. The specific value for the tested iLID variant is not explicitly stated but is described as having "very fast switch-off kinetics" [13]. |
| Spatial Confinement to Illuminated Area | Low | High | Recruitment to ER or mitochondria upon illumination of a 3μm x 3μm ROI. Cry2/CIB showed recruitment over large areas beyond the ROI [13]. |
| Protein Clustering | Yes (light-induced clusters/aggregates) | No (minimal clustering reported) | Observed after whole-cell activation of the Cry2/CIB system [13]. |
| Key Performance Trade-off | Can recruit a higher total amount of protein | Higher spatial resolution is achieved at the expense of the total amount of dimer formed [13] |
Beyond the standard CRY2PHR/CIBN pair, engineered CRY2 variants with altered photocycles can modify its performance. For instance, the CRY2(L348F) mutation creates a long-cycling variant with a dissociation half-life of approximately 24 minutes, while the CRY2(W349R) mutation creates a short-cycling variant with a half-life of about 2.5 minutes [25]. However, even the faster CRY2(W349R) variant is slower than the fastest iLID variants.
Analysis of Spatial Confinement Performance
The fundamental difference in off-kinetics between the two systems leads to their stark contrast in spatial performance.
- Mechanism of Poor Confinement in Cry2/CIB: The slow spontaneous dissociation of CRY2 from CIB1 (half-life of ~5 minutes) means that once a CRY2-preY complex is activated within the illuminated region, it can diffuse away and bind to its bait partner in non-illuminated areas of the cell [13]. This leads to the recruitment of protein over large portions of an organelle like the ER or mitochondrial network, despite highly localized illumination. Furthermore, the light-induced oligomerization of CRY2 can exacerbate this effect and create large, delayed aggregates [13].
- Mechanism of High Confinement in iLID: The fast off-kinetics of the iLID system ensure that the activated state is short-lived. If an iLID-preY complex diffuses out of the illuminated region, it rapidly reverts to its inactive state before it can bind to the bait partner elsewhere. This effectively traps the dimerization event within the illuminated volume, providing high spatial precision [13].
Experimental Protocols for Performance Assessment
The following methodology details a standardized assay used to directly compare the spatial confinement capabilities of different optogenetic dimerizers, as described in the benchmarking data [13].
Experimental Workflow for Assessing Spatial Confinement
Key Reagents and Experimental Setup
Table 4: Essential Research Reagent Solutions
| Reagent / Material | Function / Description | Example Constructs |
|---|---|---|
| Bait Plasmid | Encodes the membrane-anchored partner to define the recruitment site. | CIBN-CAAX: For Cry2/CIB system. CIBN (aa 1-170 of CIB1) fused to a plasma membrane localization signal (e.g., CAAX) [45].iLID-CAAX: For iLID system. iLID fused to a plasma membrane localization signal [45]. |
| Prey Plasmid | Encodes the cytosolic, light-sensitive partner fused to a fluorescent reporter. | CRY2PHR-mCherry: For Cry2/CIB system. CRY2PHR (aa 1-498) fused to mCherry [13] [45].SspB-RFP: For iLID system. SspB binding partner fused to RFP or mCherry [45]. |
| Cell Line | Mammalian cells suitable for imaging. | Fibroblastic cells, HEK293T, or other adherent cell lines [13]. |
| Confocal Microscope | For high-resolution imaging and targeted illumination. | Must be equipped with a 488nm laser for activation and appropriate lasers/channels for imaging fluorescent reporters (e.g., 561nm for mCherry/RFP). |
| Photo-stimulation System | To define and illuminate a specific Region of Interest (ROI). | A system capable of illuminating a small (e.g., 3μm x 3μm) ROI with 488nm light, typically controlled by software [13]. |
Detailed Procedural Steps
- Construct Design: Express the bait and prey constructs in the chosen cell line. The bait (e.g., CIBN-CAAX or iLID-CAAX) is targeted to the plasma membrane or specific organelles like the endoplasmic reticulum (ER) or mitochondria. The prey (e.g., CRY2PHR-mCherry or SspB-RFP) is expressed in the cytosol [13].
- Cell Culture & Transfection: Culture cells on imaging-grade dishes or coverslips. Transfect with the bait and prey plasmids. Allow sufficient time for protein expression (typically 24-48 hours).
- Microscope Setup: Place the sample on a confocal microscope maintained at 37°C and 5% CO₂. Use settings to image the fluorescent reporter (e.g., mCherry) without activating the optogenetic system.
- Localized Illumination & Imaging:
- Select a small, defined Region of Interest (ROI), such as a 3μm x 3μm square, on a part of the cell containing the bait-localized membrane or organelle.
- Acquire a pre-stimulation image.
- Illuminate the ROI with pulsed blue light (e.g., 200-ms pulses every 2 seconds for 5 minutes at 488 nm with an irradiance of ~7 W/cm²) [13].
- Simultaneously, acquire time-lapse images of the entire cell to monitor the redistribution of the cytosolic prey protein.
- Image Analysis & Quantification: Analyze the time-lapse data to quantify:
- Recruitment Speed: The rate at of prey accumulation in the ROI.
- Spatial Spread: The area over which the prey protein is recruited, specifically measuring signal intensity outside the illuminated ROI over time.
- Dissociation Kinetics: After stopping illumination, monitor the decay of the prey signal at the recruitment site to calculate the half-life.
The Scientist's Toolkit: Research Reagent Solutions
The following table expands on the key reagents and tools available for implementing these optogenetic systems, including advanced engineered variants.
Table 5: Research Reagent Solutions for Cry2/CIB and iLID Systems
| Category | Reagent / Tool | Key Features / Function | System |
|---|---|---|---|
| Core Components | CRY2PHR (1-498) | The most widely used photosensory module. Prone to clustering and has slow off-kinetics [13]. | Cry2/CIB |
| CIBN (1-170) | A common, truncated binding partner for CRY2PHR [25] [14]. | Cry2/CIB | |
| CRY2(535) | A truncation (1-535) showing reduced dark-state self-interaction compared to CRY2PHR [25]. | Cry2/CIB | |
| iLID | The engineered photosensory module (LOV2-SsrA) with fast off-kinetics [13]. | iLID | |
| SspB (or SspB R73Q) | The binding partner for iLID. The R73Q mutation is often used for improved affinity [45]. | iLID | |
| Engineered Variants | CRY2(L348F) | A long-cycling mutant with a ~24 min dissociation half-life; useful for sustained signaling [25]. | Cry2/CIB |
| CRY2(W349R) | A short-cycling mutant with a ~2.5 min dissociation half-life; offers improved temporal resolution [25]. | Cry2/CIB | |
| Fast iLID Variants | Engineered iLID versions with the fastest switch-off kinetics, enabling the highest spatial confinement [13]. | iLID | |
| Experimental Tools | CAAX Tag | A peptide motif from K-Ras used to target bait proteins to the plasma membrane [45]. | Universal |
| Organelle Targeting Sequences | Specific sequences (e.g., for mitochondria, ER) to localize bait proteins to subcellular compartments [13]. | Universal | |
| Addgene Plasmids | CIBN-CAAX / pLJM1 | Ready-to-use plasmid for mammalian expression of membrane-anchored CIBN (Addgene #201749) [45]. | Cry2/CIB |
| CRY2PHR-mCherry / pPB-bsr2 | Plasmid for expression of the photosensory prey (Addgene #201754) [45]. | Cry2/CIB | |
| pLL7.0-Venus-iLID-CAAX | Plasmid for membrane-anchored iLID (Addgene #60411) [45]. | iLID | |
| pLL7.0-tgRFPt-SspB R73Q | Plasmid for the binding partner prey (Addgene #60416) [45]. | iLID |
Optogenetic tools have revolutionized the biological sciences by enabling precise, light-controlled manipulation of cellular processes. Among the most versatile of these tools are blue-light-inducible systems, with the Cry2/CIB1 pair and various Light-Oxygen-Voltage (LOV) domain-based systems representing two dominant platforms. The kinetic properties of these systems—specifically their activation (on) and deactivation (off) rates—directly determine their temporal resolution and suitability for specific applications. This guide provides a comprehensive comparison of these systems, focusing on quantitative kinetic profiling to inform researchers and drug development professionals in selecting the optimal tool for their experimental needs.
Cry2/CIB1 System
The Cry2/CIB1 system is derived from Arabidopsis thaliana cryptochrome 2. The core component, Cry2, is a photolyase-like photoreceptor that undergoes light-induced conformational changes and oligomerization. Upon blue light illumination, the N-terminal photolyase homology region of Cry2 (amino acids 1-498) interacts with its binding partner, the 170 N-terminal amino acids of the CIB1 transcription factor. This system operates as a heterodimerization tool where light triggers binding between two distinct protein components. A significant kinetic limitation of this system is the propensity of activated Cry2 to form clusters both in solution and on target membranes, which can complicate experimental interpretation [13].
LOV Domain Systems
LOV domains are a subclass of PAS domains that bind flavin cofactors (FMN, FAD, or riboflavin) and sense blue light through a conserved cysteine residue. Unlike Cry2/CIB1, certain LOV-based systems function as single-component optogenetic tools [10]. The primary systems include:
- EL222: From Erythrobacter litoralis, consists of an N-terminal LOV sensor and a C-terminal helix-turn-helix DNA-binding domain. Light absorption triggers undocking of the LOV domain, releasing the HTH domain for dimerization and DNA binding [10] [35].
- AsLOV2: From Avena sativa phototropin 1, widely used in engineered systems like iLID, where light exposure releases a C-terminal Jα helix that can be fused to effector domains [35].
- Magnets: Engineered from the Neurospora crassa photoreceptor Vivid, features two photoreceptor components that must both be activated by light to achieve dimerization [13].
The fundamental photocycle is conserved across LOV domains: blue light promotes formation of a covalent adduct between a conserved cysteine and the C4a atom of the flavin cofactor, creating a signaling state with characteristic 390 nm absorption. This adduct then thermally reverts to the ground state in the dark [35].
Quantitative Kinetic Comparison
The temporal resolution of optogenetic tools is fundamentally governed by their kinetic properties. The table below summarizes the key kinetic parameters for major Cry2/CIB and LOV-based systems.
Table 1: Kinetic Parameters of Blue-Light Optogenetic Systems
| System | τ₁/₂ ON (s) | τ₁/₂ OFF (s) | Thermal Reversion Lifetime (s) | Key Kinetic Features |
|---|---|---|---|---|
| Cry2/CIB1 | 3.7 ± 0.9 [13] | 290 ± 30 [13] | - | Slow OFF kinetics limits temporal and spatial resolution [13] |
| EL222 | Seconds [10] | ~50 [10] | ~30 [35] | Fast cycling enables dynamic control |
| AsLOV2 | - | - | ~80 [35] | Intermediate cycling |
| iLID | Fast [13] | Fast [13] | - | Engineered for rapid OFF kinetics |
| Magnets | - | - | 25-17000 [35] | Tunable OFF kinetics; high spatial confinement [13] |
| PhoBIT1 | - | 8.5 (dissociation) [12] | - | Light-OFF switch with rapid dissociation |
Table 2: Performance Characteristics and Applications
| System | Dynamic Range (Fold Induction) | Spatial Confinement | Primary Applications |
|---|---|---|---|
| Cry2/CIB1 | Varies by construct | Low [13] | Transcriptional control, protein localization [13] |
| EL222 | Up to 570 [10] | High (single-component) [10] | Light-induced gene expression, bioproduction [10] |
| AsLOV2-based | Varies by design | Moderate to High [13] | Protein trafficking, caspase activation [35] |
| Magnets | ~40 [35] | Highest among blue-light systems [13] | Subcellular manipulation, high-precision recruitment [13] |
The kinetic data reveals a fundamental trade-off: systems with faster off kinetics (e.g., iLID, Magnets, EL222) achieve superior spatial confinement but may produce lower total recruitment of target proteins, while systems with slower off kinetics (e.g., Cry2/CIB1) enable greater accumulation but with diminished spatial control [13].
Experimental Methodologies for Kinetic Profiling
Subcellular Confinement Assay
This method quantifies the spatial precision of optogenetic tools by measuring protein recruitment to specific organelles:
- Construct Design: Express a cytosolic prey fusion (e.g., mCherry-Cry2) with an organelle-targeted bait (e.g., ER-CIBN or CIBN-Mito) [13].
- Localized Illumination: Irradiate a 3μm × 3μm region of interest for 5 minutes using 200-ms blue-light pulses every 2 seconds (488 nm; 7.07 W/cm²) via confocal microscopy [13].
- Quantitative Imaging: Monitor prey redistribution during and after illumination. Systems with fast off kinetics (iLID, Magnets) show restricted recruitment, while slow systems (Cry2/CIB1) exhibit widespread diffusion away from the illuminated area [13].
Transcriptional Activation Kinetics
For gene regulation systems like EL222, kinetic profiling involves:
- Reporter System: Transfert cells with the optogenetic construct and a reporter gene (e.g., firefly luciferase or mCherry) under a light-responsive promoter (C120-minP) [10].
- Pulsed Illumination: Apply blue light pulses of varying durations and frequencies.
- Expression Monitoring: Quantify reporter expression via fluorescence or luminescence measurements. DEL-VPR, an EL222-VPR fusion, demonstrates rapid induction with minimal basal activity [10].
PhoBIT1 Dissociation Kinetics
For light-OFF switches like PhoBIT1:
- Mitochondrial Translocation Assay: Anchor ssrA tag to mitochondria and express sspB(LOV2) variant S5 [12].
- Continuous Imaging: Monitor co-localization before, during, and after blue light stimulation (470 nm).
- Exponential Fitting: Calculate dissociation (t₁/₂ = 8.5 s) and re-association (t₁/₂ = 28.1 s) half-lives from fluorescence recovery curves [12].
Mechanism Diagrams
Research Reagent Solutions
Table 3: Essential Research Reagents for Optogenetic Kinetic Studies
| Reagent/Solution | Function | Example Applications |
|---|---|---|
| CIBN-ER/CIBN-Mito | Bait constructs for subcellular localization | Spatial confinement assays; measures recruitment precision to organelles [13] |
| mCherry-Cry2 | Prey construct for visualizing recruitment | Monitoring Cry2/CIB1 interaction dynamics and oligomerization [13] |
| C120-minP Promoter Reporter | Light-responsive reporter construct | Quantifying transcriptional activation kinetics (e.g., with EL222) [10] |
| DEL-VPR (EL222-VPR) | High-potency optogenetic transactivator | Strong light-induced gene expression; biotherapeutic production [10] |
| sspB(LOV2) Variant S5 | Engineered PhoBIT1 dissociation module | Light-OFF control of protein interactions; CRISPRi regulation [12] |
| Mitochondrial ssrA Anchor | Target for translocation assays | Measuring PhoBIT1 dissociation/re-association kinetics [12] |
Kinetic profiling reveals that LOV domain systems generally outperform Cry2/CIB1 for applications requiring high temporal and spatial resolution. The fast off kinetics of tools like EL222 (~30 s), iLID, and Magnets enable precise subcellular manipulation and dynamic control of cellular processes. In contrast, the slow off kinetics of Cry2/CIB1 (τ₁/₂OFF ≈ 290 s) limits its spatial resolution despite potentially achieving high recruitment yields. For transcriptional control, the EL222-based DEL-VPR system offers exceptional induction (up to 570-fold) with minimal basal activity. Recent engineering advances such as PhoBIT1 (t₁/₂OFF = 8.5 s) further demonstrate how LOV domains can be optimized for specific kinetic performance, providing researchers with an expanding toolkit for precision optogenetics. Selection criteria should prioritize temporal requirements first, then match system kinetics to the specific biological process under investigation.
Comparative Analysis of Oligomerization Propensity and Clustering Behavior
The precise control of protein-protein interactions using light has revolutionized cell biology, enabling the manipulation of signaling pathways with high spatiotemporal resolution. Among the most widely utilized optogenetic systems are those based on Arabidopsis thaliana Cryptochrome 2 (Cry2) and various Light-Oxygen-Voltage (LOV) domains, which offer distinct mechanisms for inducing protein oligomerization and clustering. Cry2 systems exhibit a unique dual functionality, capable of both light-induced heterodimerization with its binding partner CIB1 and homo-oligomerization into clusters, a phenomenon first observed in plant cells where Cry2 forms photobodies [3] [8]. In parallel, LOV domain-based systems, particularly those derived from Avena sativa Phototropin 1, have been engineered to create versatile tools such as iLID and Magnets, which utilize conformational changes to control protein interactions [13]. Understanding the oligomerization propensity and clustering behavior of these systems is crucial for selecting the appropriate tool for specific experimental applications, particularly in studies of cellular signaling, phase separation, and neurodegenerative disease mechanisms [46] [47].
This comparative analysis examines the fundamental mechanisms, kinetic properties, and experimental considerations for Cry2/CIB and LOV domain systems, providing researchers with a framework for selecting optimal tools for manipulating cellular processes. We present quantitative data on their performance characteristics and outline detailed protocols for implementing these systems in live-cell experiments, with particular emphasis on their divergent clustering behaviors and applications in probing protein function and biomolecular condensation.
System Fundamentals and Molecular Mechanisms
Cry2/CIB System: Dual Interaction Capabilities
The Cry2/CIB optogenetic system exhibits a remarkable dual functionality that enables both heterodimerization and homo-oligomerization. The molecular basis for these interactions has been elucidated through structural studies revealing that CRY2-CIB1 and CRY2-CRY2 interactions are governed by well-separated protein interfaces at the two termini of Cry2 [3]. Specifically, N-terminal charges are critical for Cry2-CIB1 interaction, while C-terminal residues 489 and 490 dramatically affect light-induced Cry2 homo-oligomerization, with positive charges facilitating oligomerization and negative charges inhibiting it [3]. This mechanistic understanding has enabled the engineering of Cry2 variants with tuned oligomerization properties, including CRY2high with elevated oligomerization and CRY2low with suppressed oligomerization [3].
A key advantage of the Cry2 system is its ability to function without exogenous cofactors, as it uses ubiquitously expressed flavin molecules as its chromophore to absorb blue light in the 430-490 nm range [8]. Upon blue light activation, photoexcited Cry2 undergoes a conformational change that enables rapid heterodimerization with CIB1 within subseconds in mammalian cells [8]. The Cry2-CIB1 interaction dissociates with a half-life of approximately 5.5 minutes after light withdrawal, enabling repeated induction over multiple cycles [8]. Concurrently, light-activated Cry2 undergoes self-oligomerization, forming clusters that can be harnessed for optogenetic applications [8].
LOV Domain Systems: Engineering Conformational Control
LOV domain-based optogenetic systems operate on a fundamentally different principle, utilizing a light-sensitive module that undergoes reversible conformational changes to control protein function. The most widely used LOV domain derives from Avena sativa Phototropin 1 (AsLOV2), which contains a conserved flavin-binding core that responds to blue light exposure [37]. In the dark state, the C-terminal Jα helix of AsLOV2 remains bound to the core domain, while light activation disrupts this interaction, leading to helix undocking and conformational rearrangement [37]. This molecular mechanism has been harnessed to develop several engineered systems, including iLID (improved Light-Inducible Dimer) and Magnets, which offer distinct oligomerization properties.
The iLID system utilizes the AsLOV2 domain fused to the Escherichia coli peptide SsrA, which binds to its interacting partner SspB upon light activation [13]. This system exhibits relatively fast kinetics, with the light-induced interaction reversing rapidly in darkness. In contrast, the Magnets system was developed through multidirectional engineering of the Neurospora crassa photoreceptor Vivid, resulting in a unique configuration where both components of the dimer are photoreceptors that must be activated by light to achieve dimerization [13]. This design contributes to faster off-kinetics compared to Cry2-based systems.
A significant engineering advancement in LOV domain technology is the development of opto-nanobodies (OptoNBs), which incorporate the LOV domain into solvent-exposed loops of nanobodies to create light-switchable binding proteins capable of recognizing untagged protein targets [37]. This innovation expands the utility of LOV domain systems beyond engineered protein pairs to enable control of endogenous cellular proteins, opening new possibilities for perturbing native signaling pathways.
Quantitative Comparison of System Properties
Kinetic Parameters and Performance Metrics
The operational characteristics of Cry2/CIB and LOV domain systems differ significantly in their kinetic parameters, oligomerization propensity, and spatial control capabilities. The table below summarizes key performance metrics based on experimental measurements from comparative studies:
Table 1: Quantitative Comparison of Optogenetic Oligomerization Systems
| Parameter | Cry2/CIB1 | CRY2olig (E490G) | iLID | Magnets |
|---|---|---|---|---|
| Activation Kinetics (t½ ON) | 3.7 ± 0.9 s [13] | <15-75 s (concentration-dependent) [48] | Seconds range [13] | Seconds range [13] |
| Deactivation Kinetics (t½ OFF) | 290 ± 30 s [13] | 23.1 min [48] | Fastest variants: ~10-30 s [13] | Fastest variants: ~10-30 s [13] |
| Oligomerization Propensity | Moderate (6 ± 3% cytosolic protein clusters) [48] | High (70 ± 15% cytosolic protein clusters) [48] | Minimal homo-oligomerization [13] | Minimal homo-oligomerization [13] |
| Spatial Confinement | Low (diffusion-mediated spread) [13] | High (localized clustering) [48] | High (fast off-kinetics limit spread) [13] | Highest (fast off-kinetics limit spread) [13] |
| Cluster Dissolution | ~5.5 min half-life [8] | ~23 min half-life [48] | Rapid (seconds to minutes) [13] | Rapid (seconds to minutes) [13] |
| Two-Photon Compatibility | Yes (850 nm) [48] | Yes (850 nm) [48] | Limited data | Limited data |
The data reveal fundamental trade-offs between these systems. Cry2 variants, particularly CRY2olig, enable robust clustering with high efficiency but suffer from slow off-kinetics that limit temporal resolution and spatial confinement [13] [48]. In contrast, optimized LOV domain systems like iLID and Magnets offer superior spatial precision due to their faster off-kinetics but may produce lower total dimerization levels [13]. The enhanced oligomerization capability of CRY2olig, which contains an E490G mutation, is particularly notable—it redistributes 70 ± 15% of cytosolic protein into clusters compared to only 6 ± 3% for wild-type Cry2PHR under identical conditions [48].
Spatial Control and Subcellular Localization
The spatial precision of optogenetic oligomerization is critically dependent on the system's off-kinetics and cellular localization. Studies comparing Cry2/CIB1, iLID, and Magnets for subcellular confinement have demonstrated that efficient spatial restriction of dimer formation requires both membrane tethering of the photosensitive component and fast switch-off kinetics [13]. When attempting to confine dimerization to a 3×3 μm region using ER-targeted baits, Cry2/CIB1 showed substantial spread beyond the illumination area due to its slow dissociation kinetics (t½ OFF = 290 ± 30 s), allowing activated prey to diffuse and bind throughout the organelle network [13].
Furthermore, Cry2 exhibits dramatically enhanced oligomerization when tethered to cellular membranes compared to its cytoplasmic form [8]. While cytoplasmic Cry2 forms clusters in only about 20% of cells with limited clusters per cell, membrane-bound Cry2 (targeted to ER, plasma membrane, or mitochondria) undergoes rapid and extensive oligomerization within seconds of blue light exposure, forming hundreds to thousands of bright clusters that deplete the diffuse pool [8]. This membrane-enhanced clustering behavior has important implications for experimental design, particularly for applications requiring precise spatial control.
Experimental Applications and Methodologies
Implementation Guidelines and Protocols
Successful implementation of Cry2/CIB and LOV domain systems requires careful consideration of experimental parameters and appropriate controls. Below are detailed protocols for key applications of these optogenetic tools:
Light-Induced Co-clustering (LINC) Assay with CRY2olig: The LINC assay enables rapid detection of protein-protein interactions in live cells with spatial and temporal control [48]. The experimental workflow involves:
- Construct Design: Fuse the bait protein to CRY2olig (containing the E490G mutation) and tag the prey protein with a fluorescent marker (e.g., GFP).
- Cell Preparation: Co-express CRY2olig-bait and fluorescent prey constructs in mammalian cells (HEK293 or COS-7 cells work well).
- Image Acquisition: Capture baseline images of prey distribution before blue light stimulation.
- Light Activation: Expose cells to brief pulses of blue light (200 ms pulses at 2-s intervals, 488 nm laser at 5% power) to induce CRY2olig clustering.
- Post-Activation Imaging: Monitor prey distribution after light application—co-clustering with CRY2olig-bait indicates interaction.
- Specificity Controls: Include pairs of non-interacting proteins to validate assay specificity.
This method has been successfully applied to study both cytosolic and membrane protein interactions, including the homodimerization of homer1c and the interaction between stargazin and PSD95 [48]. The assay can also monitor interaction dynamics in response to cellular stimuli, as demonstrated for the Ca²⁺-dependent interaction between CaMKIIα and calmodulin [48].
Subcellular Recruitment with iLID System: For precise subcellular manipulation of protein activity using the iLID system:
- Component Design: Fuse the iLID photoswitch (SsrA-AsLOV2) to the protein of interest (prey) and target the binding partner (SspB) to specific subcellular compartments (bait).
- Cell Line Generation: Establish stable cell lines expressing membrane-targeted SspB (e.g., ER-SspB or mitochondrial-SspB) to serve as recruitment sites.
- Localized Illumination: Apply spatially restricted blue light illumination (200-ms pulses at 488 nm) using confocal microscopy to define the activation region.
- Kinetic Monitoring: Track recruitment dynamics with high temporal resolution, noting the rapid reversal of interaction upon light cessation.
- Optimization: Adjust illumination parameters (duration, intensity, duty cycle) based on expression levels and desired recruitment efficiency.
This approach has been successfully used to recruit cytosolic proteins to organelles including the endoplasmic reticulum, mitochondria, and plasma membrane with higher spatial confinement than Cry2/CIB1 [13].
Troubleshooting Common Experimental Issues
Several technical challenges may arise when implementing these optogenetic systems:
Insufficient Clustering with Cry2:
- Cause: Low expression levels or cytoplasmic localization of Cry2.
- Solution: Enhance clustering by tethering Cry2 to cellular membranes or using the CRY2olig mutant. Membrane-bound Cry2 exhibits dramatically enhanced oligomerization compared to cytoplasmic forms [8].
Excessive Spatial Spread with Cry2/CIB1:
- Cause: Slow off-kinetics (t½ OFF = 290 ± 30 s) allow diffusion of activated molecules.
- Solution: For applications requiring high spatial precision, consider using iLID or Magnets instead, or confine Cry2 to membranes to limit diffusion [13] [8].
Unintended Nuclear Export with LOV Fusions:
- Cause: Exposure of cryptic nuclear export sequences (NES) in engineered LOV constructs.
- Solution: Use truncated "sLOV" domains (residues 408-543 of AsLOV2) to eliminate unintended NES activity, as demonstrated in opto-nanobody development [37].
Phototoxicity During Prolonged Illumination:
- Cause: Extended blue light exposure can damage cellular components.
- Solution: Optimize illumination protocols using minimal intensity and duration, and consider two-photon activation (850 nm for Cry2) for reduced phototoxicity in thick samples [48].
Research Reagent Solutions
Table 2: Essential Reagents for Optogenetic Oligomerization Studies
| Reagent Category | Specific Examples | Key Features & Applications |
|---|---|---|
| Cry2 Variants | CRY2PHR (1-498 aa) [3] [13] | Standard photolyase homology region for basic optogenetic applications |
| CRY2olig (E490G) [48] | Enhanced clustering mutant for robust oligomerization (70% cytosolic protein clustered) | |
| CRY2high & CRY2low [3] | Engineered variants with tuned oligomerization propensity | |
| LOV Domain Systems | iLID [13] | Fast-responding heterodimerization system with superior spatial confinement |
| Magnets [13] | Bidirectional photoswitch system with rapid off-kinetics | |
| OptoNBs [37] | Light-switchable nanobodies for targeting endogenous proteins | |
| Binding Partners | CIBN (1-170 aa of CIB1) [13] [8] | Truncated Cry2 interaction partner for reduced background interactions |
| SspB [13] | iLID binding partner for recruitment applications | |
| Fluorescent Tags | mCherry-Cry2 fusions [13] [8] | Standard visualization with minimal interference to clustering |
| GFP-CIBN fusions [8] | Bait visualization for recruitment assays | |
| Targeting Sequences | Sec61TM [3] [8] | ER membrane targeting for enhanced Cry2 clustering |
| CAAX motif [8] | Plasma membrane targeting | |
| Miro1TM [8] | Mitochondrial outer membrane targeting |
Application Case Studies
Signaling Pathway Activation
Optogenetic oligomerization tools have been particularly valuable for dissecting signaling pathway mechanisms. The CRY2olig system has enabled precise control over the Raf/MEK/ERK cascade, demonstrating that light-induced clustering of Raf kinase domains can trigger downstream signaling and drive neurite outgrowth in PC12 cells in the absence of growth factors [8]. Similarly, CRY2-based clustering has been applied to activate Wnt/β-catenin signaling by inducing receptor oligomerization [46], and to control Rho-family GTPase pathways through cytoskeletal remodeling [3].
A recent innovative application employed an eGFP-specific nanobody fused to Cry2 variants to cluster eGFP-tagged IKKα and IKKβ, achieving potent and reversible activation of NF-κB signaling without requiring upstream stimuli [46]. This approach demonstrated that graded clustering strength directly correlates with signaling output, with CRY2olig-NbGFP generating the strongest NF-κB activation due to its enhanced oligomerization capability [46]. This system provides a generalizable strategy for controlling diverse signaling pathways by targeting eGFP-tagged proteins, bypassing the need for custom receptor engineering.
Biomolecular Condensates and Neurodegenerative Disease
The study of biomolecular phase separation has been transformed by optogenetic clustering tools, particularly for investigating protein aggregation in neurodegenerative diseases. The OptoDroplet system, which combines Cry2 with the intrinsically disordered region (IDR) of FUS (FUsed in Sarcoma), enables light-induced formation of liquid-like condensates that can mature into pathological aggregates [47]. This approach has revealed that small oligomeric aggregates, rather than large inclusions, are key drivers of toxicity in models of amyotrophic lateral sclerosis (ALS) [47].
Similarly, optogenetic control of syntaxin clustering has illuminated the role of liquid-liquid phase separation in neuronal exocytosis, demonstrating that Munc18 modulates syntaxin phase separation to promote efficient synaptic vesicle fusion [49]. These applications highlight how Cry2-based clustering tools can probe the functional consequences of controlled protein condensation in live cells, providing insights into both physiological and pathological processes.
The comparative analysis of Cry2/CIB and LOV domain optogenetic systems reveals complementary strengths suited to different experimental requirements. Cry2-based systems, particularly the CRY2olig variant, excel in applications requiring robust oligomerization and the formation of large, stable clusters, such as in signaling pathway activation and biomolecular condensate studies [3] [48]. The dual functionality of Cry2—capable of both heterodimerization with CIB1 and homo-oligomerization—provides versatility but also introduces potential complications from unintended interactions in complex experimental setups [3] [8].
In contrast, LOV domain systems like iLID and Magnets offer superior spatial and temporal precision due to their faster off-kinetics, making them ideal for applications requiring subcellular manipulation with minimal spread beyond the illumination area [13]. The development of opto-nanobodies further expands LOV domain applications to include targeting of endogenous proteins without genetic modification [37].
System selection should be guided by experimental priorities: Cry2 variants for maximal clustering efficiency and pathway activation, or LOV domain systems for high spatial precision and rapid reversibility. Future developments will likely focus on engineering improved variants with optimized kinetics, reduced phototoxicity, and expanded spectral properties, further enhancing the capabilities of these powerful tools for manipulating cellular processes with light.
Optogenetics provides unparalleled spatiotemporal control over biological processes by using light to manipulate protein interactions and functions. For researchers aiming to control signaling pathways, transcription, or subcellular sequestration, two primary blue-light-sensing systems are widely adopted: the CRY2/CIB system from Arabidopsis thaliana and various Light-Oxygen-Voltage (LOV) domain-based systems. The CRY2/CIB system is a heterodimerizer; the cryptochrome 2 (CRY2) protein undergoes a light-induced conformational change that enables binding to its partner, CIB1 [25] [50]. In contrast, many LOV-based systems, such as EL222, function as light-regulated homodimers or allosteric switches [10]. Framing your choice within the context of your specific experimental goal—be it probing fast signaling dynamics, controlling gene expression, or achieving robust protein sequestration—is critical to success. This guide provides a direct, data-driven comparison of these systems to help you build a decisive selection framework.
Performance & Characteristics: A Quantitative Comparison
The table below summarizes the core performance characteristics of CRY2/CIB and LOV-based systems, highlighting key quantitative differences that influence tool selection.
Table 1: Performance Comparison of CRY2/CIB and LOV-based Optogenetic Systems
| Characteristic | CRY2/CIB System | LOV-based Systems (e.g., EL222) | Experimental Implications |
|---|---|---|---|
| Core Mechanism | Light-induced heterodimerization [25] | Light-induced homodimerization or allosteric unfolding [10] | CRY2/CIB is ideal for bringing two different proteins together; LOV is simpler for self-association or controlling a fused effector. |
| Activation Kinetics | Very fast (seconds) [50] | Fast (seconds) [10] | Both systems suitable for rapid, second-scale interventions. |
| Deactivation Kinetics (Half-life) | ~2.5 to 24 minutes (tunable with mutants) [25] | ~50 seconds (for EL222) [10] | CRY2/CIB is suited for processes requiring sustained activity; LOV is superior for rapid, pulsed interventions. |
| Dynamic Range (Transcriptional Activation) | Up to 570-fold induction (with optimized designs) [10] | Up to 570-fold induction (with DEL-VPR design) [10] | Both can achieve high dynamic ranges with advanced engineering. |
| Key Mutants/Tools | CRY2(535) truncation (reduced dark activity), L348F (longer half-life), CRY2olig (E490G, enhanced clustering) [25] [50] | DEL-VPR (high-potency transcriptional activator) [10] | A suite of optimized variants exists for both systems to fine-tune performance. |
| Clustering Behavior | Yes (native CRY2 and enhanced in CRY2olig) [50] | Not typically for transcriptional systems | CRY2's clustering can be harnessed for sequestration but may complicate some quantitative applications. |
Decision Matrix: Matching the Tool to the Experimental Goal
Use the following decision matrix to select the optimal system based on your primary experimental goal. The recommendations are based on key performance characteristics and established use cases from the literature.
Table 2: Decision Matrix for Selecting an Optogenetic System
| Your Experimental Goal | Recommended System | Rationale and Key Tools |
|---|---|---|
| Controlling Signaling Pathways | CRY2/CIB | The reversible, minute-scale deactivation is well-suited to many intracellular signaling kinetics. Mutants like short-cycling W349R (t½ ~2.5 min) allow for matching the tool's kinetics to the pathway's [25]. |
| High-Level Transcription Activation | LOV (EL222-VPR) or CRY2/CIB | For the highest possible expression, the single-component LOV system EL222 fused to the potent VPR transactivation domain (DEL-VPR) achieves robust, 570-fold induction with very low background, ideal for bioproduction [10]. CRY2/CIB can achieve similar highs but may require more complex circuit engineering. |
| Protein Sequestration & Clustering | CRY2olig | The engineered CRY2olig (E490G) variant is the superior choice. It shows rapid, robust, and reversible clustering, redistributing up to 70% of cytosolic protein into puncta within seconds of light exposure [50]. |
| Reversible Control with Minimal Residual Activity | LOV (EL222) | The fast spontaneous reversion of EL222 (~50 s) in the dark ensures minimal residual activity after light is off, ideal for pulsed stimulation [10]. |
| Tunable Signaling State Lifetime | CRY2/CIB | The availability of well-characterized photocycle mutants like long-lived L348F (t½ ~24 min) and short-lived W349R (t½ ~2.5 min) allows you to precisely match the tool's lifetime to your process [25]. |
Experimental Protocols for Key Applications
Protocol 1: Light-Activated Transcription Using CRY2/CIB or LOV Systems
This protocol outlines a general setup for achieving light-induced transcription in mammalian cells (e.g., HEK293T), adaptable for both CRY2/CIB and LOV-based systems [10] [51].
- Key Reagents:
- Effector Plasmid: For CRY2/CIB, this is typically a fusion of a DNA-binding domain (e.g., Gal4BD) to CRY2 and a transactivation domain (e.g., VP64) to CIB1. For LOV, use a construct like DEL-VPR (EL222-VPR-NLS) [10].
- Reporter Plasmid: A plasmid containing a minimal promoter upstream of a reporter gene (e.g., luciferase, GFP), driven by the corresponding response elements (e.g., UAS for Gal4-based systems, C120 for EL222) [10].
- Detailed Workflow:
- Cell Seeding and Transfection: Seed HEK293T cells in multi-well plates. The following day, co-transfect the effector and reporter plasmids using a standard transfection reagent.
- Dark Adaptation: After transfection (e.g., 4-6 hours), wrap control plates in aluminum foil to maintain dark conditions.
- Light Stimulation: Place experimental plates under blue light (e.g., 450-490 nm, 1-10 mW/cm²). A regimen of pulsed light (e.g., 1 sec ON / 99 sec OFF) is often more effective and less toxic than continuous illumination [10].
- Harvest and Assay: After 24-48 hours of stimulation, harvest cells and quantify reporter gene expression (e.g., measure luciferase luminescence or GFP fluorescence).
- Data Interpretation: Calculate the dynamic range (fold induction) as the ratio of reporter activity in light-stimulated cells versus dark-adapted controls. A successful experiment will show high induction in light with minimal background activity in the dark.
Protocol 2: Probing Protein Interactions with LINC (Light-Induced Co-clustering)
This protocol uses the CRY2olig module to detect and visualize protein-protein interactions in live cells [50].
- Key Reagents:
- "Bait" Construct: A fusion of your protein of interest with CRY2olig (E490G).
- "Prey" Construct: A fusion of your putative interacting protein with a fluorescent tag (e.g., GFP).
- Detailed Workflow:
- Cell Preparation: Co-transfect the bait and prey constructs into your target cells (e.g., COS-7, HEK293).
- Pre-Illumination Imaging: Use a confocal microscope to capture a baseline image of the prey fluorescence (e.g., using 488 nm laser at low power to avoid activation).
- Photoactivation: Apply a brief pulse of blue light (e.g., 458 nm or 488 nm laser at 5-10% power for a few seconds) to a specific region of interest (ROI) to induce clustering of the CRY2olig-bait.
- Post-Illumination Imaging: Immediately image the same ROI to monitor the localization of the fluorescently tagged prey.
- Data Interpretation: A positive interaction is confirmed if the prey protein redistributes and co-clusters with the CRY2olig-bait puncta after light application. A negative control with a non-interacting protein should show no redistribution of the prey.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and resources required for implementing the protocols and tools discussed in this guide.
Table 3: Essential Research Reagents for CRY2/CIB and LOV Optogenetics
| Reagent / Resource | Function / Description | Example Source / Identifier |
|---|---|---|
| CRY2/CIB Plasmids | Core components for heterodimerization systems. Includes CRY2, CIB1, and their truncations/mutants (e.g., CRY2(535), CRY2olig). | Addgene (e.g., #79569, #79570) [25] [50] |
| LOV Plasmids | Core components for LOV-based systems. Includes EL222 and its fusions (e.g., DEL-VPR). | Addgene (e.g., #133726, #133727) [10] |
| Reporter Plasmids | Plasmids to measure the output of the optogenetic system (e.g., UAS-Luciferase, C120-minP-GFP). | Addgene (e.g., #33020) [10] [51] |
| Blue LED Light Source | Provides uniform, cool illumination for plate-based experiments. Wavelength: 450-490 nm. | Commercial vendors (e.g., Thorlabs, Lumencor) [52] [53] |
| Confocal Microscope | For live-cell imaging and spatially precise activation (e.g., using a 405/458/488 nm laser). | Commercial vendors (e.g., Zeiss, Nikon, Olympus) [50] |
| HEK293T Cells | A widely used, easily transfected mammalian cell line for testing and optimization. | ATCC (CRL-3216) [10] [51] |
Conclusion
The choice between Cry2/CIB and LOV domain systems is not one of superiority but of suitability. Cry2/CIB offers robust, high-level recruitment and is powerful for applications where some diffusion from the illumination site is acceptable. In contrast, LOV-based tools like iLID and Magnets, with their faster off-kinetics, provide unparalleled subcellular spatial precision, albeit sometimes with a lower total yield of dimerization. The ongoing engineering of variants with tailored oligomerization tendencies and kinetics, such as CRY2high and CRY2low, provides an expanding toolkit for precise biological control. Future directions will focus on the development of novel photoreceptors responsive to longer, tissue-penetrating wavelengths, multi-color optogenetics for parallel control of several pathways, and the increased translation of these precise tools to guide the development of next-generation, spatially informed therapeutic interventions.
References
- 1. Constitutively active Arabidopsis cryptochrome two alleles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12212125/]
- 2. The Arabidopsis blue-light photoreceptor CRY2 is active in ... [https://www.sciencedirect.com/science/article/abs/pii/S0092867424012133]
- 3. Understanding CRY2 interactions for optical control of ... [https://www.nature.com/articles/s41467-017-00648-8]
- 4. Optogenetic activation of intracellular signaling based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8451544/]
- 5. Archaeal LOV domains from Lake Diamante: first functional ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1572269/full]
- 6. Regulation of Arabidopsis photoreceptor CRY2 by two ... [https://www.nature.com/articles/s41467-021-22410-x]
- 7. Strategies for development of optogenetic systems and ... [https://www.sciencedirect.com/science/article/pii/S1389556716300612]
- 8. The Dual Characteristics of Light-Induced Cryptochrome 2, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5061508/]
- 9. The Dual Characteristics of Light-Induced Cryptochrome 2 ... [https://pubmed.ncbi.nlm.nih.gov/25985220/]
- 10. Potent optogenetic regulation of gene expression in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12207406/]
- 11. Optimal fluorescent protein tags for quantifying ... [https://www.nature.com/articles/s41598-018-28858-0]
- 12. Engineering of photo-inducible binary interaction tools for ... [https://www.nature.com/articles/s41467-025-61710-4]
- 13. Light-activated protein interaction with high spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5877946/]
- 14. Light-dependent modulation of protein localization and ... [https://www.nature.com/articles/s41467-024-54974-9]
- 15. Constitutively active Arabidopsis cryptochrome 2 alleles ... [https://www.sciencedirect.com/science/article/pii/S0021925825021155]
- 16. Optobiochemistry: Genetically Encoded Control of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11464261/]
- 17. A structural view of plant CRY2 photoactivation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7440854/]
- 18. Structural insights into photoactivation of plant ... [https://www.nature.com/articles/s42003-020-01531-x]
- 19. Unraveling the Mechanism of a LOV Domain Optogenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7572778/]
- 20. Blue light-induced LOV domain dimerization enhances the ... [https://elifesciences.org/articles/11860]
- 21. Electron transfer pathways in a light, oxygen, voltage (LOV) ... [https://www.nature.com/articles/s41598-017-13420-1]
- 22. Opening New Frontiers in Optogenetic Research, Sensitive ... [https://science.xyz/news/new-frontiers-in-optogenetic-research/]
- 23. Illuminating cell signalling with optogenetic tools - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4145075/]
- 24. Rapid blue light induction of protein interactions in living cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC3059133/]
- 25. Optimized second generation CRY2/CIB dimerizers and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4871718/]
- 26. CRY2/CIB1 [https://www.optobase.org/switches/Cryptochromes/CRY2-CIB1/]
- 27. Light-dependent modulation of protein localization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11685620/]
- 28. Optogenetic tools for dissecting complex intracellular ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X20301005]
- 29. Optogenetically controlled protein kinases for regulation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5882534/]
- 30. Optogenetic dissection of RET signaling reveals robust ... [https://molecularbrain.biomedcentral.com/articles/10.1186/s13041-023-01046-6]
- 31. Optogenetic activation of EphB2 receptor in dendrites induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5769660/]
- 32. Benchmarking of optical dimerizer systems - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/25350266/]
- 33. Optogenetic Regulation of EphA1 RTK Activation and Signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC10871282/]
- 34. CRISPR/Cas System and Factors Affecting Its Precision ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8652214/]
- 35. LOV-based optogenetic devices: light-driven modules to ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2015.00018/full]
- 36. Optogenetic approaches to drug discovery in neuroscience ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5495001/]
- 37. Optogenetic control of protein binding using light ... [https://www.nature.com/articles/s41467-020-17836-8]
- 38. Optogenetic Approaches for the Spatiotemporal Control of ... [https://www.mdpi.com/1422-0067/22/10/5300]
- 39. Spatiotemporal Control of Protein Activity through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12199233/]
- 40. Genetically-stable engineered optogenetic gene switches ... [https://www.nature.com/articles/s41467-024-54350-7]
- 41. Blue Light‐Induced, Dosed Protein Expression of Active BDNF in... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11671672/]
- 42. New optogenetic screening platform for drug discovery [https://www.ddw-online.com/new-optogenetic-screening-platform-for-drug-discovery-35658-202507/]
- 43. Optogenetic platform illuminates new antiviral strategies [https://news.ucsb.edu/2025/021955/optogenetic-platform-illuminates-new-antiviral-strategies]
- 44. Engineering of photo-inducible binary interaction tools for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12304284/]
- 45. Optogenetic modulation of guanine nucleotide exchange ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2023.1195806/full]
- 46. Activation of NF‐κB Signaling by Optogenetic Clustering of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447120/]
- 47. Optogenetics to biomolecular phase separation in ... [https://www.sciencedirect.com/science/article/pii/S1016847825000718]
- 48. An optimized optogenetic clustering tool for probing protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4170572/]
- 49. Munc18 modulates syntaxin phase separation to promote ... [https://www.nature.com/articles/s41593-025-02140-9]
- 50. An optimized optogenetic clustering tool for probing protein ... [https://www.nature.com/articles/ncomms5925]
- 51. Bidirectional approaches for optogenetic regulation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5714224/]
- 52. Optogenetics in the Real World: 5 Uses You'll Actually See ... [https://www.linkedin.com/pulse/optogenetics-real-world-5-uses-youll-actually-see-2025-qbsjf/]
- 53. Key Drivers for Optogenetics Equipment Market Growth [https://www.datainsightsmarket.com/reports/optogenetics-equipment-952598]