Efficient CRISPR-Cas9 Tp53 Knockout Protocol for Rapid MEF Immortalization
This article provides a comprehensive guide for researchers on an optimized protocol for immortalizing Mouse Embryonic Fibroblasts (MEFs) using CRISPR-Cas9-mediated deletion of the Tp53 gene.
Efficient CRISPR-Cas9 Tp53 Knockout Protocol for Rapid MEF Immortalization
Abstract
This article provides a comprehensive guide for researchers on an optimized protocol for immortalizing Mouse Embryonic Fibroblasts (MEFs) using CRISPR-Cas9-mediated deletion of the Tp53 gene. Covering foundational principles, step-by-step methodology, troubleshooting, and validation, it details a highly efficient technique that generates immortalized MEF (iMEF) lines within 2-3 weeks. The protocol overcomes the limitations of traditional methods like serial passaging (3T3) or oncogene transformation, which are often inefficient, time-consuming, or alter cellular physiology. The resulting iMEFs closely resemble parent primary cells, enabling long-term genetic manipulation, cloning, and functional studies, with broad applications in gene function analysis, disease modeling, and drug development.
Why Target Tp53? The Scientific Foundation for Reliable MEF Immortalization
The Challenge of Primary MEF Senescence in Long-Term Studies
Mouse Embryonic Fibroblasts (MEFs) are a fundamental model system in biological research, particularly for studying gene function, regulation, and cellular processes. However, primary MEFs present a significant challenge for long-term studies due to their inherent limited lifespan in culture. After several rounds of passaging (typically 5-7 passages under standard conditions), primary MEFs undergo replicative senescence, a state of permanent growth arrest also known as the "Hayflick limit" [1]. This senescence barrier drastically restricts the amount of experimentation that can be performed, limits the generation of biological replicates, and prevents the accumulation of sufficient protein lysate for comprehensive analysis [1].
The transition to senescence is characterized by specific morphological changes, including cells becoming large and flat, and the expression of senescence-associated β-galactosidase (SA-β-gal) [2]. At the molecular level, this process involves upregulation of cell cycle inhibitors such as p16INK4a and p21, which enforce the growth arrest [3] [2]. Standard cell culture conditions, particularly exposure to atmospheric oxygen levels (20%), impose significant oxidative stress that contributes to DNA damage and accelerates the onset of senescence [4]. This fundamental limitation necessitates strategies to overcome cellular senescence for long-term studies requiring stable, proliferative cell populations.
Established Immortalization Strategies and Their Limitations
Several traditional approaches have been employed to immortalize primary MEFs, each with distinct advantages and drawbacks. The table below summarizes the key characteristics of these established methods:
Table 1: Comparison of Traditional MEF Immortalization Methods
| Method | Key Features | Time Required | Efficiency | Major Limitations |
|---|---|---|---|---|
| Serial Passaging (Spontaneous) | Cultivation at 3% O₂; may use ROCK inhibitor (Y-27632) [1] | 20-25 passages (weeks to months) [1] | Variable and inconsistent [1] [5] | Unpredictable; relies on spontaneous mutations; time-consuming [5] |
| SV40 Large T-Antigen | Oncoprotein inactivates p53 and pRb tumor suppressors [1] | ~2-3 weeks [1] | High and reliable [1] | Alters physiology; induces cancer-like phenotypes; resistance to apoptosis [1] [5] |
| Telomerase (TERT) Overexpression | Prevents telomere shortening [5] | Varies | Effective for human cells [5] | Generally ineffective for mouse cells [5] |
The serial passaging method, often called the "3T3 protocol," is inefficient and time-consuming, taking many weeks with no guarantee of success [5]. While culturing at physiological oxygen (3% O₂) can delay the DNA damage response and extend the proliferative phase, it does not reliably prevent senescence [1]. Methods involving oncogene overexpression, such as SV40 Large T-antigen, efficiently immortalize cells but significantly alter their physiological properties, including growth factor requirements, metabolism, and signaling pathways, potentially compromising their relevance to normal cellular function [1] [5]. These limitations highlight the need for a more controlled, efficient, and physiologically relevant immortalization strategy.
CRISPR-Mediated Tp53 Deletion: A Novel Protocol for Efficient Immortalization
The discovery that MEFs immortalized through serial passaging frequently harbor loss-of-function mutations in the Tp53 gene (encoding the p53 protein) revealed a key molecular target for a more directed approach [5]. p53 is a critical tumor suppressor that regulates cell cycle arrest and senescence in response to stress and DNA damage. CRISPR-mediated deletion of Tp53 effectively recapitulates this spontaneous immortalizing event in a controlled and highly efficient manner.
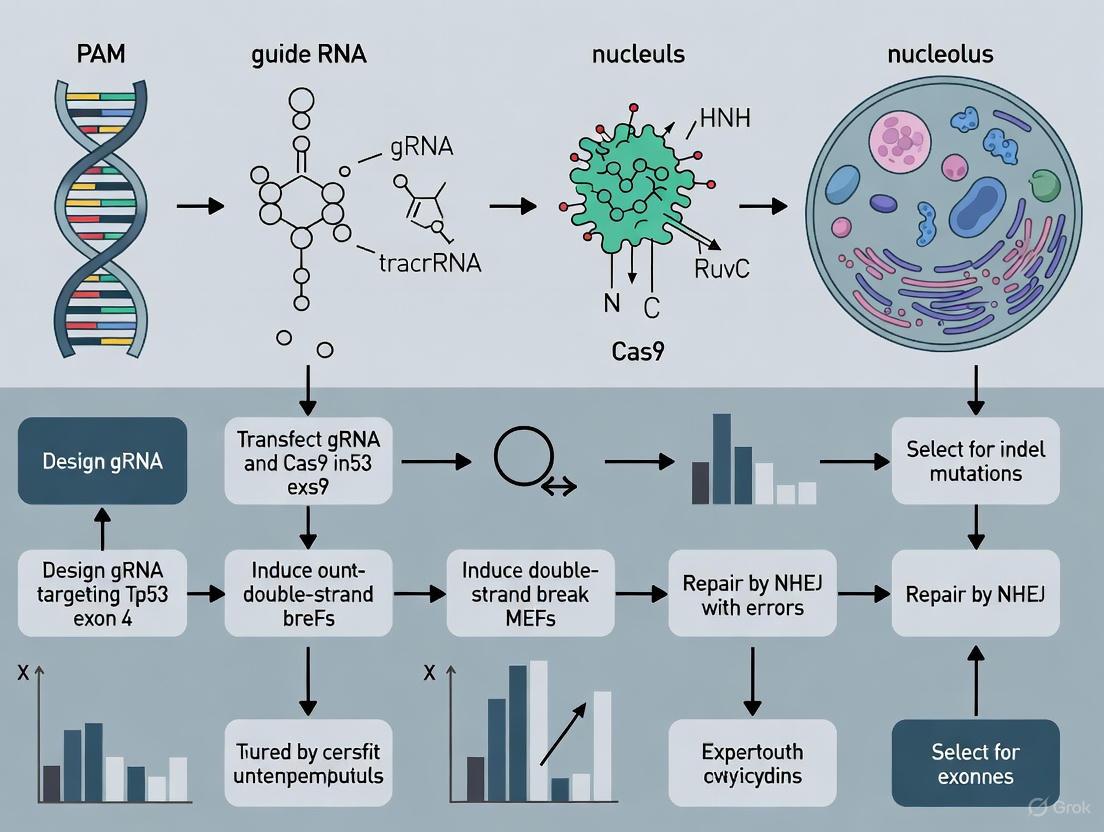
This protocol enables the reliable generation of immortalized MEFs (iMEFs) within three weeks from a minimal number of primary cells (as few as 50,000), closely resembling the parent cell population without inducing neoplastic transformation [6] [5]. The workflow for this optimized protocol is illustrated below:
Detailed Experimental Protocol
Primary MEF Isolation
- Mouse Embryos: Sacrifice a pregnant mouse at embryonic day 12.5 (E12.5) to E13.5. Remove the intact uterus and isolate individual embryos [1] [5].
- Tissue Dissection: Separate the embryo body from the head and internal organs. The head can be saved for genotyping if needed.
- Tissue Dissociation: Mince the remaining embryonic tissue finely and dissociate in 0.1% trypsin solution, optionally supplemented with RNase-free DNase (1 µg/mL), for 10-15 minutes at 37°C [5].
- Plating: Inactivate trypsin with complete culture media (DMEM with 10% FBS, 1% penicillin-streptomycin, 2 mM L-glutamine). Pellet cells, resuspend, and plate in tissue culture dishes. This is considered passage 0 (P0).
Tp53 Knockout via Electroporation
- CRISPR Constructs: Use validated CRISPR plasmids targeting the Tp53 gene, such as the Px461-Cas9n-Trp53-sgRNA-alpha and -beta plasmids available from Addgene [5].
- Cell Preparation: Harvest primary MEFs at passage 3 or 4 when they are approximately 50-70% confluent. Count cells and resuspend them in Buffer R (from the Neon Transfection System kit) at a concentration of 1-5 x 10⁷ cells/mL [5].
- Electroporation: For each reaction, mix 10 µL of cell suspension with 1-3 µg of total CRISPR plasmid DNA. Electroporate using the Neon Transfection System with the following optimized parameters: 1,350 V, 20 ms width, 2 pulses [5].
- Post-Transfection Recovery: Immediately transfer the electroporated cells to pre-warmed culture media (without antibiotics) in a 6-well plate. Allow cells to recover for 3-5 days, monitoring for GFP expression if a fluorescent marker is co-transfected.
Clonal Expansion and Validation
- Clonal Isolation: After recovery, trypsinize and dilute cells to seed at low density or perform limited dilution in 96-well plates to isolate single clones.
- Expansion: Expand individual clones and cryopreserve aliquots for long-term storage.
- Validation: Confirm Tp53 knockout via:
- Western Blotting: Assess p53 protein loss.
- Genomic DNA Sequencing: Verify indels or deletions at the Tp53 locus.
- Functional Assay: Confirm resistance to senescence upon serial passaging.
Research Reagent Solutions for MEF Immortalization
The following table lists essential reagents and materials required for successful immortalization of MEFs using the CRISPR-Cas9 protocol.
Table 2: Key Research Reagents for CRISPR-mediated MEF Immortalization
| Reagent / Material | Function / Application | Example Details |
|---|---|---|
| Tp53-targeting CRISPR Plasmids | Introduction of double-strand breaks in the Tp53 gene to generate knockout mutations. | e.g., Px461-Cas9n-Trp53-sgRNA plasmids (Addgene #88846, #88847) [5]. |
| Electroporation System | High-efficiency delivery of CRISPR constructs into hard-to-transfect primary MEFs. | Neon Transfection System (Thermo Fisher) or comparable systems [6] [5]. |
| Cell Culture Media | Support growth and maintenance of MEFs before and after immortalization. | High-glucose DMEM, 10% FBS, 1% Penicillin-Streptomycin, 2 mM L-glutamine [5]. |
| Senescence Detection Kit | Validation of senescence bypass in immortalized clones. | Senescence-associated β-Galactosidase (SA-β-gal) Staining Kit [2]. |
| Antibodies for Validation | Confirmation of p53 protein loss in immortalized MEFs. | Anti-p53 antibody for Western blot analysis [2]. |
p53 Pathway and Immortalization Mechanism
The pivotal role of p53 in maintaining cellular senescence and how its deletion enables immortalization is summarized in the pathway below:
The p53 protein acts as a central integrator of stress signals, including the oxidative stress encountered in standard cell culture conditions [4]. Upon activation, p53 transcriptionally upregulates the cyclin-dependent kinase inhibitor p21, which enforces cell cycle arrest and establishes the senescent state [2]. CRISPR-Cas9 technology directly targets the source of this pathway by creating a loss-of-function mutation in the Tp53 gene. This ablation prevents the initiation of the senescence program, allowing cells to bypass the Hayflick limit and achieve immortalization while largely retaining the physiological characteristics of the primary parent cells [6] [5].
The challenge of primary MEF senescence presents a significant obstacle in biomedical research, limiting the scope and reproducibility of long-term cellular studies. While traditional immortalization methods exist, they are often hampered by low efficiency, extended timelines, and the induction of aberrant cellular phenotypes. The protocol for CRISPR-mediated Tp53 deletion represents a superior alternative, offering a controlled, efficient, and rapid solution. By directly targeting a well-defined genetic node in the senescence pathway, this method reliably generates immortalized MEF lines within three weeks, providing researchers with a consistent and physiologically relevant cellular model system. This approach not only facilitates basic research but also enhances drug discovery by enabling stable, long-term genetic manipulation and screening in a defined genetic background.
Application Note
Limitations of Traditional Immortalization Methods: 3T3, Telomerase, and Oncogenes
This application note details the principal limitations of three conventional cell immortalization techniques—serial passaging (3T3), telomerase overexpression, and viral oncogene expression—and positions CRISPR-mediated Tp53 deletion as a superior methodology for the immortalization of Mouse Embryonic Fibroblasts (MEFs). The content is framed within ongoing research for establishing a robust and phenotypically stable MEF immortalization protocol.
Critical Analysis of Traditional Immortalization Methods
The establishment of immortalized cell lines is a cornerstone of biomedical research, enabling long-term studies and consistent experimental outcomes. However, traditional methods are fraught with significant drawbacks that can compromise research validity. The table below summarizes the key limitations of three widely used approaches.
Table 1: Quantitative and Qualitative Limitations of Traditional Immortalization Methods
| Method | Key Mechanism | Typical Immortalization Time | Major Limitations | Impact on Cell Phenotype |
|---|---|---|---|---|
| Serial Passaging (3T3 Protocol) | Spontaneous mutation emergence through prolonged culture [5] [7]. | Several weeks to months [5] [7]. | Inefficient and time-consuming; relies on random, undefined mutations; difficult to reproduce consistently [5] [7]. | Frequent karyotype instability and altered physiological properties [5]. |
| Telomerase (TERT) Overexpression | Ectopic expression of telomerase to maintain telomere length and bypass replicative senescence [8] [9]. | Varies by cell type; can be several weeks. | Ineffective for many mouse cells, including MEFs, due to alternative telomere maintenance mechanisms [5] [7]. | Generally preserves normal cell phenotype and karyotype when successful in human cells [8]. |
| Viral Oncogenes (SV40 LT, HPV E6/E7) | Inactivation of tumor suppressor pathways (e.g., p53, Rb) by viral proteins [10]. | Relatively rapid (1-3 weeks). | High frequency of oncogenic transformation; aberrant karyotype; altered differentiation capacity [5] [10]. | Significant phenotypic alterations, loss of contact inhibition, and cancer-like phenotypes [5] [10]. |
Experimental Protocol: A Case Study in Oncogene-Induced Immortalization
The following protocol, adapted from studies using viral oncogenes, exemplifies a method that, while effective, introduces the phenotypic alterations noted in Table 1.
Objective: To immortalize primary human cells via transduction with lentiviral vectors expressing the HPV16 E6/E7 oncogenes. Background: The E6 and E7 proteins inactivate the p53 and retinoblastoma (Rb) tumor suppressor pathways, respectively, which is a common strategy to overcome replicative senescence [10].
Materials:
- Primary Cells (e.g., Keratinocytes or Fibroblasts)
- Lentiviral Vectors: Encoding HPV16 E6 and E7 genes.
- Polybrene (4 µg/mL): To enhance viral transduction efficiency [8].
- Puromycin (1 µg/mL): For selection of successfully transduced cells [8].
- Complete Cell Culture Media: Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, and 1% penicillin-streptomycin [5] [7].
Methodology:
- Cell Seeding: Plate primary cells at a density of 5 x 10⁴ cells per well in a 6-well plate and culture until 50-60% confluent.
- Viral Transduction: Incubate cells with the lentiviral supernatant in the presence of 4 µg/mL polybrene for 24-48 hours [8].
- Selection: Replace the medium with fresh complete media containing 1 µg/mL puromycin. Maintain selection pressure for 5-7 days, replacing the puromycin-containing media every 2-3 days until all non-transduced control cells have died.
- Expansion and Validation: Expand the resistant cell population and validate immortalization through:
- Proliferation Assay: Confirmation of extended lifespan beyond the Hayflick limit.
- Senescence-Associated β-galactosidase (SA-β-gal) Staining: A significant reduction in SA-β-gal positive cells indicates bypassed senescence [11].
- Phenotype Check: Assessment of cell-specific markers (e.g., keratins for epithelial cells) to document potential phenotypic drift [8] [10].
Visual Workflow: From Traditional Methods to a Modern Solution
The following diagram illustrates the logical progression from the limitations of traditional methods to the targeted approach of CRISPR/Cas9.
The Scientist's Toolkit: Essential Reagents for CRISPR-mediated Immortalization
The following reagents are critical for implementing the modern CRISPR/Cas9-based immortalization protocol, which directly addresses the limitations of traditional methods.
Table 2: Key Research Reagent Solutions for CRISPR-mediated Tp53 Knockout
| Reagent / Tool | Function / Application | Specific Example |
|---|---|---|
| CRISPR Plasmids | Targeted knockout of the Tp53 gene to bypass senescence. | pX461-Cas9n-Trp53-sgRNA-alpha & -beta plasmids (Addgene #88846, #88847) [5] [7]. |
| Electroporation System | High-efficiency delivery of CRISPR constructs into primary MEFs. | Neon Transfection System 10 µL Kit (Thermo Fisher) [5] [7]. |
| Primary Cell Culture Media | Supports the growth and viability of primary MEFs pre- and post-immortalization. | High-glucose DMEM, 10% FBS, L-glutamine, sodium pyruvate, non-essential amino acids [5] [7]. |
| Genotyping Assays | Confirmation of successful Tp53 gene editing. | PCR with locus-specific primers followed by Sanger sequencing to detect "indel" mutations [11]. |
| Phenotypic Validation Assays | Verify that immortalized cells (iMEFs) retain characteristics of parent cells. | Proliferation rate analysis, karyotype stability checks, and expression of cell-specific markers [8] [5]. |
Visual Guide: CRISPR Workflow for MEF Immortalization
The detailed experimental workflow for the recommended CRISPR/Cas9 method is outlined below.
Traditional immortalization techniques, while foundational, present substantial obstacles to efficient and reliable MEF line generation. The 3T3 protocol is inefficient, telomerase overexpression is often ineffective in mouse cells, and viral oncogenes profoundly disrupt normal cell physiology. In contrast, CRISPR/Cas9-mediated deletion of Tp53 emerges as a targeted, rapid, and consistent alternative. This method reliably produces immortalized MEFs (iMEFs) within three weeks, with the significant advantage of closely resembling the parent cell population, making it ideally suited for downstream applications in gene function studies and drug development [6] [5] [7].
Tp53 as a Master Regulator of Senescence and Cell Cycle Arrest
The tumor protein p53, encoded by the TP53 gene, functions as a critical tumor suppressor and master regulator of cellular homeostasis, principally through its roles in initiating senescence and cell cycle arrest [12]. As the "guardian of the genome," p53 integrates diverse stress signals—including DNA damage, oncogene activation, and oxidative stress—to determine whether a cell undergoes repair, permanent cell cycle exit (senescence), or programmed cell death [13] [12]. This decision-making capability is fundamental to preventing the proliferation of damaged cells and suppressing tumor development. In the context of experimental cell biology, the precise manipulation of Tp53 provides a powerful tool for controlling cellular lifespan. Specifically, CRISPR-mediated deletion of Tp53 in primary mouse embryonic fibroblasts (MEFs) efficiently disrupts these protective pathways, enabling cell immortalization while preserving physiological properties more closely than oncogene-based methods [5] [14]. This application note details the molecular mechanisms by which Tp53 governs senescence and cell cycle arrest and provides a detailed protocol for leveraging Tp53 deletion to generate immortalized MEFs for research applications.
Molecular Mechanisms of p53-Mediated Senescence and Cell Cycle Arrest
p53 Activation and Transcriptional Regulation
The p53 protein functions primarily as a tetrameric transcription factor that directly regulates the expression of approximately 500 target genes [15] [13]. Under normal physiological conditions, p53 levels remain low due to continuous degradation mediated by its negative regulator, MDM2 [12] [16]. Upon cellular stress, post-translational modifications (including phosphorylation and acetylation) stabilize p53, leading to its accumulation and activation [12]. The stabilized p53 protein forms homotetramers that bind specific DNA response elements, initiating transcriptional programs that determine cell fate [13].
Key Downstream Effector Pathways
The tumor suppressor activity of p53 is executed through multiple downstream pathways, with senescence and cell cycle arrest representing two critical mechanisms.
Cell Cycle Arrest: p53 orchestrates cell cycle arrest at both the G1/S and G2/M checkpoints, primarily through the transcriptional activation of p21 (CDKN1A) [12]. p21 is a potent cyclin-dependent kinase (CDK) inhibitor that binds to and inactivates cyclin-CDK complexes, preventing phosphorylation of the retinoblastoma (Rb) protein [12]. Hypophosphorylated Rb remains bound to E2F transcription factors, maintaining a stable G1 arrest [12]. For G2/M arrest, p53 activates additional targets including 14-3-3 sigma and Reprimo, which sequester cyclin B1-Cdc2 complexes essential for mitotic entry [12].
Cellular Senescence: p53-mediated senescence is a stable cell cycle arrest program that occurs in response to various stressors, including DNA damage and oncogene activation [12] [17]. This process involves the coordinated action of p53 and p16INK4A, which converge on Rb activation to enforce permanent growth arrest [12]. Senescent cells establish senescence-associated super-enhancers (SASEs) that drive the expression of pro-survival genes while maintaining the non-proliferative state [17]. Research has identified 40 common SASEs in senescent MEFs, regulating approximately 50 genes critical for core senescent properties [17].
Table 1: Key p53 Target Genes in Senescence and Cell Cycle Arrest
| Target Gene | Function | Biological Outcome |
|---|---|---|
| p21 (CDKN1A) | CDK inhibitor, prevents Rb phosphorylation | G1/S cell cycle arrest [12] |
| 14-3-3 sigma | Sequesters cyclin B1-Cdc2 complexes | G2/M cell cycle arrest [12] |
| Gadd45 | Disrupts cyclin B1/Cdc2 complex | G2 phase arrest [12] |
| Reprimo | Involved in p53-dependent G2 arrest | G2/M cell cycle arrest [12] |
| MDM2 | Negative regulator of p53; promotes survival of senescent cells [17] | Limits p53-mediated apoptosis in senescent cells [17] |
| PTPRV | Tyrosine phosphatase | Contributes to G1 arrest [12] |
Figure 1: p53-Mediated Signaling in Senescence and Cell Cycle Arrest. Cellular stress triggers p53 activation, leading to transcription of target genes like p21 that enforce cell cycle arrest and senescence. Senescent cells subsequently activate super-enhancer-driven survival genes (e.g., MDM2, Rnase4) to remain viable while non-dividing [12] [17].
Experimental Protocol: CRISPR-Mediated Tp53 Deletion for MEF Immortalization
Background and Principle
Primary MEFs undergo replicative senescence after a limited number of population doublings, restricting their utility for long-term genetic studies [5] [14]. The spontaneous immortalization of 3T3 fibroblast lines frequently involves loss-of-function mutations in Tp53 [5]. This protocol leverages CRISPR-Cas9 genome editing to specifically target and delete the Tp53 gene in primary MEFs, enabling efficient and consistent generation of immortalized MEF (iMEF) lines within 2-3 weeks while minimizing alterations to physiological cellular properties [5] [14].
Materials and Reagents
Table 2: Key Research Reagent Solutions for Tp53 Knockout
| Reagent / Material | Function / Application | Specifications / Notes |
|---|---|---|
| Px461-Cas9n-Trp53-sgRNA plasmids [5] | CRISPR genome editing | Specific sgRNAs targeting mouse Tp53 gene; available from Addgene (#88846, #88847) |
| Neon Transfection System [5] | Electroporation delivery | Efficient delivery of CRISPR constructs into MEFs; 10μL kit recommended |
| Primary MEFs | Biological starting material | Isolated from E12.5-E13.5 mouse embryos [5] |
| Electroporation Buffer R [5] | Electroporation | Component of Neon Transfection System |
| MEF Culture Medium | Cell culture | DMEM, 10% FBS, 2mM L-glutamine, 1x penicillin-streptomycin [5] |
| Trypsin-EDTA (0.25%) | Cell passaging | For harvesting and subculturing cells after initial isolation |
Step-by-Step Workflow
Figure 2: Experimental Workflow for Generating Immortalized MEFs. The process begins with isolation of primary MEFs from mouse embryos, followed by CRISPR-Cas9 electroporation to delete Tp53, recovery and expansion of edited cells, clonal isolation, and final validation of immortalized lines [5].
Step 1: Isolation of Primary Mouse Embryonic Fibroblasts (MEFs)
- Isolate MEFs from day E12.5-E13.5 mouse embryos using standard dissection protocols.
- Dissociate embryonic tissues using 0.1% trypsin solution with RQ1 RNase-free DNase.
- Culture primary MEFs in complete DMEM medium supplemented with 10% FBS, L-glutamine, non-essential amino acids, and sodium pyruvate [5].
Step 2: CRISPR-Cas9 Transfection via Electroporation
- Prepare CRISPR plasmid DNA (Px461-Cas9n-Trp53-sgRNA-alpha and beta plasmids) using standard molecular biology methods.
- Harvest early-passage MEFs (recommended: 50,000-100,000 cells) and resuspend in Buffer R.
- Electroporate using Neon Transfection System with the following optimized parameters: 1300V, 20ms, 2 pulses [5].
- Plate transfected cells in antibiotic-free culture medium and incubate at 37°C with 5% CO₂.
Step 3: Post-Transfection Recovery and Expansion
- Culture cells for 48-72 hours without disturbance to allow recovery and initial proliferation.
- Begin regular passaging (every 3-4 days) at a density of 3 × 10⁵ cells per 100mm dish.
- Monitor for emergence of rapidly dividing populations, typically occurring within 14-21 days post-transfection [5] [14].
Step 4: Clonal Selection and Expansion
- Once immortalized populations are established, isolate single cells using limited dilution or cloning rings.
- Expand individual clones in separate culture vessels.
- Cryopreserve multiple vials of each clone using freezing medium (90% culture medium, 10% DMSO) [5].
Step 5: Validation of Tp53 Knockout and Immortalization
- Confirm Tp53 deletion by genomic DNA sequencing and western blot analysis.
- Functionally validate by assessing loss of p21 induction and cell cycle arrest following DNA damage.
- Verify immortalized phenotype by continued proliferation beyond normal senescence checkpoint (typically passage 8-10 for primary MEFs).
Expected Results and Technical Validation
Quantitative Assessment of Immortalization Efficiency
Successful implementation of this protocol typically yields immortalized MEF lines with high efficiency. The key advantage of this CRISPR-based approach is its reliability and speed compared to traditional serial passaging methods.
Table 3: Expected Outcomes and Validation Criteria for Immortalized MEFs
| Parameter | Primary MEFs | Tp53-KO iMEFs | Validation Method |
|---|---|---|---|
| Proliferation lifespan | Senescence at ~P8-P10 [5] | Indefinite (>30 passages) [5] | Long-term culture monitoring |
| Tp53 protein expression | Present | Absent | Western blot, immunostaining |
| p21 induction post-DNA damage | Robust | Absent/minimal | qPCR, western blot |
| Cell cycle arrest | Intact | Deficient | EdU incorporation, flow cytometry |
| Morphology | Flattened, enlarged in senescence | Similar to primary, without senescence | Phase-contrast microscopy |
| Genetic background | N/A | Preserved from original strain | Genomic PCR |
Troubleshooting Common Technical Challenges
- Low Transfection Efficiency: Optimize electroporation parameters and ensure high-quality plasmid DNA preparation. Include a fluorescent reporter plasmid (e.g., pCAG-GFP) to monitor efficiency [5].
- Failed Immortalization: Verify CRISPR target efficacy and use early-passage MEFs (passage 2-4) with high viability.
- Altered Physiological Properties: Characterize multiple clones to identify those most closely resembling primary MEFs in key signaling pathways and responses.
Application in Drug Development and Cancer Research
The Tp53 knockout MEF model provides a valuable platform for studying cancer biology and therapeutic development. p53 is mutated in approximately 50% of all human cancers, with specific mutation patterns varying across cancer types [15] [16]. For instance, TP53 mutation frequency reaches 89% in small cell lung cancer and 73% in colorectal cancer [16]. These mutations not only abolish p53's tumor suppressor functions but often confer gain-of-function (GOF) activities that promote tumor progression, genomic instability, and therapy resistance [18] [19] [16].
Immortalized MEFs lacking Tp53 provide a controlled system for:
- Investigating mechanisms of genomic instability and chromosomal instability (CIN) driven by p53 loss [18] [19]
- Studying therapy resistance mechanisms in p53-deficient backgrounds
- Validating novel therapeutic approaches targeting p53-deficient cancers, including compounds that restore p53 function or exploit synthetic lethal interactions
- Modeling the impact of specific p53 mutations on drug sensitivity and resistance patterns
The protocol described herein enables generation of physiologically relevant cellular models that recapitulate key aspects of p53-deficient tumors, providing a bridge between basic molecular studies and preclinical drug development.
Application Notes and Protocols
The process of cellular immortalization is a critical step in oncogenesis and a necessary technique for establishing stable cell lines for biomedical research. A substantial body of evidence, accumulated over decades, identifies the loss of the tumor suppressor protein p53, encoded by the Tp53 gene, as a central and common event in the spontaneous immortalization of primary cells [20]. Primary cells, such as mouse embryonic fibroblasts (MEFs), possess intrinsic signaling pathways that trigger senescence or apoptosis after a limited number of cell divisions, presenting a major barrier to long-term culture and experimentation [5] [7]. The inactivation of p53 function bypasses these critical fail-safes, allowing cells to escape proliferation limits and achieve an immortalized state. This document details the historical evidence for this phenomenon and provides a modern, efficient protocol for achieving targeted immortalization via CRISPR-mediated deletion of Tp53.
Historical and Mechanistic Evidence
The link between p53 dysfunction and immortalization was firmly established in the early 1990s. Seminal research demonstrated that spontaneously immortalized cell lines consistently harbored alterations in the Tp53 gene.
Key Historical Findings
- Universal Alteration in Spontaneous Immortalization: A foundational study examining 11 independently established, clonally derived BALB/c murine embryo fibroblast lines found that all 11 lines had acquired mutations in at least one Tp53 allele [21]. The molecular nature of these alterations varied, including point mutations leading to an extended protein half-life, in-frame deletions, and events that resulted in no detectable p53 protein expression [21]. This diversity indicated a strong selective pressure for the loss of wild-type p53 function, rather than for one specific mutational mechanism.
- Functional Consequence: The conclusion from this and subsequent work was that p53 alteration is not a rare occurrence but a common, and likely necessary, step in the spontaneous immortalization pathway for murine fibroblasts [21] [20]. The loss of p53's normal "guardian of the genome" function allows genetically damaged cells to continue proliferating, thereby facilitating immortalization.
The following table summarizes the types of p53 alterations identified in these spontaneously immortalized cell lines:
Table 1: Spectrum of Tp53 Alterations in Spontaneously Immortalized BALB/c MEF Lines [21]
| Category of Alteration | Molecular Description | Observed Outcome |
|---|---|---|
| Missense Mutations | Point mutations within conserved domains of the p53 DNA-binding domain. | Production of a stable, mutant p53 protein with loss of tumor suppressor function. |
| Deletion Mutations | A 24-base pair in-frame deletion identified in one cell line. | Production of a truncated or dysfunctional p53 protein. |
| Loss of Expression | Various events including deletion of gene exons, introduction of a stop codon, or undetectable mRNA levels. | Complete absence of p53 protein expression in the immortalized cell line. |
From Spontaneous Mutation to Targeted Deletion
The "3T3" serial passaging protocol is a traditional method for spontaneously immortalizing MEFs, but it is inefficient and time-consuming, often taking months [5] [7]. The discovery that these spontaneously derived lines consistently harbored Tp53 mutations provided a rational genetic target for a direct and efficient immortalization strategy [5] [21] [7]. Modern CRISPR/Cas9 gene-editing technology now allows researchers to precisely recapitulate this key immortalizing event in a controlled and highly reproducible manner, moving from spontaneous and stochastic methods to a targeted and rapid protocol.
Modern Protocol: CRISPR-Mediated Tp53 Deletion for Efficient MEF Immortalization
This protocol outlines an optimized method for generating immortalized MEFs (iMEFs) within two to three weeks by using CRISPR/Cas9 to knockout the Tp53 gene [5] [6] [7].
Key Workflow and Underlying Signaling Pathway
The experimental workflow and the critical signaling pathway targeted in this protocol are summarized in the diagrams below.
Research Reagent Solutions
The following reagents are essential for the successful execution of this protocol. All materials should be sterile and of cell culture grade.
Table 2: Essential Research Reagents for CRISPR-mediated MEF Immortalization [5] [7]
| Reagent / Supply | Function / Application | Example Catalog Number |
|---|---|---|
| Biological Material | ||
| Pregnant Mice (E12.5 embryos) | Source of primary Mouse Embryonic Fibroblasts (MEFs). | N/A |
| Plasmids | ||
| Px461-Cas9n-Trp53-sgRNA-alpha | CRISPR plasmid for targeted cleavage of the Tp53 gene. | Addgene #88846 |
| Px461-Cas9n-Trp53-sgRNA-beta | Second CRISPR plasmid to enhance knockout efficiency. | Addgene #88847 |
| pCAG-GFP | Fluorescence marker plasmid for tracking transfection efficiency. | Addgene #11150 |
| Critical Reagents | ||
| 0.1% Trypsin in HBSS | Enzymatic dissociation of embryonic tissue for MEF isolation. | Worthington LS003702 |
| Complete Culture Media (DMEM + 10% FBS) | Base nutrient medium for cell growth and maintenance. | Gibco 11960-044 |
| Neon Transfection System 10 μL Kit | Electroporation system for high-efficiency delivery of CRISPR constructs into MEFs. | Thermo Fisher MPK1096 |
| Laboratory Supplies | ||
| Cell Strainers (70 μm) | Physical filtration to obtain a single-cell suspension from dissociated tissue. | Corning Falcon 352340 |
| TC-treated Culture Dishes | Treated surface for optimal attachment and growth of adherent MEFs. | Corning Falcon 353002 |
Detailed Methodological Steps
Part A: Preparation of Primary Mouse Embryonic Fibroblasts (MEFs)
- Dissection: Sacrifice a pregnant mouse at E12.5 and aseptically dissect the uterus. Isolate individual embryos and transfer to a sterile dish containing PBS.
- Tissue Dissociation: Remove the head and internal organs (liver, heart) from each embryo. Mince the remaining embryonic tissue finely with sterile scissors.
- Digestion: Add 2-3 mL of pre-warmed 0.1% trypsin solution to the minced tissue. Incubate at 37°C for 15-20 minutes, gently pipetting every 5 minutes to dissociate the tissue.
- Filtration and Plating: Neutralize trypsin by adding an equal volume of complete culture media (supplemented with sodium pyruvate and MEM NEAA). Pass the cell suspension through a 70 μm cell strainer into a new tube. Centrifuge, resuspend the cell pellet, and plate in a 100 mm culture dish.
- Expansion: Culture the primary MEFs until they reach 80-90% confluence (Passage 0). These can be cryopreserved or used directly for immortalization.
Part B: Immortalization via Tp53 Knockout
- CRISPR Transfection Complex: Prepare a DNA mixture containing the two Tp53-targeting CRISPR plasmids (Px461-Cas9n-Trp53-sgRNA-alpha and -beta) and the pCAG-GFP plasmid at a 1:1:1 mass ratio.
- Electroporation: Harvest primary MEFs at early passage (P1-P3). Use the Neon Transfection System with the recommended settings for MEFs (e.g., 1400 V, 20 ms, 2 pulses) to deliver the CRISPR plasmid mix into the cells.
- Post-Transfection Culture: Immediately plate the electroporated cells into culture media without penicillin-streptomycin. The GFP marker can be used to confirm transfection efficiency 24-48 hours post-electroporation.
- Selection and Expansion: Continue to culture the cells, passaging them as needed when they reach confluence. Within approximately 14 days, rapidly dividing, immortalized populations will emerge and overtake the culture as the non-transfected and non-immortalized cells undergo senescence.
- Clonal Isolation (Optional): To establish clonal iMEF lines, seed the polyclonal immortalized population at a very low density. Isolate individual colonies using cloning rings and expand them for further characterization.
The transition from observing Tp53 loss in spontaneous immortalization to directly engineering it with CRISPR technology represents a significant advancement in cell biology methodology. The protocol described herein is highly reliable, generating immortalized MEFs in under three weeks, a stark contrast to the months often required for the spontaneous 3T3 method [5] [7]. A key advantage is that the resulting iMEFs more closely resemble the primary parent cell population compared to those immortalized by oncogene overexpression, which often acquire aberrant cancer-like phenotypes [5]. This makes iMEFs ideal for subsequent genetic manipulations, such as gene rescue experiments, or for functional studies of other genes in a physiologically relevant cellular context. By understanding and leveraging the critical event of Tp53 inactivation, researchers can efficiently create tailored cellular models to accelerate discovery in gene function, regulatory mechanisms, and drug development.
Advantages of CRISPR-Cas9 for Precise Genetic Ablation over Random Mutagenesis
The ability to selectively inactivate gene function is a cornerstone of biological research and therapeutic development. For decades, random mutagenesis techniques were the primary tool for this task, relying on non-specific agents to induce mutations with unpredictable outcomes. The emergence of CRISPR-Cas9 genome editing has revolutionized this paradigm, offering an unprecedented level of precision and control. This application note details the distinct advantages of CRISPR-Cas9 for precise genetic ablation, particularly within the context of a common and critical cellular manipulation: the CRISPR-mediated deletion of the Tp53 gene for the immortalization of mouse embryonic fibroblasts (MEFs). We provide a comprehensive comparison of these technologies, detailed experimental protocols, and essential reagent solutions to guide researchers in implementing this powerful methodology.
Fundamental Technological Comparison
CRISPR-Cas9 and random mutagenesis represent fundamentally different approaches to genetic modification. The core distinction lies in the specificity of the targeting mechanism.
Mechanism of Random Mutagenesis
Random mutagenesis employs chemical agents (e.g., ethyl methanesulfonate, EMS) or radiation to induce mutations stochastically throughout the genome. These methods create a wide spectrum of genetic lesions, including point mutations, insertions, and deletions, at random locations. The identification of a cell with the desired mutation in a target gene requires laborious, large-scale screening. Furthermore, the potential for numerous uncharacterized secondary mutations poses a significant risk, as these can confound phenotypic interpretations [22].
Mechanism of CRISPR-Cas9
In contrast, CRISPR-Cas9 functions as a programmable DNA endonuclease. The system comprises two key components: the Cas9 enzyme, which acts as molecular "scissors" to create a double-stranded break (DSB) in the DNA, and a single guide RNA (sgRNA), which directs Cas9 to a specific genomic locus through complementary base-pairing. The cellular repair of this DSB, primarily through the error-prone non-homologous end joining (NHEJ) pathway, results in small insertions or deletions (indels) that disrupt the target gene's function with high precision [23] [24].
Table 1: Fundamental Comparison of CRISPR-Cas9 vs. Random Mutagenesis
| Feature | CRISPR-Cas9 | Random Mutagenesis |
|---|---|---|
| Targeting Mechanism | Programmable, sequence-specific guide RNA | Stochastic, genome-wide damage |
| Mutation Specificity | High (targets a predefined locus) | None (mutations occur randomly) |
| Mutation Profile | Primarily indels at the target site | Spectrum of point mutations, indels, and chromosomal rearrangements |
| Screening Requirement | Minimal; verification of on-target editing | Extensive; required to find rare cells with the desired mutation |
| Number of Off-Target Lesions | Low to moderate (predictable and screenable) | High (unpredictable and pervasive) |
| Experimental Timeline | Weeks | Months to years |
| Key Technical Challenge | Off-target effects, delivery efficiency | Genetic background noise, screening throughput |
The following diagram illustrates the fundamental workflow and key differences between these two approaches for a gene knockout objective.
Quantitative Advantages of CRISPR-Cas9
The theoretical precision of CRISPR-Cas9 translates into concrete, measurable benefits in the laboratory. The application of a high-throughput CRISPR-Cas9 pipeline in maize successfully targeted 743 candidate genes, with 412 edited sequences from 118 genes precisely identified from phenotyped individuals. This demonstrates the system's capacity for systematic, large-scale functional genomics [22]. In the specific context of MEF immortalization, the CRISPR-mediated deletion of Tp53 consistently generates immortalized MEFs (iMEFs) within 14 days, a process that is not only highly efficient but also preserves the physiological properties of the parent cells, unlike many older methods [7] [25].
Table 2: Quantitative Performance Metrics for Genetic Ablation
| Performance Metric | CRISPR-Cas9 | Random Mutagenesis |
|---|---|---|
| Typical Targeting Efficiency | High (e.g., 70-90% in MEFs for Tp53) [7] | Extremely Low (requires screening of thousands of clones) |
| Experimental Duration | ~2 weeks for MEF immortalization [7] | Several months to over a year |
| Number of Unintended Mutations | Low (limited to potential off-target sites) | Very High (hundreds to thousands per genome) |
| Success Rate for Specific Gene | High (depends on sgRNA design and delivery) | Very Low (a function of gene size and screening capacity) |
| Screening Throughput | High-throughput and scalable [22] [26] | Low-throughput and labor-intensive |
Detailed Protocol: CRISPR-Cas9 Mediated Tp53 Deletion for MEF Immortalization
The following optimized protocol for immortalizing primary MEFs via Tp53 knockout has been adapted from a recent, highly efficient method [7] [25].
Materials and Reagents
- Biological Material: Primary Mouse Embryonic Fibroblasts (MEFs) isolated from E12.5-E13.5 embryos.
- Plasmids:
- pX461-Cas9n-Trp53-sgRNA-alpha (Addgene #88846)
- pX461-Cas9n-Trp53-sgRNA-beta (Addgene #88847)
- Optional: pCAG-GFP (Addgene #11150) for monitoring transfection efficiency.
- Cell Culture Reagents:
- Dulbecco’s Modified Eagle Medium (DMEM), high glucose
- Fetal Bovine Serum (FBS)
- Non-essential amino acids (MEM NEAA)
- Sodium pyruvate
- L-glutamine
- Penicillin-Streptomycin
- Trypsin-EDTA (0.25%)
- Transfection Reagent: Neon Transfection System 10 μL Kit (Thermo Fisher Scientific, MPK1096).
Step-by-Step Workflow and Protocol
The entire workflow, from MEF isolation to the expansion of validated iMEF clones, is summarized below.
Procedure:
- MEF Preparation and Culture: Isolate MEFs from E12.5 mouse embryos using standard protocols. Culture primary MEFs in complete DMEM media supplemented with 10% FBS, 1% MEM NEAA, 1 mM sodium pyruvate, and 1% penicillin-streptomycin. Culture cells in a 37°C incubator with 5% CO₂.
- sgRNA/Cas9 Ribonucleoprotein (RNP) Complex Formation: The protocol utilizes a dual-sgRNA strategy to delete a segment of the Tp53 gene. While plasmid-based delivery is effective, using purified Cas9 protein and synthetic sgRNAs as an RNP complex can enhance efficiency and reduce off-target effects.
- Electroporation: Harvest MEFs at a early passage (P1-P2). Resuspend 5 x 10⁵ to 1 x 10⁶ cells in the provided Neon electroporation buffer. Mix the cell suspension with the pre-formed RNP complex (or with the two Tp53 sgRNA plasmids). Electroporate using the Neon Transfection System with the following optimized parameters: 1300 V, 20 ms, 2 pulses. Immediately transfer the electroporated cells to pre-warmed antibiotic-free culture media.
- Post-Transfection Culture and Immortalization: Approximately 48 hours post-electroporation, resume passaging the cells with standard trypsinization. Seed cells at a consistent density (e.g., 3 x 10⁵ cells per 60-mm dish) and passage every 3 days. Critical Step: Monitor cells closely. Primary MEFs will initially undergo senescence, but rapidly dividing, immortalized cells (iMEFs) will emerge and become the dominant population within approximately 14 days.
- Validation and Cloning:
- Validation: Confirm Tp53 knockout by western blot analysis for the p53 protein and by Sanger sequencing of the targeted genomic region after PCR amplification.
- Cloning: For a uniform cell line, isolate single-cell clones by serial dilution or using cloning rings. Expand individual clones and validate the Tp53 knockout in each.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for CRISPR-Cas9 Mediated MEF Immortalization
| Research Reagent / Solution | Function / Application | Specific Example / Note |
|---|---|---|
| Tp53-specific sgRNAs | Guides the Cas9 nuclease to the mouse Trp53 gene locus. | Use a pair of sgRNAs (e.g., from Addgene #88846 & #88847) to excise a critical exon. |
| Cas9 Nuclease | Creates a double-stranded break in the DNA at the site specified by the sgRNA. | Can be delivered as a plasmid, mRNA, or as a purified protein for RNP formation. |
| Electroporation System | Enables highly efficient delivery of CRISPR components into hard-to-transfect primary MEFs. | The Neon Transfection System is optimized for this protocol [7]. |
| MEF Culture Media | Supports the growth and viability of primary and immortalized MEFs. | DMEM + 10% FBS. For primary MEFs, supplement with NEAA and sodium pyruvate. |
| Selection Antibiotics | Used to select for cells that have stably integrated Cas9/sgRNA plasmids (if using plasmid-based delivery). | Blasticidin or Puromycin, depending on the resistance marker on the plasmid. |
| Genotyping Primers | For PCR amplification and subsequent sequencing of the edited Tp53 locus to confirm knockout. | Design primers flanking the dual sgRNA target sites to detect the deletion. |
Critical Considerations and Troubleshooting
While highly efficient, CRISPR-Cas9 editing requires careful optimization and validation.
- Minimizing Off-Target Effects: The specificity of the sgRNA is paramount. Utilize bioinformatic tools to design sgRNAs with minimal potential for off-target binding. Using a dual-sgRNA strategy for a large deletion, as described here, can reduce the likelihood of in-frame mutations that restore gene function. Furthermore, using Cas9 RNP complexes instead of plasmid vectors can reduce the time the nuclease is active in the cell, thereby lowering off-target effects [24].
- The p53 Selection Pressure: A critical consideration is that CRISPR-Cas9-induced double-strand breaks can activate a p53-mediated DNA damage response. This can create a selective pressure where cells with pre-existing dysfunctional p53 have a survival advantage. While this is leveraged in this protocol for immortalization, it is a crucial confounder in other functional genomics screens. It is essential to genotype edited cells to confirm the intended Tp53 mutation is present, rather than a pre-existing one [26].
- Troubleshooting Low Efficiency: If immortalization efficiency is low, verify the activity of the sgRNAs and the efficiency of the electroporation step. Using a fluorescent reporter plasmid (e.g., pCAG-GFP) in a test electroporation can help optimize transfection parameters.
The transition from random mutagenesis to CRISPR-Cas9 for genetic ablation represents a quantum leap in precision, efficiency, and experimental control. The detailed protocol for Tp53-mediated MEF immortalization underscores these advantages, enabling researchers to generate well-defined cellular models within a predictable timeframe. By leveraging the specific reagents and methodologies outlined in this application note, scientists and drug development professionals can accelerate their research with greater confidence in the genetic integrity of their experimental systems.
A Step-by-Step Protocol: From MEF Isolation to Immortalized Cell Line
The immortalization of mouse embryonic fibroblasts (MEFs) is a critical step for establishing stable cell lines suitable for long-term genetic studies and drug discovery applications. Primary MEFs undergo replicative senescence after a limited number of population doublings, significantly hampering extended experimental manipulation. This application note details a highly efficient protocol for MEF immortalization through CRISPR-mediated deletion of the Tp53 gene, a key regulator of the cell cycle and senescence. Unlike traditional methods such as spontaneous immortalization through serial passaging (the 3T3 protocol) or oncogene overexpression, this targeted genetic approach is rapid, reliable, and produces immortalized cells (iMEFs) that closely resemble their primary parent populations without inducing cancer-like phenotypes [27] [7]. The core principle involves the electroporation of CRISPR-Cas9 constructs specifically designed to knockout the Tp53 gene, enabling the generation of stable iMEF lines within two to three weeks [27] [6].
Required Reagents and Equipment
Research Reagent Solutions
The following table catalogs the essential biological materials, reagents, and laboratory supplies required for the successful isolation and immortalization of MEFs.
Table 1: Key Research Reagent Solutions for MEF Immortalization
| Item | Function/Application | Specifications / Example Sources |
|---|---|---|
| Biological Materials | ||
| Pregnant Mice | Source of E12.5 embryos for MEF isolation [27] [7]. | Embryonic day 12.5 (E12.5) embryos. |
| Key Plasmid Constructs | ||
| Px461-Cas9n-Trp53-sgRNA-alpha | CRISPR plasmid for targeted deletion of Tp53 [27] [7]. | Addgene Plasmid #88846. |
| Px461-Cas9n-Trp53-sgRNA-beta | Second CRISPR plasmid targeting a different Tp53 site [27] [7]. | Addgene Plasmid #88847. |
| pCAG-GFP Plasmid | Control plasmid for monitoring transfection efficiency [27] [7]. | Addgene Plasmid #11150. |
| Critical Reagents | ||
| Trypsin from Bovine Pancreas | Enzymatic dissociation of embryonic tissue for primary MEF isolation [27] [7]. | 0.1% solution in HBSS. |
| RQ1 RNase-free DNase | Prevents cell clumping during primary cell preparation by digesting DNA released from dead cells [27] [7]. | 1 U/μL. |
| Fetal Bovine Serum (FBS) | Essential growth supplement for cell culture media [27] [7]. | 10% in complete culture media. |
| Dimethyl Sulfoxide (DMSO) | Cryoprotectant for freezing down primary MEFs and immortalized lines [27] [7]. | 10% in cell freezing media. |
| Laboratory Supplies & Equipment | ||
| Neon Transfection System | Electroporation system for high-efficiency delivery of CRISPR constructs into MEFs [27]. | Thermo Fisher Scientific, Cat# MPK1096 (10 μL kit). |
| Electroporation Tips and Buffers | Specific consumables for the Neon Transfection System [27]. | Included in the Neon kit. |
| Tissue Culture Dishware | For cell culture and expansion [27] [7]. | 60 mm, 100 mm dishes; 6-well, 24-well plates. |
| Cell Strainers | Physical filtration to achieve a single-cell suspension after embryo dissection [27] [7]. | 70 μm mesh. |
Media and Solution Formulations
Precise media formulation is crucial for cell viability, proliferation, and successful transfection. The following table summarizes the required solutions and their compositions.
Table 2: Media and Solution Formulations
| Solution Name | Primary Function | Composition | Special Notes |
|---|---|---|---|
| 0.1% Trypsin for MEF Prep [27] [7] | Primary tissue dissociation. | - 40 mg Trypsin from bovine pancreas- 40 mL HBSS (without Ca2+ and Mg2+) | Filter sterilize (0.22 μm), aliquot, and store at -80°C. |
| Complete Cell Culture Media [27] [7] | Routine culture and expansion of MEFs. | - 500 mL DMEM (high glucose)- 50 mL FBS (10%)- 5.5 mL Penicillin-Streptomycin (100X, 1X)- 5.5 mL L-Glutamine (200 mM, 2 mM) | Pre-warm to 37°C before use. |
| Supplemented Culture Media [27] [7] | Culture of freshly isolated primary MEFs. | Complete Culture Media +- 1 mM Sodium Pyruvate (final)- 1X MEM Non-Essential Amino Acids | Use for the first few passages after isolation. |
| Antibiotic-Free Culture Media [27] [7] | Culture of cells immediately post-electroporation. | Complete Culture Media without Penicillin-Streptomycin. | Pre-warm before use. |
| Cell Freezing Media [27] [7] | Cryopreservation of cell stocks. | - 45 mL Complete Culture Media (90%)- 5 mL DMSO (10%) | Filter sterilize (0.22 μm) and use cold. |
Experimental Protocol
The diagram below outlines the complete experimental workflow from MEF isolation to the establishment of cloned immortalized cell lines.
Detailed Methodological Steps
Primary MEF Isolation (Duration: 2-3 hours)
- Isolate E12.5 embryos from a pregnant mouse and remove the head and internal organs.
- Mince the remaining embryonic tissue finely using sterile scissors and dissociate the tissue fragments in 0.1% trypsin solution supplemented with RQ1 DNase (1 U/μL) for 15-20 minutes at 37°C.
- Inactivate the trypsin by adding an equal volume of complete culture media. Filter the cell suspension through a 70 μm cell strainer to remove debris and collect the flow-through containing the primary MEFs.
- Centrifuge the cells, resuspend in Supplemented Culture Media, and seed into TC-treated culture dishes. Culture these P0 cells until they reach 80-90% confluence [27] [7].
CRISPR Transfection via Electroporation (Duration: 1 day)
- Harvest primary MEFs at passage 1 or 2 using 0.25% trypsin-EDTA. Resuspend the cell pellet in Buffer R (from the Neon kit) at a concentration of 1-2 x 10^7 cells/mL.
- For each electroporation reaction, mix 2 μg of both Px461-Cas9n-Trp53-sgRNA-alpha and -beta plasmids with 10 μL of the cell suspension.
- Electroporate using the Neon Transfection System with the following optimized parameters: 1300 V, 20 ms, 2 pulses [27].
- Immediately transfer the electroporated cells into pre-warmed Antibiotic-Free Culture Media and seed them into a 24-well plate.
Post-Transfection Culture and Immortalization (Duration: 2-3 weeks)
- Culture the transfected cells, refreshing the media every 2-3 days. Do not use antibiotic-containing media for at least the first 48-72 hours post-transfection.
- Within 5-7 days, actively dividing, immortalized cells (iMEFs) will become visible and begin to overgrow the senescing primary MEF population.
- Once the culture reaches confluence, passage the cells using standard trypsinization and continue to expand them. The immortalized line is typically established within 14-21 days post-transfection [27] [7].
Subcloning and Expansion (Duration: 2-3 weeks)
- To generate clonal iMEF lines, trypsinize the pooled iMEF population and seed a limited number of cells (e.g., 0.5 cells/well) into 96-well plates to facilitate the growth of single-cell clones.
- Expand individual clones and screen for the absence of p53 protein expression via western blotting to confirm successful Tp53 knockout.
Underlying Mechanism
Signaling Pathway and Logic
The following diagram illustrates the molecular mechanism by which Tp53 deletion leads to bypass of cellular senescence and enables immortalization.
The tumor suppressor protein p53, encoded by the Tp53 gene, serves as a critical "guardian of the genome." In response to cellular stress, such as the DNA damage perceived by primary cells during routine culture and passaging, p53 becomes activated and induces the expression of genes that lead to cell cycle arrest or apoptosis. This mechanism acts as a fundamental barrier to the uncontrolled proliferation of potentially damaged cells and is a primary driver of replicative senescence in primary MEFs [27] [7]. The CRISPR-mediated knockout of the Tp53 gene ablates this key protein. Without functional p53, the DNA damage checkpoint is compromised, allowing MEFs to bypass senescence and continue proliferating indefinitely, thereby achieving immortalization. This method is highly efficient because it directly targets a gene known to be mutated in spontaneously immortalized 3T3 lines [27].
Within the broader context of establishing a CRISPR-mediated Tp53 deletion protocol for the efficient immortalization of mouse embryonic fibroblasts (MEFs), the isolation and preparation of high-quality primary cells is a critical first step. Primary MEFs, derived from genetically modified mice, are a valuable resource for studying gene function and regulation [7] [6]. However, these primary cells undergo senescence after only a few passages, which severely limits long-term genetic manipulations and studies [7] [27]. The subsequent protocol parts will detail a highly efficient immortalization method using CRISPR to ablate the Tp53 gene, enabling the generation of stable, immortalized MEF (iMEF) lines within 14-21 days [6] [25]. This first protocol part provides the essential foundation: a detailed method for the reliable isolation and preparation of primary MEFs from E12.5 mouse embryos, which is a prerequisite for any successful immortalization and downstream genetic research [27] [28].
Materials and Reagents
Research Reagent Solutions
The following table lists the essential materials and reagents required for the isolation and initial culture of primary MEFs.
Table 1: Essential Reagents and Materials for MEF Isolation
| Item | Function/Application | Example Catalog Number |
|---|---|---|
| Pregnant mice (E12.5-E14.5) [27] [28] | Source of embryos for MEF isolation. | N/A |
| Phosphate Buffered Saline (PBS), without Ca²⁺ and Mg²⁺ [27] | Washing and rinsing embryos and tissues. | SH30256.01 (Cytiva) |
| Trypsin from bovine pancreas [7] [27] | Enzymatic dissociation of embryonic tissue. | LS003702 (Worthington) |
| RQ1 RNase-free DNase [7] [27] | Degrades sticky genomic DNA released from lysed cells to prevent cell clumping. | M6101 (Promega) |
| Dulbecco’s Modified Eagle Medium (DMEM), high glucose [7] [27] | Base medium for cell culture. | 11960-044 (Gibco) |
| Fetal Bovine Serum (FBS) [7] [27] | Essential growth factors and nutrients for cell culture. | 26-140-079 (Gibco) |
| Penicillin-Streptomycin (100X) [7] [27] | Antibiotic to prevent bacterial contamination. | 15-140-122 (Gibco) |
| L-Glutamine (200 mM) [7] [27] | Essential amino acid for cell growth. | 25030081 (Gibco) |
| Non-essential Amino Acids (MEM NEAA, 100X) [7] [27] | Supplements medium for optimal growth of primary cells. | 11140050 (Gibco) |
| Sodium Pyruvate (100 mM) [7] [27] | Energy source for cells. | 11360070 (Gibco) |
| Dimethyl Sulfoxide (DMSO) [7] [27] | Cryoprotectant for freezing cells. | D2650-100ML (Millipore Sigma) |
| 0.25% Trypsin-EDTA [7] [27] | For passaging cells after initial preparation. | 25200072 (Gibco) |
| Cell Strainers (70 μm or 100 μm) [27] | Filtering single-cell suspension from tissue debris. | 352340 (Falcon) |
Equipment and Laboratory Supplies
Table 2: Necessary Equipment and Supplies
| Category | Specific Items |
|---|---|
| Surgical Tools | Surgical scissors, Adson forceps, fine tip forceps (Dumont #5) [7] [27]. |
| Consumables | 60 mm and 100 mm TC-treated culture dishes, 6-well and 24-well plates, 50 mL conical tubes, 5 mL serological pipettes, cryogenic vials [7] [27]. |
| Major Equipment | Dissecting microscope, biological safety cabinet, CO₂ incubator (set at 37°C, 5% CO₂), benchtop centrifuge, water bath [7] [27] [28]. |
Solution Recipes
- 0.1% Trypsin for MEF Preparation: Dissolve 40 mg of trypsin from bovine pancreas in 40 mL of HBSS without Ca²⁺ and Mg²⁺. Filter sterilize using a 0.22 μm syringe filter, aliquot, snap-freeze in liquid nitrogen, and store at -80°C [7] [27].
- Complete Cell Culture Media: Combine 500 mL of DMEM, 50 mL of FBS (10% final), 5.5 mL of Penicillin-Streptomycin (1X final), and 5.5 mL of L-Glutamine (2 mM final). Pre-warm to 37°C before use. For freshly isolated MEFs, supplement the culture media with sodium pyruvate (1 mM final) and MEM NEAA (1X final) [7] [27].
- Cell Freezing Media: Combine 45 mL of culture media with 5 mL of DMSO (10% final). Filter sterilize with a 0.22 μm syringe filter. Keep cold and use promptly [7] [27].
Step-by-Step Protocol
The following diagram summarizes the entire workflow for the isolation and preparation of primary MEFs, from embryo dissection to the establishment of primary cultures.
Detailed Protocol Steps
Note: All procedures must be performed under sterile conditions in a biological safety cabinet using aseptic technique, in accordance with the host institution's animal care guidelines [28].
Embryo Dissection
- Euthanize a pregnant mouse at E12.5-E14.5 of gestation using an institution-approved method (e.g., CO₂ inhalation followed by cervical dislocation) [28].
- Soak the abdomen with 70% ethanol. Using sterile scissors and forceps, open the abdominal cavity and surgically remove the uterine horns containing the embryos [28].
- Place the uterine horns in a 10 cm dish with 5-10 mL of sterile, chilled PBS.
- Using a dissecting microscope and fine forceps, make an incision along the uterine horn to isolate the individual yolk sacs. Transfer the sacs to a new dish with chilled PBS [28].
- Carefully open the yolk sacs and separate the embryos from the placenta by cutting the umbilical cord. Place the embryos in a new dish with chilled PBS and keep on ice [29] [28].
Embryo Dissociation and Tissue Preparation
- Transfer the embryos to a fresh dish. Decapitate each embryo. Using fine forceps and scissors, carefully cut down the midline to open the abdomen and remove the visceral organs (heart, liver, lungs, etc.) [29] [28].
- Transfer the remaining embryo trunks (or the entire embryo if organs are not removed, though removal is recommended for cleaner fibroblast culture [29]) to a 50 mL conical tube containing 10 mL of pre-warmed 0.1% trypsin solution [27] [28].
- Using sterile scissors, mince the embryonic tissue into very fine pieces. Pipet the tissue and trypsin mixture up and down 20-30 times with a 10 mL pipette to further dissociate it [28].
Trypsin Digestion and Cell Collection
- Incubate the tube in a 37°C water bath for 20-45 minutes, swirling or gently agitating the tube every 5-10 minutes to promote digestion [29] [28].
- After incubation, add a 3x volume (approximately 30 mL) of complete culture media (supplemented with NEAA and sodium pyruvate) to neutralize the trypsin.
- Pipet the cell suspension up and down aggressively with a 25 mL pipette to break up any remaining tissue clumps. To reduce viscosity from released DNA, add DNase to a final concentration of 100 µg/mL and incubate for an additional 5-15 minutes [27] [29].
- Pass the cell suspension through a sterile 70 µm or 100 µm cell strainer into a new 50 mL tube to remove any remaining tissue debris and obtain a single-cell suspension [27].
Plating and Culturing Primary MEFs (Passage 0)
- Centrifuge the filtered cell suspension at 200-300 × g for 5 minutes. Aspirate the supernatant.
- Resuspend the cell pellet in a sufficient volume of complete culture media (with supplements). Plate the cells onto appropriate tissue culture-treated dishes.
- Label the plates as Passage 0 (P0). Culture the cells in a 37°C incubator with 5% CO₂.
- Change the media the next day to remove non-adherent cells and debris. The primary MEFs should attach and appear growing and confluent within 2-4 days [29] [28].
Harvesting and Cryopreservation
- Once the P0 cultures reach confluence (typically 80-90%), they can be passaged or cryopreserved.
- To passage, wash the cells with PBS and harvest using 0.25% Trypsin-EDTA. Neutralize with culture media and split the cells at a recommended ratio (e.g., 1:6 for expansion) [29].
- For cryopreservation, harvest the cells as above, resuspend the pellet in cold freezing media (e.g., 90% FBS/10% DMSO), aliquot into cryovials, and freeze slowly (e.g., using an isopropanol freezing container) at -80°C before transferring to liquid nitrogen for long-term storage [27].
Troubleshooting and Notes
Table 3: Common Issues and Solutions During MEF Isolation
| Problem | Potential Cause | Solution / Recommendation |
|---|---|---|
| Low Cell Yield | Incomplete tissue digestion; embryos too young. | Ensure thorough mincing of tissue and adequate trypsinization time. Use E12.5-E14.5 embryos. |
| Excessive Cell Clumping | Release of genomic DNA from lysed cells. | Use DNase during cell suspension preparation. Pipet more aggressively to dissociate clumps [29]. |
| Poor Cell Attachment | Serum quality; over-trypsinization. | Use qualified FBS. Avoid prolonged trypsin exposure when neutralizing. |
| High Contamination | Non-sterile technique during dissection. | Use ample PBS washes during dissection. Sterilize tools properly and work carefully in a biosafety cabinet. |
| Premature Senescence | High oxidative stress; over-confluence. | Do not let cells become over-confluent. Passage promptly when 80-90% confluent. Use early passage cells (P2-P4) for immortalization [7]. |
Critical Notes:
- The genetic background of the mice (e.g., C57BL/6, 129J) can influence MEF behavior and immortalization efficiency. This should be documented and considered for experimental consistency [7] [27].
- Primary MEFs have a finite lifespan and will undergo senescence, typically after 4-7 passages [29]. For the subsequent CRISPR-Tp53 immortalization protocol, it is recommended to use low-passage MEFs (e.g., P2-P4) to ensure high cell viability and transfection efficiency [7] [27].
Within the broader methodology for CRISPR-mediated deletion of the Tp53 gene to generate immortalized mouse embryonic fibroblasts (iMEFs), efficient delivery of CRISPR constructs into primary cells is a critical determinant of success. Electroporation leverages electrical pulses to create transient pores in the cell membrane, facilitating the intracellular transfer of nucleic acids or ribonucleoproteins (RNPs) with high efficiency. This protocol section details optimized electroporation procedures, specifically using the Neon Transfection System, to achieve high knockout efficiency while preserving cell viability, enabling the reliable establishment of iMEF lines within three weeks [6] [5].
Key Reagent Solutions
The table below catalogues the essential reagents and materials required for the electroporation protocol.
Table 1: Key Research Reagent Solutions for CRISPR Electroporation
| Item Name | Function/Description | Source/Example |
|---|---|---|
| Neon Transfection System | Electroporation device optimized for high efficiency and viability in hard-to-transfect cells, including primary MEFs. | Thermo Fisher Scientific |
| Neon Transfection System 10 µL Kit | Includes specialized tips, electroporation tubes, and buffers (Buffer R and Buffer E) required for the Neon system. | Thermo Fisher Scientific, Cat# MPK1096 [5] |
| PX461 Cas9n Plasmids | CRISPR plasmids expressing Cas9 nickase and Tp53-targeting sgRNAs (alpha and beta). Critical for generating DSBs and Tp53 knockout. | Addgene, #88846 & #88847 [5] |
| pCAG-GFP Plasmid | Reporter plasmid for visually monitoring transfection efficiency via GFP expression. | Addgene, #11150 [5] |
| Primary MEFs | Mouse Embryonic Fibroblasts isolated from E12.5 embryos, the primary cells for immortalization. | Isolated in-house per protocol [5] |
| DMEM, High Glucose | Base medium for cell culture. | Gibco, Cat# 11960-044 [5] |
| Fetal Bovine Serum (FBS) | Serum supplement for cell growth medium. | Qualified grade, e.g., Gibco [5] |
Methodology
Plasmid Preparation and Cell Harvest
Effective electroporation begins with the preparation of high-quality DNA and a healthy, single-cell suspension of primary MEFs.
- CRISPR Construct Preparation: The protocol utilizes a two-plasmid system for robust Tp53 targeting. The plasmids pX461-Cas9n-Trp53-sgRNA-alpha and pX461-Cas9n-Trp53-sgRNA-beta should be purified using an endotoxin-free maxiprep kit to ensure high purity and cell viability [5]. A pCAG-GFP plasmid is included as a transfection reporter.
- MEF Preparation and Harvesting: Primary MEFs should be cultured in complete medium (DMEM with 10% FBS) without antibiotics for at least one passage prior to electroporation. On the day of transfection, harvest cells using trypsin-EDTA, quench with serum-containing medium, and pellet by centrifugation. Resuspend the cell pellet in the provided Buffer R to a final density of 1.0-1.5 x 10^7 cells/mL to achieve a single-cell suspension critical for consistent electroporation [5].
Neon Electroporation Procedure
The core electroporation steps must be followed precisely to balance high transfection efficiency with cell survival.
- Electroporation Mix Assembly: For a standard 10 µL Neon tip, combine the following in a sterile microcentrifuge tube:
- 1.5 µg of each pX461-Tp53-sgRNA plasmid (3 µg total)
- 0.5 µg of pCAG-GFP plasmid
- 1.0 x 10^5 to 1.5 x 10^5 MEFs (in 10 µL of Buffer R)
- Bring the total volume to 20 µL with Buffer R.
- Electroporation Execution: Draw the entire 20 µL mixture into a Neon pipette tip. Insert the tip into the Neon pipette station and apply the following pre-optimized electrical pulse conditions [5]:
- Voltage: 1,400 V
- Pulse Width: 20 ms
- Pulse Number: 2
- Immediately after pulsing, transfer the electroporated cells into pre-warmed, antibiotic-free complete medium in a tissue culture plate. The workflow for this entire process is summarized in the diagram below.
Post-Transfection Processing and Culture
Proper handling of cells following electroporation is essential for their recovery and expansion.
- Cell Recovery and Expansion: After electroporation, seed the cells into 6-well or 24-well plates pre-filled with pre-warmed, antibiotic-free culture medium. The initial seeding density should be optimized to facilitate rapid recovery; a density of 1.0 x 10^5 cells per well of a 6-well plate is recommended. After 24 hours, replace the medium with fresh complete medium containing penicillin-streptomycin to prevent contamination.
- Monitoring and Validation: Transfection efficiency can be estimated 48 hours post-electroporation by visualizing GFP fluorescence using a standard fluorescence microscope. Monitor cells daily for the emergence of rapidly dividing, immortalized populations, which typically outgrow senescing primary cells within 1-2 weeks. Successful Tp53 knockout in the resulting iMEF lines must be confirmed via genomic DNA PCR, followed by Sanger sequencing and/or tracking of indels by decomposition (TIDE) analysis.
Optimization and Troubleshooting
Achieving an optimal balance between editing efficiency and cell viability often requires parameter adjustment. The table below synthesizes key experimental data from similar studies to guide optimization.
Table 2: Electroporation Parameter Optimization and Outcomes from Comparative Studies
| Cell Type / System | Electroporation Parameters | Key Outcome Metrics | Citation |
|---|---|---|---|
| Primary MEFs (Neon) | 1400 V, 20 ms, 2 pulses | Reliable iMEF generation in <3 weeks; high viability and efficiency. | [6] [5] |
| Bovine Zygotes (Neon) | 700 V, 20 ms, 1 pulse | Editing Efficiency: 65.2% (highest). Blastocyst Rate: 10% (low). | [30] [31] |
| Bovine Zygotes (NEPA21) | Increasing voltage/length/number of pulses | Editing Efficiency: Up to 47.6%. Blastocyst Rate: 18% (compromised). | [30] [31] |
| Bovine Zygotes (Lipofection) | Lipofectamine CRISPRMAX | Editing Efficiency: ~30%. Blastocyst Rate: 39% (high, no equipment needed). | [30] [31] |
The relationship between the aggressiveness of electrical parameters and its impact on efficiency versus viability is a key principle, illustrated below.
Troubleshooting Common Issues
- Low Transfection Efficiency: Confirm the quality and concentration of the DNA. Ensure the cell suspension is a pure single-cell solution without clumps. Verify the electroporation buffer is fresh and at room temperature.
- Poor Cell Viability: Avoid leaving cells in Buffer R for extended periods pre- or post-pulse. Titrate the DNA amount downward. Consider slightly reducing the pulse voltage or width and assess the impact on viability and efficiency.
- Lack of Immortalization: Verify the activity and specificity of the Tp53 sgRNAs. Ensure primary MEFs are used at low passage number (recommended P2-P3). Re-assess the transfection efficiency via the GFP control plasmid and confirm knockout via genotyping.
Following the successful CRISPR-mediated deletion of the Tp53 gene in Mouse Embryonic Fibroblasts (MEFs), the subsequent culture and expansion phase is critical for establishing stable, immortalized MEF (iMEF) lines. This section details the standardized protocols for the post-transfection culture, handling of emerging immortalized colonies, and their expansion into clonal cell lines suitable for downstream applications. The procedures are framed within the broader thesis of creating reliable and physiologically relevant cellular models for studying gene function and for drug development research.
The diagram below outlines the complete post-transfection workflow, from initial recovery to the establishment of frozen stocks.
Detailed Post-Transfection Protocol
Immediate Post-Transfection Culture (Day 0-3)
Objective: To ensure cell viability and facilitate recovery after electroporation.
- Culture Vessel: Plate the transfected MEFs in a 6-well tissue culture plate [7] [27].
- Culture Medium: Use complete culture medium without penicillin-streptomycin immediately after electroporation to maximize cell recovery [7] [27]. Antibiotics can be re-introduced after the first 24-48 hours.
- Cell Density: Plate cells at a density sufficient to support recovery and growth, typically between ( 1 \times 10^5 ) to ( 5 \times 10^5 ) cells per well, depending on transfection efficiency and viability.
- Incubation: Maintain cells in a humidified incubator at 37°C with 5% CO₂.
- Monitoring: Check cells daily for attachment, morphology, and viability. The successful delivery of CRISPR constructs can be monitored via GFP fluorescence if a co-transfected reporter plasmid (e.g., pCAG-GFP) is used [27].
Serial Passaging and Monitoring for Immortalization (Day 4-21)
Objective: To sustain the culture until emergent, immortalized clones overcome the senescence crisis.
- Passaging: Once cells reach approximately 80-90% confluence, passage them using 0.25% trypsin-EDTA [7] [27].
- Seeding Density: Maintain a relatively high seeding density during the initial phases (e.g., ( 3 \times 10^5 ) cells per 100 mm dish) to support cell survival [7].
- Observation: Primary MEFs will typically undergo proliferation arrest and enter a senescence crisis around passages 3-5. Following Tp53 knockout, rapidly dividing, morphologically distinct foci will emerge from the senescent background within 14 to 21 days [7] [6] [27].
- Medium Management: Continuously use complete culture medium, replenishing it every 2-3 days.
Isolation and Expansion of Clonal iMEF Lines
Objective: To establish pure, clonal immortalized lines from emergent foci.
- Clonal Isolation: Once stable, proliferative foci are identified, they can be isolated using trypsin-EDTA and cloning rings, or by performing serial dilution in 96-well plates to obtain single-cell-derived clones.
- Expansion: Transfer individual clones to separate wells of a 24-well plate and subsequently to 6-well plates and larger culture vessels as cell numbers increase [7] [27].
- Cryopreservation: Preserve early-passage stocks of each clonal line. Harvest cells, resuspend in freezing medium (e.g., 90% culture medium + 10% DMSO), and freeze slowly (e.g., using a isopropanol-jacketed freezing container) at -80°C before transferring to liquid nitrogen for long-term storage [7] [27].
Key Experimental Parameters and Data Presentation
The table below summarizes critical quantitative parameters for monitoring successful immortalization.
Table 1: Key Parameters for Post-Transfection Culture and Immortalization
| Parameter | Typical Value / Observation | Significance / Notes |
|---|---|---|
| Transfection Efficiency | >90% (with optimized protocol) [32] | Indicates successful delivery of CRISPR constructs. |
| Time to Emergence | 14 - 21 days [7] [6] [27] | Period post-transfection before proliferative foci are observed. |
| Senescence Crisis | Occurs around passages 3-5 [7] | Critical phase; only Tp53-null cells will continue to proliferate. |
| Cell Viability Post-Transfection | >75% (in OptiMEM-GlutaMAX) [32] | Crucial for ensuring sufficient cells recover from electroporation. |
| Seeding Density (Post-Transfection) | ( 3 \times 10^5 ) cells/100mm dish [7] | A high density supports cell survival and growth post-transfection. |
| Clonal Expansion | From 24-well to 6-well to dish [7] [27] | Standard progression for scaling up a clonal population. |
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential Reagents for Post-Transfection Culture of iMEFs
| Reagent / Supply | Function / Application | Example (from Protocol) |
|---|---|---|
| DMEM, High Glucose | Base culture medium providing essential nutrients and energy. | Gibco, cat# 11960-044 [7] [27] |
| Fetal Bovine Serum (FBS) | Supplements medium with growth factors, hormones, and lipids critical for cell growth. | Qualified Grade, 10% final concentration [7] [27] |
| 0.25% Trypsin-EDTA | Proteolytic enzyme solution for dissociating adherent cells during passaging. | Gibco, cat# 25200072 [7] [27] |
| Dimethyl Sulfoxide (DMSO) | Cryoprotective agent used in freezing medium to prevent ice crystal formation during cryopreservation. | Sterile, tissue culture grade [7] [27] |
| L-Glutamine | Essential amino acid required for protein synthesis and cell metabolism. | 200 mM solution, 2 mM final [7] [27] |
| OptiMEM-GlutaMAX | Electroporation medium; enhances cell viability and transfection efficiency post-pulse. | Used during electroporation [32] |
| TC-Treated Culture Dishes | Surface-treated polystyrene vessels for optimal cell attachment and growth. | 60 mm, 100 mm, 6-well, 24-well plates [7] [27] |
The ability to generate immortalized mouse embryonic fibroblast (MEF) lines is a cornerstone technique for studying gene function, cancer biology, and for developing consistent in vitro model systems. Primary MEFs undergo replicative senescence after a finite number of divisions, limiting long-term experiments. While spontaneous immortalization through serial passaging is possible, it is inefficient, time-consuming, and unpredictable. The advent of CRISPR-Cas9 technology has revolutionized this process, enabling targeted, efficient, and rapid immortalization. This Application Note details a refined protocol for achieving MEF immortalization in 14-21 days through CRISPR-mediated deletion of the Tp53 gene, a critical tumor suppressor that governs cell cycle arrest and senescence [6] [5]. This protocol provides a reliable alternative to traditional methods such as overexpression of oncogenes or the serial passaging "3T3" protocol.
Scientific Background: The Role of p53 in Senescence and Immortalization
Cellular senescence is a fundamental barrier to unlimited proliferation. The p53 protein, encoded by the Tp53 gene, sits at the heart of this pathway, acting as a key sensor of cellular stress.
- p53 Pathway Activation: In response to various stresses, including the DNA double-strand breaks (DSBs) introduced by CRISPR-Cas9, the p53 pathway is activated [33] [34]. This leads to cell cycle arrest, primarily through the transcriptional upregulation of p21, which prevents the cell from progressing through the cycle until the damage is repaired.
- Bypassing Senescence: The introduction of Cas9 nuclease alone can activate the p53 pathway, potentially selecting for cells that have inactivated p53 to stably express Cas9 and survive editing [34]. By directly knocking out Tp53, this protocol preemptively disables this primary senescence and cell cycle arrest pathway, allowing edited cells to proliferate indefinitely.
- Genomic Stability: It is important to note that p53 loss can lead to genomic instability, including aneuploidy, as its role in ensuring faithful chromosome segregation is compromised [35] [36]. This is a recognized characteristic of many immortalized cell lines.
The following diagram illustrates the core molecular pathway targeted by this protocol.
Quantitative Timeline and Milestones
The protocol is designed for speed and efficiency, with immortalized polyclonal populations typically emerging within three weeks. The table below outlines the key milestones and their associated timeframes.
Table 1: Key Milestones in the 21-Day Immortalization Protocol
| Day | Milestone | Key Actions and Expected Outcomes |
|---|---|---|
| -5 to -1 | MEF Isolation | Isolate MEFs from E12.5-E13.5 mouse embryos. Plate and expand to achieve sufficient cell numbers for transfection. |
| 0 | CRISPR Transfection | Electroporate MEFs with CRISPR/Cas9 constructs targeting the Tp53 gene. |
| 1-3 | Recovery & Selection | Allow cells to recover from electroporation. Apply puromycin selection if using lentiviral delivery or sorted based on a co-transfected fluorescent marker. |
| 4-14 | Emergence of Proliferating Clones | Observe a period of reduced proliferation followed by the emergence of rapidly dividing, immortalized polyclonal populations. |
| 15-21 | Expansion & Validation | Expand the immortalized polyclonal cell population. Validate Tp53 knockout via genotyping (e.g., T7E1 assay, sequencing) and functional assays (e.g., loss of p53/p21 protein expression). |
Detailed Experimental Protocol
A. Materials and Reagents
Table 2: Essential Research Reagent Solutions
| Reagent / Tool | Function / Description | Example Source / Reference |
|---|---|---|
| Px461-Cas9n-Trp53-sgRNA Plasmids | CRISPR plasmids expressing Cas9 nuclease and guide RNAs targeting the Tp53 gene. | Addgene #88846, #88847 [5] |
| Neon Transfection System | Electroporation device for high-efficiency delivery of CRISPR constructs into MEFs. | Thermo Fisher Scientific [6] [5] |
| MEF Culture Media | High-glucose DMEM, supplemented with 10% FBS, L-glutamine, and antibiotics. | - |
| Puromycin | Selection antibiotic for cells transduced with lentiviral CRISPR constructs. | - |
| Senescence-associated β-galactosidase (SA-β-gal) Staining Kit | To detect and quantify senescent cells before and after editing. | Cell Signaling Technology [37] |
B. Step-by-Step Workflow
The following diagram and corresponding text outline the core experimental workflow.
- MEF Preparation: Isolate MEFs from E12.5 mouse embryos and culture in complete media. It is critical to use low-passage (passage 1-3) MEFs to ensure robust proliferation potential prior to editing [5].
- CRISPR Delivery: On day 0, electroporate approximately 50,000-100,000 MEFs using the Neon Transfection System with 2-5 µg of the Tp53-targeting CRISPR/Cas9 plasmid(s). The use of a plasmid encoding a fluorescent marker (e.g., GFP) can help monitor transfection efficiency.
- Post-Transfection Culture: Immediately after electroporation, plate the cells in pre-warmed antibiotic-free culture media. After 24-48 hours, if a selection marker is present, begin puromycin selection to eliminate non-transfected cells.
- Expansion and Monitoring: Culture the cells, passaging them as they reach confluence. A period of slowed growth may be observed initially, followed by the emergence of rapidly proliferating, immortalized MEFs (iMEFs) within two weeks.
- Validation:
- Genotypic Analysis: Extract genomic DNA and perform a T7 Endonuclease I (T7E1) assay or sequence the target region to confirm the presence of insertion/deletion (indel) mutations in the Tp53 gene [37].
- Phenotypic Analysis: Perform SA-β-gal staining. The immortalized population should show a significant decrease in SA-β-gal-positive cells compared to senescent, primary MEF controls [38] [35].
- Cloning and Cryopreservation: For clonal lines, isolate single cells and expand them. Create master and working cell banks of the polyclonal or clonal iMEF populations.
Troubleshooting and Key Considerations
- Low Immortalization Efficiency: Ensure high transfection efficiency by optimizing electroporation parameters. Using a combination of two sgRNAs against Tp53 can improve the frequency of biallelic knockout.
- p53-Mediated Cell Cycle Arrest: As Cas9 binding and cleavage can induce a p53-dependent cell cycle arrest, the rapid knockout of Tp53 is essential to circumvent this barrier and improve the recovery of edited cells [33] [34].
- Genomic Instability: Acknowledge that Tp53 knockout leads to genomic instability. Regularly monitor the karyotype of iMEFs in long-term culture, especially if genome integrity is critical for downstream applications [35] [36].
This detailed protocol demonstrates that CRISPR/Cas9-mediated knockout of Tp53 is a highly efficient and rapid method for generating immortalized MEFs. By disabling the primary cellular senescence pathway, researchers can reliably produce iMEF lines within a 14-21 day timeframe. These iMEFs provide a consistent and invaluable tool for a wide array of genetic and functional studies, accelerating research in drug discovery and molecular biology.
Subcloning and Cryopreservation of Clonal iMEF Lines for Downstream Applications
The generation of immortalized mouse embryonic fibroblast (iMEF) lines via CRISPR/Cas9-mediated knockout of the Tp53 gene provides a stable, consistent cell system for a wide range of downstream applications, including drug screening, studies of cellular senescence, and protein production. A critical step following immortalization is the isolation and preservation of genetically identical clonal populations. This process of subcloning ensures that subsequent experimental data are derived from a uniform cell population, thereby enhancing reproducibility and reliability. This application note provides a detailed, step-by-step protocol for the efficient subcloning of clonal iMEF lines and their subsequent cryopreservation to establish a long-term, stable cell stock.
Subcloning of Clonal iMEF Lines
Subcloning by limiting dilution is the preferred method for establishing monoclonality, which is crucial for validating the success of the Tp53 knockout and for functional studies.
Materials and Reagents
- Cell Line: A heterogeneous population of Tp53-targeted iMEFs, generated via CRISPR/Cas9 and enriched but not yet clonally isolated.
- Growth Medium: Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin.
- Sterile PBS: Dulbecco's Phosphate Buffered Saline (DPBS), without calcium or magnesium.
- Trypsin Solution: 0.25% Trypsin-EDTA.
- 96-well Cell Culture Plates, tissue culture-treated.
Protocol: Limiting Dilution for Monoclonal Isolation
- Preparation: Pre-warm growth medium and trypsin in a 37°C water bath. Irradiate or mitotically inactivate a feeder layer of MEFs if required for your specific iMEF line, and seed them into a 96-well plate at a low density 24 hours prior to subcloning.
- Harvesting Cells: Wash the heterogeneous iMEF population in a T-25 or T-75 flask with sterile PBS. Add enough trypsin to cover the cell layer (e.g., 2 mL for a T-25 flask) and incubate at 37°C for 2-5 minutes. Neutralize the trypsin with 2-3 volumes of complete growth medium.
- Cell Counting and Dilution: Centrifuge the cell suspension at approximately 100–400 × g for 5 minutes. Aspirate the supernatant and resuspend the cell pellet in a known volume of fresh medium. Perform a viable cell count using an automated cell counter or hemocytometer with Trypan Blue exclusion [39].
- Serially dilute the cell suspension to a final concentration of 10 cells/10 mL of growth medium. This aims for a statistical probability of depositing 0.5-1 cell per well.
- Plating: Aliquot 100 µL of the diluted cell suspension into each well of the prepared 96-well plate. Gently shake the plate to ensure even distribution.
- Culture and Monitoring: Place the plate in a 37°C, 5% CO₂ incubator. Do not disturb the plate for 48-72 hours to allow single cells to settle and begin dividing.
- Microscopic Screening: After 3-5 days, screen every well using an inverted microscope. Flag wells that contain a single, isolated colony originating from a single progenitor cell. Discard wells with multiple colonies or no growth.
- Expansion: Once a flagged colony reaches 70-90% confluency, trypsinize the well and expand the cells sequentially into a 48-well plate, then a 12-well plate, and finally a T-25 flask, maintaining careful labeling to track each clone.
Workflow Visualization
The following diagram illustrates the complete workflow from the mixed cell population to the cryopreservation of validated monoclonal lines.
Cryopreservation of Validated Clonal iMEFs
Cryopreservation maintains the genetic and phenotypic stability of the validated clonal lines for future use and prevents cellular aging and transformation in continuous culture [39].
Materials and Reagents
- Log-phase Cells: The validated clonal iMEF line at 80-90% confluency and high viability (>90%).
- Freezing Medium: Complete growth medium supplemented with 10% DMSO. Alternatively, use a commercially available, serum-free, chemically defined cryopreservation medium like Gibco Synth-a-Freeze [39]. Chill the freezing medium to 2°–8°C before use.
- Cryogenic Vials: Sterile, internally threaded vials.
- Isopropanol Chamber (e.g., "Mr. Frosty") or a Controlled-Rate Freezer.
- Liquid Nitrogen Storage Dewar.
Protocol: Cryopreservation
- Preparation: Harvest the cells from the culture vessel using standard trypsinization as described in Step 2.2.
- Centrifugation and Resuspension: Centrifuge the cell suspension at approximately 100–400 × g for 5 minutes. Aspirate the supernatant completely and resuspend the cell pellet in cold freezing medium to achieve a high cell density. A final concentration of 1-3 x 10^6 cells/mL is generally recommended for fibroblasts [39].
- Aliquoting: Quickly dispense 1 mL aliquots of the cell suspension into labeled cryovials. Tighten the caps securely.
- Slow Freezing: Place the cryovials immediately into an isopropanol chamber pre-cooled to 4°C, and transfer the chamber to a -80°C freezer for 24 hours. This apparatus slows the cooling rate to approximately -1°C per minute, which is critical for high post-thaw viability [39]. Alternatively, use a controlled-rate freezer.
- Long-Term Storage: After 24 hours, promptly transfer the frozen cryovials to a liquid nitrogen storage tank for long-term preservation in the gas phase (below -135°C) to minimize the risk of explosion associated with liquid-phase storage [39].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and materials essential for the successful subcloning and cryopreservation of iMEF lines.
Table 1: Essential Research Reagents for iMEF Subcloning and Cryopreservation
| Item | Function/Application | Example/Notes |
|---|---|---|
| CRISPR/Cas9 System | Mediates targeted knockout of the Tp53 gene for MEF immortalization. | Can be delivered via lentivirus for high efficiency in hard-to-transfect cells [40]. |
| DMSO (Cell Culture Grade) | Cryoprotective agent that prevents lethal ice crystal formation within cells during freezing [39]. | Use a bottle dedicated for cell culture; handle in a laminar flow hood. |
| Controlled-Rate Freezer | Provides a consistent, slow cooling rate (~-1°C/min), critical for maintaining high cell viability during cryopreservation [39]. | An isopropanol chamber is a cost-effective alternative for slow freezing. |
| Trypsin-EDTA Solution | A dissociation reagent used to detach adherent cells (like iMEFs) from the culture surface for passaging or harvesting [39]. | TrypLE Express is a recombinant alternative that is gentler on some sensitive cell types. |
| Serum-Free Freezing Medium | A chemically defined, ready-to-use solution for cryopreservation; eliminates batch-to-batch variability of serum [39]. | Gibco Synth-a-Freeze is suitable for stem and primary cells, including iMEFs. |
Validation and Quality Control
To ensure the integrity of your clonal iMEF bank, perform the following quality control checks.
Genotypic Validation
Confirm the Tp53 knockout in expanded clonal lines.
- Genomic DNA Extraction: Isolate genomic DNA from a portion of the cells before cryopreservation.
- PCR Amplification: Design primers flanking the CRISPR target site in the Tp53 gene and amplify the region.
- Analysis: Analyze the PCR product by gel electrophoresis for size shifts indicative of indels. Confirm the specific mutation by Sanger sequencing of the amplified fragment [40].
Phenotypic and Functional Validation
Common validation steps include:
- Growth Curve Analysis: Compare the proliferation rate of the knockout line to wild-type MEFs. iMEFs often exhibit extended proliferative capacity and avoidance of senescence.
- Western Blotting: Confirm the absence of p53 protein expression in the clonal line [40].
Table 2: Key Quantitative Parameters for Subcloning and Cryopreservation
| Parameter | Target Value / Specification | Purpose / Rationale |
|---|---|---|
| Cell Density for Limiting Dilution | 0.5 - 1 cell/100 µL (10 cells/10 mL) | Maximizes probability of monoclonality per well. |
| Freezing Cell Density | 1 - 3 x 10^6 cells/mL | Ensures sufficient cell mass for recovery upon thawing. |
| DMSO Concentration | 10% (in complete medium) | Standard cryoprotectant concentration for many mammalian cell types [39]. |
| Cooling Rate | ~ -1°C / minute | Optimal rate to minimize intracellular ice crystal formation [39]. |
| Post-Thaw Viability Benchmark | > 80% | Indicator of a successful and optimized cryopreservation process. |
Table 3: Troubleshooting Common Issues
| Problem | Potential Cause | Solution |
|---|---|---|
| No single-cell colonies after dilution | Cell concentration too low; low clonogenicity. | Re-optimize dilution factor; ensure cells are in log-phase growth; consider using conditioned medium. |
| Low post-thaw viability | Suboptimal freezing rate; outdated DMSO; cell damage during handling. | Ensure use of a controlled-rate freezer or isopropanol chamber; use fresh, high-quality DMSO; avoid holding cells in freezing medium at RT for >10 min. |
| Contamination in cryovial | Non-sterile technique during aliquoting. | Perform all steps in a laminar flow hood using sterile technique. |
Solving Common Problems: A Troubleshooting Guide for CRISPR MEF Immortalization
In the context of CRISPR-mediated deletion of the Tp53 gene for mouse embryonic fibroblast (MEF) immortalization, achieving high editing efficiency is paramount to successfully generating stable, immortalized MEF (iMEF) lines. Low efficiency can lead to prolonged cell culture periods, increased senescence, and inconsistent experimental results. This application note provides a detailed, evidence-based framework for optimizing the two most critical factors: guide RNA (gRNA) design and electroporation delivery. By systematically addressing these components, researchers can significantly improve the success rate of their MEF immortalization protocols, reducing the time required to generate iMEF lines from months to under three weeks [27] [6].
Optimizing gRNA Design for Enhanced On-Target Activity
The selection of a highly efficient and specific gRNA is the foundational step for a successful CRISPR experiment. Computational prediction tools, grounded in large-scale experimental data, are indispensable for this process.
Key Principles for gRNA Selection
When designing gRNAs for Tp53 knockout, several sequence-specific features significantly influence cleavage efficiency. The table below summarizes the primary features to consider, based on an overview of gRNA efficiency prediction studies [41].
Table 1: Nucleotide Features Influencing gRNA Efficiency
| Category | Features Associated with High Efficiency | Features Associated with Low Efficiency |
|---|---|---|
| Overall Nucleotide Usage | High adenosine (A) count; AG, CA, AC, UA dinucleotides | High uracil (U) and guanine (G) count; GG, GGG repeats; UU, GC dinucleotides |
| Position-Specific Nucleotides | Guanine (G) or Adenine (A) in position 19; Cytosine (C) in positions 16 & 18 | Cytosine (C) or Uracil (U) in position 20; U in positions 17-20; Thymine (T) in PAM (TGG) |
| Motifs & Composition | GC content between 40%-60%; NGG PAM (especially CGG); TT, GCC at the 3' end | GC content >80%; poly-N repeats (especially GGGG) |
Utilizing Computational Prediction Tools
Several online tools incorporate these principles into user-friendly algorithms to score gRNAs for on-target efficiency and off-target risk. The following table compares the most widely used tools and their underlying scoring methods [42].
Table 2: Comparison of gRNA Design and Scoring Tools
| Tool Name | On-Target Scoring Method(s) | Off-Target Scoring Method(s) | Key Characteristics |
|---|---|---|---|
| CRISPick | Rule Set 2, Rule Set 3 | Cutting Frequency Determination (CFD) | Developed by the Broad Institute; provides a simple interface and uses updated logic from large-scale experiments. |
| CHOPCHOP | Rule Set 1, CRISPRscan | Homology Analysis | A versatile tool supporting various CRISPR-Cas systems beyond Cas9; provides visual off-target representations. |
| CRISPOR | Rule Set 2, CRISPRscan, Lindel | MIT Score, CFD | Offers detailed off-target analysis with position-specific mismatch scoring and practical experimental considerations. |
| GenScript Tool | Rule Set 3 | CFD | Uses the latest Rule Set 3 logic, which considers the tracrRNA sequence for improved accuracy; integrates with ordering. |
For critical applications like Tp53 knockout, it is advisable to check gRNA designs across multiple tools and select candidates that are consistently ranked highly. A recent independent evaluation in human pluripotent stem cells found that tools like Benchling (which incorporates several of these algorithms) provided the most accurate predictions, though performance can vary by cell type [43].
Electroporation Optimization for Efficient RNP Delivery
Once high-quality gRNAs are designed, their efficient delivery into cells is crucial. Electroporation of ribonucleoprotein (RNP) complexes—pre-assembled Cas9 protein and gRNA—is a highly effective method for primary cells like MEFs, as it minimizes off-target effects and enables rapid genome editing without the need for vector delivery.
A Protocol for CRISPR-Mediated MEF Immortalization
The following optimized protocol for immortalizing MEFs via Tp53 knockout has been demonstrated to reliably generate iMEF lines within three weeks [27] [6].
Key Materials & Reagents:
- Biological Material: Primary MEFs isolated from E12.5 mouse embryos.
- CRISPR Components:
- Cas9 protein (e.g., IDT Alt-R S.p. Cas9 Nuclease V3)
- Synthetic crRNAs targeting Tp53 and tracrRNA (e.g., IDT Alt-R CRISPR-Cas9 crRNA and tracrRNA)
- Electroporation System: Neon Transfection System (Thermo Fisher Scientific) or comparable device.
- Cell Culture Media: Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2mM L-glutamine, and antibiotics.
Step-by-Step Procedure:
- gRNA Complex Formation: Resuspend synthetic crRNA and tracrRNA to 100 µM in nuclease-free buffer. To form the gRNA complex, mix equimolar ratios of crRNA and tracrRNA, heat to 95°C for 5 minutes, and allow to cool slowly to room temperature.
- RNP Complex Assembly: Combine 4 µM Cas9 protein with 4 µM of the assembled gRNA complex in 20 µL of Opti-MEM medium. Incubate for 10-20 minutes at room temperature to form the active RNP complex.
- MEF Preparation: Harvest primary MEFs and resuspend the cell pellet in Resuspension Buffer R (Neon System) at a concentration of 2-5 x 10⁶ cells/mL.
- Electroporation: Mix 100 µL of cell suspension with 10 µL of the prepared RNP complex. Aspirate the mixture into a Neon Pipette Tip and electroporate using the following optimized parameters: 1,350 V, 20 ms, 2 pulses. These parameters have been specifically optimized for MEFs and result in high viability and editing efficiency [27].
- Post-Transfection Recovery: Immediately transfer the electroporated cells into pre-warmed complete culture media (without antibiotics) in a tissue culture dish.
- Cell Expansion and Cloning: Culture the cells, passaging them as needed. Immortalized, rapidly dividing cells typically emerge within 2-3 weeks. These polyclonal iMEF populations can be used directly, or individual clones can be isolated and expanded for downstream characterization and experiments.
The workflow for this entire process, from design to validation, is summarized in the diagram below.
Critical Factors for Electroporation Success
- RNP vs. Other Formats: Using synthetic, chemically modified gRNAs in RNP complexes can significantly enhance stability and editing efficiency compared to in vitro transcribed (IVT) sgRNA or plasmid-based delivery [43].
- Cell Health and Concentration: The viability of the starting MEF population is critical. Using low-passage cells and optimizing the cell concentration for electroporation (typically 2-5 x 10⁶ cells/mL) helps ensure high post-transfection survival.
- Parameter Optimization: While the parameters above are effective for MEFs, slight optimization may be needed for different electroporation systems. For example, the NEPA21 system has been used successfully for bovine embryos [31], and the EEZy protocol for mouse zygotes uses a square wave electroporator at 30V [44].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for CRISPR-mediated MEF Immortalization
| Item | Function / Application | Examples / Notes |
|---|---|---|
| Synthetic crRNA & tracrRNA | Defines target specificity; synthetic RNAs offer high purity and consistency, and can be chemically modified for enhanced stability. | IDT Alt-R CRISPR-Cas9 crRNA and tracrRNA. |
| Recombinant Cas9 Nuclease | The effector protein that creates the double-strand break at the genomic target site. | IDT Alt-R S.p. Cas9 Nuclease V3; can be wild-type or high-fidelity variants. |
| Electroporation System | Enables efficient delivery of RNP complexes into hard-to-transfect primary cells. | Neon Transfection System (Thermo Fisher Scientific); other comparable systems can be optimized. |
| Cell Culture Media & Supplements | Supports the growth and viability of primary MEFs before and after the stressful electroporation process. | High-glucose DMEM, Qualified FBS, L-Glutamine. Avoid antibiotics immediately after electroporation. |
Achieving high-efficiency CRISPR editing for Tp53 deletion in MEFs is a systematic process that hinges on two pillars: intelligent gRNA design and optimized electroporation delivery. By leveraging modern computational tools to select gRNAs with high predicted on-target activity and low off-target risk, and by employing a robust RNP electroporation protocol, researchers can reliably generate immortalized MEF lines. This optimized approach streamlines the creation of critical cellular models, accelerating subsequent functional studies in gene regulation and disease modeling.
Minimizing CRISPR-Cas9 Genotoxicity and Off-Target Effects
CRISPR-Cas9 technology has revolutionized genetic engineering, offering unprecedented precision in genome editing for research and therapeutic applications. Within the specific context of CRISPR-mediated Tp53 deletion for Mouse Embryonic Fibroblast (MEF) immortalization, controlling genotoxicity and off-target effects becomes paramount for generating reliable, stable cell lines that accurately reflect the intended genetic modification. Off-target effects refer to unintended edits at genomic loci with sequence similarity to the target site, while genotoxicity encompasses broader DNA damage responses, including persistent double-strand breaks and large structural variations that can compromise genomic integrity and experimental validity [45] [46].
The immortalization of MEFs through Tp53 deletion creates a valuable system for studying gene function and performing long-term cellular assays. However, the fidelity of this model depends entirely on the specificity of the CRISPR editing process [6] [27]. This Application Note provides detailed protocols and strategic frameworks to minimize these risks, ensuring the generation of high-quality immortalized MEF (iMEF) lines suitable for downstream research and drug development applications.
Understanding and Quantifying CRISPR Edits
Accurate measurement of editing outcomes is fundamental to assessing both on-target efficiency and off-target activity. Multiple methodological approaches exist, each with distinct advantages, sensitivity thresholds, and technical requirements.
Method Comparison for Edit Quantification
Table 1: Comparison of Methods for Quantifying CRISPR-Cas9 Editing Efficiency
| Method | Working Principle | Reported Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Targeted Amplicon Sequencing (AmpSeq) [47] | High-throughput sequencing of PCR-amplified target regions | ~0.1% | Considered the "gold standard"; provides nucleotide-resolution data; highly sensitive and quantitative | Higher cost; longer turnaround time; requires specialized bioinformatics analysis |
| T7 Endonuclease 1 (T7E1) Assay [47] | Detection of heteroduplex DNA formed by wild-type and edited alleles | ~1-5% | Rapid and inexpensive; no specialized equipment required | Lower sensitivity; semi-quantitative; does not provide sequence information |
| PCR-Restriction Fragment Length Polymorphism (RFLP) [47] | Loss of a restriction site due to successful editing | ~1-5% | Simple and cost-effective; clear yes/no readout for specific edits | Requires a restriction site to be ablated by the edit; not suitable for all targets |
| Droplet Digital PCR (ddPCR) [47] | Partitioning of PCR reactions into thousands of droplets for absolute quantification | ~0.1-1% | High sensitivity and precision; absolute quantification without standard curves | Requires specific probe design; higher cost per sample than traditional PCR |
| PCR-Capillary Electrophoresis/IDAA [47] | Fragment analysis to detect small insertions and deletions (indels) | ~0.5-1% | Good balance of sensitivity and throughput; provides indel spectrum | Limited ability to detect complex edits or single-nucleotide variants |
| Sanger Sequencing + Deconvolution Tools (ICE, TIDE) [47] | Sequencing of bulk PCR product followed by computational decomposition of chromatograms | ~1-5% | Provides sequence context and indel information from accessible technology | Lower sensitivity for detecting low-frequency edits; accuracy depends on the decomposition algorithm |
In a comprehensive benchmarking study, AmpSeq, PCR-CE/IDAA, and ddPCR were identified as the most accurate methods when benchmarked against AmpSeq, which serves as the reference standard due to its sensitivity and reliability [47]. For the initial screening of Tp53-edited iMEF clones, T7E1 or RFLP assays offer a practical balance of speed and cost. However, for final clone validation and comprehensive off-target assessment, AmpSeq is strongly recommended to achieve the sensitivity required for confident results [47].
Protocol: Targeted Amplicon Sequencing for iMEF Clone Validation
This protocol details the steps for validating Tp53 edits in immortalized MEF clones using AmpSeq.
- Step 1: Genomic DNA Extraction. Harvest putative immortalized MEF clones expanded from single cells. Extract high-quality genomic DNA using a silica-column-based or magnetic-bead-based kit. Quantify DNA using a fluorometer to ensure accurate quantification.
- Step 2: PCR Amplification of Target Loci. Design primers to amplify ~300-500 bp regions flanking the intended Tp53 cut site(s) and all potential off-target sites identified by bioinformatic prediction (e.g., via CRISPOR). Use a high-fidelity DNA polymerase to minimize PCR errors. Perform PCR with 20-50 ng of genomic DNA.
- Step 3: Library Preparation and Barcoding. Purify the PCR amplicons. For Illumina platforms, use a library preparation kit that attaches dual-index barcodes to each sample, enabling multiplexing of multiple clones and targets in a single sequencing run.
- Step 4: Sequencing. Pool the barcoded libraries in equimolar ratios and sequence on an Illumina MiSeq or NovaSeq platform, aiming for a minimum depth of 50,000x coverage per amplicon to robustly detect low-frequency edits.
- Step 5: Data Analysis. Process the raw sequencing data through a standardized pipeline:
- Demultiplex reads by their barcodes.
- Align reads to the reference mouse genome (mm10) using tools like BWA or Bowtie2.
- Quantify editing efficiency and characterize the spectrum of insertion and deletion (indel) mutations at each target site using specialized software such as CRISPResso2.
Strategic Guide RNA Design and Tool Selection
The single most critical factor in minimizing off-target effects is the rational design of the guide RNA (gRNA). A well-designed gRNA maximizes on-target activity while minimizing homology to other genomic sites.
Key Parameters for gRNA Design
- On-Target Efficiency: Predictive algorithms score gRNAs based on features correlated with high activity. Key scoring systems include:
- Rule Set 3: An updated model that considers the tracrRNA sequence, recommended for its accuracy based on large-scale training datasets [42].
- CRISPRscan: A model trained on in vivo data from zebrafish, useful for predicting performance in biological systems [42].
- Lindel: A logistic regression model that predicts the likelihood of frameshift-indels, which is crucial for effective gene knockout strategies like Tp53 deletion [42].
- Off-Target Specificity: A thorough genome-wide analysis is non-negotiable. Key evaluation methods include:
- Cutting Frequency Determination (CFD) Score: A weighted scoring system that more accurately predicts the impact of mismatches on off-target activity compared to simple mismatch counting [42].
- MIT Specificity Score: An earlier but still relevant algorithm that counts potential off-target sites with a defined number of mismatches, assigning higher risk to sites with fewer mismatches, particularly those distal to the PAM [42].
- Genomic Context: The target site should be checked for proximity to repetitive elements, common polymorphisms, and within a region of open chromatin to ensure accessibility.
Online gRNA Design Tools
Table 2: Comparison of Guide RNA Design Tools
| Tool Name | On-Target Score(s) | Off-Target Score(s) | Key Features |
|---|---|---|---|
| CRISPick [42] | Rule Set 3 | CFD Score | User-friendly interface from the Broad Institute; provides a ranked list of gRNAs with comprehensive scores. |
| CHOPCHOP [42] | Rule Set, CRISPRscan | MIT Score, Homology Analysis | Versatile tool supporting multiple CRISPR systems; provides visual guides and primer design. |
| CRISPOR [42] | Rule Set 2, CRISPRscan | MIT Score, CFD Score | Detailed off-target analysis with position-specific mismatch scoring; includes experimental advice. |
| GenScript gRNA Tool [42] | Rule Set 3 | CFD Score | Integrates on/off-target scores with transcript coverage and cutting position; direct ordering capability. |
For the design of gRNAs targeting Tp53 for MEF immortalization, it is recommended to use at least two of these tools in concert. Select the top 2-3 gRNAs that consistently rank highly for both on-target efficiency (high Rule Set 3 score) and specificity (low CFD scores for all potential off-target sites) across platforms [42].
Experimental Workflow for Genotoxicity-Reduced iMEF Generation
The following workflow integrates the principles of careful gRNA design, efficient delivery, and rigorous validation to minimize genotoxicity during the immortalization of MEFs. This process reliably generates immortalized MEF lines (iMEFs) within three weeks [6] [27].
Protocol: Tp53 Knockout via Electroporation in MEFs
This detailed protocol is adapted from an established method for efficient MEF immortalization [6] [27].
Materials and Reagents:
- Biological Material: Primary Mouse Embryonic Fibroblasts (MEFs) isolated from E12.5 embryos [27].
- CRISPR Plasmids: px461-Cas9n-Trp53-sgRNA-alpha and -beta plasmids (Addgene #88846, #88847), which express Cas9 nickase and Tp53-targeting sgRNAs, and a pCAG-GFP plasmid for tracking transfection efficiency [27].
- Equipment: Neon Transfection System or comparable electroporator.
- Culture Media: Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine. For fresh MEFs, also add 1 mM sodium pyruvate and 1x MEM Non-Essential Amino Acids. Use media without antibiotics immediately after electroporation [27].
Procedure:
- MEF Preparation: Isolate MEFs from E12.5 mouse embryos using standard protocols. Culture primary MEFs in complete media for 1-2 passages.
- Electroporation Complex Preparation: For a 10 µL Neon tip, mix 1-2 µg of each Tp53 CRISPR plasmid and 0.5 µg of pCAG-GFP plasmid with 1-2 million MEFs resuspended in Buffer R.
- Electroporation: Electroporate using the Neon system with the following optimized parameters: 1300 V, 20 ms, 2 pulses [27]. Immediately transfer the electroporated cells into pre-warmed antibiotic-free media in a 6-well plate.
- Post-Transfection Culture: After 24-48 hours, assess GFP fluorescence to estimate transfection efficiency (~50-70% is typical). Continue culturing the cells, passaging them when they reach 80-90% confluence. Observe for the emergence of rapidly dividing, immortalized cells, which typically occurs after 3-4 weeks.
- Cloning and Expansion: Once a polyclonal immortalized population is established, dissociate the cells and seed them at low density (e.g., 10 cells/mL) in 96-well plates to isolate single-cell clones. Expand positive clones for validation.
Table 3: Research Reagent Solutions for CRISPR-mediated MEF Immortalization
| Item Category | Specific Example(s) | Function/Application |
|---|---|---|
| CRISPR Reagents | px461-Cas9n-Trp53-sgRNA-alpha/beta (Addgene) [27] | CRISPR plasmids for targeted Tp53 gene deletion. Using a nickase version of Cas9 (Cas9n) can further reduce off-target effects. |
| Delivery System | Neon Transfection System 10 µL Kit (Thermo Fisher) [27] | Electroporation system optimized for high-efficiency, low-toxicity delivery of CRISPR constructs into MEFs. |
| Cell Culture | High-glucose DMEM, Qualified FBS, Trypsin-EDTA [27] | Essential components for the isolation, culture, and passaging of primary and immortalized MEFs. |
| Validation Kits | T7 Endonuclease I, Gel Electrophoresis System [47] | For initial, cost-effective screening of editing efficiency in putative iMEF clones. |
| Validation Services | Targeted Amplicon Sequencing Services (e.g., Illumina) [47] | For definitive, high-sensitivity confirmation of on-target edits and screening for off-target effects. |
| Design Tools | CRISPOR, CRISPick [42] | Free, web-based software for comprehensive gRNA design and off-target prediction. |
Advanced Considerations: AI and Novel Editors
The field of CRISPR technology is advancing rapidly, with new developments offering promising paths to further reduce genotoxicity.
- AI-Designed Editors: Large language models (LLMs) trained on vast datasets of natural CRISPR sequences are now being used to generate novel, highly functional gene editors. These AI-designed proteins, such as OpenCRISPR-1, can exhibit comparable or improved activity and specificity relative to SpCas9 while being vastly different in sequence, potentially bypassing evolutionary constraints and optimizing properties for use in mammalian cells [48].
- High-Fidelity Cas Variants: While not covered in the supplied results, it is widely documented in literature that engineered "high-fidelity" Cas9 variants (e.g., SpCas9-HF1, eSpCas9) contain mutations that reduce off-target editing by strengthening the binding requirements between the gRNA and target DNA.
- Clinical Translation Perspective: For therapeutic development, a comprehensive risk-benefit assessment framework is essential. This involves weighing the potential for genome-related adverse events against the severity of the target disease, acknowledging that 'perfect' therapeutics with zero off-targets do not exist. Advanced in vitro assays like CIRCLE-seq and CHANGE-seq provide sensitive methods to profile genome-wide off-target activity for critical applications [45].
The successful application of CRISPR-Cas9 for generating immortalized MEF lines via Tp53 deletion hinges on a multi-faceted strategy to minimize genotoxicity. This involves the meticulous bioinformatic design of gRNAs using modern scoring algorithms, selection of efficient and specific gRNAs through comparative tool analysis, careful experimental execution using optimized delivery methods like electroporation, and rigorous validation of edited clones with a combination of rapid screening and definitive, high-sensitivity sequencing technologies. By adhering to the detailed protocols and principles outlined in this Application Note, researchers can consistently produce high-quality, genomically stable iMEFs, thereby ensuring the reliability of downstream research findings in cell biology and drug development.
Managing Cell Toxicity and Poor Survival Post-Transfection
The CRISPR-mediated deletion of the Tp53 gene has emerged as a highly efficient method for generating immortalized mouse embryonic fibroblasts (iMEFs), reliably yielding immortalized cells within two to three weeks [6] [7] [5]. However, a significant bottleneck in this protocol remains the poor cell survival and toxicity frequently encountered during the transfection step, where CRISPR constructs are delivered into primary MEFs. This cytotoxicity can drastically reduce the yield of viable immortalized cells and compromise subsequent experimental results. Transfection-related toxicity stems from multiple factors, including the inherent sensitivity of primary cells, the method of delivery, and the cellular stress responses triggered by introducing foreign nucleic acids [49] [50]. Addressing these challenges is paramount for researchers aiming to establish reliable iMEF lines for studying gene function, disease-causing variants, and long-term cellular characterization.
Understanding the Causes of Transfection-Related Toxicity
Transfection-induced cytotoxicity is a multifaceted problem. Recognizing the root causes is the first step in developing an effective mitigation strategy.
- Membrane Damage and Charge Interactions: Many transfection reagents, particularly cationic lipids and polymers, function by forming positively charged complexes with negatively charged nucleic acids. These complexes can fuse with or disrupt the cell membrane, leading to loss of membrane integrity, leakage of cellular contents, and ultimately, cell lysis or necrosis [49] [51].
- Activation of Cellular Stress and Immune Pathways: Cells possess innate immune mechanisms to detect foreign nucleic acids. The introduction of CRISPR plasmids or ribonucleoproteins (RNPs) can trigger these pathways, leading to a stress response and the upregulation of inflammatory cytokines, which can induce apoptosis or alter the physiological state of the cell [50].
- Burden on Cellular Machinery and Oxidative Stress: The process of endocytosis and lysosomal processing of transfection complexes places a significant metabolic burden on cells. Furthermore, some delivery methods can induce mitochondrial dysfunction and a burst of reactive oxygen species (ROS), causing oxidative damage to lipids, proteins, and DNA [51].
- Method-Specific Cytotoxicity: The choice of delivery method itself is a critical factor.
- Lipofection: While convenient, lipid-based reagents can be highly cytotoxic, with toxicity levels varying significantly between different commercial formulations [49] [50].
- Electroporation: This physical method uses electrical pulses to permeabilize the cell membrane. However, improper optimization of voltage, pulse width, or number of pulses can cause significant cell damage and high mortality rates [52] [53].
The following diagram illustrates the primary pathways through which transfection causes cellular toxicity.
Detecting and Assessing Cytotoxicity
Before mitigating toxicity, researchers must accurately assess its presence and severity. Several methods can be employed, often in combination.
- Morphological Assessment: The simplest and most immediate method is visual inspection under a microscope. Healthy, adherent MEFs should appear spread and fibroblastic. Signs of toxicity include cell rounding, detachment, granulation, and a general reduction in cell density [49] [50].
- Viability and Cytotoxicity Assays:
- Dye Exclusion (Trypan Blue, Propidium Iodide): These dyes enter cells with compromised membranes, allowing for the quantification of dead cells [50] [54].
- Metabolic Activity (MTT, CCK-8): These assays measure the metabolic activity of cells, which correlates with viability. A decrease in signal indicates reduced cell health or number [50] [51].
- LDH Release: Lactate dehydrogenase (LDH) is a cytosolic enzyme that leaks into the culture medium upon membrane damage, serving as a direct marker of cytotoxicity [50] [51].
- Advanced Molecular Methods: For a deeper understanding, gene expression profiling can reveal the upregulation of stress-related genes (e.g., those involved in oxidative stress or apoptosis) long before morphological changes become apparent [50].
Table 1: Methods for Assessing Transfection-Induced Cytotoxicity
| Method | Principle | Key Advantage | Typical Readout |
|---|---|---|---|
| Morphological Inspection | Visual observation of cell health | Fast, inexpensive, no specialized equipment | Qualitative (e.g., rounded vs. adherent cells) |
| Trypan Blue/Propidium Iodide | Membrane integrity | Quantitative, easy to combine with cell counting | Percentage of dead cells |
| MTT/CCK-8 Assay | Cellular metabolic activity | Quantitative, high-throughput compatible | Absorbance/Fluorescence proportional to live cells |
| LDH Release Assay | Cytosolic enzyme release upon membrane damage | Quantifies membrane leakage directly | Absorbance proportional to cytotoxicity |
| Gene Expression (qPCR) | Stress pathway gene activation | Highly sensitive, detects early stress | Fold-change in stress-related mRNA levels |
A Strategic Workflow to Minimize Toxicity
Implementing a systematic approach to optimize transfection conditions is crucial for maximizing cell survival. The following workflow provides a roadmap for mitigating toxicity while maintaining high editing efficiency in your MEF immortalization experiments.
Pre-Transfection: Ensuring Optimal Cell Health
The foundation of a successful transfection is healthy starting material.
- Use Low-Passage, Log-Phase Cells: Primary MEFs should be used at low passage numbers and harvested during their logarithmic growth phase when they are most robust and receptive to transfection [52].
- Optimal Seeding Density: Seed cells to reach a confluency of 70–90% at the time of transfection. This density promotes healthy cell-cell contacts and ensures cells are in an optimal metabolic state [52] [51]. For the 3T3-style passaging used in the referenced protocol, this translates to a precise density of 3 x 10^5 cells per 50-mm dish [5].
Delivery Method and Parameter Optimization
The choice and execution of the delivery method are critical.
- Select a Lower-Toxicity Reagent/Method: The referenced iMEF protocol successfully uses electroporation with a Neon Transfection System [6] [7] [5]. As an alternative to traditional lipid-based reagents, consider polymer-based transfection reagents, which are designed to enter cells via endocytosis and rupture inside the cytoplasm, minimizing damage to the cell membrane [49].
- Empirically Optimize Electroporation Parameters: If using electroporation, parameters like voltage, pulse width, and pulse number must be optimized for your specific primary MEFs [53]. Using the recommended settings for primary fibroblasts from the manufacturer is a necessary starting point.
Transfection Complex and Incubation Optimization
The amount and exposure time of transfection complexes directly impact toxicity.
- Dose Optimization: Systematically titrate the amounts of both the CRISPR construct (e.g., Px461-Cas9n-Trp53-sgRNA plasmids) and the transfection reagent. The goal is to find the lowest effective dose that still achieves high knockout efficiency, as toxicity often increases with dose more than efficiency [50] [52] [51].
- Minimize Incubation Time: For lipid-based transfections, reducing the incubation time of complexes with cells can significantly improve viability. While the iMEF protocol uses electroporation (a short pulse), if switching to lipofection, a time-course experiment (e.g., 4-8 hours) can identify the shortest sufficient exposure time [51].
Post-Transfection Care to Support Recovery
The steps immediately following transfection are crucial for cell recovery.
- Timely Medium Change: After a sufficient period for complex uptake (as early as 4-6 hours post-transfection for lipids), replace the transfection medium with fresh, complete culture media. This removes residual complexes and replenishes nutrients, aiding cell recovery [50] [54]. Note that the referenced iMEF protocol uses media without penicillin-streptomycin immediately after electroporation to support recovery [5].
- Consider Cytoprotective Additives: The addition of antioxidants like N-acetylcysteine (NAC) to the culture media can help mitigate oxidative stress induced by the transfection process [51].
The Scientist's Toolkit: Essential Reagents for iMEF Generation
Table 2: Key Research Reagent Solutions for Tp53 Knockout iMEF Generation
| Reagent/Equipment | Function in Protocol | Example & Catalog Number |
|---|---|---|
| CRISPR Plasmids | Targets & knocks out the Tp53 gene for immortalization. | Px461-Cas9n-Trp53-sgRNA-alpha (Addgene #88846); Px461-Cas9n-Trp53-sgRNA-beta (Addgene #88847) [7] [5] |
| Electroporation System | Physically delivers CRISPR constructs into primary MEFs. | Neon Transfection System 10 μL Kit (Thermo Fisher, MPK1096) [7] [5] |
| Control Plasmid | Monitors transfection efficiency. | pCAG-GFP (Addgene #11150) [7] [5] |
| Primary Cells | The starting biological material for immortalization. | Mouse Embryonic Fibroblasts (MEFs) from E12.5 embryos [7] [5] |
| Culture Media | Supports growth and maintenance of MEFs/iMEFs. | DMEM + 10% FBS + L-Glutamine + Penicillin-Streptomycin [7] [5] |
| Polymer-Based Transfection Reagent (Alternative) | Lower-toxicity chemical delivery method for nucleic acids. | jetOPTIMUS / jetPRIME (Polyplus) or TransIT-LT1 (Mirus Bio) [49] [5] |
Concluding Remarks
Managing cell toxicity is not merely about increasing the number of surviving cells; it is about ensuring the physiological relevance and reliability of the resulting immortalized mouse embryonic fibroblasts. By understanding the causes of cytotoxicity, systematically assessing its impact, and implementing a strategic optimization workflow, researchers can robustly apply the CRISPR-mediated Tp53 deletion protocol. This enables the consistent generation of high-quality iMEFs that closely resemble their primary parent populations, thereby providing a robust and valuable tool for long-term genetic studies and therapeutic development.
Overcoming Contamination and Culture Health Issues
Mouse Embryonic Fibroblasts (MEFs) derived from genetically modified mice represent a valuable resource for studying gene function and regulation, particularly when combined with rescue studies to characterize mutant genes/proteins, including disease-causing variants [6] [27]. However, primary MEFs face a significant limitation: they undergo replicative senescence soon after isolation and passaging, making long-term genetic manipulations challenging [6] [27]. While previous immortalization methods existed, they often suffered from inconsistencies or altered the physiological properties of the cells, compromising their utility for downstream applications [27].
The advent of CRISPR-mediated gene editing has revolutionized this process, enabling precise genetic modifications that overcome these limitations. This application note details an optimized protocol for CRISPR-mediated deletion of the Tp53 gene to efficiently generate immortalized MEFs (iMEFs) while specifically addressing the critical challenges of maintaining culture health and preventing contamination throughout the process. This method reliably produces iMEFs within three weeks while ensuring the resulting cells closely resemble their parent populations, preserving their biological relevance for subsequent experimental applications [6] [27].
Methodology: Tp53 Knockout via CRISPR-Cas9
The core methodology centers on using electroporation to deliver CRISPR constructs specifically targeting the Tp53 gene, achieving efficient immortalization in under three weeks [6]. This approach offers distinct advantages over traditional methods:
- Precision Engineering: Unlike serial passaging (3T3 protocol) or oncogene overexpression, CRISPR specifically targets the Tp53 gene, a known gatekeeper in cellular senescence pathways, minimizing unintended phenotypic changes [27] [7].
- Efficiency and Speed: The protocol consistently generates immortalized MEF lines (iMEFs) within 14-21 days, significantly faster than spontaneous immortalization methods [27] [7].
- Preservation of Physiological Properties: iMEFs generated through this method closely resemble parent cell populations, enabling more biologically relevant downstream applications [6].
Detailed Experimental Procedure
MEF Isolation and Primary Culture
The initial stage involves careful isolation and establishment of primary MEF cultures, where aseptic technique is paramount:
- Embryo Source: Utilize pregnant mice carrying embryonic day 12.5 (E12.5) embryos [27] [7].
- Dissection and Processing: Under sterile conditions, dissect embryos, carefully removing extraembryonic tissues. Mechanically and enzymatically dissociate embryonic bodies using 0.1% trypsin solution with RQ1 RNase-free DNase [27].
- Primary Plating: Plate resulting cell suspensions in complete culture media (high-glucose DMEM supplemented with 10% FBS, 2mM L-glutamine, 1mM sodium pyruvate, 1× MEM NEAA, and 1× penicillin-streptomycin) [27]. Pre-warm all media to 37°C before use to minimize thermal shock to primary cells.
- Culture Conditions: Maintain cells at 37°C in a 5% CO₂ incubator throughout the process [55]. Monitor cultures daily for signs of contamination or poor cell health, including medium cloudiness, unexpected pH shifts, or cellular debris.
CRISPR Transfection via Electroporation
The critical genetic modification step requires precise execution:
- CRISPR Constructs: Use Px461-Cas9n-Trp53-sgRNA-alpha and Px461-Cas9n-Trp53-sgRNA-beta plasmids (available through Addgene as numbers 88846 and 88847, respectively) [27] [7].
- Electroporation System: Employ the Neon Transfection System with the 10 μL kit for optimal delivery efficiency [6] [27].
- Transfection Parameters: Follow manufacturer recommendations for primary fibroblast electroporation. Critical note: Use culture media without penicillin-streptomycin immediately after electroporation to maximize cell recovery [27].
- Control Elements: Include a pCAG-GFP plasmid to monitor transfection efficiency through fluorescent marker expression [27].
Post-Transfection Culture and Selection
Following genetic modification, careful culture management ensures successful outgrowth of immortalized cells:
- Initial Recovery: Allow cells to recover for 48-72 hours post-electroporation before initiating any selection procedures.
- Clonal Expansion: Subculture cells at appropriate dilutions to facilitate the isolation of clonal populations with successful Tp53 knockout.
- Validation Phase: Monitor cultures for emergence of rapidly dividing populations, typically evident within 14-21 days [7]. Confirm Tp53 knockout through genomic sequencing or functional assays.
Contamination Control and Culture Health Management
Aseptic Technique and Sterility Maintenance
Maintaining sterile conditions throughout the protocol is fundamental to success, particularly during vulnerable stages like primary cell isolation and post-electroporation recovery:
- Dissection Best Practices: Sterilize surgical instruments (fine scissors, Adson forceps, fine tip forceps) before use and between samples when processing multiple embryos [56]. Work quickly but methodically to minimize tissue exposure to non-sterile environments.
- Reagent Management: Aliquot all reagents and supplements into smaller sterile containers to reduce contamination risk from repeated opening of main stocks [56]. Filter-sterilize enzymatic solutions like trypsin using 0.22 μm syringe filters before use [27].
- Environmental Control: Perform all manipulations within a certified biological safety cabinet using proper aseptic technique [7]. Regularly monitor incubator environments for fungal and bacterial contamination.
Culture Health Monitoring and Quality Control
Vigilant monitoring of cell health and early problem detection prevents culture loss and ensures experimental consistency:
- Morphological Assessment: Actively monitor cultures for typical fibroblast morphology - spindle-shaped cells forming organized, pavement-like arrangements [56]. Be alert for signs of senescence (enlarged, flattened cells with irregular appearances) or contamination (unexpected pH changes, cloudiness, or fungal hyphae) [56].
- Mycoplasma Screening: Routinely test all cultures for mycoplasma contamination using PCR-based detection methods or commercial kits [55] [56].
- Passage Management: Use early-passage MEFs (P0-P2) for immortalization experiments, as they exhibit optimal proliferation capacity and minimal senescence markers [56]. Split early-passage MEFs at 1:4 to 1:6 ratios when they reach 80-90% confluency [56].
Troubleshooting Common Culture Issues
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Microbial Contamination | Non-sterile techniques, contaminated reagents | Discard culture, review aseptic technique, test reagents [56] |
| Poor Cell Viability Post-Electroporation | Electroporation parameters too harsh, poor post-transfection care | Optimize voltage/pulse duration, use antibiotic-free media during recovery [27] |
| Slow Proliferation Post-Transfection | Inefficient Tp53 editing, early senescence | Verify CRISPR efficiency, use low-passage cells, ensure optimal seeding density [56] |
| Senescence Despite Transfection | Incomplete Tp53 knockout, high passage number | Check knockout at clonal level, use earliest passage cells possible [56] |
Quantitative Data and Experimental Outcomes
Protocol Efficiency and Timeline
The CRISPR-mediated Tp53 deletion method demonstrates significant advantages in efficiency and reproducibility compared to traditional approaches:
Table 1: Immortalization Efficiency Comparison
| Method | Time to Immortalization | Success Rate | Key Advantages |
|---|---|---|---|
| CRISPR-Tp53 Deletion | 14-21 days [27] [7] | High and consistent [6] | Preserves physiological properties, reproducible |
| Traditional 3T3 Protocol | Several weeks to months [27] | Inefficient and variable [27] | Gentle process, no neoplastic transformation [27] |
| Oncogene Overexpression | Varies | Efficient but alters cell physiology [27] | Rapid, but induces cancer-like phenotypes [27] |
This method has proven effective for generating iMEF lines from both wild-type and genetically modified embryos across different genetic backgrounds (C57BL/6 and mixed C57BL/6;129J) [27]. The resulting iMEFs maintain the ability to be subcloned and further genetically manipulated, making them suitable for downstream applications including gene rescue experiments [27].
Essential Research Reagent Solutions
Table 2: Key Research Reagents for CRISPR-Mediated MEF Immortalization
| Reagent/Supply | Function | Specifications |
|---|---|---|
| Px461-Cas9n-Trp53-sgRNA Plasmids | CRISPR constructs for Tp53 targeting | Addgene #88846 & #88847; express Cas9n and Tp53-specific sgRNAs [27] |
| Neon Transfection System | Electroporation delivery | Enables efficient plasmid delivery into primary MEFs [6] [27] |
| DMEM, High Glucose | Base culture medium | Supports robust MEF growth; supplement with FBS and additives [27] |
| Fetal Bovine Serum (FBS) | Culture supplement | Provides essential growth factors and nutrients; use 10% final concentration [27] |
| Trypsin-EDTA (0.25%) | Cell dissociation | Enzymatically detaches adherent cells for passaging and harvesting [27] |
| Cell Strainers | Tissue filtration | Removes tissue aggregates and clumps during primary MEF preparation [27] |
Workflow Visualization and Procedural Integration
The following workflow diagram illustrates the integrated process of MEF isolation, CRISPR immortalization, and critical contamination control points:
MEF Immortalization and Contamination Control Workflow
This integrated workflow emphasizes the critical control points where contamination risks are highest and where culture health monitoring is most essential. The process flows systematically from embryo isolation through to validated immortalized cell lines, with specific attention to the stages most vulnerable to culture health complications.
The CRISPR-mediated Tp53 deletion protocol represents a significant advancement in MEF immortalization methodology, offering researchers a reliable, efficient approach for generating immortalized cell lines while maintaining physiological relevance. The success of this technique depends not only on precise genetic editing but equally on vigilant contamination control and culture health management throughout the process.
By implementing the sterile techniques, monitoring practices, and troubleshooting approaches outlined in this application note, researchers can consistently generate high-quality iMEFs suitable for a wide range of downstream applications in gene function studies, disease modeling, and drug development. The standardized protocols and quality control measures ensure reproducible results while minimizing the experimental variables introduced by contamination or compromised cell health.
In the field of cell biology, the ability to generate immortalized cell lines is a cornerstone for long-term functional studies. CRISPR-mediated deletion of the Tp53 gene has emerged as a powerful and efficient method for the immortalization of primary cells, such as mouse embryonic fibroblasts (MEFs), overcoming the limitations of spontaneous senescence. However, the success of this technique hinges on a critical factor: the completeness of Tp53 knockout. Incomplete ablation can lead to residual p53 activity, which compromises immortalization, alters cellular physiology, and introduces confounding variables in downstream experiments. This Application Note details a rigorous framework for validating complete Tp53 knockout and ensuring the quality of resulting immortalized MEFs (iMEFs), providing essential guidance for researchers in drug development and basic science.
Validation Strategies for Complete Tp53 Knockout
A multi-tiered approach, moving from bulk population assessment to clonal analysis, is essential to confirm the successful and complete knockout of Tp53.
Genotypic Validation
The initial step involves confirming the presence of insertions or deletions (INDELs) at the DNA level.
- PCR Amplification and Sequencing: The genomic region surrounding the CRISPR target site(s) should be amplified by PCR. Sanger sequencing of the PCR products, followed by analysis with tools like ICE (Inference of CRISPR Edits) or TIDE (Tracking of Indels by Decomposition), can provide an initial estimate of the editing efficiency within the bulk cell population. [43]
- Next-Generation Sequencing (NGS): For a more comprehensive and quantitative assessment, NGS of the amplified target locus provides a detailed profile of all induced mutations and their frequencies, confirming the presence of frameshift mutations in both Tp53 alleles. [57]
Table 1: Genotypic and Phenotypic Analysis of Edited MEFs
| Analysis Type | Method | Key Outcome Measures | Interpretation of Success |
|---|---|---|---|
| Genotypic (DNA) | ICE Analysis / TIDE | INDEL frequency (%) | High efficiency (>80%) in bulk population. [43] |
| Next-Generation Sequencing | Mutation spectrum and zygosity | Presence of biallelic frameshift mutations in clonal lines. | |
| Phenotypic (Protein) | Western Blot | p53 protein expression | Absence of p53 protein, both at baseline and after stress (e.g., Nutlin-3a treatment). [57] |
| Immunofluorescence | p53 protein expression and localization | No nuclear p53 signal post-DNA damage. | |
| Functional Assay | Response to MDM2 inhibition (e.g., Nutlin-3a) | Cell proliferation / Cell cycle arrest | Continued proliferation where wild-type cells arrest. [57] |
| p21/CDKN1A expression | Lack of p21 induction upon p53 pathway activation. [57] |
Phenotypic and Functional Validation
The absence of genetic code does not guarantee the absence of functional protein. Therefore, validation must proceed to the protein and functional levels.
- Protein-Level Analysis: Western blotting is a crucial tool for confirming the absence of p53 protein. It is imperative to perform this analysis both under steady-state conditions and following p53 pathway activation, such as with MDM2 inhibitors like Nutlin-3a or upon induction of DNA damage. The complete lack of p53 protein stabilization under these conditions is a strong indicator of successful knockout. [57] This step is also vital for identifying "ineffective sgRNAs" that create INDELs but fail to abolish protein expression. [43]
- Functional Cellular Assays: The ultimate test of a successful Tp53 knockout is the loss of p53 function. This can be assessed by treating the edited cells with Nutlin-3a. Wild-type MEFs will undergo cell-cycle arrest and upregulate p53 target genes like p21 (CDKN1A). In contrast, Tp53-knockout iMEFs should continue to proliferate and show no induction of p21. [57] The failure to activate this pathway upon challenge confirms the functional loss of p53.
The diagram below illustrates this multi-layered validation workflow and the logical decision points for ensuring a complete knockout.
A Protocol for Tp53 Knockout and Immortalization of MEFs
The following optimized protocol, adapted from peer-reviewed methods, reliably generates immortalized MEFs (iMEFs) within three weeks. [6] [5]
Materials and Reagents
Table 2: Research Reagent Solutions for Tp53 Knockout
| Item | Function/Description | Example/Catalog Number |
|---|---|---|
| Tp53 CRISPR Plasmids | Express Cas9 and sgRNAs targeting the Tp53 gene. | Px461-Cas9n-Trp53-sgRNA-alpha (Addgene #88846); Px461-Cas9n-Trp53-sgRNA-beta (Addgene #88847). [5] |
| Electroporation System | High-efficiency delivery of CRISPR constructs into primary MEFs. | Neon Transfection System (Thermo Fisher Scientific) or comparable. [6] [5] |
| Positive Editing Control | Validated sgRNA to confirm transfection and editing efficiency. | sgRNA targeting a common locus (e.g., ROSA26). [58] |
| Negative Editing Control | Establishes baseline phenotype from transfection stress. | Cells transfected with "scramble" sgRNA or Cas9 only. [58] |
| p53 Pathway Activator | Used in functional assays to stress the p53 pathway. | Nutlin-3a (an MDM2 inhibitor). [57] |
| MEF Culture Media | Supports growth of primary and immortalized MEFs. | High-glucose DMEM, 10% FBS, 2mM L-glutamine, 1mM sodium pyruvate, 1x MEM NEAA. [5] |
Step-by-Step Workflow
- MEF Isolation and Culture: Isolate primary MEFs from E12.5-E13.5 mouse embryos. Culture in complete MEF media. Early-passage cells (P2-P4) are ideal for transfection.
- CRISPR Delivery via Electroporation: Use the Neon Transfection System to deliver a mix of the two Tp53-targeting CRISPR plasmids (Px461-Cas9n-Trp53-sgRNA-alpha and -beta) into MEFs. Critical: Include positive and negative control transfections in parallel. [5] [58]
- Recovery and Expansion: Plate transfected cells and culture for 3-5 days. Subsequently, passage cells at a consistent density (e.g., 1:4 or 1:6) every 3 days. Observe for the emergence of rapidly dividing cells, a hallmark of escaping senescence.
- Single-Cell Cloning: Once a proliferative population is established, seed cells at low density to isolate single clones. Expand individual clones for validation.
- Validation of Clones: Subject expanded clonal lines to the validation cascade described in Section 1:
- Genotype clonal lines via sequencing to confirm biallelic knockout.
- Perform Western blot to confirm absence of p53 protein, with and without Nutlin-3a treatment.
- Conduct a functional assay by treating cells with Nutlin-3a; successful iMEFs will not arrest.
The end-to-step workflow, from isolation to validated clone, is summarized below.
Critical Quality Control Considerations
The Critical Importance of Experimental Controls
Appropriate controls are non-negotiable for interpreting the outcomes of CRISPR experiments correctly. [58]
- Positive Editing Control: Transfecting cells with a validated, highly efficient sgRNA (e.g., targeting ROSA26) confirms that your transfection and editing workflow is functioning optimally. If this control fails, the problem lies with the method, not the Tp53-specific reagents. [58]
- Negative Editing Controls: These include cells transfected with a non-targeting "scramble" sgRNA or with Cas9 protein alone. These controls are vital for distinguishing true phenotypic consequences of Tp53 loss (e.g., immortalization) from artifacts caused by the cellular stress of transfection and clonal selection. [58]
Addressing Cas9-Induced p53 Pathway Activation
A critical and often overlooked aspect of CRISPR editing in primary cells is that the act of introducing Cas9 and creating double-strand breaks can itself activate the p53 pathway. [34] This is particularly problematic when knocking out Tp53, as cells that have a robust p53 response may be selectively disadvantaged during the editing process. This can lead to a bias where the successfully edited cells that proliferate are those that already had compromised p53 function, rather than those where CRISPR cleanly knocked out the gene.
- Mitigation Strategy: Be aware of this potential bias. Using high-efficiency delivery methods (like electroporation) and highly active sgRNAs can help ensure rapid and complete editing, minimizing the window for p53-mediated selection. [6] [43] Analyzing editing outcomes early and in bulk populations, before extensive expansion, can also provide a less biased view.
The CRISPR-mediated Tp53 knockout protocol provides a robust and efficient path to MEF immortalization. However, the reliability of any subsequent research using these iMEFs is entirely dependent on the rigor of the validation process. By implementing the multi-tiered strategy outlined here—combining genotypic, protein-level, and functional assays with stringent quality controls—researchers can confidently generate and utilize high-quality, fully characterized Tp53-null iMEFs, thereby ensuring the integrity and reproducibility of their scientific findings.
Beyond Immortalization: Validating iMEF Phenotype and Functional Utility
The tumor suppressor gene TP53 is a critical regulator of cell cycle arrest, senescence, and apoptosis, making its functional knockout a cornerstone technique for cellular immortalization in biological research. Within the context of a broader thesis on CRISPR-mediated Tp53 deletion for mouse embryonic fibroblast (MEF) immortalization, robust validation of a successful knockout is paramount. Unvalidated edits can lead to misinterpretation of experimental results, wasting significant time and resources [59]. This application note provides detailed protocols for confirming Tp53 knockout through integrated genotypic and protein-level analysis, ensuring researchers can reliably generate immortalized MEFs (iMEFs) for downstream applications.
Genotypic Analysis: Confirming Deletion at the DNA Level
Genotypic validation confirms that the CRISPR/Cas9 system has successfully introduced indels (insertions or deletions) into the TP53 gene locus, disrupting its coding sequence.
Target Site Amplification and Sequencing
The initial step involves PCR amplification of the genomic region targeted by the CRISPR guide RNAs, followed by sequencing to identify mutations.
- Primer Design: Design primers that flank the target site(s) within the Tp53 gene. The ideal amplicon size for Sanger sequencing is between 500 bp and 1200 bp [60]. For the MEF immortalization protocol, the target sites are defined by the specific sgRNAs (e.g., those in plasmids Px461-Cas9n-Trp53-sgRNA-alpha and -beta) [5].
- PCR and Sequencing: Perform PCR using a high-fidelity polymerase to minimize errors. Purify the PCR product and submit it for Sanger sequencing.
- Data Analysis: Analyze the sequencing chromatograms from edited cell populations and compare them to wild-type controls. The presence of overlapping peaks downstream of the cut site indicates a heterogeneous mixture of indels. For clearer results, especially with mixed populations, subclone the PCR product into a sequencing vector before sequencing individual clones [60].
Analysis of Sequencing Data
Several analytical methods can interpret the sequencing data to quantify editing efficiency.
- Tracking of Indels by Decomposition (TIDE): This widely used method decomposes the complex Sanger sequencing chromatogram from a pooled population of edited cells. It rapidly quantifies the spectrum and frequency of indel mutations without the need for subcloning [59].
- Next-Generation Sequencing (NGS): For the deepest and most quantitative analysis, NGS of the amplified target region provides a comprehensive profile of all induced mutations at single-base resolution. This is the gold standard for characterizing heterogeneous editing outcomes [57] [60].
The diagram below outlines the primary workflow for genotypic confirmation.
Western Blot Analysis: Confirming Knockout at the Protein Level
The definitive confirmation of a successful knockout is the absence of the p53 protein. Western blotting is the standard method for this protein-level validation.
Protein Extraction and Quantification
Harvest wild-type and CRISPR-edited MEFs and lyse them using a suitable RIPA buffer supplemented with protease and phosphatase inhibitors. Quantify the total protein concentration for each sample using a standardized assay (e.g., BCA or Bradford) to ensure equal loading across gels.
Immunoblotting Protocol
- Gel Electrophoresis and Transfer: Load 20-40 µg of total protein per lane on an SDS-PAGE gel. After electrophoresis, transfer the proteins to a PVDF or nitrocellulose membrane [61].
- Antibody Probing:
- Primary Antibodies: Use a validated anti-p53 antibody (e.g., clone DO-7 targeting an N-terminal epitope) to detect total p53 protein [62]. To confirm knockout, also probe for a loading control, such as GAPDH or β-actin.
- Secondary Antibodies: Use an appropriate HRP-conjugated secondary antibody.
- Detection: Develop the blot using enhanced chemiluminescence (ECL) reagents and image it.
Expected Results and Interpretation
In wild-type MEFs, p53 levels are typically low but detectable. Upon cellular stress (e.g., γ-irradiation), p53 protein levels stabilize and increase markedly [62]. In a confirmed Tp53 knockout, no p53 protein should be detectable under either basal or stressed conditions. The presence of a band in the wild-type lane and its absence in the knockout lane, with equivalent loading control signals, confirms a successful protein knockout.
Table 1: Key Reagents for Western Blot Analysis of p53
| Reagent / Resource | Function / Target | Example Specifications |
|---|---|---|
| Anti-p53 Antibody | Detection of total p53 protein | Clone DO-7, epitope AA 37-45 [62] |
| Anti-GAPDH Antibody | Loading control | Housekeeping gene protein |
| HRP-conjugated Secondary Antibody | Signal generation | Species-specific |
| ECL Substrate | Chemiluminescent detection | Enhanced sensitivity kits |
| PVDF Membrane | Protein immobilization | 0.45 µm pore size |
Advanced and Multiplexed Validation Strategies
While genotypic analysis and Western blotting are foundational, advanced techniques can provide deeper, single-cell resolution.
Mass Cytometry for Single-Cell Protein Analysis
Mass cytometry (CyTOF) allows for the simultaneous detection of p53 and its post-translational modifications alongside multiple cell surface and intracellular signaling markers at the single-cell level. This is particularly useful for heterogeneous cell populations [62].
- Methodology: Conjugate antibodies against p53 and related signaling proteins to rare metal isotopes. Stain the cells and analyze them by time-of-flight mass spectrometry.
- Advantages: This method eliminates the issue of spectral overlap found in fluorescence flow cytometry, allowing for a much higher number of parameters (up to 50) to be measured per cell [62].
Functional Validation in MEF Immortalization
A successful Tp53 knockout confers a clear phenotypic advantage: escape from senescence. The core protocol for MEF immortalization relies on this principle [6] [5].
- Protocol Outline: Isolate primary MEFs from E12.5 embryos. Electroporate the cells with CRISPR constructs targeting Tp53 using a system like the Neon Transfection System. Culture the cells and passage them continuously.
- Expected Outcome: Primary MEFs undergo senescence within a few passages. In contrast, Tp53 knockout MEFs will continue to proliferate, allowing for the establishment of a stable, immortalized cell line (iMEF) within approximately three weeks [6] [5].
Table 2: Comparison of Tp53 Knockout Validation Methods
| Method | What It Measures | Key Strength | Throughput |
|---|---|---|---|
| Sanger + TIDE | Spectrum and frequency of indels | Rapid, cost-effective for initial screening | Medium |
| NGS Amplicon Seq | Comprehensive mutation profile | Gold standard for depth and quantification | High |
| Western Blot | Presence/Absence of p53 protein | Confirms functional knockout at protein level | Low |
| Mass Cytometry | p53 protein and signaling in single cells | Multiplexed analysis of heterogeneous populations | Medium |
The relationship between Tp53 loss, downstream signaling, and the resulting cellular phenotype is summarized below.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CRISPR-mediated Tp53 Knockout and Validation
| Reagent / Resource | Function | Source / Example |
|---|---|---|
| Px461-Cas9n-Trp53-sgRNA plasmids | CRISPR constructs for targeting Tp53 | Addgene #88846, #88847 [5] |
| Neon Transfection System | Electroporation-based delivery of CRISPR constructs | Thermo Fisher Scientific [5] |
| Anti-p53 Antibody (DO-7) | Detection of total p53 protein in Western blot | [62] |
| p53 Mutant Cell Line (e.g., NB4) | Positive control for mutant p53 expression | [62] |
| p53 Null Cell Line (e.g., HL-60) | Negative control for p53 expression | [62] |
A multi-tiered validation strategy is essential for confirming a successful Tp53 knockout in MEF immortalization protocols. Initial genotypic screening with TIDE analysis or NGS should be conclusively supported by Western blot analysis demonstrating the absence of the p53 protein. Incorporating functional assays that demonstrate escape from senescence provides the ultimate biological validation. This rigorous, integrated approach ensures the reliability of your immortalized MEF models, forming a solid foundation for all subsequent research on gene function, regulation, and therapeutic candidate screening in a physiologically relevant cellular context.
The immortalization of mouse embryonic fibroblasts (MEFs) is a critical step for establishing sustainable in vitro models for biological research and drug development. Traditional methods, including spontaneous immortalization through serial passaging (the 3T3 protocol) or oncogene overexpression, often result in cells with altered physiological properties that poorly reflect primary cell characteristics. The emergence of CRISPR-mediated gene editing has revolutionized this process, with Tp53 deletion emerging as a highly efficient strategy. However, the utility of resulting immortalized MEF (iMEF) lines depends entirely on rigorous phenotypic characterization to confirm they maintain the essential biological properties of their primary counterparts. This application note details the necessary validation methodologies to ensure physiological relevance.
Core Immortalization Protocol and Rationale
The foundational protocol for generating iMEFs via Tp53 knockout involves a highly efficient CRISPR/Cas9 system delivered via electroporation, reliably producing immortalized lines within 14 to 21 days [6] [27] [7].
Key Advantages of Tp53 Knockout
- Genetic Fidelity: Unlike oncogenic transformation, Tp53 deletion does not introduce foreign oncogenes, preserving a more native genetic landscape [27] [7].
- Efficiency and Speed: This method bypasses the inefficiency and extended timeline (several months) of the spontaneous 3T3 method [7].
- Physiological Preservation: iMEFs generated via this method closely resemble parent primary cell populations, enabling more reliable downstream applications like gene rescue studies [27].
The workflow for creating and validating iMEFs is outlined below.
Comprehensive Phenotypic Characterization Framework
A multi-parameter approach is essential for demonstrating that iMEFs retain the phenotypic hallmarks of primary MEFs. The following characterization assays provide a comprehensive assessment.
Growth and Senescence Profiling
Continuous monitoring of growth parameters is fundamental to establishing immortalization success and population stability.
Methodology:
- Population Doubling Time: Calculate over consecutive passages using cell counts from standardized seeding densities [27] [7].
- Senescence-Associated β-Galactosidase (SA-β-Gal) Staining: Quantify the percentage of SA-β-Gal-positive cells in primary MEFs at passage 3-4 versus iMEFs at equivalent and later passages (e.g., passage 10+). Primary MEFs should exhibit high senescence, while valid iMEFs show minimal activity [7].
- Saturation Density: Assess contact inhibition by measuring the maximum cell density achievable per cm² in a culture vessel.
Table 1: Expected Outcomes for Growth and Senescence Parameters
| Parameter | Primary MEFs (Early Passage) | Validated iMEFs (Post-Immortalization) |
|---|---|---|
| Population Doubling Time | ~24-30 hours | Consistent ~24-30 hours, stable over >10 passages |
| SA-β-Gal Staining | High (>60% positive cells at P4) | Low (<5% positive cells at P10+) |
| Saturation Density | Defined, contact-inhibited | Similar to primary MEFs, without significant overgrowth |
| Long-Term Proliferation | Senescence after 5-10 passages | Sustained proliferation beyond 30 passages |
Genomic and Telomeric Integrity Assessment
CRISPR editing and the loss of p53, a guardian of the genome, necessitate rigorous checks for genomic stability.
Methodology:
- Karyotyping: Perform G-banding chromosome analysis at passage 5 and 15 to confirm a stable, diploid karyotype without major chromosomal rearrangements.
- Telomere Length and Function Analysis: Utilize Telomere Fluorescence in Situ Hybridization (FISH) to measure telomere length. As highlighted in recent research, ensure proper telomere end-protection mechanisms are functional to prevent spurious DNA repair events, a key consideration in p53-deficient cells [63].
- TP53 Sequencing: Confirm complete knockout of the Tp53 gene via Sanger sequencing or next-generation sequencing of the targeted region.
Stress Response and Pathway Functionality
A critical test for iMEFs is their response to stressors and activation of key pathways, mirroring primary cell behavior.
Methodology:
- Serum Starvation and Apoptosis Assay: Monitor apoptosis via flow cytometry (Annexin V/PI staining) following 48-hour serum starvation. iMEFs should exhibit an appropriate apoptotic response, albeit modulated by Tp53 knockout.
- Oxidative Stress Challenge: Treat cells with hydrogen peroxide and measure cell viability and the induction of antioxidant response genes.
- DNA Damage Response: Expose cells to low-dose gamma radiation or genotoxic drugs (e.g., Etoposide) and assess the phosphorylation of H2AX (γH2AX) as a marker of DNA damage repair, recognizing that the p53-mediated apoptotic arm will be impaired.
The p53 protein sits at the center of a complex network of stress responses. Its deletion alters, but does not completely abolish, these pathways as illustrated below.
Table 2: Key Reagent Solutions for Characterization Workflow
| Research Reagent / Material | Function in Characterization | Example from Protocol |
|---|---|---|
| Tp53 CRISPR Plasmids (e.g., Px461-Cas9n-Trp53-sgRNA-alpha/beta) | Mediates targeted knockout of the Tp53 gene for immortalization. | [27] [7] |
| Neon Transfection System | Electroporation system for high-efficiency delivery of CRISPR constructs into primary MEFs. | [27] |
| Senescence-Associated β-Galactosidase Staining Kit | Histochemical detection of senescent cells to confirm bypass of senescence. | [7] |
| Telomere FISH Kit | Fluorescent labeling of telomeres to assess telomere length and integrity. | [63] |
| Annexin V Apoptosis Kit | Flow cytometry-based quantification of apoptotic cells in stress assays. | (Standard Assay) |
| Phospho-H2AX (γH2AX) Antibody | Immunofluorescence detection of DNA double-strand breaks. | (Standard Assay) |
The CRISPR-mediated Tp53 deletion protocol provides a robust and rapid method for generating immortalized MEFs. However, the value of these iMEFs for physiologically relevant research is contingent upon a thorough and multi-faceted phenotypic characterization strategy. By implementing the standardized framework of growth kinetic analyses, genomic stability checks, and functional stress response assays detailed herein, researchers can confidently validate their iMEF lines. This rigorous approach ensures that immortalized cells serve as a reliable and powerful tool for modeling development, disease, and therapeutic responses, thereby supporting the critical work of scientists and drug development professionals.
Within the broader context of developing a robust CRISPR-mediated Tp53 deletion protocol for generating immortalized Mouse Embryonic Fibroblasts (iMEFs), this application note addresses a critical comparative question: how do the transcriptomic profiles of these engineered iMEFs compare to those of primary MEFs and MEFs immortalized via the traditional spontaneous method? Spontaneous immortalization, typically achieved through serial passaging using a modified 3T3 protocol, is a stochastic process that serves as a common but genetically undefined benchmark [64]. Understanding the transcriptomic differences between these models is essential for researchers and drug development professionals who must select the most appropriate cell system for their specific applications, whether for modeling disease, studying gene function, or performing high-throughput drug screening. This analysis leverages RNA sequencing (RNA-Seq) data to systematically identify differentially expressed genes (DEGs), altered biological pathways, and key hub genes that distinguish these cellular states, providing a molecular rationale for cell model selection.
Key Methodologies and Protocols
Immortalization Techniques
A. CRISPR-Mediated Tp53 Knockout for Efficient Immortalization
This optimized protocol enables the reliable generation of immortalized MEFs (iMEFs) within three weeks by targeting the Tp53 gene, a critical tumor suppressor [6] [27].
- Key Features: The method uses CRISPR/Cas9 and electroporation to create biallelic knockouts of Tp53. The resulting iMEFs closely resemble the parent cell population, can be subcloned, and are suitable for subsequent genetic manipulations like gene rescue experiments [27].
- Procedure Overview:
- MEF Isolation: Isolate primary MEFs from E12.5 mouse embryos.
- Electroporation: Transfect primary MEFs using a Neon electroporator with two CRISPR constructs (e.g., Px461-Cas9n-Trp53-sgRNA-alpha and -beta plasmids) to target Tp53.
- Culture and Expansion: Plate transfected cells in complete culture media (without antibiotics immediately post-electroporation) and culture until immortalized, proliferating cells emerge.
- Validation: Confirm immortalization and Tp53 knockout via functional assays and genotyping.
B. Spontaneous Immortalization via Modified 3T3 Protocol
This traditional method involves the serial passaging of primary MEFs until mutations enabling immortality spontaneously arise [64].
- Key Features: The process is considered "gentle" and less likely to induce neoplastic transformation compared to oncogene overexpression. However, it is inefficient, time-consuming (taking several weeks), and the underlying genetic alterations can vary between cell lines [64] [27].
- Procedure Overview:
- Serial Passaging: Seed primary MEFs at a defined density (e.g., 3 x 10^5 cells per 50-mm dish).
- Regular Transfer: Re-seed cells at the same density every 3 days.
- Selection: Monitor cultures for the emergence of rapidly dividing, immortalized cell populations.
Transcriptomic Profiling and Data Analysis
A standardized RNA-Seq analysis pipeline is crucial for comparing transcriptomes across different MEF types [65] [66]. The following workflow outlines the key steps from raw data processing to biological interpretation.
- Data Collection and Preprocessing: RNA-seq libraries (e.g., poly-A selected mRNA-seq) are prepared with a minimum of three biological replicates. Publicly available data can be sourced from databases like GEO (Gene Expression Omnibus) [65] [66]. Initial quality control is performed using FastQC and MultiQC, followed by adapter trimming and quality filtering with Trimmomatic or Cutadapt [65] [66].
- Alignment and Quantification: Quality-controlled reads are aligned to a reference genome (e.g., mouse mm10) using splice-aware tools like STAR or HISAT2. Gene-level counts are then generated using featureCounts or RSEM [65] [66].
- Differential Expression and Functional Analysis: DESeq2 or edgeR are used to identify DEGs with thresholds such as |Log2FC| ≥ 1 and adjusted p-value (FDR) < 0.01 [64] [65]. Subsequent Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses reveal altered biological processes and pathways. Protein-protein interaction (PPI) networks constructed using the STRING database and analyzed with Cytohubba and MCODE can pinpoint functionally important hub genes [64].
Comparative Transcriptomic Analysis
Spontaneous immortalization triggers extensive transcriptomic reprogramming. A representative study identified 1,096 differentially expressed genes (DEGs) when comparing spontaneously immortalized MEFs to their primary counterparts [64]. A striking pattern is the predominant downregulation of genes, with 1,001 genes decreased and only 95 genes increased in expression [64]. This suggests a massive shutdown of specific genetic programs is a hallmark of the spontaneous immortalization process.
Table 1: Summary of Differentially Expressed Genes in Spontaneously Immortalized MEFs
| Category | Number of Genes | Examples of Altered Genes |
|---|---|---|
| Total DEGs | 1,096 | |
| Upregulated | 95 | Alklal1, Pax8, Il6, Mapk11, Dusp10, Vegfa |
| Downregulated | 1,001 | Cd68, C1qa, Tlr13, Cdh1, Icam1, Itgb2, Itgam |
Altered Biological Pathways and Functions
Gene ontology enrichment analysis of DEGs reveals coherent shifts in biological processes and molecular functions.
Table 2: Enriched Biological Processes in Spontaneously Immortalized MEFs
| Direction | Biological Process | Key Associated Genes |
|---|---|---|
| Upregulated | Regulation of epithelial cell proliferation | Mapk11, Dusp10, Il6, Vegfa |
| Angiogenesis and vasculature development | Pax8, Aldh1a1, Grem1 | |
| Downregulated | Cell-cell adhesion | Cdh1, Cdh19, Pcdh10, Pcdh17 |
| Immune and inflammatory response | Nlrp3, Fcgr2b, Fcgr3, Tnf | |
| Leukocyte migration and activation | Itgam, Ptprc, Icam1 |
These changes reflect a phenotype geared towards sustained growth and evasion of microenvironmental controls. The downregulation of adhesion and immune pathways may facilitate escape from contact inhibition and immune surveillance, while upregulated proliferative and angiogenic signals support a growth-advantaged state [64].
Core Regulatory Hub Genes
Protein-protein interaction network analysis of the 1,096 DEGs identified several hub genes considered highly influential in the network topology. Most of these hub genes were downregulated, with Il6 being a notable exception [64].
Table 3: Key Hub Genes Identified in Spontaneously Immortalized MEFs
| Hub Gene | Description | Expression Change |
|---|---|---|
| Itgam | Integrin Subunit Alpha M | Down |
| Ptprc | Protein Tyrosine Phosphatase Receptor Type C | Down |
| Tnf | Tumor Necrosis Factor | Down |
| Cxcr4 | C-X-C Motif Chemokine Receptor 4 | Down |
| Itgb2 | Integrin Subunit Beta 2 | Down |
| Il6 | Interleukin 6 | Up |
| Icam1 | Intercellular Adhesion Molecule 1 | Down |
The concurrent downregulation of Itgb2, Itgam, and Icam1 strongly underscores the loss of adhesive and immune-regulatory functions. In contrast, the upregulation of Il6, a pro-inflammatory cytokine, suggests it may act as a novel driver of proliferation in the immortalized state [64]. A proposed gene regulatory network for spontaneous immortalization also involves other key players like Mapk11, Cdh1, Zic1, and Hoxd10 [64].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials essential for performing the MEF immortalization and transcriptomic analysis protocols described in this note [27].
Table 4: Essential Research Reagents and Materials
| Item | Function/Application | Example Catalog Number |
|---|---|---|
| Px461-Cas9n-Trp53-sgRNA-alpha/beta | CRISPR plasmids for targeted Tp53 knockout. | Addgene #88846, #88847 |
| Neon Transfection System 10 μL Kit | Electroporation system for efficient plasmid delivery. | Thermo Fisher MPK1096 |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | Base medium for MEF cell culture. | Gibco 11960-044 |
| Fetal Bovine Serum (FBS) | Serum supplement for cell culture media. | Gibco 26-140-079 |
| 0.25% Trypsin-EDTA | Reagent for detaching and passaging adherent cells. | Gibco 25200072 |
| Penicillin-Streptomycin (100X) | Antibiotic to prevent bacterial contamination in culture. | Gibco 15-140-122 |
| FastQC | Software for quality control of raw sequencing data. | [Online Resource] |
| Trimmomatic | Tool for trimming adapter sequences and low-quality bases. | [Online Resource] |
| STAR Aligner | Spliced aligner for RNA-seq reads against a reference genome. | [Online Resource] |
| DESeq2 | R package for differential expression analysis of count data. | Bioconductor |
The comparative transcriptome analysis reveals that spontaneous immortalization drives a profound and skewed transcriptomic shift, largely characterized by the suppression of genes governing cell adhesion and immune responses, alongside selective upregulation of proliferative and pro-survival signals. The identified hub genes, particularly the downregulated adhesion molecules (Itgb2, Icam1) and the upregulated cytokine Il6, provide compelling candidates for further functional validation as drivers of the immortalized phenotype.
When framed within the broader thesis of optimizing a CRISPR-mediated Tp53 deletion protocol, this comparison highlights key advantages of the genetic engineering approach. The CRISPR method directly targets a known and consistent immortalizing event—Tp53 loss—leading to a more uniform and predictable iMEF population within a short, defined timeframe [6] [27]. In contrast, the transcriptomic heterogeneity inherent in spontaneous immortalization, resulting from stochastic mutational events, can complicate experimental reproducibility and interpretation [64]. Therefore, for researchers requiring defined genetics, rapid generation, and a transcriptomic profile closer to the primary state (minus senescence pathways), the CRISPR-generated iMEFs represent a superior and more reliable model system for most downstream applications in basic research and drug development.
Application Notes
This document details the core functional assays for characterizing immortalized mouse embryonic fibroblasts (iMEFs) generated via CRISPR-mediated deletion of the Tp53 gene. These assays are essential for confirming successful immortalization, assessing the stability of resulting cell lines, and validating their suitability for downstream applications in basic research and drug development [67] [27].
The ablation of Tp53, a critical tumor suppressor, enables cells to bypass senescence; however, thorough functional characterization is required to ensure that the immortalized cells exhibit the desired physiological properties without uncontrolled transformation [67] [11]. The assays outlined below—evaluating proliferation capacity, stress response, and genetic manipulability—provide a comprehensive profile of the iMEF lines.
Proliferation and Senescence Escape
The primary indicator of successful immortalization is the acquisition of an infinite cellular lifespan. Primary MEFs undergo senescence after a few passages, whereas Tp53-knockout iMEFs exhibit sustained proliferation [67] [11]. Key quantitative metrics for proliferation capacity are summarized in Table 1.
Table 1: Quantitative Proliferation and Senescence Assay Data
| Assay Type | Measured Parameter | Primary MEFs (Senescent) | Tp53-KO iMEFs (Immortalized) | Measurement Method/Tools |
|---|---|---|---|---|
| Growth Kinetics | Cumulative Population Doublings | Plateaus at low passages (e.g., <10) [11] | Maintained for over 15 passages and beyond [11] | Cell counting, cumulative growth curve [11] |
| Senescence Assay | SA-β-gal Positive Cells [68] | High percentage (>70%) at senescence [11] | Rarely detected (<5%) even at late passages [11] | Senescence-associated β-galactosidase (SA-β-gal) staining [11] |
| Cell Cycle Analysis | S-phase Fraction | Low | High | BrdU incorporation, flow cytometry with DNA staining (e.g., Hoechst dye) [68] |
Stress Response Profiling
The loss of p53 function profoundly alters how cells respond to genotoxic and proteotoxic stress. Characterizing these responses is crucial for understanding the phenotype of iMEFs and their utility in toxicity studies.
- Genotoxic Stress Resistance: A hallmark of TP53-deficient cells is acquired resistance to DNA-damaging agents. This is evidenced by a blunted induction of p21, a key p53-target gene, upon treatment. As shown in canine TP53-knockout cells, treatment with the alkylating agent temozolomide (TMZ) robustly induces p21 protein in control cells but fails to do so in TP53-knockout cells, confirming the loss of this canonical p53-p21 pathway function [11].
- Monitoring Proteotoxic Stress Pathways: Activation of specific stress response pathways can be monitored using fluorescent transcriptional reporters. While demonstrated in other model organisms, this principle can be adapted for mammalian cells. For instance, a multi-color reporter system can simultaneously monitor pathways like the heat shock response (cytotoxicity, often via Hsp promoters) and the unfolded protein response [69] [70]. This allows for a multimodal assessment of how iMEFs handle proteotoxic insults.
Genetic Manipulability and Stability
A key advantage of the CRISPR-generated iMEF system is its compatibility with further genetic engineering for functional rescue studies or to introduce additional disease-relevant mutations [67] [27]. The genetic stability of these lines is a critical consideration.
- Karyotype Stability: TP53 deficiency can lead to genomic instability. Karyotype analysis of TP53-knockout canine fibroblasts revealed an aberrant chromosome status, which is a known consequence of disrupting TP53-governed genome protective mechanisms [11]. Regular karyotyping is recommended to monitor for gross chromosomal abnormalities in iMEF lines.
- Transformation Potential: While Tp53 knockout immortalizes cells, it is often insufficient for full transformation without additional oncogenic activation [11]. Assays such as anchorage-independent growth in soft agar can distinguish non-tumorigenic immortalized cells from transformed ones. For example, one study showed that only a specific TP53-knockout canine cell line (TP53KO#30) did not grow in soft agar, whereas another (TP53KO#39) did, potentially due to off-target effects during CRISPR editing [11].
Experimental Protocols
Protocol 1: Senescence-Associated β-Galactosidase (SA-β-gal) Staining
This protocol detects β-galactosidase activity at pH 6.0, a biomarker characteristic of senescent cells [11] [68].
Materials:
- Cells: iMEFs and primary MEFs (positive control).
- Fixation Solution: 2% Formaldehyde and 0.2% Glutaraldehyde in PBS.
- Staining Solution: 1 mg/mL X-gal, 5 mM Potassium Ferrocyanide, 5 mM Potassium Ferricyanide, 150 mM NaCl, 2 mM MgCl₂ in 40 mM Citric Acid/Sodium Phosphate buffer (pH 6.0).
Procedure:
- Plate Cells: Seed cells at a low density in a multi-well plate and culture for 24-48 hours.
- Wash: Rinse cells once with room temperature PBS.
- Fix: Incubate cells with Fixation Solution for 5 minutes at room temperature.
- Wash: Rinse cells twice with PBS.
- Stain: Add fresh Staining Solution to cover the cells. Incubate at 37°C in a dry incubator (without CO₂) for 4-16 hours, protected from light.
- Analyze: Examine cells under a standard brightfield microscope. Senescent cells will display blue cytoplasmic staining. Count the percentage of blue cells in multiple random fields.
Protocol 2: Genotoxic Stress Challenge and p21 Induction Assay
This protocol evaluates the functional loss of the p53 pathway by measuring p21 protein levels after genotoxic stress.
Materials:
- Treatment Compound: Temozolomide (TMZ) or Nutlin-3a (Mdm2 inhibitor) [57] [11].
- Lysis Buffer: RIPA buffer supplemented with protease and phosphatase inhibitors.
- Antibodies: Anti-p21 and anti-β-actin (or GAPDH) for loading control.
Procedure:
- Seed Cells: Plate iMEFs and wild-type control MEFs in 6-well plates.
- Treat: Once cells are 70-80% confluent, treat with an appropriate concentration of TMZ (e.g., 100-500 µM) or Nutlin-3a (e.g., 10 µM). Include a vehicle control (e.g., DMSO).
- Incubate: Incubate cells for 24-48 hours [11].
- Lyse Cells: Aspirate media, wash with cold PBS, and lyse cells in RIPA buffer on ice.
- Immunoblotting:
- Determine protein concentration.
- Separate equal amounts of protein by SDS-PAGE and transfer to a PVDF membrane.
- Block the membrane and probe with anti-p21 and loading control antibodies.
- Develop and quantify band intensities. Tp53-KO iMEFs should show significantly attenuated p21 induction compared to wild-type controls.
Protocol 3: Anchorage-Independent Growth Assay (Soft Agar)
This assay tests for transformation potential by assessing the ability of cells to proliferate without adhering to a solid substrate.
Materials:
- Agarose: Low-melting-point agarose.
- Base Agar Layer: 0.6% agarose in complete culture media.
- Top Agar Layer: 0.3% agarose in complete culture media containing the cells.
Procedure:
- Prepare Base Layer: Melt and cool the 0.6% agarose solution, then add 1.5 mL to each well of a 6-well plate. Allow it to solidify at room temperature.
- Prepare Cell Layer: Trypsinize, count, and resuspend cells in culture media. Mix the cell suspension with an equal volume of 0.6% agarose solution (pre-warmed to 40°C) to yield a final 0.3% agarose layer with 5,000-10,000 cells/mL.
- Plate Cell Layer: Add 1.5 mL of the cell-agarose mixture on top of the base layer. Allow it to set.
- Feed Cells: Once the top layer is solid, add 1 mL of fresh culture media on top. Feed the cells with fresh media twice a week.
- Incubate and Score: Incubate plates for 2-4 weeks. Score colonies larger than 50-100 µm under a microscope. Non-tumorigenic iMEFs will form few to no colonies, while transformed cells will form significant colonies [11].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Materials
| Item | Function/Application | Example/Specification |
|---|---|---|
| Neon Transfection System | Electroporation-based delivery of CRISPR constructs into primary MEFs [67] [27]. | Thermo Fisher Scientific, Neon Transfection System 10 μL Kit (MPK1096) [67] [27]. |
| Tp53-Targeting CRISPR Plasmids | CRISPR/Cas9 system for targeted knockout of the Tp53 gene. | Px461-Cas9n-Trp53-sgRNA-alpha/beta plasmids (Addgene #88846, #88847) [67] [27]. |
| Senescence Detection Kit | Histochemical detection of senescent cells. | SA-β-gal Staining Kit [11] [68]. |
| p21 (CDKN1A) Antibody | Immunoblotting to confirm loss of p53 pathway function. | Used in western blot to assess p21 induction after genotoxic stress [11]. |
| Mdm2 Inhibitor (e.g., Nutlin-3a) | Small molecule activator of the p53 pathway; used to challenge cells in stress response assays [57]. | Induces p53 stabilization and transcriptional activity in wild-type cells [57]. |
| DNA Alkylating Agent (e.g., Temozolomide) | Genotoxic stressor to challenge the p53 pathway. | Used to test for resistance and blunted p21 response in TP53-KO cells [11]. |
Visualized Workflows and Pathways
Immortalized Mouse Embryonic Fibroblasts (iMEFs) generated through CRISPR-mediated deletion of the Tp53 gene represent a transformative tool for studying gene function and disease mechanisms. Primary MEFs undergo senescence rapidly after isolation, severely limiting long-term genetic manipulations and experimental scalability. The CRISPR-based immortalization method overcomes this critical limitation by producing stable, physiologically relevant cell lines within three weeks, enabling sophisticated gene rescue studies and disease modeling applications that were previously challenging or impossible with primary cells [6] [5].
This application note details specific case studies and protocols for utilizing iMEFs in gene rescue experiments and disease modeling, providing researchers with practical methodologies to investigate gene function and disease-associated variants. The Tp53 knockout iMEF platform offers distinct advantages over traditional immortalization methods, including preservation of physiological properties, elimination of spontaneous mutations associated with serial passaging, and compatibility with subsequent genetic manipulations through clonal expansion [6] [25].
Technical Applications and Experimental Data
Gene Rescue Applications
Gene rescue experiments in iMEFs enable researchers to characterize the functional consequences of disease-causing genetic variants and test potential therapeutic interventions. The Tp53-deleted iMEF platform provides a stable, consistent cellular background for introducing mutant genes and their wild-type counterparts to assess functional differences.
Functional Characterization of Mutant Proteins: The MEF system can be combined with rescue studies to characterize the function of mutant genes/proteins, including disease-causing variants. In practice, researchers introduce the mutant gene of interest into Tp53-knockout iMEFs and compare its activity to wild-type versions introduced in parallel experiments [6] [5]. This approach allows for direct assessment of how specific mutations alter protein function without the confounding factor of cellular senescence.
Stable Expression System: Unlike primary MEFs that senesce quickly, iMEFs maintain viability over numerous passages, enabling long-term studies of gene expression and function. Individual iMEF clones can be expanded after genetic manipulation, providing consistent biological material for replicated experiments and reducing inter-experimental variability [6].
Compatibility with Diverse Genetic Backgrounds: The Tp53 deletion method has successfully generated iMEF lines from wild-type and genetically modified embryos of different genetic backgrounds, including C57BL/6 and mixed C57BL/6;129J strains, enhancing the flexibility and applicability of this platform for studying genotype-specific effects [5].
Disease Modeling Capabilities
The iMEF platform serves as a valuable model for investigating disease mechanisms, particularly in cancer biology and genetic disorders where p53 pathway alterations play a central role.
Cancer Biology Studies: TP53 is the most frequently mutated gene in human cancer, with approximately 50% of all cancers carrying TP53 mutations [71] [72]. iMEFs with CRISPR-inactivated Tp53 provide a controlled system for investigating how p53 loss contributes to oncogenic transformation and cancer progression. Research using similar TP53 knockout models in other cell types has demonstrated that p53 ablation significantly increases cell migration rates and alters expression of genes associated with invasion, migration, and epithelial-mesenchymal transition (EMT) [71].
Signal Transduction Analysis: iMEFs serve as a platform for studying intracellular signaling pathways in a consistent cellular environment. For example, they have been utilized to characterize the structure and function of the ROR2 cysteine-rich domain in vertebrate noncanonical WNT5A signaling, revealing insights into developmental and disease processes [25].
Multi-Gene Interaction Studies: Emerging CRISPR technologies like CRISPR-Cas12a enable researchers to assess multiple genetic interactions simultaneously in mouse models [73]. This advancement facilitates more complex disease modeling in iMEFs, allowing investigators to study how Tp53 deletion interacts with other genetic alterations in driving disease phenotypes.
Table 1: Quantitative Assessment of iMEF Applications in Disease Modeling
| Application Area | Key Measurable Parameters | Experimental Readouts | Significance in Disease Research |
|---|---|---|---|
| Oncogenic Transformation | Migration rate, Invasion capacity, EMT markers | Increased migration in TP53-KO cells [71], Altered expression of invasion/migration genes [71] | Models early events in cancer metastasis and progression |
| Pathway Analysis | Gene expression changes, Protein localization, Signaling activity | Transcriptomic and proteomic profiling [71], Pathway-specific reporter assays | Elucidates molecular mechanisms in disease development |
| Therapeutic Screening | Drug sensitivity, Gene expression normalization, Cell viability | Response to candidate therapeutics, Rescue of normal gene expression patterns | Identifies potential treatments for genetic disorders |
Experimental Protocols and Workflows
Tp53 Knockout iMEF Generation Protocol
The foundational protocol for generating iMEFs through Tp53 deletion enables researchers to establish their own immortalized cell lines for subsequent gene rescue and disease modeling applications.
CRISPR Construct Delivery: Utilize electroporation with the Neon transfection system to deliver CRISPR constructs targeting the Tp53 gene. The protocol specifically uses Px461-Cas9n-Trp53-sgRNA-alpha and Px461-Cas9n-Trp53-sgRNA-beta plasmids (available from Addgene as #88846 and #88847, respectively), which were generated and deposited by the Massagué lab [5] [74].
Critical Reagent Note: The electroporation solution should be Ingenio Electroporation Solution (Mirus Bio, catalog number: MIR 50114), as specified in the correction to the original protocol [74].
Immortalization Timeline: The protocol reliably generates immortalized MEFs within three weeks through efficient Tp53 ablation, significantly faster than traditional 3T3 serial passaging methods which rely on spontaneous mutations [6] [5].
Validation Requirements: Confirm Tp53 knockout through sequencing and functional assays demonstrating loss of p53 activity. Verify that iMEFs maintain resemblance to parent cell populations to ensure physiological relevance [6].
Gene Rescue Experimental Workflow
The following detailed protocol enables researchers to perform gene rescue experiments in iMEFs to characterize gene/protein function.
Clone Expansion: Isolate and expand individual iMEF clones following Tp53 deletion to establish stable, homogeneous cell lines for subsequent genetic manipulation [6].
Rescue Construct Design: Create expression vectors containing the gene of interest (wild-type or mutant variants). For precise gene editing approaches, consider newer CRISPR tools like Cas12a which enables more seamless gene editing and assessment of multiple genetic interactions [73].
Stable Line Generation: Introduce rescue constructs into iMEFs using appropriate transfection methods followed by selection with antibiotics or fluorescence-activated cell sorting (FACS). The original iMEF generation protocol uses electroporation, but other methods such as lipid-based transfection may be suitable for this step depending on the construct size and cell viability requirements [5].
Functional Characterization: Assess the functional consequences of gene rescue through transcriptomic analysis, proteomic profiling, migration assays, and other phenotype-specific tests. Use DESeq2 for differential gene expression analysis and ClusterProfiler for geneset enrichment analysis as described in similar TP53 knockout studies [71].
Diagram 1: Experimental workflow for generating and utilizing Tp53 knockout iMEFs in gene rescue and disease modeling studies, highlighting key steps from primary cell isolation to functional characterization.
Signaling Pathway Analysis in iMEFs
Understanding the signaling consequences of Tp53 deletion and subsequent gene rescue requires careful pathway analysis, as p53 interacts with multiple critical cellular signaling networks.
Transcriptomic Profiling: Conduct RNA sequencing to identify differentially expressed genes between experimental conditions. Use high-throughput paired-end sequencing (e.g., Illumina NovaSeq 6000) with appropriate replication (n=3 recommended) to ensure statistical power [71].
Proteomic Approaches: Perform label-free quantitative proteomics using LC-MS/MS analysis to complement transcriptomic data. Process raw proteomic data with MaxQuant software and deposit in the ProteomeXchange Consortium via the PRIDE partner repository for data validation and sharing [71].
Bioinformatic Integration: Utilize biological knowledge bases including Wikipathways and PROGENy for pathway analysis, and Gene Ontology (GO) for functional enrichment of proteomic results. Estimate EMT scores using gene expression signatures and two-sample Kolmogorov-Smirnov tests [71].
Validation Methods: Confirm key findings using RT-qPCR with the ΔΔCt method (using GAPDH and ACTB as reference genes) and functional assays such as migration rate assessments to connect molecular changes to phenotypic outcomes [71].
Diagram 2: Signaling pathways affected by Tp53 deletion in iMEFs and potential rescue points, showing the molecular consequences of p53 loss and opportunities for therapeutic intervention through gene rescue approaches.
Research Reagent Solutions
Table 2: Essential Research Reagents for iMEF Generation and Applications
| Reagent Category | Specific Product/Model | Application Purpose | Technical Notes |
|---|---|---|---|
| CRISPR Plasmids | Px461-Cas9n-Trp53-sgRNA-alpha (Addgene #88846) [5] | Tp53 gene targeting | Used with Cas9n D10A mutant nickase for enhanced specificity |
| Px461-Cas9n-Trp53-sgRNA-beta (Addgene #88847) [5] | Tp53 gene targeting | Second sgRNA for improved knockout efficiency | |
| Electroporation System | Neon Transfection System 10 μL kit (Thermo Fisher) [5] | CRISPR construct delivery | Optimal for primary MEF transfection |
| Electroporation Solution | Ingenio Electroporation Solution (Mirus Bio, MIR 50114) [74] | Electroporation buffer | Critical correction from original protocol [74] |
| Cell Culture Media | DMEM high glucose + 10% FBS + 2mM L-glutamine [5] | iMEF culture | Supplement with NEAA and sodium pyruvate for fresh MEFs |
| Validation Tools | DESeq2 package [71] | Differential gene expression | For transcriptomic analysis validation |
| ClusterProfiler [71] | Geneset enrichment analysis | Pathway analysis of omics data | |
| MaxQuant software [71] | Proteomic data processing | Label-free quantitation of proteomic changes |
iMEFs generated through CRISPR-mediated Tp53 deletion provide a robust and physiologically relevant platform for gene rescue studies and disease modeling. This application note demonstrates how this innovative approach addresses critical limitations of primary MEFs while enabling sophisticated experimental designs for characterizing gene function and disease mechanisms. The detailed protocols and analytical frameworks presented here offer researchers comprehensive methodologies to implement this powerful technology in their investigation of disease-associated genetic variants and potential therapeutic interventions.
Conclusion
The CRISPR-Cas9-mediated Tp53 knockout protocol represents a significant advancement over traditional MEF immortalization techniques, offering a rapid, efficient, and controlled method. By directly targeting a key senescence pathway, this approach generates immortalized cell lines that retain physiological properties crucial for reliable research. The streamlined timeline of under three weeks and the ability to work with small cell numbers make this protocol highly accessible. Future directions include adapting this strategy for immortalizing other primary murine cell types and integrating it with complex disease modeling. For biomedical research, this robust tool accelerates the study of gene function, signaling pathways, and the characterization of disease variants, solidifying its role as a cornerstone technique in basic science and pre-clinical drug development.
References
- 1. A comparison of strategies for immortalizing mouse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6706133/]
- 2. Haplo-insufficiency of both BubR1 and SGO1 accelerates cellular... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-016-0238-5]
- 3. IL-23R is a senescence-linked circulating and tissue ... [https://www.nature.com/articles/s43587-024-00752-7]
- 4. Spontaneous and photosensitization-induced mutations in primary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7380187/]
- 5. An Efficient Method for Immortalizing Mouse Embryonic ... [https://bio-protocol.org/en/bpdetail?id=5159&type=0]
- 6. An Efficient Method for Immortalizing Mouse Embryonic ... [https://pubmed.ncbi.nlm.nih.gov/39872719/]
- 7. An efficient method for immortalizing mouse embryonic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11230376/]
- 8. Review of hTERT-Immortalized Cells: How to Assess ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11641324/]
- 9. Cell Immortality: In Vitro Effective Techniques to Achieve and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10971253/]
- 10. Cell Immortalization - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/cell-immortalization]
- 11. Establishment of TP -knockout canine cells using optimized... 53 [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-018-0491-5]
- 12. Targeting p53 pathways: mechanisms, structures ... - Nature [https://www.nature.com/articles/s41392-023-01347-1]
- 13. Tumor-Suppressor Functions of the TP53 Pathway - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4852799/]
- 14. An efficient method for immortalizing mouse embryonic... | CoLab [https://colab.ws/articles/10.1101%2F2024.06.27.601088]
- 15. Of the many cellular responses activated by TP53, which ... [https://pubmed.ncbi.nlm.nih.gov/35396345/]
- 16. Mutant p53 in cancer: from molecular mechanism ... - Nature [https://www.nature.com/articles/s41419-022-05408-1]
- 17. Senescent cells limit p53 activity via multiple mechanisms ... [https://www.nature.com/articles/s41467-022-31239-x]
- 18. Links Between Mutant p53 and Genomic Instability - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4407809/]
- 19. Mutant p53 gains oncogenic functions through a ... [https://www.nature.com/articles/s41467-023-44239-2]
- 20. p53 loss of function: implications for the processes ... [https://pubmed.ncbi.nlm.nih.gov/1365909/]
- 21. p53 alteration is a common event in the ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/1752433/]
- 22. High-Throughput CRISPR/Cas9 Mutagenesis Streamlines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7203946/]
- 23. CRISPR/Cas9 therapeutics: progress and prospects | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-023-01309-7]
- 24. CRISPR-Cas9 Technology: Mechanism & Gene Editing Solutions | Danaher Life Sciences [https://lifesciences.danaher.com/us/en/library/crispr-cas9.html]
- 25. An efficient method for immortalizing mouse embryonic ... [https://pubmed.ncbi.nlm.nih.gov/38979230/]
- 26. A systematic genome-wide mapping of oncogenic mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8586238/]
- 27. An Efficient Method for Immortalizing Mouse Embryonic ... [https://bio-protocol.org/pdf/Bio-protocol5159.pdf]
- 28. Reprogramming Mouse Embryonic Fibroblasts with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5226406/]
- 29. Primary Mouse Embryonic Fibroblasts or Feeders | PMIT [https://ki-sbc.mit.edu/preclinical-modeling/methods/primary-MEFs]
- 30. Optimizing the delivery of CRISPR/Cas9 ... [https://pubmed.ncbi.nlm.nih.gov/40780629/]
- 31. Optimizing the delivery of CRISPR/Cas9 ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925005049]
- 32. A versatile bulk electrotransfection protocol for murine embryonic fibroblasts and iPS cells | Scientific Reports [https://www.nature.com/articles/s41598-020-70258-w]
- 33. CRISPR/Cas9 treatment causes extended TP53 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7498335/]
- 34. Cas9 activates the p53 pathway and selects for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7343612/]
- 35. Immortalization of MEF is characterized by the deregulation ... [https://www.aging-us.com/article/100353/text]
- 36. Essential role for centromeric factors following p53 loss and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5393061/]
- 37. Immortalization of primary marmoset skin fibroblasts by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9809370/]
- 38. Genome-wide CRISPR Screen to Identify Genes that ... [https://www.nature.com/articles/s41598-019-53572-w]
- 39. Cell Freezing Protocols | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/freezing-cells.html]
- 40. Protocol for Generation of Single-Gene Knockout in Hard- ... [https://pubmed.ncbi.nlm.nih.gov/40655415/]
- 41. CRISPR–Cas9 gRNA efficiency prediction: an overview of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9023298/]
- 42. A Guide to Efficient CRISPR gRNA Design: Principles and ... [https://www.genscript.com/a-guide-to-efficient-crispr-grna-design-principles-and-design-tools.html]
- 43. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 44. An optimized electroporation approach for efficient CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5933690/]
- 45. Measurement and clinical interpretation of CRISPR off- ... [https://www.nature.com/articles/s41588-025-02428-3]
- 46. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 47. Comprehensive benchmarking of genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144422/]
- 48. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 49. How to reduce cytotoxicity during cell transfection [https://westburg.eu/knowledge/cell-culture/cell-transfection-cytotoxicity]
- 50. Toxic Transfections [https://www.mirusbio.com/toxic-transfections/?srsltid=AfmBOoolMAG-LBdhDGalTBfSmPiEucT-G3ie11obj9uPwzhnK2g-vx8Z]
- 51. Reduce Toxicity in Transfection Without Sacrifice [https://www.bocsci.com/resources/how-to-reduce-cytotoxicity-while-maintaining-high-transfection-efficiency.html?srsltid=AfmBOooY1xNmkhIe_7XR2fV7Y1p8rDlj0IlueWK4Q1bGjT65t5RU_wXs]
- 52. Optimizing Cell Transfection Conditions in "Four Steps" [https://www.linkedin.com/pulse/optimizing-cell-transfection-conditions-four-steps-say-goodbye-07mqc]
- 53. Prevent cell death after CRISPR RNA transfection | IDT [https://www.idtdna.com/pages/support/faqs/i-am-seeing-cell-death-after-crispr-rna-transfection-what-can-i-do-to-prevent-this]
- 54. Estimating transfection efficiency in differentiated and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6466792/]
- 55. Generation of immortalized mouse embryonic fibroblasts ... [https://bio-protocol.org/exchange/minidetail?id=82170&type=30]
- 56. Mouse embryonic fibroblast isolation, culture, and ... [https://www.protocols.io/view/mouse-embryonic-fibroblast-isolation-culture-and-m-gzrbbx52p.html]
- 57. Deep CRISPR mutagenesis characterizes the functional ... [https://www.nature.com/articles/s41588-024-02039-4]
- 58. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]
- 59. How to Validate a CRISPR Knockout [https://biognosys.com/how-to-validate-crispr-knockout/]
- 60. Guidelines for Performing CRISPR/Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10610170/]
- 61. Preparation of a phosphotyrosine-protein standard for use in ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0234645]
- 62. Single Cell Detection of the p53 Protein by Mass Cytometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC7764694/]
- 63. Chromosome end protection by RAP1-mediated inhibition ... [https://www.nature.com/articles/s41586-025-08896-1]
- 64. Transcriptome Profiling of Mouse Embryonic Fibroblast ... [https://www.mdpi.com/1422-0067/25/15/8116]
- 65. Brief guide to RNA sequencing analysis for nonexperts in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11091515/]
- 66. A Guide to Basic RNA Sequencing Data Processing and ... [https://en.bio-protocol.org/en/bpdetail?id=5295&type=0]
- 67. An Efficient Method for Immortalizing Mouse Embryonic Fibroblasts by CRISPR-mediated Deletion of the Tp53 Gene - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11769752/]
- 68. Cell Proliferation Assays [https://www.creative-biolabs.com/drug-discovery/therapeutics/cell-proliferation-assessment.htm]
- 69. A three-colour stress biosensor reveals multimodal ... [https://www.nature.com/articles/s41522-023-00424-1]
- 70. Measurements of physiological stress responses in C. elegans [https://pmc.ncbi.nlm.nih.gov/articles/PMC7840273/]
- 71. Exploring the Functions of Mutant p53 through TP53 ... [https://www.mdpi.com/1467-3045/46/2/94]
- 72. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7349023/]
- 73. New CRISPR tool enables more seamless gene editing [https://news.yale.edu/2025/03/20/new-crispr-tool-enables-more-seamless-gene-editing-and-improved-disease-modeling]
- 74. An Efficient Method for Immortalizing Mouse Embryonic ... [https://bio-protocol.org/en/bpdetail?id=5320&type=0]