Fresh vs Frozen Cells for Embryo scRNA-seq: A Comprehensive Guide for Reproductive Research and Clinical Applications
Single-cell RNA sequencing has revolutionized our understanding of cellular heterogeneity in embryonic development and fertility research.
Fresh vs Frozen Cells for Embryo scRNA-seq: A Comprehensive Guide for Reproductive Research and Clinical Applications
Abstract
Single-cell RNA sequencing has revolutionized our understanding of cellular heterogeneity in embryonic development and fertility research. This article provides a comprehensive analysis of the critical decision between using fresh or frozen cells and tissues for embryo-related scRNA-seq studies. We examine the foundational biological principles underlying cryopreservation effects on transcriptomes, methodological considerations for sample preparation and platform selection, troubleshooting strategies for common technical challenges, and validation approaches for data quality assurance. Drawing from recent studies including direct comparisons of fresh and frozen human oocytes, this resource equips researchers and clinicians with evidence-based strategies to optimize experimental design, maximize data quality, and advance both basic reproductive science and clinical applications in assisted reproductive technologies.
Understanding Cryopreservation Effects on Embryonic Cell Transcriptomes
Fundamental Principles of scRNA-seq Technology and Embryo Research Applications
Single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology by enabling unprecedented resolution in analyzing gene expression at the individual cell level. This technology is particularly transformative for human embryo research, where ethical and technical limitations restrict access to precious samples [1]. By capturing transcriptomic profiles of individual cells, scRNA-seq allows researchers to decipher cellular heterogeneity, identify rare cell populations, and reconstruct developmental trajectories during critical stages of embryogenesis [2]. In the specific context of comparing fresh versus cryopreserved embryonic cells, scRNA-seq provides the sensitive, quantitative data necessary to evaluate the transcriptomic impact of cryopreservation protocols, a crucial consideration for fertility preservation and developmental biology research [3].
Fundamental Principles and Protocols of scRNA-seq
Core Technological Principles
The fundamental power of scRNA-seq lies in its ability to resolve cellular heterogeneity by measuring the complete set of RNA transcripts in each individual cell, rather than providing a population average [4]. The core workflow involves isolating single cells, capturing their mRNA, converting RNA to complementary DNA (cDNA), amplifying the cDNA, and preparing sequencing libraries [5]. Two critical innovations ensure accurate digital quantification of transcripts: Cell Barcodes (short nucleotide sequences that tag all mRNAs from the same cell, allowing pooled sequencing of multiple cells) and Unique Molecular Identifiers (UMIs) (random sequences that label individual mRNA molecules, correcting for amplification bias and enabling precise transcript counting) [5].
Different scRNA-seq protocols offer distinct advantages depending on experimental goals, particularly in balancing throughput and transcriptome coverage [4]. The table below summarizes key characteristics of common methods.
Table 1: Comparison of Common scRNA-seq Protocols
| Protocol | Isolation Strategy | Transcript Coverage | UMI | Amplification Method | Key Applications in Embryo Research |
|---|---|---|---|---|---|
| Smart-Seq2 [4] | FACS | Full-length | No | PCR | Detection of low-abundance transcripts; isoform and allele-specific expression in early embryos [1]. |
| Drop-Seq [4] | Droplet-based | 3'-end | Yes | PCR | High-throughput analysis of thousands of cells; identifying diverse cell populations in gastrulae [6]. |
| inDrop [4] | Droplet-based | 3'-end | Yes | IVT | Similar to Drop-Seq; efficient barcode capture for large cell numbers. |
| CEL-Seq2 [4] | FACS | 3'-only | Yes | IVT | Linear amplification reduces bias; suitable for smaller-scale, focused studies. |
| MATQ-Seq [4] | Droplet-based | Full-length | Yes | PCR | High accuracy in quantifying transcripts and detecting variants; useful for characterizing genomic activation. |
Figure 1: A generalized scRNA-seq wet-lab and computational workflow, from sample preparation to biological interpretation.
Essential Computational Workflow
Following sequencing, the "digital life" of mRNA begins with primary analysis, where raw sequencing data (BCL files) are converted to FASTQ files and processed through pipelines like Cell Ranger to generate a cell-feature matrix [5]. This matrix forms the foundation for all subsequent analyses. Secondary analysis includes quality control to remove low-quality cells or data representing multiple cells, normalization, and dimensionality reduction using techniques like Principal Component Analysis, t-SNE, and UMAP to visualize high-dimensional data in 2D or 3D plots [5]. Subsequent clustering groups cells with similar expression profiles, and differential expression analysis identifies genes that vary between conditions or cell types [2] [5].
Application in Human Embryo Research and Model Validation
Charting Human Embryogenesis
scRNA-seq has been instrumental in creating high-resolution transcriptomic maps of human development. A landmark effort integrated six published datasets to build a comprehensive reference of human embryogenesis from the zygote to the gastrula stage, encompassing 3,304 cells [6]. This reference enables precise annotation of cell lineages—such as epiblast, hypoblast, and trophectoderm—and their developmental trajectories, capturing key events like the maternal-to-zygotic transition and lineage specification [6] [1]. The analysis further revealed dynamic activity of critical transcription factors, including DUXA in morula, VENTX in the epiblast, and OVOL2 in the trophectoderm, driving lineage decisions [6].
Benchmarking Stem Cell-Derived Embryo Models
A critical application of scRNA-seq is validating the fidelity of stem cell-derived embryo models, such as blastoids and gastruloids [1]. These models are essential for studying early human development while navigating ethical constraints. By projecting the scRNA-seq profiles of these models onto the integrated human embryo reference, researchers can benchmark their molecular and cellular similarity to real embryos [6]. This process is vital for authenticating models and identifying potential misannotations, ensuring they faithfully represent in vivo development [6]. For example, a recent post-implantation model termed "hematoids" was shown to contain a definitive hematopoietic niche, including SOX17+RUNX1+ hemogenic buds, which was characterized using scRNA-seq [7].
Table 2: Key Lineage Markers Identified via scRNA-seq in Early Human Embryos
| Developmental Stage | Cell Lineage / Type | Key Marker Genes | Functional Role |
|---|---|---|---|
| Morula [6] | - | DUXA | Transcription factor involved in zygotic genome activation. |
| Blastocyst | Inner Cell Mass (ICM) [6] | PRSS3 | Characterizes pluripotent inner cell mass cells. |
| Epiblast (EPI) [6] [1] | POU5F1 (OCT4), NANOG, SOX2 | Pluripotency markers; maintain the progenitor population for the embryo proper. | |
| Primitive Endoderm (Hypoblast) [6] [1] | GATA4, GATA6, SOX17, PDGFRA | Key transcription factors for hypoblast specification. | |
| Trophectoderm (TE) [6] [1] | GATA2, GATA3, CDX2 | Specifies the lineage giving rise to placental tissues. | |
| Gastrula | Primitive Streak (PriS) [6] | TBXT (Brachyury) | Marks the primitive streak and the onset of gastrulation. |
| Amnion [6] | ISL1, GABRP | Characterizes the developing amniotic epithelium. | |
| Definitive Endoderm [6] | SOX17 | Specifies the definitive endoderm lineage. | |
| Extraembryonic Mesoderm [6] | LUM, POSTN | Identifies mesoderm derived from extraembryonic tissues. |
Figure 2: A simplified lineage tree of early human development, showing key stages and marker genes identified through scRNA-seq studies.
Application Note: scRNA-seq for Evaluating Cryopreservation Impact
Experimental Protocol for Fresh vs. Frozen Comparison
A pivotal study demonstrates the application of scRNA-seq to directly compare the transcriptomes of oocytes from fresh and slow-frozen/thawed human ovarian cortex [3]. The methodology provides a template for similar investigations on embryonic cells.
1. Sample Preparation and Cryopreservation:
- Human ovarian cortex from three donors was cut into small squares.
- Test samples were slow-frozen and thawed, while control samples were processed fresh [3].
2. Single-Cell Isolation:
- A novel method employing a tissue chopper and enzymatic digestion was used to isolate live oocytes from primordial and primary follicles.
- Oocytes were mechanically denuded under a dissection microscope and individually placed into lysis buffer [3].
3. Library Preparation and Sequencing:
- Single oocytes were processed using the seqWell PlexWell rapid single-cell RNA protocol.
- Pooled libraries were sequenced (150-bp paired-end) on an Illumina NovaSeq6000 platform.
- In total, 144 oocytes were sequenced (24 fresh and 24 frozen/thawed from each of three donors) [3].
4. Data Analysis:
- Standard scRNA-seq analysis pipelines were used for quality control, normalization, and dimensionality reduction.
- Differential expression analysis was conducted to compare fresh and frozen/thawed groups.
- Gene ontology (GO) term enrichment analysis was performed on differentially expressed genes to identify affected biological processes [3].
Key Findings and Biological Interpretation
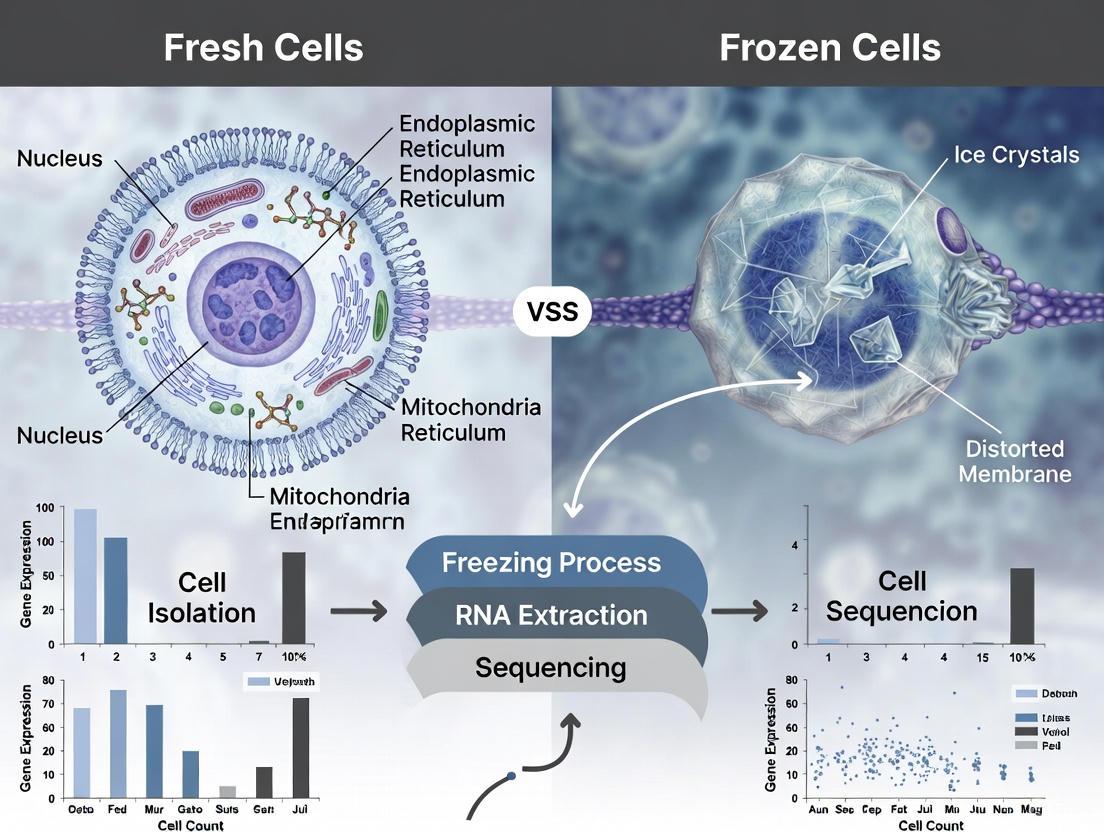
The scRNA-seq analysis revealed that the global transcriptional profiles of fresh and frozen/thawed oocytes did not form separate clusters, indicating that cryopreservation did not induce major, detectable shifts in the transcriptome relative to the inherent heterogeneity between donors [3]. However, at the group mean level, a small but consistent shift was observed. GO analysis indicated that fresh oocytes were enriched for terms related to chromosome segregation and mitosis, whereas frozen/thawed oocytes were enriched for terms linked to wound response, cAMP signaling, and extracellular matrix organization [3]. This suggests that cryopreservation may transiently stress cells, activating repair and signaling pathways, while potentially briefly dampening core cell cycle processes. This study underscores the sensitivity of scRNA-seq in detecting subtle, protocol-induced transcriptomic changes that might be missed by other methods.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for scRNA-seq in Embryo Research
| Item / Reagent | Function / Application | Example Use Case |
|---|---|---|
| seqWell PlexWell [3] | A library preparation kit for rapid single-cell RNA sequencing. | Used for building libraries from individual fresh and frozen oocytes [3]. |
| 10x Genomics Chromium [5] | A droplet-based system for high-throughput single-cell partitioning and barcoding. | Suitable for generating large-scale cell-feature matrices from complex embryo model tissues. |
| Smart-Seq2 Reagents [4] | A full-length transcript protocol for high-sensitivity analysis of individual cells. | Ideal for sequencing preimplantation embryos to detect low-abundance transcripts and splice variants [1]. |
| Reference Transcriptome (e.g., GRCh38) [6] [5] | A standardized genome FASTA and annotation GTF file for read alignment. | Essential for mapping sequencing reads and ensuring consistent analysis across studies, as done in the integrated human embryo reference [6]. |
| Cell Ranger Pipeline [5] | A software package for demultiplexing, alignment, filtering, and UMI counting. | The primary analysis workflow to generate cell-feature matrices from raw FASTQ files. |
| Seurat R Package [2] [8] | A comprehensive toolkit for the secondary and tertiary analysis of scRNA-seq data. | Used for quality control, integration, clustering, and differential expression analysis in comparative studies [2]. |
Comparative Analysis of Cryopreservation Impact on Cellular Integrity and RNA Quality
Cryopreservation serves as a cornerstone technique in biomedical research, enabling the long-term storage of cells and tissues for a wide range of applications, including single-cell RNA sequencing (scRNA-seq) in embryo research. The central challenge lies in balancing the need for long-term sample preservation with the maintenance of cellular integrity and molecular fidelity. For sensitive applications like scRNA-seq, where the full transcriptome of individual cells is analyzed, even minor cryopreservation-induced alterations can compromise data quality and biological interpretations. This application note provides a comparative analysis of cryopreservation effects across different biological systems and presents optimized protocols to maximize cellular and RNA integrity for downstream single-cell applications.
The impact of cryopreservation varies significantly across cell types, preservation methods, and assessment parameters. The tables below summarize key quantitative findings from recent studies.
Table 1: Impact of Cryopreservation on RNA Integrity Across Sample Types
| Sample Type | Preservation Method | Key Condition | RNA Integrity Number (RIN) | Reference |
|---|---|---|---|---|
| Frozen EDTA Blood | Traditional Thawing | No stabilizer | < 5 | [9] |
| Frozen EDTA Blood | EmN Protocol | Nucleospin lysis buffer during thawing | 8.0 ± 0.21 | [9] |
| Rabbit Kidney Tissue | RNALater, thawed on ice | Small aliquots (≤ 30 mg) | ≥ 8 | [10] |
| Rabbit Kidney Tissue | No preservative, RT thaw | Large aliquots (250-300 mg) | 5.25 ± 0.24 | [10] |
| Rabbit Kidney Tissue | RNALater, -20°C thaw | Large aliquots (250-300 mg) | 7.13 ± 0.69 | [10] |
Table 2: Impact of Cryopreservation on Cellular Integrity and Transcriptome
| Cell/Tissue Type | Cryopreservation Method | Storage Duration | Key Finding | Impact Level |
|---|---|---|---|---|
| Human Oocytes | Slow-freezing | N/A | No separate clustering of transcriptional profiles from fresh oocytes | Minimal [3] |
| Human PBMCs | Optimized Controlled-Rate | 12 months | No substantial transcriptome perturbation; reduced cell capture efficiency (~32%) | Low [11] |
| General Cell Types | Suboptimal Protocol | N/A | Viability loss, altered biological function | High [12] |
Detailed Experimental Protocols
Protocol 1: High-Quality RNA Extraction from Frozen EDTA Blood
This protocol, adapted from Scientific Reports, enables the extraction of high-quality RNA (RIN ≥ 7) from frozen blood stored in conventional EDTA tubes, which was previously considered challenging [9].
Applications: Transcriptomic profiling, RNA sequencing, biomarker discovery from legacy blood samples.
Reagents and Materials:
- Nucleospin Blood RNA Kit (Macherey-Nagel)
- Frozen whole blood collected in EDTA tubes
- Centrifuge
- RNase-free microcentrifuge tubes
Procedure:
- Pre-thaw Lysis: Prior to thawing frozen EDTA blood, add the recommended volume of Nucleospin lysis buffer from the blood RNA kit directly to the frozen blood sample.
- Thawing: Allow the sample to thaw completely at room temperature in the presence of the lysis buffer. This critical step stabilizes RNA immediately upon thawing.
- Homogenize: Mix the lysate thoroughly by vortexing or pipetting to ensure complete lysis.
- RNA Extraction: Continue with the standard RNA purification procedure as described in the Nucleospin Blood RNA Kit protocol, including DNase digestion steps.
- Quality Control: Assess RNA concentration and purity using spectrophotometry (e.g., Nanodrop) and determine RNA Integrity Number (RIN) using a Bioanalyzer or TapeStation.
Validation: This protocol yields RNA with an average RIN of 8.0 ± 0.21 and provides a 5-fold higher RNA yield compared to the PAXgene PreAnalytix method, with nearly identical gene expression profiles for tested genes (18S, ACTB, MCP1, TNFa, TXNIP) [9].
Protocol 2: Optimized Thawing of Cryopreserved Tissues for RNA Preservation
This protocol addresses the challenge of RNA degradation during the thawing of cryopreserved tissues, which is a critical issue for biobanked samples [10].
Applications: RNA extraction from cryopreserved tissue aliquots, biobanking quality control.
Reagents and Materials:
- RNALater Stabilization Solution (e.g., Beyotime Biotechnology)
- TRIzol Reagent (Thermo Fisher Scientific)
- RL Lysis Buffer (Magen Biotechnology)
- Cryogenic mortar and pestle
- Liquid nitrogen
Procedure:
- Cryogenic Pulverization: For large tissue samples, use a liquid nitrogen-pre-cooled mortar and pestle to gently smash the frozen tissue into small aliquots (10-30 mg) under liquid nitrogen [10].
- Preservative Application: Add 750 µL of an appropriate preservative (RNALater, TRIzol, or RL lysis buffer) to sterile 2 mL microcentrifuge tubes before adding tissue aliquots.
- Optimized Thawing:
- Processing: After thawing is complete (confirmed by tissue softening), proceed immediately with RNA extraction using a kit compatible with the chosen preservative.
Validation: Tissues treated with RNALater and thawed on ice showed significantly greater RNA integrity compared to room temperature thawing (p < 0.01). RNALater performed best in maintaining high-quality RNA (RIN ≥ 8), particularly for small aliquots (≤ 30 mg) [10].
Protocol 3: Controlled-Rate Freezing of PBMCs for scRNA-seq
This optimized protocol for cryopreserving Peripheral Blood Mononuclear Cells (PBMCs) minimizes transcriptomic alterations, making it suitable for sensitive downstream applications like scRNA-seq [11].
Applications: Immunological research, scRNA-seq of immune cells, long-term biobanking of PBMCs.
Reagents and Materials:
- Recovery Cell Culture Freezing Medium (Gibco, Thermo Fisher Scientific)
- Controlled-rate freezer (CryoMed or equivalent)
- CryoELITE cryogenic vials (Wheaton)
- Liquid nitrogen storage tank
- RP10 medium: RPMI1640 with 10% FBS, 10 mM HEPES, 0.1 mg/mL Gentamycin
Freezing Procedure:
- Cell Preparation: Resuspend PBMCs (100 × 10^6 cells/mL) in Recovery Cell Culture Freezing Medium [11].
- Aliquoting: Dispense 1 mL of cell suspension into cryogenic vials.
- Controlled-Rate Freezing: Use the following optimized freezing cycle in a controlled-rate freezer:
- 1.0°C/min to -4°C
- 25.0°C/min to -40°C
- 10.0°C/min to -12.0°C
- 1.0°C/min to -40°C
- 10.0°C/min to -90°C [11]
- Storage: Transfer frozen vials to liquid nitrogen tank (-161°C) for long-term storage.
Thawing Procedure:
- Rapid Thaw: Remove vials from storage and thaw in a 37°C water bath until a small ice crystal remains.
- Gentle Transfer: Transfer cell suspension to a 15 mL tube containing 10 mL of pre-warmed RP10 medium.
- Washing: Centrifuge at 500 × g for 5 minutes, remove supernatant, and resuspend in fresh RP10 medium. Repeat washing step once.
- Viability Assessment: Determine cell viability using trypan blue exclusion or propidium iodide staining before proceeding to scRNA-seq.
Validation: PBMCs cryopreserved using this method showed minimal transcriptome perturbation after 6 and 12 months of storage, with stable cell viability and population composition across major immune cell types (monocytes, DCs, NK cells, CD4+ T cells, CD8+ T cells, B cells) [11].
Workflow and Decision Diagrams
Diagram 1: Comprehensive workflow for cryopreserving different sample types for scRNA-seq applications, incorporating optimized protocols for each sample type.
Diagram 2: Relationship between cryopreservation parameters and their effects on cellular and molecular integrity, highlighting key factors that influence scRNA-seq outcomes.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Cryopreservation Studies
| Product/Reagent | Primary Function | Application Notes |
|---|---|---|
| RNALater Stabilization Solution | RNA stabilizer that permeates tissues to inhibit RNases | Ideal for tissue aliquots; performs best in maintaining RIN ≥8 when added during thawing [10] |
| Nucleospin Blood RNA Kit | Integrated lysis and RNA purification system | Enables high-quality RNA (RIN ~8) from frozen EDTA blood when lysis buffer is added pre-thaw [9] |
| Recovery Cell Culture Freezing Medium | Complete, ready-to-use cryopreservation medium | Optimized for PBMCs; maintains viability and transcriptome profile during long-term storage [11] |
| Bambanker DMSO-Free | Serum-free, DMSO-free cryomedium | Eliminates DMSO toxicity concerns; ideal for sensitive cells like stem cells and for clinical applications [13] |
| CryoStor CS10 | cGMP-manufactured, serum-free freezing medium | Chemically defined formulation; provides consistent performance for cell therapy and research applications [14] |
| Controlled-Rate Freezer | Programmable freezing apparatus | Enables precise -1°C/min cooling rate; critical for reproducible cryopreservation with minimal ice crystal damage [15] |
The comparative analysis presented in this application note reveals that cryopreservation outcomes are highly dependent on both the sample type and the specific protocols employed. The key finding across studies is that protocol optimization can minimize, and in some cases render undetectable, the impact of cryopreservation on cellular integrity and RNA quality.
For embryo and oocyte research, the slow-freezing method shows remarkable promise, with transcriptomic profiles of frozen-thawed oocytes showing no separate clustering from fresh controls [3]. This indicates that cryopreservation-induced changes were minor compared to inherent biological variability between donors.
For tissue samples, the critical factors are aliquot size and thawing conditions. Small aliquots (≤30 mg) thawed on ice with RNALater maintain excellent RNA integrity (RIN ≥8), while larger aliquots require different thawing strategies [10]. The practice of adding stabilization buffers during thawing rather than after represents a significant methodological advancement for recovering quality RNA from suboptimally stored samples [9].
For PBMCs and cell suspensions, controlled-rate freezing with optimized media maintains cell viability, population composition, and transcriptomic profiles even after 12 months of storage, though reduced scRNA-seq capture efficiency highlights the need for further optimization [11].
The movement toward DMSO-free cryomedium addresses cytotoxicity concerns while maintaining cell viability and functionality [13]. Furthermore, the integration of automation and AI in cryopreservation workflows promises enhanced reproducibility and quality control, particularly as the field moves toward larger-scale applications [12].
In conclusion, while cryopreservation does present challenges for scRNA-seq and other sensitive applications, the protocols and insights presented here demonstrate that through careful optimization of freezing parameters, cryoprotectant choice, and thawing conditions, researchers can reliably preserve cellular integrity and molecular information for high-quality downstream analysis.
Within the context of single-cell RNA sequencing (scRNA-seq) research on embryos, the decision to use fresh or cryopreserved cells is paramount. This application note assesses the impact of cryopreservation on cellular transcriptomes, framing the findings within a broader thesis on embryo research. Cryopreservation is an inevitable process for scaling experiments and synchronizing analyses in multi-center trials, yet its effects on key biological processes must be thoroughly evaluated to ensure data integrity [16] [17]. This document summarizes current data on transcriptomic stability, provides detailed protocols for stability assessment, and highlights critical pathways affected by freezing procedures to guide robust experimental design in embryo scRNA-seq studies.
Quantitative Impact of Cryopreservation on scRNA-seq Metrics
The following tables consolidate quantitative findings from key studies investigating cryopreservation effects on single-cell transcriptomic metrics across various cell types, providing a reference for expected experimental outcomes.
Table 1: scRNA-seq Quality Metrics from Fresh vs. Cryopreserved PBMCs and T Cells
| Sample Type | Condition | Median Genes/Cell | Median UMI/Cell | Mitochondrial % | Key Observations | Source |
|---|---|---|---|---|---|---|
| PBMC Tregs | Fresh | 3,503 | 986 | 2.1% | Baseline metrics | [16] |
| PBMC Tregs | Cryopreserved | 1,435 | 600 | 2.3% | ~32-39% reduction in genes/UMI; specific HSP+ cluster | [16] |
| CSF Cells | Fresh | 1,532 | 4,396 | 2.6% | Baseline metrics | [17] |
| CSF Cells | Cryopreserved | 1,199 | 3,368 | 3.0% | Significant but acceptable reduction | [17] |
| PBMCs | 12-month cryopreservation | Not specified | Not specified | Not specified | ~32% reduction in cell capture efficiency | [11] |
Table 2: Effects of Cryostorage Duration on Embryos and Oocytes
| Cell Type | Storage Duration | Differentially Expressed Genes | Key Findings | Source |
|---|---|---|---|---|
| Human 8-cell embryos | 3 vs. 8 years | 0 mRNAs, 0 lncRNAs | Long-term storage does not affect transcriptomes | [18] |
| Human MII oocytes | 1, 2, 3, 12 months | 0 between storage groups | Vitrification procedure, not storage, affects transcriptomes | [19] |
| Human 8-cell embryos | Fresh vs. vitrified (any duration) | 128 mRNAs, 365 lncRNAs | Vitrification-warming alters metabolism, stress, apoptosis pathways | [18] |
| Human MII oocytes | Fresh vs. vitrified | 1,987 genes | Aberrant genes related to oogenesis and development | [19] |
Experimental Protocols for Transcriptomic Stability Assessment
PBMC Cryopreservation and Thawing Protocol
This protocol, adapted from fundamental immunology studies, provides a standardized approach for peripheral blood mononuclear cell processing [16] [11].
Freezing Medium Preparation:
- Combine 70% FBS, 20% RPMI 1640 media, and 10% DMSO [16]
- Prepare fresh and keep at 4°C until use
Cell Freezing Procedure:
- Isolate PBMCs via Ficoll-Paque density gradient centrifugation
- Resuspend cell pellet in freezing medium at 10×10⁶ cells/mL
- Aliquot 1 mL into cryogenic vials
- Freeze using controlled rate freezing apparatus:
- Cool at 1.0°C/min to -4°C
- Further cool at 25.0°C/min to -40°C
- Cool at 10.0°C/min to -12°C
- Final cooling at 1.0°C/min to -40°C
- Rapid cool at 10.0°C/min to -90°C
- Transfer vials to liquid nitrogen for long-term storage (-161°C to -196°C) [11]
Thawing and Recovery:
- Rapidly thaw cryovial in 37°C water bath until small ice crystal remains
- Transfer cell suspension to 15 mL tube containing 10 mL pre-warmed RP10 medium (RPMI1640 with 10% FBS, 10 mM HEPES, and 0.1 mg/mL Gentamycin)
- Centrifuge at 500 × g for 5 minutes at room temperature
- Gently resuspend pellet in 10 mL fresh RP10 medium
- Repeat washing step twice to ensure complete DMSO removal [11]
scRNA-seq Library Preparation and Quality Control
Cell Processing for scRNA-seq:
- Determine cell viability and concentration using trypan blue exclusion or automated cell counters
- Target cell recovery of >75% viability post-thaw [11]
- For 10X Genomics Chromium platform:
- Load viable cells at appropriate concentration (700-1,200 cells/μL)
- Generate libraries using Chromium Single Cell 3' Library & Gel Bead Kit v2 [16]
- Sequence to depth of approximately 50,000 reads per cell on Illumina platforms
Quality Control Metrics:
- Exclude cells with <100 unique genes or >25% mitochondrial gene percentage [17]
- Monitor proportion of mitochondrial transcripts (should remain <5% in quality samples)
- Calculate cell capture efficiency compared to fresh controls
- Assess sample quality independently of RNA Integrity Number (RIN) when possible, as RIN may not capture freeze-thaw-induced degradation [20]
Data Analysis Pipeline:
- Demultiplex fastq files using cellranger mkfastq (10X Genomics)
- Align reads to appropriate reference genome (GRCh38 for human)
- Generate feature-barcode matrices using cellranger count
- Normalize and log-transform gene expression data
- Perform clustering and dimensionality reduction (UMAP/t-SNE)
- Identify differentially expressed genes using Wilcoxon rank sum test [16]
Key Biological Processes Affected by Cryopreservation
Analysis of multiple studies reveals consistent effects of cryopreservation on specific biological processes across cell types, providing crucial insights for embryo scRNA-seq research.
Stress Response Pathways
The most consistently observed transcriptomic change involves heat shock protein upregulation, forming a conserved stress response signature:
- Heat Shock Proteins: Cryopreserved PBMC Tregs show a specific cluster with significant upregulation of HSPA1A, HSPA1B, HSPA6, HSPB1, HSPE1, HSPH1, and HSP90AA1 [16]
- Oxidative Stress: Vitrified-warmed human embryos exhibit alterations in genes involved in oxidative stress response and ROS elimination [18] [21]
- Transcription Factor Activation: Norway spruce embryogenic tissues show differential expression of TFs within MYB, AP2/ERF, NAC, and WRKY families in response to cryoprotectant-induced osmotic stress [21]
Metabolic and Developmental Pathways
Cryopreservation procedures consistently impact metabolic and developmental processes:
- Metabolic Reprogramming: Vitrified human embryos show significant alterations in metabolic pathways, potentially affecting developmental competence [18]
- Cell Cycle Regulation: Cryoprotectant treatment induces G1 to G2 cell cycle shift in Norway spruce embryogenic tissues [21]
- Developmental Processes: Vitrified human oocytes exhibit differential expression of genes closely related to oogenesis and embryonic development [19]
Apoptosis and Cell Survival Mechanisms
The balance between cell survival and programmed cell death pathways is significantly affected:
- Apoptotic Signaling: Vitrified-warmed human embryos show alterations in apoptosis-related pathways [18]
- Cell Survival Mechanisms: Upregulation of PaMYB11 in Norway spruce embryogenic tissues is necessary for survival under cryoprotectant-induced osmotic stress [21]
- Cell Death Execution: Extensive necrosis detected after suboptimal cryoprotectant pretreatment conditions [21]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Cryopreservation and scRNA-seq Studies
| Reagent/Catalog Number | Function | Application Notes |
|---|---|---|
| DMSO (Cryoprotectant) | Prevents intracellular ice crystal formation | Use at 10% concentration in freezing medium; cell-penetrating [22] |
| Recovery Cell Culture Freezing Medium | Commercial optimized freezing medium | Alternative to lab-prepared DMSO mixtures [11] |
| Lymphocyte Separation Medium | PBMC isolation from whole blood | Density gradient centrifugation for immune cell isolation [16] [11] |
| EasySep Human Treg Isolation Kit | Cell type-specific sorting | Negative selection for untouched Tregs [16] |
| Chromium Single Cell 3' Kit v2 | scRNA-seq library preparation | 10X Genomics platform; high-throughput droplet-based [16] |
| Smart-Seq2 Reagents | Full-length scRNA-seq | Lower throughput but superior transcript coverage [23] |
| Trypan Blue/Live-Dead Stains | Viability assessment | Critical for post-thaw quality control [11] [22] |
| CryoELITE Cryogenic Vials | Sample storage | Wheaton brand specified for optimal preservation [11] |
This transcriptomic stability assessment demonstrates that while cryopreservation inevitably induces specific stress response pathways, optimized protocols can maintain core transcriptomic profiles across diverse cell types. For embryo scRNA-seq research, the vitrification procedure itself rather than storage duration appears to be the primary factor influencing transcriptomic alterations. Key biological processes consistently affected include heat shock response, metabolic pathways, and apoptotic signaling. By implementing the standardized protocols and quality metrics outlined herein, researchers can confidently integrate cryopreserved samples into embryo scRNA-seq studies while accounting for specific, defined transcriptomic changes induced by freezing procedures.
The cryopreservation of ovarian tissue has become a vital fertility preservation strategy, particularly for prepubertal cancer patients and those who cannot delay cancer treatment [3]. While histological assessments have traditionally shown no significant differences in follicle morphology between fresh and slow-frozen/thawed ovarian cortex, clinical outcomes have revealed lower, though not statistically significant, rates of pregnancies and live births from cryopreserved tissue [3]. This discrepancy underscores the critical need to investigate the molecular impacts of cryopreservation beyond structural preservation.
This case study examines the immediate transcriptomic effects of slow-freezing and thawing on early-stage human oocytes through single-cell RNA-sequencing (scRNA-seq) analysis. The research is situated within the broader thesis of fresh versus frozen cells for embryo single-cell research, addressing a significant gap in understanding how cryopreservation affects the most fundamental level of cellular function—gene expression. The findings provide crucial insights for assisted reproductive technologies (ART) and fertility preservation protocols.
Key Findings and Data Analysis
Histological Analysis Reveals Preserved Follicle Integrity
Comprehensive histological analysis using hematoxylin and eosin staining demonstrated that the slow-freezing and thawing process did not adversely affect overall follicle morphology or stage distribution across donors [3]. The quantitative data, summarized in Table 1, confirm the structural preservation of ovarian tissue following cryopreservation.
Table 1: Histological Analysis of Fresh and Frozen-Thawed Ovarian Cortex
| Donor | Condition | Normal Follicles (%) | Follicle Density (per mm³) | Stromal Cell Density |
|---|---|---|---|---|
| Donor 1 (16 years) | Fresh | 86.7 | 279.4 | 0.014 |
| Donor 1 (16 years) | Frozen-Thawed | 91.0 | 235.8 | 0.014 |
| Donor 2 (18 years) | Fresh | 91.7 | 662.2 | 0.014 |
| Donor 2 (18 years) | Frozen-Thawed | 92.5 | 553.5 | 0.016 |
| Donor 3 (27 years) | Fresh | 96.1 | 55.8 | 0.013 |
| Donor 3 (27 years) | Frozen-Thawed | 91.1 | 71.4 | 0.014 |
The percentage of normal follicles remained consistently high across both conditions for all three donors (16, 18, and 27 years old), with no pattern of cryopreservation-induced morphological damage [3]. Although a statistically significant difference in follicle density was observed for Donor 1 (P = 0.017), this pattern was not consistent across all donors, suggesting donor-specific variation rather than a systematic effect of cryopreservation [3].
scRNA-seq Analysis Shows Minimal Transcriptomic Impact
The core transcriptomic analysis compared 144 human oocytes isolated from cadaver ovaries—comprising 24 fresh and 24 frozen-thawed oocytes from each of the three donors [3]. The transcriptional profiles of fresh and frozen-thawed oocytes did not cluster separately, indicating that the differences introduced by the cryopreservation process were undetectable compared to the inherent biological heterogeneity between donors [3].
Despite the overall transcriptional similarity, analysis at the group mean level revealed a small, consistent shift between fresh and frozen-thawed oocytes across all three donors [3]. Differential gene expression analysis identified distinct biological processes enriched in each group, as detailed in Table 2.
Table 2: Transcriptomic Differences Between Fresh and Frozen-Thawed Oocytes
| Oocyte Condition | Enriched Biological Processes | Key Functional Implications |
|---|---|---|
| Fresh Oocytes | Chromosome segregation, Mitosis [3] | Cell division and genomic integrity mechanisms |
| Frozen-Thawed Oocytes | Wound response, cAMP signaling, Extracellular matrix organization [3] | Cellular stress response and signaling adaptation |
The enrichment of wound response pathways in frozen-thawed oocytes suggests activation of cellular repair mechanisms following the freezing and thawing process [3]. The alteration in cAMP signaling—a critical pathway in oocyte maturation and metabolic regulation—indicates potential subtle functional adaptations, while changes in extracellular matrix organization genes may reflect modifications to the oocyte's immediate microenvironment [3].
Experimental Protocol
Ovarian Tissue Processing and Cryopreservation
The experimental workflow, detailed in Figure 1, began with the collection of human ovarian cortex from three healthy premenopausal donors (16, 18, and 27 years old) [3]. The cortex was cut into standardized squares measuring 10 × 10 × 1 mm³ and subjected to either immediate processing or slow-freezing protocols [3].
Figure 1: Experimental Workflow for Oocyte Isolation and Processing
Oocyte Isolation and Single-Cell RNA Sequencing
A novel methodological approach was developed to isolate live oocytes from primordial and primary follicles in both fresh and frozen-thawed human ovarian cortex [3]. The protocol involved:
- Tissue Fragmentation: Ovarian cortex squares were fragmented using a McIlwain tissue chopper for mechanical disruption [3].
- Enzymatic Digestion: Tissue fragments underwent enzymatic digestion to dissociate cellular components [3].
- Oocyte Isolation: Individual oocytes were mechanically isolated under a dissection microscope and manually denuded of surrounding somatic cells [3].
- Single-Cell Processing: Denuded oocytes were placed individually into wells containing lysis buffer for subsequent scRNA-seq analysis [3].
- Library Preparation and Sequencing: Lysed single oocytes underwent library preparation using the seqWell PlexWell rapid single-cell RNA protocol. Pooled libraries were sequenced with 150-bp paired-end sequencing on the NovaSeq6000 Illumina platform [3].
This innovative isolation technique enabled the first transcriptomic comparison between individual fresh and slow-frozen/thawed human oocytes from early-stage follicles [3].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials
| Item | Specification/Model | Application |
|---|---|---|
| McIlwain Tissue Chopper | Standard model | Precise mechanical fragmentation of ovarian cortex [3] |
| seqWell PlexWell Kit | Rapid single-cell RNA protocol | High-throughput scRNA-seq library preparation [3] |
| Sequencing Platform | NovaSeq6000 Illumina | 150-bp paired-end sequencing with high depth [3] |
| Dissection Microscope | Standard stereomicroscope | Visual identification and mechanical denuding of oocytes [3] |
| Enzymatic Digestion Cocktail | Cell-specific composition | Dissociation of ovarian tissue while preserving oocyte viability [3] |
Biological Pathway Analysis
The transcriptomic shifts observed between fresh and frozen-thawed oocytes revealed specific alterations in key biological pathways, as illustrated in Figure 2.
Figure 2: Signaling Pathway Alterations in Frozen-Thawed Oocytes
The pathway analysis demonstrates how cryopreservation stress triggers a coordinated cellular response involving wound healing mechanisms, cAMP signaling modification, and extracellular matrix reorganization [3]. These adaptations appear to facilitate functional compensation, allowing oocytes to maintain overall transcriptomic stability despite the rigors of freezing and thawing.
Discussion and Clinical Implications
This case study provides compelling evidence that slow-freezing cryopreservation of human ovarian cortex preserves the fundamental transcriptomic integrity of early-stage oocytes. The minimal transcriptomic alterations observed immediately post-thawing support the clinical use of this fertility preservation method [3]. However, the subtle but consistent enrichment of stress response pathways in frozen-thawed oocytes warrants consideration.
The clinical context of these findings is particularly relevant given the increasing use of frozen donor oocytes in assisted reproduction. Recent national surveillance data from the United States has shown that while frozen donor oocytes are associated with slightly reduced live birth rates compared to fresh oocytes (46.2% vs. 55.9% for fresh embryo transfers; 41.3% vs. 45.8% for frozen embryo transfers), the rates of term, normal birthweight neonates among singleton live births were comparable between donor oocyte states [24]. This suggests that any molecular perturbations introduced by cryopreservation may not translate to significant adverse perinatal outcomes.
Several considerations must be acknowledged when interpreting these findings. The study focused exclusively on early-stage oocytes and did not investigate transcriptomic changes in the surrounding somatic cells, which play crucial roles in follicle development and oocyte maturation [3]. Additionally, the analysis captured only immediate transcriptomic effects; investigations of longer-term culture or implantation in animal models might reveal additional manifestations of the freeze-thaw process [3].
This comprehensive single-cell analysis demonstrates that the slow-freezing cryopreservation of human ovarian cortex has minimal impact on the transcriptome of early-stage oocytes. The development of a novel method for isolating live denuded oocytes from both fresh and frozen-thawed tissue represents a significant technical advancement in reproductive biology [3]. The findings reinforce the safety and efficacy of ovarian tissue cryopreservation for fertility preservation while highlighting the resilience of oocytes at the molecular level.
Future research directions should include investigation of transcriptomic changes in follicular somatic cells, analysis of later-stage follicles, and longitudinal studies examining molecular recovery after transplantation. Such investigations will further refine cryopreservation protocols and enhance outcomes for patients relying on these fertility preservation strategies.
The transition to single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology, offering unprecedented resolution to decipher cellular heterogeneity during embryogenesis [1] [4]. For researchers studying human embryos, a fundamental practical challenge arises: the necessity to balance the optimal biological fidelity of fresh cells against the logistical practicality of preserved or frozen specimens [1] [3]. This framework examines the trade-offs between preservation-induced artifacts and the experimental flexibility afforded by cryopreservation and other stabilization methods, with a specific focus on embryonic scRNA-seq research.
Quantitative Comparison of Preservation Impacts
The decision between fresh and preserved tissue hinges on understanding the quantifiable impacts of preservation on downstream data quality. The table below summarizes key comparative metrics from relevant studies.
Table 1: Quantitative Impacts of Tissue Preservation on scRNA-seq Data Quality
| Metric | Fresh Tissue (Reference) | Cryopreserved / Stabilized Tissue | Experimental Context |
|---|---|---|---|
| Transcriptional Profile | Reference transcriptome [3] | "No significant differences" in clustering; profiles "did not cluster separately" from fresh [3] | Human oocytes from ovarian cortex [3] |
| Median Genes per Cell | Varies by protocol | ~301 genes/cell (IQR 235-456) [25] | Skeletal muscle in ATR preservative [25] |
| Cell Type Identification | Full expected diversity [25] | Recapitulates 8 major skeletal muscle cell types [25] | Skeletal muscle in ATR preservative [25] |
| Mitochondrial RNA % | Baseline level | 7.87% (IQR 6.49-8.89) [25] | Skeletal muscle in ATR preservative [25] |
| Doublet Rate | Protocol-dependent [5] | 95-99% singlets (simulated detection) [25] | Skeletal muscle in ATR preservative [25] |
| Key Advantages | Highest RNA integrity, no preservation artifacts [26] | Enables multicenter studies, less temperature-sensitive archiving [25] [3] | General experimental design |
Detailed Experimental Protocols
Protocol for snRNA-seq from Chemically Stabilized Tissue
This protocol, adapted for embryonic tissues, is based on a validated workflow for human skeletal muscle archived in Allprotect Tissue Reagent (ATR) [25].
Workflow Overview
Step-by-Step Methodology
Tissue Preservation and Storage
- Immediately following dissection, immerse embryonic tissue fragments in a nucleic acid stabilizing agent such as Allprotect Tissue Reagent.
- Incubate at 4°C for 24-48 hours as per manufacturer's instructions, after which tissue can be archived at -80°C for long-term storage [25].
Tissue Dissociation and Nuclei Preparation
- Wash preserved tissue samples in cold, nuclease-free phosphate-buffered saline (PBS) to remove the preservative.
- Mechanically dissociate tissue using a McIlwain tissue chopper or similar instrument, followed by enzymatic digestion tailored to embryonic tissue composition [25] [3].
- Isolate nuclei via homogenization in a lysis buffer (e.g., NP-40 or Igepal-based) supplemented with RNase inhibitors, followed by sequential filtration through 40μm and 20μm strainers [25].
Quality Control and Enrichment
- Confirm nuclear integrity and membrane presence by staining with antibodies against Nuclear Pore Complex (NPC) proteins.
- Optionally, perform Fluorescence-Activated Cell Sorting (FACS) to enrich for intact, NPC-positive nuclei. Note: While FACS effectively depletes debris, it may significantly reduce final yield [25].
- Validate sample quality by staining with DAPI and imaging with fluorescence microscopy [25].
Library Preparation and Sequencing
- Load the nuclear suspension onto the 10x Genomics Chromium platform per manufacturer's guidelines for 3’ or 5’ gene expression libraries.
- Use standard 10x Genomics protocols for GEM generation, barcoding, and cDNA amplification.
- Sequence libraries on an Illumina platform (e.g., NovaSeq 6000) to a minimum depth of 20,000 paired-end reads per cell [26] [3] [5].
Protocol for scRNA-seq of Cryopreserved Embryonic Cells
This protocol is informed by studies on cryopreserved human oocytes, which show minimal transcriptomic impact from the freeze-thaw process [3].
Workflow Overview
Step-by-Step Methodology
Slow-Freezing Process
- Prepare tissue fragments or dissociated cells in a cryoprotectant solution containing Dimethyl Sulfoxide (DMSO).
- Use a controlled-rate freezer to slowly cool samples to -80°C before transferring to liquid nitrogen for long-term storage [3].
Thawing and Cell Isolation
- Rapidly thaw cryovials in a 37°C water bath.
- For ovarian cortex or similar tissues, fragment thawed tissue squares using a tissue chopper, followed by gentle enzymatic digestion to isolate live target cells (e.g., oocytes) [3].
- Mechanically denude cells under a dissection microscope and transfer individual, viable cells to lysis buffer.
Library Construction and Sequencing
- For low-input samples, use a library preparation method such as the seqWell PlexWell rapid single-cell RNA protocol.
- Perform 150-bp paired-end sequencing on an Illumina NovaSeq 6000 platform [3].
- Focus initial analysis on immediate transcriptomic changes post-thaw; consider longer-term culture experiments to capture delayed effects [3].
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of the above protocols relies on specific reagents and tools. The following table details essential solutions for navigating the fresh versus preserved tissue dilemma.
Table 2: Essential Reagents and Tools for scRNA-seq with Preserved Tissues
| Tool/Reagent | Function | Application Context |
|---|---|---|
| Allprotect Tissue Reagent (ATR) | Chemical stabilizer for DNA/RNA/proteins; allows storage at 4°C to -80°C [25]. | Multicenter studies; field collection; when immediate processing is impossible [25]. |
| Dimethyl Sulfoxide (DMSO) | Cryoprotectant that prevents ice crystal formation during slow-freezing [3]. | Standard cryopreservation of dissociated cells or small tissue fragments [3]. |
| Nuclear Pore Complex (NPC) Antibodies | Cell surface staining for FACS enrichment of intact nuclei [25]. | Quality control and debris removal for single-nucleus RNA-seq (snRNA-seq) [25]. |
| 10x Genomics Chromium | Droplet-based microfluidics platform for high-throughput single-cell capture [25] [5]. | Standardized, scalable scRNA-seq and snRNA-seq library generation. |
| seqWell PlexWell | Low-input library prep technology for single-cells [3]. | Scenarios with limited cell numbers, such as after FACS sorting or from rare samples [3]. |
| Cell Ranger Pipeline | Primary analysis software for demultiplexing, alignment, and count matrix generation [5]. | Standard first-step computational analysis of 10x Genomics data. |
Decision Framework and Analysis Considerations
The choice between preservation methods is not one-size-fits-all but should be guided by the experimental question, logistical constraints, and acceptable levels of technical artifact.
Pathway to Experimental Design
Key Analytical Considerations:
- Data Integration: When combining datasets from fresh and preserved samples, use robust batch correction tools available in platforms like Nygen, BBrowserX, or Seurat to mitigate technical variation [27].
- Quality Control (QC): Apply stringent QC filters based on unique molecular identifier (UMI) counts, genes detected per cell, and mitochondrial RNA percentage. The filtered cell-feature matrix is crucial for reliable downstream analysis [5].
- Biological Interpretation: Be aware that preservation can induce subtle, consistent shifts in gene expression. For example, cryopreserved oocytes show enrichment for pathways like "wound response" and "cAMP signaling," while fresh oocytes show enrichment for "chromosome segregation" [3]. These patterns should be acknowledged in biological interpretations.
Optimized Workflows for Fresh and Frozen Embryo scRNA-seq Experiments
The choice between using fresh or frozen samples is a critical strategic decision in single-cell RNA sequencing (scRNA-seq) experiments, profoundly influencing data quality, cellular composition, and biological interpretation. This dilemma is particularly acute in embryo research, where sample acquisition is often limited and unpredictable. Fresh samples provide the full cytoplasmic RNA content but require immediate processing, posing significant logistical challenges [28] [29]. Frozen samples, including cryopreserved tissues or isolated nuclei, offer scheduling flexibility and enable batch processing across multiple experiments but may incur some RNA loss [28] [4]. This application note establishes a structured decision framework tailored to embryo scRNA-seq research, providing validated protocols and analytical tools to guide researchers in selecting the optimal sample preparation method for their specific experimental requirements and constraints.
Quantitative Comparison: Fresh vs. Frozen Samples
Table 1: Experimental Performance Metrics Between Fresh and Frozen Sample Types
| Performance Parameter | Fresh Samples (scRNA-seq) | Frozen Samples (snRNA-seq) | Research Implications |
|---|---|---|---|
| Transcriptomic Coverage | Full-length transcript coverage possible [4] | 3'-end or 5'-end focused; nominal loss of cytoplasmic RNA [29] | Fresh preferred for isoform analysis; frozen sufficient for cell typing |
| Cell Type Recovery | Protocol-dependent; can recover fragile cell types [28] | Can recover cell types resistant to dissociation [28] [29] | Frozen preserves sensitive cell populations lost during fresh dissociation |
| Viability Requirements | High viability critical (70-90% recommended) [29] | Less dependent on membrane integrity [28] | Frozen more forgiving for challenging tissues |
| Logistical Flexibility | Requires immediate processing (hours) [29] | Enables biobanking and batch processing [28] [29] | Frozen enables multi-institutional studies |
| Technical Variability | Stress responses without immediate processing [29] | Reduced batch effects through synchronized processing [29] | Frozen superior for time-course experiments |
| Transcriptome Fidelity | Potential stress gene induction during processing [29] | Minimal cryopreservation artifacts in transcriptome [3] | Frozen preserves in vivo states more reliably |
Table 2: Applications-Specific Considerations for Embryo Research
| Research Application | Recommended Approach | Justification | Protocol Considerations |
|---|---|---|---|
| Developmental Atlas Construction | snRNA-seq on frozen samples [28] | Enables comprehensive sampling across developmental stages | Combinatorial barcoding for scalability [4] |
| Rare Cell Population Identification | Comparative (both approaches) | Validation across methods strengthens findings [28] | Targeted enrichment protocols may be needed |
| Clinical Fertility Studies | snRNA-seq on frozen ovarian cortex [3] | Direct clinical relevance; enables biobanking | Minimal manipulation to preserve viability |
| Time-Course Experiments | Fixed or frozen samples [29] | Eliminates batch effects; enables synchronized analysis | Fixation preserves temporal snapshots accurately |
| Pilot Studies | Fresh samples when readily available | Rapid turnaround; established protocols | Focus on viability maintenance during dissociation |
Decision Framework Workflow
The following computational workflow formalizes the decision process for selecting between fresh and frozen sample approaches:
Detailed Experimental Protocols
Protocol for Fresh Embryo scRNA-Seq
Principle: Maintain cellular viability and transcriptional fidelity from sample acquisition to library preparation through rapid processing and temperature control [29].
Workflow:
Key Considerations:
- Temperature Control: Maintain samples at 4°C throughout processing to arrest metabolic activity and prevent stress gene induction [29].
- Time Optimization: Complete processing within 3 hours of acquisition to maintain transcriptomic fidelity [29].
- Enzyme Selection: For embryonic tissues, use gentle enzyme cocktails (e.g., Liberase TM at low concentrations) to preserve cell surface receptors [28].
- Quality Metrics: Target cell viability between 70-90% with minimal debris and aggregation (<5%) [29].
Protocol for Frozen Embryo snRNA-Seq
Principle: Preserve nuclear RNA through controlled freezing and thawing cycles, enabling transcriptomic analysis without immediate processing constraints [28] [3].
Workflow:
Key Considerations:
- Freezing Protocol: Employ controlled-rate freezing for embryonic tissues using appropriate cryoprotectants (e.g., DMSO/sucrose solutions) [3].
- Thawing Optimization: Rapid thaw at 37°C with immediate transfer to cold, RNase-free buffers [3].
- Nuclear Integrity: Assess nuclear morphology and RNA integrity (RIN >7.0) before proceeding to library preparation [28].
- Protocol Validation: For embryo research, validate against fresh samples as demonstrated in ovarian cortex studies showing minimal transcriptomic impact [3].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Critical Reagents for Fresh and Frozen Sample Preparation
| Reagent/Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Dissociation Enzymes | Liberase TM, Papain, Collagenase, DNase I [28] | Breakdown extracellular matrix | Tumor-dependent optimization; DNase reduces viscosity [28] |
| Cryopreservation Media | DMSO-based solutions, Sucrose cryoprotectants [3] | Prevent ice crystal formation | Controlled-rate freezing critical for embryonic cells [3] |
| Viability Markers | Trypan blue, Propidium iodide, Calcein AM [29] | Distinguish live/dead cells | Trypan blue exclusion target >70% viability [29] |
| Nuclei Isolation Kits | Commercial nuclei purification kits [28] | Isolate intact nuclei | Density gradient centrifugation removes myelin debris [29] |
| RNA Stabilizers | RNAlater, DNA/RNA Shield | Preserve RNA integrity | Particularly important during prolonged experiments |
| Single-Cell Platforms | 10x Chromium, Parse Biosciences, Seq-Well [4] [3] | Partition individual cells | Choice affects gene detection sensitivity and cell throughput |
Analytical Considerations and Quality Control
Computational Tools for Data Analysis
The selection of analytical tools must align with the sample preparation method. For fresh scRNA-seq data with potential stress gene expression, tools like Seurat offer robust normalization approaches [4]. For frozen snRNA-seq data with potentially sparser gene detection, methods like scVI may be more appropriate [30]. Emerging transformer-based models like scGraphformer show promise for identifying subtle cellular patterns across both sample types by learning cell-cell relationships directly from the data without predefined graphs [30].
Quality Control Metrics
Establish rigorous QC thresholds specific to each approach. For fresh samples, monitor the percentage of mitochondrial reads (indicator of cell stress) and exclude samples exceeding 20% [28]. For frozen nuclei, focus on the number of genes detected per nucleus (aim for >500 genes/nucleus) and the correlation with matched fresh samples when available [28] [3]. For embryo-specific work, include developmental competence markers appropriate to the stage being analyzed.
The decision between fresh and frozen samples represents a fundamental experimental choice that should align with research objectives, logistical constraints, and analytical capabilities. Fresh samples remain the gold standard for full transcriptome characterization when rapid processing is feasible. Frozen approaches offer compelling advantages for clinical applications, biobanking, and experimental designs requiring batch processing. The emerging evidence from embryo research indicates that cryopreservation has minimal impact on transcriptional profiles, supporting the use of frozen samples particularly in fertility and developmental studies [3]. By implementing this structured decision framework and associated protocols, researchers can optimize their experimental designs to maximize biological insights while accommodating practical constraints.
The foundation of any successful single-cell RNA sequencing (scRNA-seq) experiment lies in the quality of the initial single-cell suspension. This process of tissue dissociation and cell preparation directly determines the reliability and biological accuracy of all subsequent data, influencing cell viability, recovery rates, and transcriptional fidelity. Within the specific context of embryo research, where material is often precious and irreplaceable, the choice between using fresh or cryopreserved samples carries significant implications for experimental design and outcomes. This application note details standardized protocols for generating high-quality single-cell suspensions from both fresh and frozen starting materials, providing a framework for robust experimental design in embryonic scRNA-seq studies.
The decision to use fresh or cryopreserved cells is not merely logistical; it involves balancing experimental feasibility with data integrity. Cryopreservation enables complex study designs by disconnecting the time and place of sampling from subsequent processing steps, which is particularly valuable for embryonic studies where developmental time points must be captured precisely but processed consistently [23]. Evidence confirms that with proper techniques, the cryopreservation process does not substantially alter transcriptional profiles, allowing frozen samples to be confidently incorporated into studies profiling freshly processed material [23] [31].
Quantitative Comparison: Fresh vs. Cryopreserved Sample Performance
Systematic evaluations of fresh and cryopreserved samples across multiple cell types provide critical quantitative metrics to guide experimental planning. The following table summarizes key performance characteristics based on empirical studies.
Table 1: Performance Metrics of Fresh vs. Cryopreserved Cells in scRNA-seq
| Performance Metric | Fresh Samples | Cryopreserved Samples | Notes |
|---|---|---|---|
| Cell Viability | High (Baseline) | Variable (Reduction of 20-60%) | Viability decreases with freezing but viable cells maintain transcriptomes [23] [31] |
| Transcripts Detected per Cell | High | Comparable to Fresh | Linear relationship between reads and transcripts conserved [23] |
| Genes Detected per Cell | High | Comparable to Fresh | Equal sequencing depth identifies similar gene numbers [23] |
| Transcriptome Profile Correlation | Reference | High (Pearson Correlation) | Expression profiles highly correlated between conditions [23] |
| Differentially Expressed Genes | Reference | Minimal (Often 1 or None) | No systematic bias introduced [23] |
| Cell Type Composition | Reference | Accurately Preserved | Similar proportions of cellular subtypes identified [23] |
| Experimental Flexibility | Low (Immediate Processing) | High (Batch Processing Possible) | Enables time-course studies and centralized processing [23] [31] |
Comprehensive Workflow for Sample Processing
The journey from tissue to sequencing data involves a series of critical steps that must be optimized for each sample type. The following diagram maps the core decision pathway and experimental workflow for processing both fresh and frozen tissues for single-cell RNA sequencing.
Diagram 1: Sample Processing Workflow
Specialized Protocol: Single-Cell Dissociation from Zebrafish Embryos
For embryonic research, zebrafish embryos represent a powerful model system. The following protocol is optimized for generating high-quality single-cell suspensions from individual zebrafish embryos at 96 and 120 hours post-fertilization (hpf) [32].
Table 2: Research Reagent Solutions for Embryo Dissociation
| Reagent/Buffer | Key Components | Function in Protocol |
|---|---|---|
| Supplemented HBSS | HBSS, 15mM HEPES, 0.5nM CaCl₂ | Base solution maintaining physiological conditions |
| Dissociation Buffer | Suppl. HBSS, 1X TrypLE, 2mg/mL Collagenase/Dispase, 0.04% BSA | Enzymatic breakdown of extracellular matrix and tissues |
| Washing Buffer | DMEM/F-12, 0.04% BSA | Removes enzymes and stops digestion |
| Quenching Buffer | DMEM/F-12, 10% Calf Serum, 0.04% BSA | Neutralizes protease activity |
| Dilution Buffer | HBSS, 0.04% BSA | Diluting cell concentration for counting |
Step-by-Step Procedure:
Sample Collection: Obtain zebrafish larvae from a 28°C incubator. Do not place larvae on ice as chilling reduces dissociation efficiency. Per sample, transfer five larvae into fresh 1.5 mL LowBind SafeLockTubes with cultivation water. Keep tubes at 28°C in a Thermomixer [32].
Buffer Preparation: Prepare all buffers fresh as specified in Table 2. Pre-warm the Thermomixer to 28°C and pre-cool a centrifuge to 4°C. Keep enzymes on ice until ready for use [32].
Initial Processing: Remove cultivation water completely. Add 1 mL of unsupplemented HBSS to wash the larvae. Remove HBSS completely after washing.
Enzymatic Dissociation: Add 200 μL of pre-warmed (28°C) Dissociation Buffer to the larvae. Gently pipet to mix. Incubate at 28°C for 3 minutes in the Thermomixer with shaking at 300 rpm. After each incubation, gently pipet the suspension 10 times with a 1,000 μL wide-bore filter tip to aid mechanical dissociation. Repeat this 3-minute incubation and pipetting cycle five times (total of 15 minutes enzymatic digestion) [32].
Quenching Reaction: Add 200 μL of pre-warmed (28°C) Quenching Buffer to stop the enzymatic reaction. Mix gently by pipetting.
Filtration and Washing: Pass the cell suspension through a 70 μm FlowMi pipette tip cell strainer into a new 1.5 mL tube. Rinse the original tube with 200 μL of Washing Buffer and pass it through the same strainer. Then, pass the filtered suspension through a 40 μm FlowMi pipette tip cell strainer. Centrifuge the filtered suspension at 300 rcf for 5 minutes at 4°C. Carefully remove the supernatant and resuspend the cell pellet in 100 μL of ice-cold Washing Buffer [32].
Quality Control: Assess cell viability and concentration using Trypan Blue staining and a Neubauer counting chamber. The protocol should yield a cell suspension with >80% viability and concentration >400 cells/μL, which is essential for successful loading on microfluidic platforms like the 10X Genomics Chromium [32].
Quality Control and Troubleshooting
Rigorous quality control is essential before proceeding to library preparation and sequencing. The following table addresses common challenges and solutions in the single-cell suspension process.
Table 3: Troubleshooting Guide for Single-Cell Preparation
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Low Cell Viability (<80%) | Over-digestion during enzymatic dissociation; Overly vigorous pipetting | Optimize enzyme concentration and incubation time; Use wide-bore tips and gentle pipetting [32] [28] |
| Low Cell Yield | Incomplete tissue dissociation; Enzyme inactivation | Use fresh enzyme aliquots (do not refreeze); Mechanically dissociate during incubation; Tailor enzyme cocktail to tissue type [32] [28] |
| High Ambient RNA | Excessive cell lysis during processing | Reduce processing time; Maintain high viability; Use computational tools (SoupX, CellBender) for background correction [33] |
| Cell Clumping | Inadequate filtration; DNA release from dead cells | Perform sequential filtration (70μm then 40μm); Include DNase I in dissociation mixtures to reduce viscosity [32] [28] |
| High Mitochondrial RNA | Unhealthy cell states or broken cells | Filter cells with high mtRNA content (e.g., >10% for PBMCs); Note: some cell types (e.g., cardiomyocytes) naturally have high mtRNA [33] |
Quality Control Metrics for scRNA-seq Data
After sequencing, initial data quality assessment is performed using the output from processing pipelines (e.g., Cell Ranger's web_summary.html). Key metrics to evaluate include [33]:
- Cell Recovery: The number of cells recovered should align with expectations (e.g., close to targeted cell numbers for the platform used).
- Sequencing Metrics: A high percentage of "Confidently mapped reads in cells" (e.g., >90%) indicates good library quality.
- Transcript Detection: "Median genes per cell" should be within the expected range for the specific sample type.
- Barcode Rank Plot: Should display a characteristic "cliff-and-knee" shape, indicating good separation between cells and background.
- Mitochondrial Read Percentage: High levels can indicate poor cell quality, though this varies by cell type.
The protocols and data presented herein provide a roadmap for generating high-quality single-cell suspensions from embryonic and other biological samples. The demonstrated feasibility of using cryopreserved material without compromising transcriptional fidelity [23] represents a paradigm shift in experimental design, offering researchers unprecedented flexibility. For embryonic scRNA-seq research, this means critical developmental time points can be captured and preserved for subsequent batched analysis, significantly reducing technical variability and enhancing study robustness.
When designing your experiment, consider that the dissociation protocol must be tailored to the specific tissue characteristics [28], while cryopreservation offers a viable path for biobanking and complex study designs [23]. By adhering to these standardized protocols, implementing rigorous quality control, and understanding the comparative performance of fresh versus frozen samples, researchers can ensure that their single-cell genomics data is built upon a reliable foundation from the very first step.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the profiling of transcriptomes at the individual cell level, revealing cellular heterogeneity that is often masked in bulk RNA-seq analyses [34] [35]. This technology is particularly valuable in embryo research, where understanding the precise molecular programs of individual cells is crucial for deciphering developmental processes, identifying rare cell types, and evaluating the effects of experimental manipulations such as cryopreservation. The choice of scRNA-seq platform significantly impacts data quality and interpretability, making platform selection a critical decision in experimental design.
This guide provides a comprehensive comparison of leading scRNA-seq technologies, with a specific focus on their application to embryo research. We examine the technical foundations, performance characteristics, and practical considerations for 10x Genomics Chromium, SMART-seq2, SMART-seq3xpress, and other emerging methods, providing a framework for selecting the most appropriate technology for investigating fresh versus frozen embryonic cells.
10x Genomics Chromium Platform
The 10x Genomics Chromium system utilizes droplet-based microfluidics to partition thousands of single cells into nanoliter-scale reaction vesicles called GEMs (Gel Beads-in-emulsion) [35]. Within each GEM, a single cell is lysed, and the released polyadenylated mRNA is barcoded with unique molecular identifiers (UMIs) during reverse transcription. This process ensures that all cDNA molecules from the same cell share the same barcode, allowing computational demultiplexing after sequencing. The platform has evolved through several iterations, including the current GEM-X technology, which generates twice as many GEMs at smaller volumes, thereby reducing multiplet rates and increasing throughput capabilities [35]. The Chromium Flex assay extends this technology to accommodate a wider range of sample types, including fixed samples and FFPE tissues, which is relevant for longitudinal embryo studies.
SMART-seq Technologies
SMART-seq2 and its successor SMART-seq3 represent the plate-based approach to scRNA-seq, focusing on full-length transcript coverage. These methods operate in microtiter plates where individual cells are sorted into separate wells [34] [36]. The core technology relies on template-switching during reverse transcription, which enables the synthesis of full-length cDNA with universal primer binding sites at both ends. SMART-seq3xpress is a recently miniaturized and streamlined version that substantially reduces reagent use and increases cellular throughput while maintaining full-transcript coverage [36]. This advancement addresses one of the major limitations of plate-based methods—throughput capacity—while preserving their key advantage: the ability to detect isoform-level variation.
Alternative scRNA-seq Platforms
Several other platforms offer unique capabilities for specific research scenarios. The Fluidigm C1 system uses integrated fluidic circuits (IFCs) for automated cell capture and processing, providing high read depth per cell but with lower throughput (typically 100-800 cells per run) [37]. The Bio-Rad ddSEQ and Wafergen ICELL8 systems represent intermediate options, with ddSEQ employing a droplet-based approach similar to 10x Genomics but with lower throughput, and ICELL8 utilizing a nanowell-based system with imaging confirmation for precise single-cell capture [37]. More recent additions to the field include HIVE, VASA-seq, and FLASH-seq, which have demonstrated competitive performance in benchmark studies [38].
Technical Comparison and Performance Benchmarking
Key Performance Metrics Across Platforms
Table 1: Comprehensive Performance Comparison of Major scRNA-seq Platforms
| Platform | Throughput (Cells) | Sensitivity (Genes/Cell) | Transcript Coverage | Multiplet Rate | Cost per Cell | strengths | Limitations |
|---|---|---|---|---|---|---|---|
| 10x Genomics 3' v3 | 1,000-80,000 [37] | ~4,800 genes [39] | 3' counting only | ~5% [39] | $$ [37] | High throughput, efficient cell capture | Limited to 3' end sequencing |
| 10x Genomics 5' v1 | 1,000-80,000 [37] | ~4,500 genes [39] | 5' counting only | ~5% [39] | $$ [37] | Compatible with immune receptor profiling | Higher dropout for low-expression genes |
| SMART-seq2 | 96-384 per plate [34] | ~6,000 genes [34] | Full-length | Minimal | $$$$ | High sensitivity, isoform detection | Low throughput, high mitochondrial reads |
| SMART-seq3xpress | Up to 26,000 [36] | Higher than SMART-seq2/SMART-seq3 [36] | Full-length | Minimal | $$$ | Full-length with improved throughput | Requires specialized equipment |
| ddSEQ | 1,000-10,000 [37] | ~3,600 genes [39] | 3' counting only | ~5% [39] | $$$ [37] | User-friendly, accessible workflow | Lower gene detection sensitivity |
| ICELL8 | 500-1,800 [37] | Varies by protocol | 3' counting | Low (imaging-confirmed) | $$$$ [37] | Precise cell capture, flexible cell types | Lower correlation with bulk sequencing |
Biological Implications for Embryo Research
The technical differences between platforms have direct consequences for data interpretation in embryo research. SMART-seq2 detects significantly more genes per cell, particularly low-abundance transcripts and alternatively spliced isoforms, making it suitable for detecting subtle transcriptional differences between embryonic cell states [34]. However, it captures a higher proportion of mitochondrial genes (averaging ~30%), which can complicate data analysis unless properly accounted for [34]. In contrast, 10x Genomics data exhibits more severe dropout effects, especially for genes with lower expression levels, but can profile thousands of cells, enabling the detection of rare cell types within heterogeneous embryonic populations [34].
Non-coding RNA profiles also differ between platforms. Approximately 10%-30% of all detected transcripts from both platforms derive from non-coding genes, with long non-coding RNAs (lncRNAs) accounting for a higher proportion in 10x data (6.5%-9.6%) compared to SMART-seq2 (2.9%-3.8%) [34]. This distinction may be relevant for studying epigenetic regulation in early development.
Application to Fresh vs. Frozen Embryo scRNA-seq Research
Experimental Considerations for Cell State Preservation
The critical decision between using fresh or frozen cells in embryo research involves balancing practical logistics with biological preservation. A 2025 study comparing early-stage oocytes from fresh and slow-frozen/thawed human ovarian cortex demonstrated that cryopreservation had minimal impact on the oocyte transcriptome when analyzed using scRNA-seq [3]. The transcriptional profiles of fresh and frozen/thawed oocytes did not cluster separately, indicating that freeze-thaw effects were minor compared to inherent donor heterogeneity [3]. However, at the group mean level, small but consistent shifts were observed: fresh oocytes were enriched for gene ontology terms related to chromosome segregation and mitosis, whereas frozen/thawed oocytes showed enrichment for terms related to wound response, cAMP signaling, and extracellular matrix organization [3].
These findings suggest that platform selection should align with the specific research questions regarding cryopreservation effects. For detecting subtle, consistent transcriptomic shifts across cell populations, SMART-seq technologies offer higher sensitivity per cell. For capturing the full spectrum of cellular heterogeneity in precious clinical samples, 10x Genomics provides the necessary throughput.
Protocol Optimization for Embryonic Cells
Cell preparation is particularly critical when working with embryonic materials. The 10x Genomics platform requires a suspension of viable single cells or nuclei as input, with minimal presence of cellular aggregates, dead cells, and non-cellular nucleic acids [40]. Similar requirements apply to SMART-seq protocols, though the plate-based nature allows for visual confirmation of cell integrity before processing. For frozen samples, the Chromium Flex assay offers compatibility with fixed samples, providing additional flexibility for embryo research logistics [35].
Table 2: Essential Research Reagent Solutions for scRNA-seq in Embryo Research
| Reagent/Category | Function | Platform Compatibility | Considerations for Embryo Research |
|---|---|---|---|
| Cell Suspension Buffer | Maintains cell viability and prevents aggregation | Universal | Optimal osmolarity for embryonic cells is critical |
| Lysis Buffer | Releases RNA while maintaining integrity | Platform-specific | Gentle lysis preserves RNA quality from delicate embryonic cells |
| Reverse Transcriptase | Synthesizes cDNA from RNA templates | Platform-specific | High efficiency crucial for low-input embryonic samples |
| Template-Switching Oligo | Enables full-length cDNA synthesis | SMART-seq series | Critical for detection of full-length transcripts |
| UMI Barcoded Beads | Labels mRNA with cell and molecule barcodes | 10x Genomics | Enables accurate molecular counting |
| Tagmentation Enzyme | Fragments and tags cDNA for sequencing | SMART-seq3, 10x (in library prep) | Optimization needed for different cDNA qualities |
| PCR Amplification Reagents | Amplifies cDNA for sufficient sequencing material | Universal | Limited cycles prevent bias in rare embryonic samples |
Experimental Protocols for Platform Evaluation
Standardized Cell Line Benchmarking Protocol
To systematically compare platform performance, we recommend a standardized benchmarking approach using well-characterized cell lines:
- Cell Culture and Preparation: Grow K562 human myeloma cells and mouse embryonic stem cells (mESCs) to mid-log phase [38]. Count cells with an automated counter and assess viability (>90% required).
- Cell Mixture Preparation: Create a 1:1 mixture of K562 and mESCs in PBS at a concentration of 1 million cells/mL [38]. Include species-mixing controls to assess multiplet rates.
- Parallel Processing: Split the cell mixture and process identical aliquots through each platform being evaluated (10x, SMART-seq, etc.) following manufacturer protocols.
- Library Preparation and Sequencing: Prepare libraries according to platform-specific methods. Sequence all libraries to a standardized depth (e.g., 50,000 reads per cell) to enable fair comparisons [39].
- Data Analysis: Process data through uniform bioinformatic pipelines. Key metrics include: genes detected per cell, UMI counts per cell, mitochondrial read percentage, doublet rate, and concordance with bulk RNA-seq signatures.
Embryo-Specific Validation Protocol
For embryo-focused applications, we recommend this additional validation:
- Sample Preparation: Split pooled embryonic cells into fresh and frozen aliquots. For frozen aliquots, employ slow-freezing protocols similar to those used in clinical settings [3].
- Cell Isolation: For ovarian cortex studies, fragment tissue with a tissue chopper and enzymatically digest to isolate live oocytes from primordial and primary follicles [3]. Mechanically denude oocytes under a dissection microscope.
- Platform Comparison: Process matched fresh and frozen samples across platforms. Include additional validation methods such as histological analysis of follicle morphology, density, and stage distribution [3].
- Data Analysis Focus: Specifically assess detection of developmental marker genes, cell cycle regulators, and stress response genes that may be affected by cryopreservation.
Platform Selection Guidelines
Selecting the appropriate scRNA-seq platform requires careful consideration of research priorities:
Choose 10x Genomics Chromium when: Your research question requires characterization of cellular heterogeneity in complex embryonic tissues, identification of rare cell types, or analysis of thousands to tens of thousands of cells. This platform is ideal for constructing comprehensive cellular atlases of developing embryos.
Choose SMART-seq2/SMART-seq3 when: Your research focuses on detecting subtle transcriptional differences, alternative splicing variants, or low-abundance transcripts in limited cell numbers. This is particularly relevant for comparing fine-grained cellular states in early development or evaluating the effects of cryopreservation on full transcriptomes.
Choose SMART-seq3xpress when: You need full-length transcript information at higher throughput, balancing the sensitivity of plate-based methods with improved scalability. This emerging technology is excellent for medium-scale studies requiring isoform-level resolution.
Consider alternative platforms (ICELL8, ddSEQ) when: Working with specialized sample types that benefit from imaging-based cell selection or when integrating with existing laboratory workflows.
Emerging Trends and Future Directions
The field of single-cell genomics continues to evolve rapidly. Recent advancements include the development of methods that preserve cellular spatial information, multi-omic approaches that simultaneously profile transcriptomes and epigenomes, and improved compatibility with fixed and frozen samples. The ongoing innovation in both droplet-based and plate-based technologies promises even greater sensitivity, throughput, and accessibility in the near future. For embryo research specifically, these advancements will enable more detailed investigations into developmental processes and more rigorous evaluations of assisted reproductive technologies.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the investigation of transcriptomic profiles at the cellular level. When designing scRNA-seq experiments comparing fresh and frozen cells for embryo research, careful consideration of replication, sample size, and power analysis is paramount to generate statistically robust and biologically relevant conclusions. The inherent technical noise and biological variability in scRNA-seq data necessitate rigorous experimental design to adequately detect meaningful differences between experimental conditions.
This application note provides a comprehensive framework for designing scRNA-seq experiments in the context of fresh versus frozen embryo research, incorporating practical protocols, quantitative benchmarks, and analytical tools to optimize research outcomes. Proper experimental design ensures that studies have sufficient sensitivity to detect true biological effects while minimizing false discoveries, thereby maximizing the return on substantial investments required for scRNA-seq workflows.
Key Considerations for Replication and Sample Size
Replication Strategies
Biological versus Technical Replication
- Biological replicates: Cells collected from different biological sources (different embryos, different donors) are essential for drawing conclusions that generalize across the population. A study comparing fresh and frozen oocytes utilized ovarian cortex from three different donors (ages 16, 18, and 27), enabling assessment of biological variability [3].
- Technical replicates: Multiple measurements of the same biological sample help quantify technical noise but cannot replace biological replicates for inferring population-level effects.
Sample Size in Practice The appropriate number of replicates depends on the expected effect size, technical variability, and biological heterogeneity. In a comparative analysis of oocyte cryopreservation, 24 oocytes per condition (fresh and frozen) from each of three donors were sequenced (total n=144 oocytes), providing sufficient data to address within-donor and between-condition variability [3]. For studies with continuous cell populations or trajectory inference, increased cell numbers may be necessary to adequately capture the continuum of states.
Quantitative Benchmarks from Published Studies
Table 1: Sample Size and Replication Benchmarks in scRNA-seq Studies
| Study Type | Biological Replicates | Cells per Condition | Key Findings | Reference |
|---|---|---|---|---|
| Oocyte cryopreservation | 3 donors | 24 oocytes per donor per condition | Sufficient to detect that transcriptional profiles did not cluster separately by condition | [3] |
| Protocol comparison | Not specified | 583 mouse embryonic stem cells | Enabled comprehensive evaluation of 6 scRNA-seq methods | [41] |
| Power analysis | Variable | 10-1,000 cells | Cost-efficiency depends on study goals; Drop-seq more efficient for large cell numbers | [42] [41] |
Power Analysis for scRNA-seq Experiments
Fundamental Concepts
Power analysis determines the probability that a study will detect an effect of a specified size when it exists. For scRNA-seq experiments, key parameters include:
- Effect size: The minimum biologically relevant difference in gene expression between conditions
- Significance level: Typically set at α = 0.05 after multiple testing correction
- Power: Generally targeted at 80% or higher
- Technical variability: Capture efficiency, amplification bias, and sequencing depth
- Biological variability: Natural heterogeneity between cells and donors
Practical Implementation
Sensitivity and Accuracy Considerations scRNA-seq protocols vary significantly in their sensitivity (minimum number of input RNA molecules required for detection) and accuracy (closeness of estimated expression to true abundance). These technical performance characteristics directly influence power calculations [42].
Table 2: Power and Protocol Selection Guidelines
| Research Objective | Recommended Approach | Advantages | Limitations |
|---|---|---|---|
| Large-scale cell quantification | Drop-seq | Cost-efficient for thousands of cells | Lower genes detected per cell |
| Detailed transcriptome characterization | Smart-seq2 | Detects more genes per cell | Less cost-effective for large cell numbers |
| Digital quantification with UMIs | CEL-seq2, MARS-seq, SCRB-seq | Reduced amplification noise | Protocol-specific bias potential |
| Continuous trajectory analysis | scDART | Preserves developmental trajectories | Complex computational requirements |
Sequencing Depth Considerations Power simulations at different sequencing depths reveal trade-offs between the number of cells sequenced and the depth of sequencing per cell. Deeper sequencing increases the detection of low-abundance transcripts, while sequencing more cells better characterizes population heterogeneity [41].
Experimental Protocols
Protocol for Single-Cell RNA Sequencing of Frozen-Thawed Oocytes
Based on: Single-cell analysis comparing early-stage oocytes from fresh and slow-frozen/thawed human ovarian cortex [3]
Reagents and Materials
- Human ovarian cortex samples (10 × 10 × 1 mm³ squares)
- McIlwain tissue chopper
- Enzymatic digestion solution
- Lysis buffer
- scRNA-seq library preparation kit (seqWell PlexWell)
- NovaSeq6000 Illumina platform
Stepwise Procedure
- Tissue Preparation: Cut ovarian cortex into squares measuring 10 × 10 × 1 mm³ using a McIlwain tissue chopper.
- Cryopreservation: Slow-freeze tissue squares using controlled-rate freezing, then thaw for processing alongside fresh controls.
- Oocyte Isolation: Fragment tissue squares and perform enzymatic digestion to isolate live oocytes from primordial and primary follicles.
- Mechanical Denuding: Manually remove surrounding somatic cells under a dissection microscope.
- Single-Cell Capture: Individually transfer oocytes to wells containing lysis buffer.
- Library Preparation: Use seqWell PlexWell rapid single-cell RNA protocol for library prep.
- Sequencing: Perform 150-bp paired-end sequencing on Illumina NovaSeq6000 platform.
Quality Control Considerations
- Histological analysis of parallel tissue samples using hematoxylin and eosin staining
- Assessment of follicle density, morphology, and stage distribution
- Stromal cell density quantification
Protocol Adaptation for Yeast Cells
Based on: A new protocol for single-cell RNA-seq reveals stochastic gene expression during carbon source shift [43]
Modification for Cell Wall Digestion
- Add zymolyase (cell-wall digestion enzyme) to reverse transcription master mix
- Temperature regimen: initial storage on ice, room temperature for droplet generation (~6 min), incubation at 53°C (45 min) for lysis and reverse transcription
- Validate lysis efficiency through cell counting throughout transition steps
Protocol for Nucleus Isolation from Frozen Tissues
Based on: Optimized nucleus isolation protocol from frozen mouse tissues [44]
Reagents
- Lysis buffer (with NP-40)
- Wash buffers
- Bovine serum albumin (BSA)
- DAPI solution
- RNaseOut
- DPBS
Procedure
- Tissue Collection: Rapidly dissect tissue and flash-freeze in liquid nitrogen.
- Homogenization: Process frozen tissue in lysis buffer using mechanical homogenization.
- Filtration: Pass homogenate through 40 μm cell strainers.
- Centrifugation: Pellet nuclei through series of washing and centrifugation steps.
- Sorting: Isolate nuclei using flow cytometry with DAPI staining.
- Quality Control: Assess nucleus integrity and RNA quality before sequencing.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for scRNA-seq Experiments
| Reagent/Material | Function | Example Application | Considerations |
|---|---|---|---|
| PlexWell kit (seqWell) | Library preparation | Single-cell RNA sequencing | Used for oocyte transcriptome study [3] |
| Zymolyase | Cell wall digestion | Yeast scRNA-seq adaptation | Enables in-droplet lysis of yeast cells [43] |
| ERCC spike-in RNAs | Technical controls | Protocol sensitivity assessment | Quantify technical variability and sensitivity [42] |
| UMI barcodes | Digital quantification | Unique molecular identifiers | Reduces amplification noise [42] [41] |
| DAPI | Nuclear staining | Nucleus isolation and sorting | Flow cytometry validation [44] |
| RNaseOut | RNase inhibition | Preserves RNA integrity | Critical for nucleus isolation protocol [44] |
| NP-40 | Detergent | Nucleus isolation | Cell membrane lysis [44] |
Data Analysis and Visualization Frameworks
Quantitative Visualization with scBubbletree
For large and complex scRNA-seq datasets, visualization approaches that avoid overplotting and distortion of high-dimensional relationships are essential. The scBubbletree method provides a quantitative visualization framework that:
- Identifies clusters of transcriptionally similar cells
- Visualizes clusters as "bubbles" at dendrogram tips
- Stacks bubble trees with cluster-associated information
- Handles datasets with over 1.2 million cells [45]
Evaluation of Dimensionality Reduction
A quantitative framework for evaluating single-cell data structure preservation by dimensionality reduction techniques defines metrics of global and local structure preservation. This evaluation is crucial as the choice of dimensionality reduction method significantly impacts the interpretation of cellular relationships [46].
Integrated Experimental Design Workflow
Figure 1: Integrated scRNA-seq Experimental Workflow
Special Considerations for Fresh vs. Frozen Experimental Designs
When comparing fresh and frozen samples, several technical factors require careful control:
- Post-thaw recovery time: Transcriptional changes immediately after thawing may differ from long-term effects
- Batch effects: Process all samples using identical reagents and equipment where possible
- Sequencing batches: Balance conditions across sequencing runs
Analytical Validation
The study comparing fresh and frozen oocytes implemented multiple validation approaches:
- Histological analysis of follicle morphology and density
- Stromal cell density quantification
- Comparison of transcriptional profiles using unsupervised clustering
- Gene ontology enrichment analysis of differentially expressed genes [3]
Robust experimental design for scRNA-seq studies comparing fresh and frozen cells requires integrated consideration of replication strategy, sample size determination, and power analysis. Implementation of appropriate protocols for cell processing, library preparation, and data analysis ensures that conclusions about cryopreservation effects reflect biological reality rather than technical artifacts. The frameworks and benchmarks provided here offer researchers a foundation for designing statistically sound studies that effectively address their specific research questions in embryo scRNA-seq research.
As single-cell technologies continue to evolve, ongoing refinement of experimental design principles will be necessary to accommodate new protocols, increased throughput, and more complex analytical approaches. The iterative relationship between experimental design and analytical validation remains crucial for advancing our understanding of cellular responses to cryopreservation in embryological contexts.
The integration of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) has emerged as a transformative approach for studying complex biological systems, offering a window into cellular heterogeneity while preserving crucial spatial context. Within embryo research, this multi-omics strategy is particularly vital for benchmarking stem cell-based embryo models against in vivo counterparts, enabling unbiased transcriptional profiling across developmental stages from zygote to gastrula [6]. The fundamental challenge in this field stems from the inherent limitations of each technology when used independently: scRNA-seq provides high-resolution gene expression profiling of individual cells but requires tissue dissociation, which irrevocably destroys native spatial architecture [47] [48]. Conversely, spatial transcriptomics technologies preserve spatial localization information but have historically faced resolution limitations, often capturing gene expression from spots containing multiple cells [47]. This application note details standardized protocols for the effective integration of these powerful technologies, with special consideration for the unique demands of embryonic research involving fresh versus frozen cellular specimens.
Comparative Platform Analysis for Embryonic Research
Technology Selection and Performance Metrics
Selecting appropriate platforms is foundational to experimental success. Table 1 summarizes the key characteristics of major commercially available imaging-based spatial transcriptomics platforms, benchmarked using formalin-fixed paraffin-embedded (FFPE) samples, providing critical performance metrics for platform selection [49].
Table 1: Comparison of Imaging-Based Spatial Transcriptomics Platforms
| Platform | Panel Size (Genes) | Resolution | Transcripts/Cell | Unique Genes/Cell | Key Strengths | Considerations for Embryonic Research |
|---|---|---|---|---|---|---|
| CosMx SMI | 1,000-plex | Single-cell | Highest (p<2.2e−16) | Highest (p<2.2e−16) | Highest transcript detection | Field of view limitation (545μm × 545μm FOV) |
| MERFISH | 500-plex | Single-cell | Variable (lower in older samples) | Variable | Whole-tissue coverage | Lack of negative control probes |
| Xenium (Unimodal) | 339-plex (289 standard + 50 custom) | Single-cell | Higher than multimodal | Higher than multimodal | Whole-tissue coverage | Multimodal segmentation reduces counts |
| Xenium (Multimodal) | 339-plex | Single-cell | Lower than unimodal | Lower than unimodal | Integrated morphology | Higher background potential |
For embryonic research, the age and preservation method of samples significantly impact data quality. Studies show consistently higher numbers of transcripts and uniquely expressed genes per cell in more recently constructed tissue microarrays across platforms [49]. When comparing segmentation approaches, Xenium's unimodal assay demonstrated significantly higher transcript and gene counts per cell than its multimodal counterpart (p < 2.2e−16) [49], an important consideration for embryonic tissues where detecting low-abundance developmental markers is critical.
Specimen Preservation Considerations
The choice between fresh and frozen specimens represents a critical decision point in experimental design. While fresh samples are ideal for high-quality scRNA-seq, single-nucleus RNA sequencing is generally preferable for frozen specimens [50]. For spatial transcriptomics, FFPE samples have been successfully utilized across major platforms, though with noted variations in performance based on tissue age [49]. The construction of a comprehensive human embryo reference dataset has incorporated multiple preservation methods, including cultured preimplantation embryos, 3D cultured postimplantation blastocysts, and a Carnegie Stage 7 human gastrula isolated in vivo [6].
Experimental Protocols for Integrated Multi-omics
Workflow for Integrated scRNA-seq and Spatial Transcriptomics
The following diagram illustrates the comprehensive workflow for integrating single-cell and spatial transcriptomic data in embryonic research:
Detailed scRNA-seq Protocol for Embryonic Samples
Sample Preparation and Quality Control
- Fresh Embryo Processing: Mechanically dissociate embryo tissues using gentle enzymatic digestion (e.g., Accutase or TrypLE) for minimal disruption. For preimplantation embryos, individual cells can be directly captured without dissociation [6].
- Frozen Specimen Processing: For frozen embryos, prioritize single-nucleus RNA sequencing. Isolate nuclei using Dounce homogenization in lysis buffer followed by density gradient centrifugation [50].
- Quality Control: Assess cell viability using fluorescent dye exclusion (e.g., propidium iodide). For embryonic cells, target viability >90% for optimal library preparation.
Library Preparation and Sequencing
- Cell Capture: Utilize microfluidic platforms (10X Genomics Chromium) for high-throughput capture, or plate-based methods (SMART-seq2) for full-length transcriptome profiling of individual embryonic cells [50] [51].
- cDNA Synthesis and Amplification: Perform reverse transcription with template-switching oligonucleotides to add universal adapter sequences, followed by PCR amplification with unique molecular identifiers (UMIs) to distinguish biological signals from amplification artifacts [50].
- Library Construction: Fragment amplified cDNA, then add sample indices and sequencing adapters using ligation or tagmentation approaches. Quality control using capillary electrophoresis (e.g., Bioanalyzer) is critical before sequencing.
- Sequencing: Platform-specific recommendations: Illumina NovaSeq for 10X libraries (minimum 20,000 reads/cell); Illumina NextSeq for plate-based methods (minimum 1 million reads/cell).
Computational Analysis Pipeline
- Preprocessing: Process raw sequencing data through alignment (STARsolo, Cell Ranger) or alignment-free (Kallisto-BUStools) methods to generate gene expression matrices [50].
- Quality Filtering: Remove low-quality cells (<200 genes/cell or >15% mitochondrial reads), doublets (DoubletFinder), and ambient RNA (SoupX) [51].
- Downstream Analysis: Normalize data (SCTransform), identify highly variable genes, perform dimensionality reduction (PCA, UMAP), and cluster cells (Louvain, Leiden) [6]. Annotate cell types using reference datasets (e.g., human embryo reference) [6] and perform trajectory inference (Monocle3, PAGA) to reconstruct developmental pathways.
Detailed Spatial Transcriptomics Protocol for Embryonic Samples
Sample Preparation and Sectioning
- Tissue Preservation: For optimal morphology, fresh-frozen processing is preferred. Embed embryos in OCT compound, flash-freeze in liquid nitrogen-cooled isopentane, and store at -80°C.
- Sectioning: Cut cryosections at 5-10μm thickness using a cryostat and transfer to specialized ST slides (Visium, NanoString). Maintain cryostat temperature at -20°C to -22°C.
- Fixation and Staining: Fix sections in pre-chilled methanol or 4% PFA, then stain with hematoxylin and eosin for histological assessment. For FFPE samples, follow standard pathological processing with potential antigen retrieval [49].
Spatial Library Preparation
- Permeabilization Optimization: Titrate permeabilization enzyme concentration and incubation time using test slides to maximize RNA release while maintaining tissue architecture. Embryonic tissues typically require gentler permeabilization.
- Spatial Barcoding: For Visium and Slide-seq platforms, mRNA molecules hybridize to spatially barcoded oligo-dT primers on the array surface [52]. For imaging-based platforms (MERFISH, CosMx), hybridize with gene-specific probes with fluorescent barcodes [49].
- Library Construction: Synthesize cDNA, then amplify with library-specific primers. Include platform-specific negative controls and spike-ins to monitor technical variability.
Data Acquisition and Processing
- Sequencing-Based Platforms: Sequence libraries on Illumina platforms following manufacturer's recommendations. Process data through spaceranger (10X Visium) or custom pipelines to generate spatial gene expression matrices.
- Imaging-Based Platforms: Perform multiple rounds of hybridization and imaging (MERFISH: 16+ rounds; CosMx: 12+ cycles) [49]. Reconstruct gene identities from combinatorial barcodes using manufacturer software suites.
- Image Processing: Align fluorescence images, segment cells based on nuclear or membrane stains, and assign transcripts to individual cells using automated algorithms with manual curation.
Data Integration Methodology
Computational Integration Framework
The SIMO (Spatial Integration of Multi-Omics) computational method provides a robust framework for integrating scRNA-seq with spatial transcriptomics data [53]. The sequential mapping process involves:
- Initial Transcriptomic Mapping: Integrate ST data with scRNA-seq data using k-nearest neighbor (k-NN) algorithms to construct spatial graphs and modality maps. Calculate mapping relationships using fused Gromov-Wasserstein optimal transport [53].
- Coordinate Refinement: Fine-tune cell coordinates based on transcriptome similarity between mapped cells and their surrounding spots.
- Multi-omics Expansion: Extend integration to non-transcriptomic single-cell data (e.g., scATAC-seq) using gene activity scores as a linkage point. Calculate Pearson Correlation Coefficients between cell groups and determine alignment probabilities through Gromov-Wasserstein transport calculations [53].
Embryo-Specific Integration Considerations
When working with embryonic data, leverage existing human embryo references [6] to:
- Project query datasets onto the reference space using stabilized UMAP
- Annotate cell identities with prediction confidence scores
- Authenticate stem cell-based embryo models by comparing with in vivo counterparts across developmental stages
Essential Research Tools and Reagents
Research Reagent Solutions
Table 2: Essential Research Reagents for Integrated Multi-omics in Embryonic Research
| Category | Reagent/Kit | Function | Specimen Compatibility |
|---|---|---|---|
| Cell Isolation | 10X Genomics Chromium Next GEM | Single-cell partitioning and barcoding | Fresh, frozen (nuclei) |
| Accutase enzyme | Gentle tissue dissociation | Fresh embryonic tissues | |
| MACS Nuclear Isolation Kit | Nuclei isolation from frozen specimens | Frozen tissues | |
| Spatial Transcriptomics | 10X Visium Spatial Gene Expression | Spatial barcoding on slides | Fresh-frozen, FFPE |
| CosMx Human Universal Cell Characterization Panel | 1,000-plex RNA imaging | FFPE [49] | |
| MERFISH Immuno-Oncology Panel | 500-plex RNA imaging | FFPE [49] | |
| Library Preparation | SMART-seq HT Plus Kit | Full-length scRNA-seq | Plate-based, low input |
| Nextera XT DNA Library Preparation Kit | Tagmentation-based library prep | All specimen types | |
| Sequencing | Illumina NovaSeq 6000 S4 Flow Cell | High-throughput sequencing | Large-scale projects |
| Illumina NextSeq 500/550 High Output | Moderate-throughput sequencing | Pilot studies | |
| Computational Tools | SIMO Algorithm | Multi-omics spatial integration [53] | All data types |
| Seurat (R) | scRNA-seq analysis and integration | All specimen types | |
| Scanpy (Python) | Scalable scRNA-seq analysis | All specimen types | |
| STalign | Multi-slice spatial alignment [54] | Spatial data |
Analysis and Interpretation Framework
Multi-omics Data Integration Workflow
The following diagram illustrates the computational workflow for integrating and analyzing multi-omics data:
Key Analytical Approaches for Embryonic Development
- Spatial Domain Identification: Apply clustering algorithms (Leiden, Louvain) to integrated data to identify transcriptionally distinct spatial regions. In embryonic development, this reveals emerging tissue compartments and patterning centers [6].
- Cell-Cell Communication Analysis: Infer ligand-receptor interactions using tools like CellChat or NicheNet, identifying signaling hubs that orchestrate developmental processes [47].
- Developmental Trajectory Reconstruction: Utilize pseudotime algorithms (Monocle3, PAGA) on integrated data to reconstruct lineage bifurcation events and identify key transcriptional regulators driving cell fate decisions [6].
- Spatial Mechano-Transcriptomics: For advanced applications, integrate transcriptional data with mechanical force inference to explore relationships between gene expression and physical forces during morphogenesis [55].
Concluding Remarks
The integrated multi-omics approach detailed in this application note provides a powerful framework for advancing embryonic research. By systematically combining scRNA-seq with spatial transcriptomics, researchers can overcome the inherent limitations of each individual technology, enabling comprehensive analysis of developmental processes with both cellular resolution and spatial context. The protocols and methodologies outlined here—particularly the considerations for fresh versus frozen specimen processing and the computational integration strategies—provide a solid foundation for generating biologically meaningful insights into human embryogenesis. As these technologies continue to evolve, with improvements in resolution, throughput, and multi-omics capabilities, they promise to further transform our understanding of early human development and enhance the fidelity of stem cell-based embryo models.
Solving Technical Challenges in Embryo scRNA-seq Sample Processing
In single-cell RNA sequencing (scRNA-seq) studies of embryonic development, the choice between using fresh or frozen cells introduces significant technical challenges. Two of the most pervasive confounders are the induction of stress response genes during sample preparation and the introduction of batch effects during experimental processing. These artifacts can obscure true biological signals, particularly when comparing embryos across different developmental stages or experimental conditions. This document outlines the sources of these technical variabilities and provides detailed protocols for their minimization, specifically framed within embryonic scRNA-seq research.
Understanding and Controlling for Dissociation-Induced Stress
The Impact of Tissue Dissociation on Transcriptional Profiles
The process of dissociating tissue into single cells is a major source of stress-induced transcriptional artifacts. During dissociation, cells are removed from their native microenvironment and subjected to enzymatic and mechanical stress, triggering a rapid transcriptional stress response that can confound true biological signals [56]. This is particularly critical in embryo research, where developmental gene programs may share similarities with stress pathways.
A novel RNA labeling strategy, scSLAM-seq, has been developed to directly measure this dissociation response. By adding the uridine analog 4-thiouridine (4sU) to the dissociation reaction, transcripts synthesized during the procedure can be specifically labeled and later identified via T-to-C substitutions in sequencing data [56]. This approach allows for the precise identification and subsequent computational removal of dissociation-related transcripts.
Experimental Strategies to Minimize Dissociation Stress
Cold Dissociation Protocol: Comparative studies have demonstrated that performing dissociation procedures at 4°C rather than 37°C reduces the induction of heat shock genes and other stress responders, although some stress responses persist [56]. The following protocol is adapted for embryonic tissues:
- Workflow Diagram: Minimizing Dissociation-Induced Stress
- Preparation: Pre-chill all solutions and equipment to 4°C. Prepare cold dissociation buffer appropriate for embryonic tissue.
- Dissociation: Mince embryonic tissue in cold buffer using sharp dissection scissors. Transfer tissue to a digestion tube with pre-chilled enzymes.
- Incubation: Place the tube on a rocking platform in a cold room (4°C) for 30-90 minutes, depending on embryo stage and tissue type.
- Mechanical Dissociation: Gently triturate the tissue every 15 minutes using a wide-bore pipette tip to avoid shear stress.
- Reaction Stopping: Add excess cold quenching buffer to stop enzymatic activity.
- Filtration and Wash: Pass the cell suspension through a pre-chilled 40-μm cell strainer. Centrifuge at 4°C and resuspend in cold PBS with 0.04% BSA.
- Optional 4sU Labeling: For scSLAM-seq, include 10 mM 4sU in the dissociation buffer to label nascent transcripts [56].
Consideration for Alternative Methods: Single-nuclei RNA sequencing (snRNA-seq) bypasses cellular dissociation stress by using isolated nuclei, making it applicable to frozen archived embryos. However, it introduces a transcriptomic bias towards nascent, nuclear RNA and may miss key cytoplasmic transcripts [57].
Table 1: Comparison of scRNA-seq and snRNA-seq for Embryo Research
| Feature | scRNA-seq (Fresh Cells) | snRNA-seq (Frozen Cells) |
|---|---|---|
| Transcript Coverage | Full-length, cytoplasmic and nuclear | Primarily nuclear, nascent transcripts |
| Dissociation Stress | High risk | Avoided |
| Compatibility with Biobanks | No | Yes |
| Cell Type Representation | May under-represent fragile cells | Can over-represent robust nuclei |
| Data Quality | High gene detection per cell | Lower gene detection per nucleus |
| Ideal Application | Active transcriptional states, splicing analysis | Archival samples, nuclear regulators |
Managing Batch Effects in scRNA-seq Study Design
Batch effects are technical variations introduced when samples are processed in different batches (e.g., different sequencing runs, dates, or operators). In scRNA-seq, these effects are pronounced because each cell is a separate sample processed in a single, unrepeatable batch [58]. Sources include:
- Variation in mRNA capture efficiency between cells
- Amplification bias during library preparation
- Differences in sequencing depth
- Day-to-day variability in reagent lots and protocols
These effects can be severe enough to drive the primary separation of samples in a PCA plot, completely obscuring biological signals of interest, such as those between embryo developmental stages [58].
Experimental Design and Correction Strategies
Effective Study Design Framework: To control for batch effects, the design must include technical replication.
- Workflow Diagram: Batch-Effect Controlled scRNA-seq Study
- Replication: Include multiple biological replicates per condition (e.g., multiple embryos from different litters per genotype).
- Balanced Processing: Distribute replicates from all experimental conditions across each processing batch (e.g., C1 microfluidic plate or 10X Chromium chip) and sequencing run [58].
- Technical Controls: Use Unique Molecular Identifiers (UMIs) to account for amplification bias and spike-in RNA standards (e.g., ERCC) to monitor technical variability [58].
- Sample Multiplexing: Where possible, use cell-hashing or genetic multiplexing to pool samples from different conditions within a single processing batch.
Benchmarking of Batch Correction Methods: A 2025 systematic evaluation of eight batch correction methods revealed significant differences in their performance and calibration [59]. The study measured the degree to which methods alter data in the absence of true batch effects, creating artifacts.
Table 2: Evaluation of Common scRNA-seq Batch Correction Methods
| Method | Correction Approach | Artifact Introduction | Recommendation for Embryo scRNA-seq |
|---|---|---|---|
| Harmony | Linear correction on PCA embedding via soft k-means | Low | Highly Recommended - Consistently performs well, preserves biology [59] |
| ComBat/ComBat-seq | Empirical Bayes / Negative binomial regression | Moderate | Use with Caution - Can introduce measurable artifacts [59] |
| Seurat | Aligning canonical correlation analysis (CCA) vectors | Moderate | Use with Caution - Can introduce measurable artifacts [59] |
| BBKNN | Graph-based correction of k-NN graph | Moderate | Use with Caution - Can introduce measurable artifacts [59] |
| MNN | Mutual nearest neighbors-based linear correction | High | Not Recommended - Often alters data considerably [59] |
| SCVI | Deep learning (variational autoencoder) | High | Not Recommended - Often alters data considerably [59] |
| LIGER | Quantile alignment of factor loadings | High | Not Recommended - Often alters data considerably [59] |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Minimizing Artifacts
| Reagent/Material | Function | Application Note |
|---|---|---|
| 4-thiouridine (4sU) | Labels nascent RNA during dissociation to identify stress-response genes [56]. | Use at 10 mM for short dissociation periods. Validate it does not induce additional stress in your system. |
| Cold Dissociation Enzymes | (e.g., Accutase, Liberase). Digest extracellular matrix at low temperatures. | Pre-test enzyme cocktails and concentrations on pilot embryo samples to balance viability and yield. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences that tag individual mRNA molecules to correct for amplification bias [58]. | Integrated into most modern scRNA-seq library prep kits (e.g., 10x Genomics). |
| ERCC Spike-in RNA Controls | A set of 96 exogenous RNA transcripts of known concentration to monitor technical variation [58]. | Add a consistent volume and amount to each cell lysate during library preparation. |
| Viability Stains | (e.g., AO/PI, DAPI). Distinguish live/dead cells or nuclei. | Critical for filtering out dead cells (scRNA-seq) or confirming intact nuclei (snRNA-seq) [57]. |
| Cell Hashing Oligonucleotides | Antibody-conjugated barcodes to label cells from different samples prior to pooling. | Enables sample multiplexing within a single batch, effectively eliminating processing batch effects. |
Integrated Protocol: A Practical Guide for Embryo Researchers
This combined protocol provides a step-by-step guide from sample collection to data correction, integrating strategies to manage both stress genes and batch effects.
Sample Preparation and Library Generation
- Experimental Design: For a study comparing mutant and wild-type embryos, ensure at least N=3 litters per genotype. Plan to process cells from each litter across at least two separate library preparation batches.
- Tissue Collection & Dissociation:
- Rapidly collect embryonic tissue into cold dissection buffer.
- Follow the Cold Dissociation Protocol in Section 2.2.
- (Optional Stress Monitoring) Include 4sU in the dissociation buffer for scSLAM-seq [56].
- Quality Control: Count cells/nuclei and assess viability using an automated cell counter with AO/PI staining. For scRNA-seq, aim for >80% viability. Filter cells/nuclei through a 40-μm strainer.
- Library Preparation with Controls:
- Use a platform (e.g., 10X Genomics) that incorporates UMIs.
- Add ERCC spike-ins to the lysis buffer according to the manufacturer's instructions [58].
- Process samples from all experimental groups in a balanced design across sequencing lanes.
Computational Data Processing
- Preprocessing: Generate a count matrix using the pipeline provided by your sequencing platform (e.g., Cell Ranger for 10X data), which leverages UMIs to count molecules.
- Stress Gene Filtering (if 4sU labeled): Use computational tools (e.g., based on [56]) to identify genes with high T-to-C transition rates and regress them out of the analysis.
- Batch Correction:
- Integrate data from different batches using Harmony [59].
- Validate that the corrected data clusters by biological condition (e.g., genotype) rather than technical batch and that known cell-type markers form coherent patterns.
- Downstream Analysis: Proceed with clustering, differential expression, and trajectory inference on the corrected, high-quality data.
By rigorously applying these experimental and computational guidelines, researchers can significantly reduce technical confounders, thereby ensuring that the biological signals of development and disease are accurately captured in embryonic scRNA-seq studies.
In single-cell RNA sequencing (scRNA-seq) of embryos, the quality of the starting biological material is paramount. Research objectives increasingly require comparing developmental trajectories using both fresh and cryopreserved specimens, making rigorous quality control (QC) a critical first step. Proper QC ensures that the data generated accurately reflect the biological state of the embryo and not artifacts introduced by sample handling or inherent cellular stress. This document outlines the essential QC metrics—cell viability, RNA integrity, and library complexity—within the context of scRNA-seq studies on fresh versus frozen embryos, providing application notes and detailed protocols for researchers and drug development professionals.
Core Quality Control Metrics
For scRNA-seq experiments, particularly those involving precious embryonic material, a multi-faceted QC approach is non-negotiable. The core metrics, their significance, and acceptable thresholds are summarized in the table below.
Table 1: Essential Quality Control Metrics for Embryo scRNA-seq
| Metric Category | Specific Metric | Description & Biological Significance | Recommended Threshold(s) | Notes for Fresh vs. Frozen Embryos |
|---|---|---|---|---|
| Cell Viability | Membrane Integrity | Assesses the percentage of cells with intact membranes, crucial for cytoplasmic mRNA retention. [60] [61] | >80% (ideally >90%) | Fresh cells may show stress from dissociation; frozen cells must survive thawing process. |
| Debris and Clumps | Presence of cellular debris or cell doublets/multiplets can skew data. [60] [61] | Minimal debris; single-cell suspension | Gentle dissociation is critical for fragile embryonic cells. [61] | |
| RNA Integrity | RNA Quality Number (RQN) / RNA Integrity Number (RIN) | Measures RNA degradation. High-quality RNA is essential for full-length transcript capture. [61] | RIN/RQN ≥ 8.5 | Cryopreservation can introduce freeze-thaw cycles that degrade RNA. |
| Mitochondrial Read Fraction | Fraction of reads mapping to mitochondrial genes; high levels indicate cellular stress or broken membranes. [60] | Typically <10-20% | Varies by cell type; elevated in low-viability samples. A key metric for dissociation stress in fresh cells. | |
| Library Complexity | Number of Genes per Cell | The count of unique genes detected per cell (also known as "feature count"). [57] [62] | Varies by protocol & cell type; should be consistent across samples. | snRNA-seq (for frozen archives) typically detects fewer genes than scRNA-seq. [57] |
| Unique Molecular Identifiers (UMIs) per Cell | Quantifies the number of unique mRNA molecules captured, directly reflecting library complexity. [60] | Varies by protocol & cell type; should be consistent. | More accurate than read counts for quantifying expression. [60] | |
| Sequencing Saturation | Measures the fraction of duplicates in the library, indicating if sequencing depth is sufficient. [60] | High saturation (>50%) suggests adequate depth. | Optimal depth is crucial for identifying rare cell populations in heterogeneous embryonic samples. |
The following workflow diagram illustrates the logical sequence of QC steps from sample preparation to sequencing, highlighting critical checkpoints.
Detailed Experimental Protocols
Protocol 1: Quality Control for Fresh Embryonic Cell Suspensions
This protocol is adapted from methodologies used in pancreatic islet and oocyte research for preparing fresh single-cell suspensions. [57] [3]
- Tissue Dissociation: Mechanically fragment the embryonic tissue using a fine scalpel or tissue chopper in ice-cold, RNAse-free PBS. Follow with gentle enzymatic digestion (e.g., with Accutase) tailored to the specific embryonic tissue. Continuously monitor the dissociation to avoid over-digestion. [57] [3] [61]
- Cell Strainer Filtration: Pass the resulting cell suspension through a pre-wet 40 µm cell strainer to remove debris and clumps. [57]
- Viability Assessment:
- Mix a small aliquot of cells (e.g., 10 µL) with an equal volume of Trypan Blue or AO/PI stain.
- Count the cells using an automated cell counter (e.g., DeNovix CellDrop) or a hemocytometer.
- Calculate viability as the percentage of unstained (live) cells out of the total cells counted. Proceed only if viability exceeds 80%. [57] [61]
- RNA Integrity Check (if cells are to be lysed in bulk):
- Extract total RNA from a representative aliquot of ~1000 cells using a micro-scale kit.
- Analyze RNA quality using an Agilent Bioanalyzer with the RNA Pico Chip to obtain an RQN value. [61]
Protocol 2: Quality Control for Frozen/Thawed Embryonic Samples and Nuclei
This protocol leverages single-nuclei RNA-seq (snRNA-seq) for cryopreserved embryos or tissues, based on optimized methods for frozen ovarian cortex and brain tissue. [3] [62]
- Nuclei Isolation from Frozen Tissue:
- Place 20-50 mg of frozen embryonic tissue in ice-cold lysis buffer (e.g., from Chromium Nuclei Isolation Kit).
- Homogenize the tissue gently with a Dounce homogenizer (约15-20 strokes).
- Filter the lysate through a 40 µm flow-through cap or strainer to remove debris.
- Wash the nuclei pellet 2-3 times with lysis buffer without detergent to remove cellular debris and free RNA. [57] [62]
- Nuclei Quality Assessment:
- Key Consideration: Note that snRNA-seq primarily captures nuclear transcripts, leading to a bias towards nascent RNA and potentially fewer detected genes per nucleus compared to scRNA-seq. This is a biological difference, not a failure of QC. [57]
Protocol 3: Post-Sequencing Data Quality Control
After sequencing, computational QC is performed on the raw data using tools like Seurat or Scater. [60] [2]
- Metric Calculation: For each cell barcode, calculate:
- Number of unique genes (feature counts).
- Total number of UMIs (count depth).
- Percentage of reads mapping to mitochondrial genes. [60]
- Threshold Filtering:
- Filter out barcodes with a mitochondrial read percentage significantly above the median (indicating dead or stressed cells).
- Filter out barcodes with very low UMI/gene counts (indicating empty droplets or broken cells).
- Filter out barcodes with anomalously high UMI/gene counts (indicating potential doublets or multiplets). [60]
- Normalization and Scaling: Normalize the gene expression matrix to account for differences in sequencing depth per cell before proceeding to downstream analysis. [60] [2]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagent Solutions for Embryo scRNA-seq QC
| Item | Function/Application | Example Products/Brands |
|---|---|---|
| Gentle Dissociation Reagent | Enzymatically dissociates fresh embryonic tissue into single cells while preserving viability and RNA integrity. | Accutase [57] |
| Nuclei Isolation Kit | Provides optimized buffers for extracting intact, RNA-preserving nuclei from frozen tissues. | Chromium Nuclei Isolation Kit (10x Genomics) [57] |
| Viability Stain | Differentiates live cells (membrane-impermeant) from dead cells (membrane-permeant) for quantitative assessment. | Trypan Blue, Acridine Orange (AO)/Propidium Iodide (PI) [57] |
| Cell Strainers | Removes cellular debris and clumps to ensure a true single-cell/nuclei suspension for droplet-based platforms. | 40 µm nylon mesh strainers [57] [62] |
| RNA QC Instrument | Microfluidics-based system for quantitatively assessing RNA integrity (RIN/RQN) from minimal input. | Agilent Bioanalyzer [61] |
| Single-Cell RNA-seq Kit | All-in-one reagents for library construction from single cells or nuclei, often platform-specific. | 10x Genomics Chromium Single Cell 3' Kit [57] [62] |
| Automated Cell Counter | Provides fast and consistent quantification of cell/nuclei concentration and viability. | DeNovix CellDrop [57] |
Rigorous quality control spanning wet-lab procedures and computational checks is the foundation of any robust scRNA-seq study. For research comparing fresh and frozen embryos, understanding the distinct QC profiles of each sample type is essential. While fresh cells require careful handling to minimize dissociation stress, frozen samples offer unique advantages for biobanked materials but require specialized nuclei isolation protocols. By adhering to the detailed metrics and protocols outlined here, researchers can ensure the generation of high-quality, reliable data capable of revealing genuine biological insights into embryonic development.
Optimization Strategies for Difficult-to-Dissociate Embryonic Tissues
Within the context of a broader thesis on fresh versus frozen cells for embryo single-cell RNA sequencing (scRNA-seq), optimizing tissue dissociation is a critical prerequisite. The choice between fresh and cryopreserved starting material presents a significant trade-off: fresh tissues best preserve native transcriptional states but impose stringent logistical constraints, whereas frozen tissues offer flexibility but risk cryopreservation-induced artifacts. This application note details targeted strategies for dissociating challenging embryonic tissues, enabling researchers to make informed decisions and maximize the quality of their scRNA-seq data for both experimental pathways.
Quantitative Impact of Tissue Processing on Sample Quality
The following table summarizes key quantitative findings from recent studies on the effects of dissociation and cryopreservation on cellular properties, providing a benchmark for protocol optimization.
Table 1: Impact of Dissociation and Cryopreservation on Sample Quality Metrics
| Study Focus | Tissue / Cell Type | Key Quality Metrics | Performance of Frozen/Processed Tissue | Source |
|---|---|---|---|---|
| Cryopreservation Impact | Human ovarian cortex (early-stage oocytes) | Transcriptomic clustering, Follicle morphology | No separate clustering of fresh vs. slow-frozen/thawed oocytes; >86% normal follicle morphology in both conditions. [3] | |
| Advanced Dissociation | Bovine Liver Tissue | Dissociation Efficacy, Cell Viability | 95% ± 4% efficacy (Electrical dissociation); 72% ± 10% efficacy (Ultrasound + Enzymatic). [63] | |
| Cold Dissociation | Various Renal Cells | Cell Viability, Transcriptional Artifacts | Minimizes stress-induced artifacts; reduced dissociation efficacy and longer processing times. [64] | |
| Microfluidic Dissociation | Mouse Kidney & Heart | Cell Yield, Viability | ~20,000 cells/mg (Kidney epithelial); ~60-90% viability (Varies by cell type). [63] |
Detailed Methodologies for Embryonic Tissue Dissociation
Protocol 1: Standardized Enzymatic-Mechanical Dissociation for Fresh Embryonic Tissues
This protocol is designed to maximize cell viability and minimize stress-induced transcriptional artifacts from fresh, difficult-to-dissociate embryonic tissues.
- Reagents and Equipment: McIlwain tissue chopper, Collagenase IV, Dispase, DNase I, HBSS with calcium and magnesium, Fetal Bovine Serum (FBS), cell strainers (40µm, 70µm), centrifuge. [3] [63]
- Procedure:
- Tissue Preparation: Immediately after dissection, place embryonic tissue in cold, oxygenated buffer. Using a McIlwain tissue chopper, mince the tissue into small, uniform fragments (e.g., <1 mm³). [3]
- Enzymatic Digestion: Transfer the minced tissue to a digestion solution containing pre-warmed Collagenase IV (1-2 mg/mL) and Dispase (1-2 mg/mL) in HBSS. Gently agitate the tissue for 30-60 minutes at 37°C. [63]
- Reaction Quenching: Halt the enzymatic reaction by adding a large volume of cold HBSS supplemented with 10% FBS.
- Mechanical Disruption and Filtration: Gently pipette the tissue mixture up and down. Pass the resulting cell suspension through a series of pre-wetted cell strainers (70µm followed by 40µm) to remove debris and undissociated clumps. [63]
- Cell Washing and Resuspension: Centrifuge the filtrate and carefully resuspend the cell pellet in a suitable buffer (e.g., PBS with 0.04% BSA) for counting and quality control.
Protocol 2: Dissociation of Slow-Frozen/Thawed Embryonic Tissues
This protocol builds on the previous one, accounting for the additional fragility of cryopreserved tissues.
- Reagents and Equipment: All reagents from Protocol 1, plus cryoprotectant (e.g., DMSO), controlled-rate freezer. [3]
- Procedure:
- Thawing: Rapidly thaw the frozen vial in a 37°C water bath until only a small ice crystal remains.
- Cryoprotectant Removal: Slowly dilute the thawed cell suspension with pre-warmed buffer containing 10% FBS. Centrifuge at a low speed to pellet the cells and carefully remove the supernatant containing the cryoprotectant.
- Gentle Digestion: Proceed with the enzymatic digestion steps outlined in Protocol 1, but consider reducing the enzyme concentration or incubation time by 20-30% to account for increased cell fragility post-thaw.
- Viability Enhancement: After the final wash, resuspend the cell pellet in a recovery medium supplemented with a viability-enhancing agent like ROCK inhibitor for 30-60 minutes before proceeding to library preparation. [3]
Technical Recommendations and Visual Workflows
Decision Framework for Fresh vs. Frozen Workflows
The choice between fresh and frozen workflows depends on experimental priorities. Fresh processing is superior for capturing unperturbed transcriptomes and is ideal when the laboratory is in close proximity to the sample source. Frozen processing should be chosen when logistical flexibility is required, such as with multi-center studies or when samples cannot be processed immediately, with the understanding that it may introduce subtle transcriptomic shifts. [3] [64]
Post-Dissociation Quality Control Workflow
Rigorous quality control is non-negotiable after tissue dissociation, as it directly determines the success of downstream scRNA-seq.
Table 2: Key Research Reagent Solutions for Embryonic Tissue Dissociation
| Item | Function | Example Use Case |
|---|---|---|
| McIlwain Tissue Chopper | Provides uniform mechanical mincing of tissue, enabling consistent enzymatic penetration. | Standardized initial fragmentation of fresh or frozen-thawed embryonic tissue. [3] |
| Collagenase/Dispase Blend | Enzymatically degrades the extracellular matrix (ECM) and basal lamina to free individual cells. | General digestion of most embryonic tissues; concentration and time must be optimized. [63] |
| Cold-Active Proteases | Enzymes active at 4-10°C, minimizing the induction of stress-related genes during dissociation. | "Cold dissociation" of sensitive tissues like neural ectoderm to preserve native transcriptomes. [64] |
| ROCK Inhibitor (Y-27632) | Improves viability of sensitive cells, like cryopreserved epithelial cells, by inhibiting apoptosis. | Added to cell recovery media after thawing frozen tissues or during plating of fragile cells. [3] |
| Propidium Iodide (PI) / SYTO9 | Fluorescent live/dead staining for precise viability assessment via fluorescence microscopy or FACS. | Quantitative QC after dissociation to ensure suspension meets viability thresholds for scRNA-seq. [65] |
| PythoN i Dissociation System | Automated instrument combining optimized mechanical and enzymatic dissociation in a closed system. | Reproducible dissociation of challenging tissues with high viability (~90%) and reduced hands-on time. [65] |
Successfully navigating the dissociation of difficult-to-dissociate embryonic tissues for scRNA-seq requires a deliberate and balanced approach. The strategies outlined here—employing gentle enzymatic-mechanical techniques, implementing rigorous QC, and understanding the implications of using fresh versus frozen tissues—provide a robust foundation. By carefully selecting and optimizing these protocols, researchers can ensure that the cellular narratives revealed by scRNA-seq are a true reflection of embryonic development, rather than an artifact of the laboratory process.
Temperature Control and Processing Time Considerations
Within the context of a broader thesis comparing fresh versus frozen specimens for embryo single-cell RNA sequencing (scRNA-seq), temperature control and processing time emerge as critical technical variables. These factors directly impact cellular integrity, RNA quality, and the fidelity of resulting transcriptomic data. Optimal handling protocols are essential for minimizing stress-induced artifacts and preserving biological truth, particularly for sensitive embryonic tissues and rare cell populations. This application note synthesizes current research to provide detailed methodologies and quantitative guidelines for managing these parameters in embryo scRNA-seq workflows.
The Impact of Temperature and Processing on scRNA-seq Data Quality
Temperature-Induced Transcriptomic Changes
Temperature stress can significantly alter cellular transcriptomes. A study on zebrafish embryos revealed that elevated temperatures perturb the proportions and gene expression programs of numerous cell types, introducing asynchrony in developmental timing. Notably, the notochord was identified as particularly sensitive, with sheath cells accumulating misfolded protein at higher temperatures, leading to structural failure and anatomic defects [66]. Furthermore, RNA-Seq analysis of Atlantic tomcod embryos exposed to higher temperatures showed a marked increase in differentially expressed genes (DEGs)—from 349 DEGs at 3.5°C to 1,286 DEGs at 8.3°C—linked to stress response, cardiac health, and protein homeostasis [67].
The unfolded protein response is a key mechanism activated under temperature stress. In zebrafish, this response was required for temperature-induced developmental acceleration, demonstrating how proteostasis governs differential temperature sensitivity across cell types [66].
Processing Time and Sample Stability
Processing time directly impacts sample viability, especially for sensitive cell types. Neutrophils, for instance, have a short ex vivo half-life, and isolation methods can induce activation or apoptosis. Evidence suggests that neutrophils suitable for functional characterization can be isolated from blood stored at room temperature or 4°C for 24 hours, or for up to 72 hours when stored at 37°C [68].
For complex tissues like brain tumors, an optimized nuclear isolation protocol for long-term frozen samples has been developed that is fast (less than 30 minutes total), low-cost, and simple to execute. This protocol maintains nuclear integrity with minimal mitochondrial reads (typically under 1%) in resulting RNA data [69].
Table 1: Effects of Temperature Stress on Embryonic Development
| Organism | Temperature Condition | Key Transcriptomic Findings | Functional Consequences |
|---|---|---|---|
| Zebrafish [66] | 32°C & 34°C vs. 28°C (control) | Altered cell type proportions; asynchronous developmental timing; activated unfolded protein response. | Axial defects; notochord structural failure due to proteostasis disruption. |
| Atlantic Tomcod [67] | 3.5°C, 6.2°C, 8.3°C vs. 2.7°C (control) | Number of Differentially Expressed Genes (DEGs) increased with temperature (349 to 1286). | Accelerated heart rate and hatching; increased larval mortality; altered morphology. |
Protocols for Temperature Control and Sample Processing
Workflow for Fresh and Frozen Embryonic Tissues
The following diagram outlines a standardized workflow for handling fresh and frozen embryonic samples, incorporating critical temperature and time checkpoints to preserve RNA integrity for scRNA-seq.
Detailed Experimental Methodologies
Protocol A: Nuclei Isolation from Long-Term Frozen Tissue [69]
This protocol is optimized for pediatric glioma tissues but is applicable to other frozen embryonic tissues.
- Tissue Preparation: Cut 20–50 mg of frozen tissue on a chilled surface in ice-cold lysis buffer.
- Homogenization: Transfer tissue to a dounce homogenizer. Dounce with a loose pestle (10–15 strokes) followed by a tight pestle (10–15 strokes) to open cell walls and liberate nuclei.
- Filtration: Pass the homogenate through a cell strainer (e.g., 40 μm) to remove large debris.
- Washing: Centrifuge the filtrate and wash the nuclear pellet 2–3 times with lysis buffer without detergent to remove cellular debris and free RNA. (Note: Two washes may be preferred if starting material is low to minimize nuclear loss).
- Resuspension: Resuspend the final, clean nuclear pellet in an appropriate storage buffer. Nuclei can be used directly for snRNA-seq platforms or frozen at -80°C for a short period (maximally 2–3 days).
Protocol B: scRNA-seq of Temperature-Stressed Whole Embryos [66]
This protocol uses nuclear hashing to profile hundreds of individual zebrafish embryos under temperature stress.
- Temperature Exposure: Raise embryos at control (e.g., 28°C) and elevated temperatures (e.g., 32°C, 34°C). Include phenotypically normal and abnormal embryos from stressed conditions.
- Embryo Dissociation and Hashing: Individually dissociate whole embryos. Liberate nuclei and label them with polyadenylated DNA oligos containing unique barcodes ("hashes") for each embryo.
- Single-Cell Library Preparation: Process the hashed nuclei using a single-cell combinatorial indexing (sci-RNA-seq) protocol to profile transcriptomes.
- Bioinformatic Analysis: Demultiplex cells by their unique hash barcodes to assign them to individual embryos. This allows for the analysis of cell type composition and gene expression with individual-embryo resolution.
Protocol C: Isolation and Sequencing of Early-Stage Oocytes [3]
This protocol for fresh and slow-frozen/thawed human ovarian cortex shows minimal cryopreservation impact.
- Tissue Preparation: Cut ovarian cortex into small squares (e.g., 10 × 10 × 1 mm³). Process fresh or after slow-freezing and thawing.
- Oocyte Isolation: Fragment the cortex with a tissue chopper and subject to enzymatic digestion. Mechanically denude oocytes from primordial and primary follicles under a dissection microscope.
- Lysis and Library Prep: Place individual, denuded oocytes into lysis buffer. Perform library preparation using a platform like the seqWell PlexWell rapid single-cell RNA protocol.
- Sequencing: Pool libraries and sequence on a high-throughput platform (e.g., Illumina NovaSeq 6000).
The Scientist's Toolkit: Essential Reagent Solutions
Table 2: Key Reagents and Kits for Embryo scRNA-seq under Temperature Constraints
| Item Name | Function/Application | Specific Example / Vendor |
|---|---|---|
| Lysis Buffer (without detergent) | Washing nuclear pellets to remove debris and free RNA without damaging nuclear walls. | Custom formulation; see Protocol A [69]. |
| Nuclear Hash Oligos | Labeling nuclei from individual embryos for multiplexing and tracing cell origins. | Polyadenylated DNA oligos with unique barcodes [66]. |
| Single-Cell Combinatorial Indexing Kits | Profiling transcriptomes from hundreds of individually-hashed samples. | sci-RNA-seq kit [66]. |
| Rapid scRNA-seq Library Prep Kits | Generating libraries from low-input, sensitive samples like oocytes. | seqWell PlexWell kit [3]. |
| Density Gradient Media | Purifying nuclei or specific cell populations from complex tissue homogenates. | Ficoll-Paque [70]; OptiPrep [69]. |
| Cell Strainers | Removing large debris and cell clumps after tissue dissociation. | 40 μm nylon mesh strainers [69]. |
Comparative Analysis: Fresh vs. Frozen/Thawed Workflows
The decision to process samples fresh or after freezing involves trade-offs. A study on human oocytes found that the transcriptional profiles of fresh and frozen/thawed oocytes did not cluster separately, indicating that cryopreservation had a minimal impact on the transcriptome compared to the inherent heterogeneity between donors [3]. This supports the use of frozen samples when immediate processing is not feasible.
However, the choice of technology can influence the success of using stabilized cells. Technologies like Parse Biosciences' Evercode and 10x Genomics' Flex, which work with fixed cells, show lower levels of mitochondrial gene expression—a key indicator of cell stress—compared to methods requiring fresh cells [68]. This makes them suitable for workflows involving clinical sites where immediate processing is a challenge.
Table 3: Technical Parameter Comparison Across Sample Types & Protocols
| Parameter | Fresh Whole Cells (Zebrafish) [66] | Frozen Nuclei (Brain Tumor) [69] | Fresh vs. Frozen Oocytes [3] |
|---|---|---|---|
| Throughput (Cells/Nuclei per sample) | 5-10% of embryo cells recovered (~20k-40k total) | High-quality data from 54,402 nuclei | 144 oocytes sequenced (24 fresh & 24 frozen per donor) |
| Median Genes Detected | Varies by cell type (Atlas data) | ~1,385 genes per nucleus | Not specified; no major clustering difference |
| Mitochondrial Read % | Not specifically reported | Typically <1% (range 0.07–1.24%) | Not specified |
| Key Quality Metric | Representation of all major cell types under stress | Debris-free supernatant; low mitochondrial reads | No separate clustering of fresh vs. frozen by transcriptome |
Bioinformatic Correction of Cryopreservation-Induced Technical Variation
Within the context of single-cell RNA sequencing (scRNA-seq) research on embryos, the decision to use fresh or cryopreserved cells is pivotal. Cryopreservation enables flexibility in experimental timing and batch processing, which is particularly beneficial for large-scale studies or when working with precious, asynchronously collected clinical samples such as human embryos [71] [72]. However, the cryopreservation and thawing processes can introduce technical variation that confounds biological interpretation. This application note details the specific technical artifacts induced by cryopreservation in embryonic cells and provides a validated bioinformatic workflow for their identification and correction, ensuring data fidelity in studies comparing fresh and frozen specimens.
Benchmarking Cryopreservation Effects on Embryonic Cells
A systematic evaluation is the first step in understanding and ultimately correcting for cryopreservation-induced variation. The following protocol outlines a paired experimental design to quantify these effects.
Experimental Design for Benchmarking
Protocol: Paired Fresh vs. Cryopreserved scRNA-seq Sample Preparation
- Sample Division: Begin with a single-cell suspension from a dissociated embryo or embryonic tissue. Split the suspension into two equal aliquots.
- Fresh Processing (Control): Process one aliquot immediately for scRNA-seq according to your standard platform protocol (e.g., 10x Genomics).
- Cryopreserved Processing (Test): Cryopreserve the second aliquot.
- Cryomedium: Use a standard cryoprotectant, such as 10% DMSO in FBS, or a commercially available, serum-free alternative.
- Freezing: Employ a controlled-rate freezer, cooling at approximately -1°C per minute to -80°C, before transferring to liquid nitrogen for long-term storage.
- Thawing: Rapidly thaw the vial in a 37°C water bath. Immediately transfer cells to pre-warmed culture medium, pellet by centrifugation, and resuspend in PBS with a viability enhancer (e.g., 10% FBS). Optionally, perform a dead cell removal step to enrich viability before loading onto the scRNA-seq platform [72].
- Sequencing: Process both samples on the same scRNA-seq platform, ideally in the same sequencing run, to minimize technical batch effects.
Quantitative Assessment of Technical Effects
Data derived from paired experiments, as described above, consistently reveals specific technical differences between fresh and cryopreserved samples. The table below summarizes the key quantitative metrics affected by cryopreservation in sensitive cell types, including embryos.
Table 1: Key Quantitative Metrics Impacted by Cryopreservation in scRNA-seq
| Metric | Typical Effect of Cryopreservation | Implication for Data Analysis |
|---|---|---|
| Cell Viability | Decreased [72] | Lower cell recovery; may necessitate dead cell removal. |
| Number of Cells Recovered | Reduced or comparable [17] | Potential loss of specific, fragile cell populations. |
| UMIs per Cell | Reduced [17] | Lower sequencing depth and gene detection sensitivity. |
| Genes per Cell | Reduced [17] | Impacts ability to resolve fine-grained cell states. |
| Mitochondrial Gene Percentage | Can be elevated [68] [72] | Indicator of cellular stress; requires careful QC filtering. |
| Cell Type Proportions | Largely conserved, but minor shifts possible [72] | Overall heterogeneity is maintained, but quantitative accuracy may be affected. |
| Stress & Apoptosis Pathways | Upregulated [72] | Major confounder for biological interpretation. |
These observed effects underscore the necessity of a robust bioinformatic correction workflow to mitigate technical bias before downstream analysis.
Bioinformatic Correction Workflow
The following workflow provides a step-by-step guide for the computational identification and correction of cryopreservation-induced artifacts. The process is implemented in R using common packages from the Bioconductor and CRAN repositories.
Diagram 1: Bioinformatic Correction Workflow
Quality Control and Filtering
The first step is to perform stringent quality control to remove low-quality cells and ambient RNA, which can be more prevalent in cryopreserved samples.
Protocol: Scatter Plot for QC Thresholding
Data Integration and Batch Correction
Even after QC, a systematic difference between fresh and frozen samples often remains. Integration algorithms are used to align these datasets.
Protocol: Integrating Fresh and Frozen Datasets with Harmony
Assessing and Correcting for Technical Confounding
After integration, it is critical to verify that technical effects are not confounded with biological signals.
Protocol: Confounding Assessment and Regression
Validation of Correction Efficacy
The success of the bioinformatic correction must be validated using both technical and biological metrics.
Table 2: Metrics for Validating Correction Efficacy
| Validation Goal | Method | Expected Outcome Post-Correction |
|---|---|---|
| Cluster Mixing | UMAP visualization colored by condition (Fresh/Frozen) | Even mixing of conditions within biological clusters. |
| Statistical Independence | Chi-square test of cluster vs. condition | No significant association (p-value > 0.05). |
| Stress Gene Expression | Differential expression of known stress genes (e.g., FOS, JUN) [72] | No significant upregulation in cryopreserved cells. |
| Biological Signal Preservation | Differential expression of key developmental markers | Conservation of expected marker genes across conditions. |
The Scientist's Toolkit
The following reagents and computational tools are essential for implementing this protocol.
Table 3: Essential Research Reagent Solutions and Computational Tools
| Item | Function/Description | Example |
|---|---|---|
| Controlled-Rate Freezer | Ensures consistent and optimal cooling rate during cryopreservation to maximize cell viability. | Mr. Frosty Freezing Container [72] |
| Cryoprotectant | Protects cells from ice crystal formation and osmotic shock during freezing and thawing. | DMSO-based media (e.g., 10% DMSO in FBS) [72] |
| Dead Cell Removal Kit | Enriches viable cells from the thawed suspension, improving scRNA-seq data quality. | EasySep Dead Cell Removal (Annexin V) Kit [72] |
| scRNA-seq Platform | High-throughput platform for generating single-cell transcriptomes from fresh and frozen cells. | 10x Genomics Chromium [71] [72] |
| Data Integration Tool | Algorithm to merge fresh and frozen datasets while removing technical variation. | Harmony [68] |
| Single-Cell Analysis Suite | Comprehensive software platform for all steps of scRNA-seq analysis, from QC to DE. | Seurat (R package) [68] |
| Stress Gene Panel | A curated list of genes to monitor for successful mitigation of freeze-thaw stress. | FOS, JUN, heat shock proteins [72] |
This application note provides a standardized framework for benchmarking and correcting cryopreservation-induced technical variation in embryonic scRNA-seq studies. By implementing the paired experimental design and subsequent bioinformatic workflow detailed herein, researchers can confidently utilize cryopreserved samples, thereby enhancing experimental flexibility and power without compromising data integrity. The validated correction pipeline ensures that the biological signals of interest, such as those governing early embryonic development and cell fate decisions, are accurately discerned from technical artifacts.
Benchmarking Data Quality and Biological Fidelity in Fresh vs Frozen Samples
The choice between using fresh or cryopreserved cells in embryo single-cell RNA sequencing (scRNA-seq) research represents a critical methodological crossroad. This decision directly impacts data quality, biological interpretation, and experimental feasibility. While fresh cells often represent the ideal biological state, access to human embryonic material is severely limited by ethical considerations and scarcity. Cryopreservation enables rare sample collection and batch processing but may introduce technical artifacts. This application note provides a systematic, quantitative comparison of key performance metrics—detection efficiency, sensitivity, and specificity—across spatial transcriptomics and single-cell platforms, framing the analysis within the context of embryo research to guide experimental design.
Quantitative Platform Comparison
Performance Metrics Across Spatial Transcriptomics Platforms
Systematic benchmarking of high-throughput subcellular spatial transcriptomics platforms reveals significant variation in performance characteristics crucial for experimental planning [73].
Table 1: Performance Metrics of Subcellular Spatial Transcriptomics Platforms
| Platform | Technology Type | Resolution | Gene Panel Size | Sensitivity (Relative to scRNA-seq) | Specificity (NCP > 0.8) | Key Strengths |
|---|---|---|---|---|---|---|
| Xenium 5K | Imaging-based (ISS) | Subcellular | 5,001 genes | 1.2-1.5x higher than scRNA-seq [74] | High [74] | Superior sensitivity, high specificity |
| CosMx 6K | Imaging-based (ISH) | Subcellular | 6,175 genes | Lower than Xenium 5K [73] | Lowest among commercial platforms [74] | High-plex imaging |
| Visium HD FFPE | Sequencing-based | 2 μm | 18,085 genes | Lower than Xenium at tissue level [74] | High [74] | Whole transcriptome, FFPE optimized |
| Stereo-seq v1.3 | Sequencing-based | 0.5 μm | Unbiased whole transcriptome | High correlation with scRNA-seq [73] | High [74] | Highest spatial resolution |
Independent analysis of 25 Xenium datasets demonstrates its reproducibility in cell-type identification and high data quality, with an average of 76.8% of reads assigned to cells and only 0.21% of cells containing fewer than ten reads [74]. For sequencing-based spatial technologies, RNA capture efficiency remains a fundamental challenge, with leading methods like Decoder-seq capturing only 20-30% of target RNA, resulting in significant loss of biological signals, particularly for low-expression genes [75].
Impact of Sample Processing on RNA Capture Efficiency
Sample processing methodology significantly influences RNA capture efficiency and data quality. Formalin-fixed paraffin-embedded (FFPE) samples, while clinically practical, present substantial challenges due to nucleic acid cross-linking and fragmentation, with RNA degradation values (DV200) as low as 18 reported in some samples [75].
Table 2: Sample Processing Methods and Their Impact on RNA Quality
| Processing Method | RNA Integrity | Tissue Morphology | Key Applications | Technical Considerations |
|---|---|---|---|---|
| Fresh Frozen | High integrity | Well-preserved | Optimal for RNA sequencing | Requires stringent storage conditions [75] |
| FFPE | Severe degradation due to cross-linking [75] | Excellent preservation | Clinical archives, pathology | Requires specialized protocols (e.g., Stereo-seq V2) [75] |
| Cryopreserved Cells | Variable based on protocol | Viable after thawing | Rare samples, batch processing | Immediate processing post-thaw recommended [3] |
Innovative approaches have been developed to address these challenges. Stereo-seq V2 incorporates dedicated steps for deparaffinization, rehydration, and cross-linking reversal to improve FFPE compatibility, and uses random hexamer primers instead of poly(T) primers to achieve unbiased whole transcriptome capture, including non-coding RNAs [75]. For cryopreserved samples, a study comparing fresh and slow-frozen/thawed human oocytes from ovarian cortex found no significant transcriptomic differences, with transcriptional profiles not clustering separately between conditions [3]. This suggests that optimized cryopreservation protocols can preserve RNA integrity for scRNA-seq applications.
Experimental Protocols for Embryo scRNA-seq
Sample Preparation and Quality Control
Protocol 1: Processing of Fresh Embryo Cells for scRNA-seq
- Tissue Dissociation: Mechanically dissociate embryo tissues using a tissue chopper followed by enzymatic digestion with collagenase to generate single-cell suspensions [3].
- Cell Viability Assessment: Assess cell viability using trypan blue exclusion or automated cell counters. Proceed only with viability >70% for optimal results [76].
- Cell Sorting and Lysis: Manually isolate target cells under a dissection microscope. Transfer individual cells into wells containing lysis buffer to immediately stabilize RNA [3].
- Library Preparation: Utilize single-cell RNA library prep protocols such as seqWell PlexWell rapid single-cell RNA protocol or 10x Genomics Chromium system [76] [3].
Protocol 2: Processing of Cryopreserved Embryo Cells for scRNA-seq
- Rapid Thawing: Thaw cryopreserved cells rapidly in a 37°C water bath, then immediately transfer to pre-warmed culture medium [3].
- Viability Assessment: Assess post-thaw viability as above. Note that some reduction in viability compared to fresh samples is expected.
- Cell Processing: Process thawed cells immediately for scRNA-seq to minimize stress-induced transcriptional changes [3].
- Library Construction: Use platforms compatible with fixed or cryopreserved cells, such as 10x Genomics Chromium Fixed RNA Profiling or Parse Biosciences, which specifically support fixed single-cell or nuclear suspensions [76].
Platform-Specific Workflows
10x Genomics Chromium Workflow [76]:
- Input Requirements: 500-20,000 cells per sample for singleplex analysis; 500-5,000 cells per sample with on-chip multiplexing
- Library Construction: Single-cell RNA-seq library preparation includes cell/nucleus quality control
- Sequencing Parameters: Read length 28-10-10-90 bp; recommended depth >20,000 reads per cell
Parse Biosciences Workflow [76]:
- Input Requirements: 10,000-1,000,000 cells, accommodating up to 384 samples
- Library Construction: Utilizes fixed single-cell or nucleus suspensions
- Sequencing Parameters: Read length 74-10-10-86 bp; recommended depth >20,000 reads per cell
Visualizing Experimental Workflows
Single-Cell RNA Sequencing Decision Pathway
Spatial Transcriptomics Experimental Pipeline
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for Embryo scRNA-seq
| Category | Product/Platform | Key Features | Application Context |
|---|---|---|---|
| High-Throughput scRNA-seq | 10x Genomics Chromium iX | 500-20,000 cells/sample; supports fresh, frozen, fixed, and FFPE samples [76] | Large-scale embryo cell profiling |
| High-Throughput scRNA-seq | Illumina Single Cell Prep | 100-100,000 cells/sample; compatible with fresh or cryopreserved cells [76] | Flexible project scales |
| High-Throughput scRNA-seq | Parse Biosciences | 10,000-1,000,000 cells; supports fixed cells [76] | Massive-scale studies |
| Low-Throughput scRNA-seq | SMART-seq Technology | 1-100 cells; high sequencing depth [76] | Rare embryo cell analysis |
| Spatial Transcriptomics | Xenium In Situ | Subcellular resolution; 5001-plex gene panel; high sensitivity [73] [74] | Spatial mapping of embryo tissues |
| Spatial Transcriptomics | Stereo-seq V2 | 0.5 μm resolution; random hexamer primers; FFPE compatible [75] | Whole transcriptome spatial analysis |
| Sample Preservation | Cryopreservation Reagents | DMSO-based cryoprotectants | Long-term storage of rare embryo samples |
| Cell Isolation | Tissue Dissociation Kits | Enzyme blends for embryo tissue dissociation [3] | Single-cell suspension preparation |
The systematic comparison of detection efficiency, sensitivity, and specificity across single-cell and spatial transcriptomics platforms reveals a complex landscape where experimental goals must guide technology selection. For embryo research specifically, where sample availability is limited, cryopreservation emerges as a viable option when followed by optimized processing protocols. The minimal transcriptomic differences observed between fresh and frozen-thawed oocytes [3] suggest that properly executed cryopreservation can preserve RNA integrity for scRNA-seq applications. Platform selection should be driven by specific research questions: imaging-based spatial transcriptomics like Xenium offer superior sensitivity for targeted panels [74], while sequencing-based approaches like Stereo-seq provide unbiased whole transcriptome coverage [75]. As the field advances, integration of these multi-modal approaches with rigorous benchmarking against reference datasets [77] will be essential for unlocking the molecular mysteries of early embryonic development.
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity, proving particularly valuable in developmental biology and embryology. A critical methodological decision facing researchers is whether to use fresh or cryopreserved cells and tissues. This choice is paramount in embryo research, where sample availability is often limited and precious. This Application Note outlines validation frameworks to ensure that transcriptomic data derived from frozen samples maintain concordance with gold standard fresh samples and meet biological expectations. Evidence from recent studies indicates that with optimized protocols, cryopreservation has a minimal impact on the oocyte transcriptome, supporting the utility of frozen samples for sensitive single-cell analyses [3]. However, the process can induce a signature of freezing marked by heightened stress and activation in certain cell types, underscoring the need for rigorous validation [78].
Quantitative Framework: Validating Frozen Against Fresh Samples
A robust validation requires benchmarking frozen sample outputs against fresh controls using quantitative metrics. The following table summarizes key findings from a seminal study that performed this comparison on human ovarian cortex, a tissue relevant to embryology.
Table 1: Validation Metrics from a Comparative Study of Fresh and Slow-Frozen/Thawed Human Ovarian Cortex
| Validation Parameter | Fresh Ovarian Cortex | Slow-Frozen/Thawed Ovarian Cortex | Statistical & Biological Significance |
|---|---|---|---|
| Follicle Morphology (% Normal) | Donor 1: 86.7%Donor 2: 91.7%Donor 3: 96.1% | Donor 1: 91.0%Donor 2: 92.5%Donor 3: 91.1% | No significant difference in follicle stage distribution or morphology between conditions [3] |
| Follicle Density (per mm³) | Donor 1: 279.4Donor 2: 662.2Donor 3: 55.8 | Donor 1: 235.8Donor 2: 553.5Donor 3: 71.4 | Not significant for Donors 2 & 3; Significant (P=0.017) for Donor 1 [3] |
| Transcriptomic Profile | N/A | N/A | Transcriptional profiles did not cluster separately by condition; differences were insignificant compared to within-donor heterogeneity [3] |
| Differentially Enriched GO Terms | Chromosome segregation, Mitosis | Wound response, cAMP signaling, Extracellular matrix organization | Reflects immediate cellular response to cryopreservation stress rather than a fundamental change in cell identity [3] |
This validation framework demonstrates that while minor differences in follicle density may occur, the overall tissue architecture and global transcriptomic landscape remain intact post-cryopreservation. The core transcriptional identity of oocytes from early-stage follicles is preserved, validating the use of frozen tissue for fertility preservation and related research [3].
Experimental Protocols for scRNA-seq of Cryopreserved Tissues
Protocol A: Single-Cell RNA Sequencing from Frozen Ovarian Cortex
This protocol is adapted from a study that successfully sequenced oocytes from frozen human ovarian cortex [3].
1. Tissue Preparation and Cryopreservation:
- Obtain ovarian cortex and cut into squares measuring 10 x 10 x 1 mm.
- For slow-freezing, use a controlled-rate freezer with appropriate cryoprotectants.
- Store samples in liquid nitrogen until analysis.
2. Thawing and Oocyte Isolation:
- Rapidly thaw cryopreserved tissue squares in a 37°C water bath.
- Fragment the ovarian cortex using a McIlwain tissue chopper.
- Subject the fragmented tissue to gentle enzymatic digestion to liberate cells from the extracellular matrix.
- Under a dissection microscope, mechanically denude and manually pick individual live oocytes from primordial and primary follicles.
3. Library Preparation and Sequencing:
- Lyse single oocytes individually in lysis buffer.
- Perform library preparation using the seqWell PlexWell rapid single-cell RNA protocol.
- Pool libraries and sequence on an Illumina platform (e.g., NovaSeq6000) with 150-bp paired-end sequencing.
Limitations: This protocol focuses on the oocyte and does not efficiently capture the surrounding somatic cells. It also captures only the immediate transcriptomic effects of thawing [3].
Protocol B: Single-Nucleus RNA Sequencing (snRNA-seq) as an Alternative
For tissues that are difficult to dissociate or are rich in RNases (e.g., pancreas) or adipocytes, snRNA-seq from frozen tissue is a superior alternative [79] [64].
1. Nuclei Isolation from Frozen Tissue:
- Grind frozen tissue on dry ice.
- Homogenize tissue in a lysis buffer containing non-ionic detergents (e.g., NP-40), RNase inhibitors, and Bovine Serum Albumin (BSA) to isolate nuclei while preserving RNA integrity.
- Filter the homogenate through a 40 μm cell strainer to remove debris.
- Pellet nuclei via centrifugation and resuspend in a wash buffer.
2. Nuclei Sorting and Sequencing:
- To ensure a pure population of intact nuclei, sort nuclei using flow cytometry (e.g., gating on DAPI signal).
- Proceed with standard single-nucleus RNA-seq library preparation protocols, such as the 10x Genomics Chromium Single Cell Gene Expression solution [79].
Advantages: This method bypasses the need for viable single-cell suspensions, avoids dissociation-induced transcriptional stress artifacts, and allows for the analysis of archived frozen samples [79] [64].
Analytical Workflow and Pathway Diagram for Validation
A critical step in validating frozen samples is the use of statistically sound analytical methods for differential expression (DE) analysis. Evidence shows that methods which do not properly account for biological replicates are biased and prone to false discoveries, falsely identifying highly expressed genes as differentially expressed [80]. The recommended analytical workflow is summarized in the following diagram.
Diagram 1: Analytical workflow for validating frozen samples, emphasizing the critical pseudobulk step to avoid false discoveries.
The core principle is the use of a pseudobulk approach, where cells are aggregated within each biological replicate to form a single expression profile per replicate before DE testing. This method has been proven to outperform single-cell level DE methods, more accurately recapitulating ground-truth data from bulk RNA-seq and providing more reliable Gene Ontology term enrichment [80].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for scRNA-seq of Frozen Samples
| Item | Function/Application | Specific Example |
|---|---|---|
| Cryoprotectant Solutions | Protect cells from ice crystal formation and osmotic shock during the freeze-thaw cycle. | Not specified in results, but DMSO is commonly used. |
| RNase Inhibitors | Preserve RNA integrity during tissue dissociation and nucleus isolation procedures. | RNaseOut [79] |
| Lysis Buffer Components | Isolate nuclei from frozen tissue by lysing cell membranes while keeping nuclei intact. | NP-40 detergent [79] |
| Bovine Serum Albumin (BSA) | Reduces non-specific binding and protects cells/nuclei during washing and filtration steps. | Used in wash buffers for nucleus isolation [79] |
| Single-Cell Library Prep Kit | For generating barcoded scRNA-seq libraries from single cells or nuclei. | 10x Genomics Chromium Single Cell Gene Expression Solution [64] [81]; seqWell PlexWell [3] |
| Bioinformatics Tools | For processing, integrating, and analyzing single-cell data. | Seurat & Scanpy (general analysis) [81]; Cell Ranger (10x data processing) [81]; Harmony (batch correction) [81]; Pseudobulk methods with edgeR/DESeq2 (DE analysis) [80] |
The use of cryopreserved tissues in embryo scRNA-seq research is a valid and powerful approach when underpinned by a rigorous validation framework. Key to success is the combination of optimized wet-lab protocols for tissue freezing and single-cell/nucleus isolation with a robust analytical workflow that employs pseudobulk methods to control for false discoveries. By adhering to these guidelines, researchers can confidently utilize frozen samples to advance our understanding of embryonic development and reproductive biology, thereby leveraging the irreplaceable value of biobanked tissues.
Differential Expression Analysis Performance in Integrated Datasets
The fidelity of single-cell RNA sequencing (scRNA-seq) data is paramount in developmental biology, where precise transcriptomic measurements underpin our understanding of cell fate decisions. In embryo research, a fundamental methodological consideration is the choice between analyzing fresh versus cryopreserved cells, a decision that directly impacts the performance of downstream differential expression (DE) analysis in integrated datasets. This application note examines the performance characteristics of DE analysis when comparing transcriptomes from fresh and frozen human embryonic materials, providing validated protocols and analytical frameworks to ensure robust biological interpretation.
Recent studies have demonstrated that the slow-freezing and thawing process does not necessarily cause global transcriptomic disruption in sensitive cell types like human oocytes. A 2025 comparative study found that the transcriptional profiles of oocytes from fresh and slow-frozen/thawed human ovarian cortex did not cluster separately, indicating undetectable differences between the groups when compared to within-donor heterogeneity [3]. This finding provides crucial reassurance for fertility preservation and embryo research, suggesting that with proper protocols, cryopreserved materials can yield biologically valid DE results.
Quantitative Data Comparison: Fresh vs. Frozen Cellular Properties
Histological and Cellular Metrics
Table 1: Histological Analysis of Fresh and Frozen/Thawed Human Ovarian Cortex from Three Donors
| Donor | Condition | Normal Follicles (%) | Follicle Density (per mm³) | Stromal Cell Density |
|---|---|---|---|---|
| Donor 1 (16 years) | Fresh | 86.7% | 279.4 | 0.014 |
| Donor 1 (16 years) | Frozen/Thawed | 91.0% | 235.8 | 0.014 |
| Donor 2 (18 years) | Fresh | 91.7% | 662.2 | 0.014 |
| Donor 2 (18 years) | Frozen/Thawed | 92.5% | 553.5 | 0.016 |
| Donor 3 (27 years) | Fresh | 96.1% | 55.8 | 0.013 |
| Donor 3 (27 years) | Frozen/Thawed | 91.1% | 71.4 | 0.014 |
The histological data reveals minimal impact of cryopreservation on follicle morphology and density across donors of different ages. While some statistically significant differences were noted in follicle density for Donor 1 (P = 0.017) and stromal cell density for Donor 2 (P ≤ 0.001), these variations were not consistent across donors and likely reflect biological heterogeneity rather than systematic cryopreservation effects [3].
Transcriptomic Comparison Metrics
Table 2: Transcriptomic Analysis Performance in Fresh vs. Frozen Oocytes
| Analysis Metric | Fresh Oocytes | Frozen/Thawed Oocytes | Statistical Significance |
|---|---|---|---|
| Enriched GO Terms | Chromosome segregation, Mitosis | Wound response, cAMP signaling, Extracellular matrix organization | Distinct functional enrichment |
| Transcriptome Clustering | No separate clustering from frozen | No separate clustering from fresh | Undetectable between-group differences |
| Key Technical Factor | Baseline transcriptome | Minimal freeze-induced transcriptome shift | Within-donor heterogeneity > between-condition differences |
| Data Quality | 144 total oocytes sequenced (24 fresh + 24 frozen per donor × 3 donors) | Equivalent library prep and sequencing | NovaSeq6000, 150-bp paired-end |
The comparable performance in transcriptomic analyses between fresh and frozen specimens enables more flexible experimental designs, particularly valuable for embryonic research where sample availability is often limited [3].
Experimental Protocols for scRNA-seq of Fresh and Frozen Embryonic Cells
Protocol 1: Oocyte Isolation and Processing from Human Ovarian Cortex
Application: Isolation of live oocytes from primordial and primary follicles for scRNA-seq, compatible with both fresh and frozen tissue.
Reagents and Equipment:
- McIlwain tissue chopper
- Dissection microscope
- Enzymatic digestion solution
- Lysis buffer for scRNA-seq
- seqWell PlexWell rapid single-cell RNA library prep kit
- Illumina NovaSeq6000 platform
Methodology:
- Tissue Preparation: Cut human ovarian cortex into squares measuring 10 × 10 × 1 mm³.
- Cryopreservation (if applicable): Apply slow-freezing protocol to tissue squares, followed by thawing for processing.
- Tissue Dissociation: Fragment cortical squares using McIlwain tissue chopper followed by enzymatic digestion.
- Oocyte Isolation: Mechanically denude oocytes under dissection microscope.
- Single-Cell Capture: Place individual oocytes into wells containing lysis buffer for scRNA-seq.
- Library Preparation: Use seqWell PlexWell rapid single-cell RNA protocol with 150-bp paired-end sequencing on NovaSeq6000 Illumina platform [3].
Protocol 2: Integrated Reference-Based Cell Type Annotation
Application: Accurate cell type identification in embryonic datasets using integrated reference atlases.
Reagents and Equipment:
- Stabilized Uniform Manifold Approximation and Projection (UMAP)
- fast mutual nearest neighbor (fastMNN) integration methods
- Single-cell regulatory network inference and clustering (SCENIC)
- Published human embryo scRNA-seq datasets (zygote to gastrula)
Methodology:
- Data Integration: Employ fastMNN methods to integrate multiple embryonic datasets into a unified reference.
- Reference Building: Incorporate six published human datasets covering development from zygote to gastrula (3,304 early human embryonic cells).
- Lineage Annotation: Validate annotations with available human and nonhuman primate datasets.
- Query Projection: Project query datasets onto reference using stabilized UMAP to annotate with predicted cell identities.
- Validation: Perform SCENIC analysis to confirm lineage identities through transcription factor activities [6].
Protocol 3: Cross-Platform Validation for Sensitive Cell Types
Application: Ensuring robust DE analysis across sequencing platforms, particularly for challenging cells like neutrophils.
Reagents and Equipment:
- 10× Genomics Chromium Flex
- PARSE Biosciences Evercode
- HIVE (Honeycomb Biotechnologies) protocols
- Flow cytometry equipment for validation
Methodology:
- Sample Splitting: Divide identical cell suspensions across multiple platforms.
- Platform-Specific Processing: Follow manufacturer protocols for each scRNA-seq platform.
- Data Quality Assessment: Compare gene capture efficiency, cell recovery rates, and multiplet rates.
- Biological Validation: Confirm population identities using flow cytometry as ground truth.
- Concordance Analysis: Evaluate consistency of DE results across platforms [82].
Visualization of Experimental Workflows and Analytical Relationships
Differential Expression Analysis Workflow
Diagram 1: Differential Expression Analysis Workflow. This workflow illustrates the sequential steps from sample collection through functional validation, highlighting key computational integration steps.
Fresh vs. Frozen Experimental Design
Diagram 2: Fresh vs. Frozen Experimental Design. This diagram outlines the split-sample approach for direct comparison of preservation methods, controlling for biological variability.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Research Reagent Solutions for Embryo scRNA-seq Studies
| Category | Product/Platform | Key Features | Application in Embryo Research |
|---|---|---|---|
| scRNA-seq Platforms | 10x Genomics Chromium | High-throughput (80,000 cells/run), ~65% capture efficiency, low multiplet rate (<0.9%) | General cell atlas work, broad transcriptome coverage [83] |
| scRNA-seq Platforms | 10x Genomics Flex | FFPE compatibility, 4% PFA fixation, multiplexing (16 samples/channel) | Biobanked samples, multi-site embryo studies [84] |
| scRNA-seq Platforms | BD Rhapsody | Microwell-based, 70% capture rate, tolerant of 65% viability | Clinical samples with variable quality [83] |
| scRNA-seq Platforms | MobiDrop | Cost-effective, automated workflow, fresh/frozen/FFPE compatible | Large-scale embryo studies with budget constraints [83] |
| Analysis Tools | fastMNN Integration | Batch correction, dataset integration | Combining multiple embryo datasets [6] |
| Analysis Tools | SCENIC | Transcription factor network inference | Lineage specification analysis [6] |
| Analysis Tools | Stabilized UMAP | Dimensionality reduction, reference projection | Cell identity prediction in embryo models [6] |
| Specialized Kits | seqWell PlexWell | Rapid single-cell RNA library prep | Oocyte transcriptome analysis [3] |
The performance of differential expression analysis in integrated datasets spanning fresh and frozen embryonic cells demonstrates remarkable robustness when proper protocols are followed. The minimal transcriptomic impact of cryopreservation in oocytes [3] provides a foundation for leveraging valuable biobanked specimens in embryonic research. However, rigorous validation through integrated reference atlases [6] and cross-platform consistency checks [82] remains essential.
Best practices recommend:
- Implementing split-sample designs that directly compare fresh and frozen aliquots from the same biological source
- Utilizing integrated reference atlases for proper cell type annotation and batch correction
- Applying platform-specific validation for sensitive cell types
- Incorporating functional enrichment analyses to interpret biological significance of minimal transcriptomic shifts
These protocols enable researchers to confidently utilize both fresh and cryopreserved embryonic materials, significantly expanding the potential scale and scope of developmental studies while maintaining analytical rigor in differential expression analysis.
A primary goal of modern assisted reproductive technology (ART) is to achieve successful live births. For researchers and clinicians, this translates to a critical need to understand how laboratory techniques, such as ovarian tissue cryopreservation, impact not just cellular markers but ultimate reproductive outcomes. While embryo implantation and live birth rates are the final metrics of success, the foundational cellular and molecular events within the oocyte often dictate these results. This Application Note details the correlation between technical findings from single-cell RNA-sequencing (scRNA-seq) of fresh and cryopreserved oocytes and their implications for clinical practice. We focus on the minimal transcriptomic impact of slow-freezing protocols on early-stage human oocytes and provide a framework for validating such techniques within a clinical research setting.
Key Findings: scRNA-seq of Fresh vs. Slow-Frozen Oocytes
A recent comparative study employed scRNA-seq to investigate the immediate transcriptomic impact of the slow-freezing and thawing process on early-stage oocytes isolated from human ovarian cortex [3]. The study aimed to determine if this process negatively impacts the oocyte transcriptome compared to fresh controls.
The following tables consolidate the key quantitative results from the study, which utilized ovarian tissue from three healthy premenopausal donors (aged 16, 18, and 27) and sequenced 144 oocytes in total (24 fresh and 24 frozen/thawed from each donor) [3].
Table 1: Histological Analysis of Follicle and Stromal Integrity Post-Cryopreservation
| Donor | Condition | % Normal Follicles | Follicle Density (per mm³) | Stromal Cell Density |
|---|---|---|---|---|
| Donor 1 | Fresh | 86.7% | 279.4 | 0.014 |
| Donor 1 | Frozen/Thawed | 91.0% | 235.8 | 0.014 |
| Donor 2 | Fresh | 91.7% | 662.2 | 0.014 |
| Donor 2 | Frozen/Thawed | 92.5% | 553.5 | 0.016 |
| Donor 3 | Fresh | 96.1% | 55.8 | 0.013 |
| Donor 3 | Frozen/Thawed | 91.1% | 71.4 | 0.014 |
Table 2: Key Transcriptomic and Functional Outcomes
| Analysis Parameter | Finding |
|---|---|
| Transcriptomic Clustering | No separate clustering of fresh and frozen/thawed oocyte transcriptional profiles; differences were undetectable compared to within-donor heterogeneity [3]. |
| Group Mean Shift | A small, parallel shift was observed between fresh and frozen/thawed oocytes across all three donors [3]. |
| Fresh Oocyte Enrichment | Gene ontology terms related to chromosome segregation and mitosis [3]. |
| Frozen/Thawed Oocyte Enrichment | Gene ontology terms related to wound response, cAMP signaling, and extracellular matrix organization [3]. |
| Clinical Correlation | Historical data shows transplanted slow-frozen/thawed ovarian cortex can restore normal serum FSH levels and regular menstrual cycles within 5 months, though non-significantly lower rates of pregnancies and live births have been reported [3]. |
Detailed Experimental Protocol
This section outlines the core methodology used in the cited study to generate the findings above, providing a reproducible protocol for comparing fresh and cryopreserved human oocytes.
Ovarian Cortex Processing and Cryopreservation
- Tissue Preparation: Human ovarian cortex from cadaveric donors is cut into squares measuring 10 × 10 × 1 mm³ [3].
- Slow-Freezing Protocol: Cortex squares designated for cryopreservation are subjected to a controlled slow-freezing process using a standard cryoprotectant solution. The specific freezing parameters (cooling rate, seeding temperature) should follow an established, clinically validated protocol. After a storage period, samples are thawed using a standardized rapid warming procedure [3].
- Fresh Control Processing: Fresh cortex squares are processed directly without subjecting them to the freezing and thawing cycle [3].
Isolation of Live Oocytes from Early-Stage Follicles
- Tissue Dissociation: Both fresh and frozen/thawed ovarian cortex squares are fragmented using a McIlwain tissue chopper and subsequently subjected to a gentle enzymatic digestion to liberate intact follicles and oocytes [3].
- Oocyte Denudation: Under a dissection microscope, individual oocytes are mechanically denuded of surrounding somatic cells to obtain a pure oocyte population for sequencing [3].
- Cell Viability: Maintaining cell viability during this dissociation process is critical. The ideal sample viability should be between 70% and 90%, with intact cell morphology [29]. All manipulations should be gentle and performed on ice where possible to arrest metabolic activity and reduce the upregulation of stress response genes [29].
Single-Cell RNA-Sequencing Library Preparation and Analysis
- Single-Cell Lysis: Individual, denuded oocytes are placed into wells containing a lysis buffer to capture the entire transcriptome [3].
- Library Prep: scRNA-seq libraries are prepared from the lysed oocytes using the seqWell PlexWell rapid single-cell RNA protocol. This is a 3'-end counting, droplet-based method that incorporates Unique Molecular Identifiers (UMIs) [3] [4].
- Sequencing: Pooled libraries are sequenced using a high-throughput platform, such as the NovaSeq6000 Illumina platform, with a 150-bp paired-end sequencing configuration [3].
- Quality Control (QC): Prior to analysis, rigorous QC is performed on the scRNA-seq data. This involves filtering out low-quality libraries based on three key metrics [85] [60]:
- Library Size: The total sum of counts per cell. Cells with small library sizes indicate RNA loss or inefficient amplification.
- Number of Expressed Genes: The number of genes with non-zero counts per cell. Cells with very few genes suggest failed capture.
- Mitochondrial Read Proportion: The fraction of reads mapped to mitochondrial genes. High proportions are indicative of poor-quality, perforated cells [85] [60].
- Data Analysis: After QC, standard analysis includes normalization, data correction, feature selection, and dimensionality reduction (e.g., PCA). Downstream analysis focuses on comparing transcriptional profiles between fresh and frozen/thawed groups to identify differential expression and enriched pathways [2] [60].
Visualizing the Cellular Response to Cryopreservation
The transcriptomic analysis revealed a specific molecular signature in frozen/thawed oocytes. The following diagram illustrates the key signaling pathways and biological processes enriched in these cells, linking the technical finding to a potential cellular mechanism.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Oocyte scRNA-seq Studies
| Item | Function/Application |
|---|---|
| McIlwain Tissue Chopper | Standardized fragmentation of human ovarian cortex prior to enzymatic digestion for oocyte isolation [3]. |
| Slow-Freezing Cryoprotectant | A clinically validated solution (e.g., containing DMSO, ethylene glycol, and sucrose) to protect cells from ice crystal formation during controlled-rate freezing [3]. |
| seqWell PlexWell Kit | A droplet-based, 3'-end scRNA-seq library preparation system that incorporates UMIs for accurate transcript counting; enables multiplexing [3] [4]. |
| Unique Molecular Identifiers (UMIs) | Short nucleotide barcodes that label individual mRNA molecules during reverse transcription, allowing for the correction of amplification bias in droplet-based protocols [4] [60]. |
| HEPES-buffered Salt Solution | Cell culture media without calcium or magnesium, used during cell suspension to prevent aggregation and minimize clumping [29]. |
| Ficoll or Optiprep | Density gradient media for separating viable cells/nuclei from debris and dead cells during suspension preparation, crucial for sample purity [29]. |
Best Practice Recommendations Based on Current Evidence and Technological Capabilities
This application note provides best-practice recommendations for single-cell RNA sequencing (scRNA-seq) of embryonic tissues, with a specific focus on the critical comparison between fresh and frozen sample preparation. The choice between these approaches significantly impacts experimental outcomes, particularly for delicate samples such as embryos where cellular viability and transcriptomic integrity are paramount. We synthesize current evidence from multiple studies to guide researchers in selecting appropriate methodologies, optimizing protocols, and interpreting data within the context of embryonic development research. The recommendations emphasize practical implementation while maintaining scientific rigor for researchers, scientists, and drug development professionals working in developmental biology and reproductive medicine.
Single-cell RNA sequencing has revolutionized developmental biology by enabling unprecedented resolution in mapping cellular heterogeneity and lineage specification during embryogenesis. The fundamental decision between using fresh or frozen samples represents a critical methodological crossroads with profound implications for data quality, experimental design, and biological interpretation. While fresh tissues ideally preserve full transcriptomic profiles, practical constraints often necessitate cryopreservation, particularly for rare mutant embryos or clinically derived samples.
Recent technical advancements have substantially improved the viability and reliability of frozen samples for scRNA-seq. A landmark study on human oocytes from ovarian cortex demonstrated that slow-freezing and thawing processes resulted in minimal transcriptomic impact when properly optimized [3]. The transcriptional profiles of fresh and frozen/thawed oocytes did not cluster separately, indicating undetectable differences between the two groups when compared to within-donor heterogeneity [3]. This finding challenges previous assumptions about inevitable cryopreservation artifacts and highlights the potential of optimized frozen protocols for sensitive embryonic materials.
Quantitative Evidence: Comparative Analysis of Fresh vs. Frozen Sample Performance
Table 1: Comparative Performance Metrics for Fresh vs. Frozen Embryonic Cells in scRNA-seq
| Performance Metric | Fresh Samples | Frozen/Thawed Samples | Experimental Context |
|---|---|---|---|
| Transcriptomic Differences | Reference standard | No separate clustering from fresh; parallel shifts in group means [3] | Human oocytes from primordial/primary follicles |
| Gene Detection Sensitivity | Optimal for full-length protocols | High for 3'-end counting methods; improved with intronic read inclusion [69] | Pediatric glioma nuclei; various platforms |
| Cell Type Identification | All expected populations detected | Preserved identification of distinct cell populations [69] [3] | Brain tumors; human oocytes |
| Differential Expression Results | Baseline for comparison | Consistent directionality with fresh; some pathway enrichment differences [3] | Multi-donor oocyte study |
| Technical Noise | Lower mitochondrial genes [69] | Variable mitochondrial reads (platform-dependent) [69] | Comparison of 10X, Drop-seq, Fluidigm C1 |
| Process-Induced Transcriptional Changes | Minimal when viability maintained | Enriched for wound response, cAMP signaling, ECM organization [3] | Pathway analysis of frozen/thawed oocytes |
| Experimental Flexibility | Requires immediate processing | Enables batch processing, genotyping precedent to sequencing [86] | Mutant mouse embryo workflow |
Table 2: Platform Comparison for Embryonic scRNA-seq Applications
| Platform/Method | Transcript Coverage | Cell Throughput | Cost per Cell | Best Application for Embryonic Samples |
|---|---|---|---|---|
| 10X Genomics Chromium | 3'-end counting | High (up to 10,000 cells) | Moderate | Large-scale embryonic cell atlases; mutant screening |
| Smart-Seq2 | Full-length | Low (96-384 cells) | High | Isoform analysis; low-abundance gene detection [87] |
| Fluidigm C1 | Full-length | Medium (up to 800 cells) | High | Detailed analysis of small embryo cell numbers [69] |
| Drop-seq | 3'-end counting | High (thousands of cells) | Low | Large-scale mutant embryo studies [69] |
| snRNA-seq | 3'-end or full-length | Platform-dependent | Platform-dependent | Frozen archival embryos; difficult-to-dissociate tissues [69] |
Experimental Protocols for Embryo scRNA-seq
Optimized Protocol for Mutant Mouse Embryo scRNA-seq
The following protocol has been specifically optimized for mutant mouse embryos during gastrulation stages (E6.5-E8.0), addressing technical challenges of genotyping, sample synchronization, and cell viability [86]:
Synchronized Timed Pregnancies:
- Establish breeding trios (2 female mice: 1 male) to increase synchronized pregnancies
- Check for vaginal plugs daily before 9 AM; consider noon on plug discovery day as E0.5
- Record female age, pregnancy history, and estrous stage for optimal breeding efficiency
- Separate plugged females immediately for embryo isolation at desired developmental stage
Embryo Isolation and Same-Day Genotyping:
- Euthanize pregnant dam via CO2 followed by cervical dislocation
- Dissect uterine horn and transfer to ice-cold DPBS−/− in Petri dish
- Isolate individual decidual swellings under stereomicroscope
- Extract embryos using fine forceps and scissors
- Perform rapid genotyping protocol (3 hours) using part of embryonic tissue
- Process remaining tissue immediately for scRNA-seq to maintain viability
Cell Dissociation and Quality Control:
- For pre-gastrulation embryos (E6.5-E7.5): use enzymatic digestion with TrypLE for 5-7 minutes at 37°C
- For later embryos (E7.5-E8.0): combine enzymatic and mechanical dissociation
- Filter through 40μm flow cytometry strainer
- Assess viability via Trypan Blue exclusion (>90% viability required)
- Adjust cell concentration to 700-1,200 cells/μL for target capture
Single-Nucleus RNA-seq Protocol for Frozen Samples
For frozen embryonic tissues or when cellular dissociation proves problematic, this optimized nuclear isolation protocol provides a robust alternative [69]:
Nuclear Isolation from Frozen Tissue:
- Begin with 20-50mg frozen embryonic tissue on dry ice
- Chop tissue in ice-cold lysis buffer (without detergent) using scalpel
- Dounce tissue with loose pestle (10-15 strokes) to open cell walls
- Filter through 40μm strainer to remove debris
- Centrifuge at 500xg for 5 minutes at 4°C
- Wash pellet with lysis buffer without detergent (2-3 times)
- Resuspend in nuclear storage buffer for immediate use or short-term freezing (-80°C, 2-3 days maximum)
Quality Assessment:
- Examine nuclear integrity and debris via microscopy
- Count nuclei using hemocytometer
- Expect mitochondrial read proportion <1% in final sequencing data
- Optimal concentration: 3,500-7,000 nuclei/μL for 10X Genomics
Experimental Workflow Visualization
Technical Considerations for Experimental Design
Impact of Sample Preparation on Transcriptomic Data
The choice between whole-cell and single-nucleus approaches significantly influences data interpretation, particularly for embryonic samples:
Gene Detection Patterns:
- Whole-cell scRNA-seq: Captures cytoplasmic mRNA, providing comprehensive transcriptome coverage
- snRNA-seq: Enriched for nuclear transcripts and nascent RNA; improved detection of intronic regions [69]
- Platform-specific biases: Full-length protocols (Smart-Seq2) superior for isoform analysis; 3'-end counting (10X) provides better throughput [87]
Technical Artifacts and Mitigation:
- Frozen samples show minimal batch effects when processed consistently [3]
- Mitochondrial read percentage: Higher in whole-cell (<5%) versus nuclear (<1%) preparations [69]
- Amplification biases: Reduced using UMIs for 3'-end counting methods [87]
Cell Type Identification and Resolution
Both fresh and frozen preparation methods effectively identify major embryonic cell types, with considerations for rare populations:
- Fresh samples: Superior for detecting transient developmental states with low RNA content
- Frozen nuclei: Preserve cellular diversity but may underrepresent fragile cell types [69]
- Experimental validation: Spatial transcriptomics (MERFISH) confirms cell type localization in both preparations [88]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Critical Reagents for Embryo scRNA-seq
| Reagent/Material | Function | Application Notes | References |
|---|---|---|---|
| McIlwain Tissue Chopper | Precision cutting of embryonic tissues | Maintains tissue architecture while enabling dissociation | [86] |
| TrypLE Enzyme | Gentle cell dissociation | Superior viability for embryonic cells versus trypsin | [86] |
| Poly[T] Primers | mRNA capture during reverse transcription | Selective for polyadenylated RNA; reduces ribosomal RNA | [87] |
| Unique Molecular Identifiers (UMIs) | Correction for amplification biases | Essential for quantitative accuracy in droplet-based methods | [87] |
| Dulbecco's Modified Eagle Medium (DMEM)/10% FBS | Embryo collection medium | Maintains viability during dissection and processing | [86] |
| OptiPrep Density Gradient Medium | Nuclear purification | Alternative to sucrose cushion; slightly lower yield | [69] |
| seqWell PlexWell Kit | Low-input library preparation | Effective for limited embryonic cell numbers | [3] |
| Chromium Single Cell 3' Kit (10X Genomics) | Droplet-based library preparation | High-throughput profiling of embryonic cell populations | [69] |
Data Analysis Framework for Comparative Studies
Analytical Pipeline for Fresh vs. Frozen Comparisons
A standardized analytical approach ensures valid comparisons between sample preparation methods:
Quality Control and Filtering:
- Remove low-quality cells with <200 detected genes or >15% mitochondrial reads
- Eliminate potential doublets using DoubletFinder or similar tools
- Normalize using SCTransform to address technical variability
Comparative Analysis:
- Integrate datasets from fresh and frozen preparations using Harmony or Seurat CCA
- Assess cluster conservation across preparations
- Identify preparation-specific differentially expressed genes
- Validate findings with orthogonal methods (spatial transcriptomics, FISH)
Biological Interpretation:
- Pathway enrichment analysis of preparation-sensitive genes
- Cell type proportion comparisons across methods
- Developmental trajectory reconstruction using Slingshot or PAGA
Signaling Pathway Analysis
Based on current evidence and technological capabilities, we recommend the following best practices for embryo scRNA-seq studies:
Prioritize fresh sample processing when investigating subtle transcriptional states, conducting isoform-level analyses, or working with particularly fragile embryonic cell types.
Implement optimized freezing protocols for rare mutant embryos, clinical materials, or when experimental design requires batch processing across multiple timepoints.
Select platform appropriately: High-throughput 3'-end counting (10X Genomics) for large-scale atlases; full-length protocols (Smart-Seq2) for focused mechanistic studies.
Incorporate nuclear RNA in sequencing libraries when working with frozen samples to improve gene detection and cell type annotation.
Validate key findings with orthogonal methods such as spatial transcriptomics or multiplexed FISH to control for preparation-specific artifacts.
The evolving methodological landscape continues to bridge the gap between fresh and frozen sample performance, expanding experimental possibilities for developmental biology research while maintaining transcriptomic fidelity.
Conclusion
The decision between using fresh or frozen samples for embryo scRNA-seq requires careful consideration of research objectives, technical capabilities, and biological questions. Current evidence demonstrates that cryopreserved samples can yield high-quality transcriptomic data comparable to fresh samples when proper protocols are followed, with recent studies showing minimal transcriptome impact in frozen-thawed human oocytes. Methodological rigor in sample preparation, platform selection, and bioinformatic analysis is paramount for data quality. As single-cell technologies continue to evolve, integrating multi-omics approaches and spatial context will further enhance our understanding of embryonic development. These advances promise to accelerate discoveries in reproductive biology and improve clinical outcomes in fertility preservation and assisted reproduction. Future directions should focus on standardized protocols, larger validation studies, and developing specialized computational tools for embryonic cell types.
References
- 1. Single Cell RNA Sequencing and Its Impact on ... [https://www.mdpi.com/1422-0067/26/16/7741]
- 2. A practical guide for single-cell transcriptome data analysis ... [https://www.sciencedirect.com/science/article/pii/S0168010225000653]
- 3. Single-cell analysis comparing early-stage oocytes from fresh ... [https://pubmed.ncbi.nlm.nih.gov/39919251/]
- 4. Navigating single-cell RNA-sequencing: protocols, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 5. How single cell sequencing data analysis works [https://www.10xgenomics.com/blog/how-single-cell-sequencing-data-analysis-works]
- 6. A comprehensive human embryo reference tool using ... [https://www.nature.com/articles/s41592-024-02493-2]
- 7. A post-implantation model of human embryo development ... [https://www.sciencedirect.com/science/article/pii/S2211124725011441]
- 8. A practical guide for single-cell transcriptome data analysis ... [https://pubmed.ncbi.nlm.nih.gov/40164433/]
- 9. Extraction of high quality and high yield RNA from frozen ... [https://www.nature.com/articles/s41598-024-58576-9]
- 10. Optimizing RNA quality in cryopreserved tissues without ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584263/]
- 11. The effects of cryopreservation on PBMCs transcriptome ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1690316/full]
- 12. Cell Cryopreservation Market Size & Outlook, 2026-2034 [https://straitsresearch.com/report/cell-cryopreservation-market]
- 13. Freezing cells for success: DMSO-free redefining the future ... [https://www.news-medical.net/news/20251125/Freezing-cells-for-success-DMSO-free-redefining-the-future-of-cryopreservation.aspx]
- 14. Protocols and Best Practices for Freezing Cells [https://www.stemcell.com/cryopreservation-basics-protocols-and-best-practices-for-freezing-cells]
- 15. Cryopreservation - Key Survey Findings from the ISCT ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/05/28/cryopreservation-key-survey-findings-from-the-isct]
- 16. Effects of Cryopreservation and Thawing on Single-Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7458795/]
- 17. A structured evaluation of cryopreservation in generating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10391561/]
- 18. Long-Term Storage Does Not Affect the Expression Profiles ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.751467/full]
- 19. Effects of vitrification and cryostorage duration on single- ... [https://link.springer.com/article/10.1007/s11684-020-0792-7]
- 20. Multiple freeze-thaw cycles lead to a loss of consistency in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-07381-z]
- 21. Integrated transcriptome and physiological analysis reveal ... [https://www.sciencedirect.com/science/article/abs/pii/S0926669023002042]
- 22. DMSO cryopreservation is the method of choice to ... [https://www.nature.com/articles/s41598-019-46932-z]
- 23. Single-cell transcriptome conservation in cryopreserved cells ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1171-9]
- 24. Trends and Outcomes of Fresh and Frozen Donor Oocyte ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11560672/]
- 25. Optimized methods for scRNA-seq and snRNA- ... [https://www.nature.com/articles/s42003-024-07445-2]
- 26. Establishing single cell RNA transcriptomics: a brief guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403637/]
- 27. Powerful scRNA-seq Data Analysis Tools in 2025 [https://www.nygen.io/resources/blog/single-cell-scrna-seq-multi-omics-data-analysis-tools]
- 28. A single-cell and single-nucleus RNA-Seq toolbox for fresh ... [https://www.nature.com/articles/s41591-020-0844-1]
- 29. Getting Started with scRNA-seq: Experimental Design and ... [https://www.parsebiosciences.com/blog/getting-started-with-scrna-seq-experimental-design-and-sample-preparation/]
- 30. scGraphformer: unveiling cellular heterogeneity and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11543810/]
- 31. Single-Cell RNA Sequencing From Frozen Samples Feasible | GenomeWeb [https://www.genomeweb.com/sequencing/single-cell-rna-sequencing-frozen-samples-feasible]
- 32. Single-cell dissociation from zebrafish larvae [https://www.protocols.io/view/single-cell-dissociation-from-zebrafish-larvae-hf5cb3q2x.html]
- 33. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 34. Direct Comparative Analyses of 10X Genomics Chromium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8602399/]
- 35. Single cell RNA-seq: An introductory overview and tools for ... [https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started]
- 36. Scalable single-cell RNA sequencing from full transcripts ... [https://www.nature.com/articles/s41587-022-01311-4]
- 37. Battle of the Single-Cell Platforms: Which scRNA ... [https://bridgeinformatics.com/battle-of-the-single-cell-platforms-which-scrna-sequencing-technology-is-right-for-your-research/]
- 38. Comparison of Single Cell Transcriptome Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10743076/]
- 39. Systematic comparison of high-throughput single-cell RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-07358-4]
- 40. Cell Preparation for Single Cell Protocols [https://www.10xgenomics.com/support/universal-five-prime-gene-expression/documentation/steps/sample-prep/single-cell-protocols-cell-preparation-guide]
- 41. Comparative Analysis of Single-Cell RNA Sequencing ... [https://www.sciencedirect.com/science/article/pii/S1097276517300497]
- 42. Power Analysis of Single Cell RNA-Sequencing Experiments [https://pmc.ncbi.nlm.nih.gov/articles/PMC5376499/]
- 43. A new protocol for single-cell RNA-seq reveals stochastic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7259953/]
- 44. Optimized nucleus isolation protocol from frozen mouse ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2023.1243863/full]
- 45. computational approach for visualization of single cell RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11401305/]
- 46. A Quantitative Framework for Evaluating Single-Cell Data ... [https://www.sciencedirect.com/science/article/pii/S2211124720305258]
- 47. Single-cell and spatial transcriptomics integration [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528096/]
- 48. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 49. Comparison of imaging based single-cell resolution spatial ... [https://www.nature.com/articles/s41467-025-63414-1]
- 50. Applications of single-cell RNA sequencing in drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141847/]
- 51. Single-cell sequencing to multi-omics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11438019/]
- 52. Scientists have developed a way to scale up spatial ... [https://www.broadinstitute.org/news/scaling-up-spatial-genomics]
- 53. Spatial integration of multi-omics single-cell data with SIMO [https://www.nature.com/articles/s41467-025-56523-4]
- 54. A comprehensive review of spatial transcriptomics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12199153/]
- 55. A computational pipeline for spatial mechano-transcriptomics [https://www.nature.com/articles/s41592-025-02618-1]
- 56. A single‐cell RNA labeling strategy for measuring stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9912023/]
- 57. Evaluating cell-specific gene expression using single ... [https://www.nature.com/articles/s41598-025-21595-1]
- 58. Batch effects and the effective design of single-cell gene expression studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5206706/]
- 59. Batch correction methods used in single-cell RNA sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315870/]
- 60. Current best practices in single‐cell RNA‐seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 61. A Beginner's Guide to Single-cell RNA Sequencing [https://rna.cd-genomics.com/resource/guide-single-cell-rna-sequencing.html]
- 62. A simplified preparation method for single-nucleus RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11997191/]
- 63. Recent advancements in tissue dissociation techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622302/]
- 64. Current Methodological Challenges of Single-Cell and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8455274/]
- 65. Quality controls after tissue dissociation for single cell RNA ... [https://singleron.bio/quality-controls-after-tissue-dissociation-for-single-cell-rna-sequencing/]
- 66. Proteostasis governs differential temperature sensitivity across ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11178971/]
- 67. Damaging effects and altered gene expression from ... [https://www.sciencedirect.com/science/article/pii/S0048969725017693]
- 68. Comparison of single-cell RNA-seq methods to enable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539239/]
- 69. A simplified preparation method for single-nucleus RNA ... [https://www.nature.com/articles/s41598-025-97053-9]
- 70. Optimizing single cell RNA sequencing of stem ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1590889/full]
- 71. Single-cell ultra-high-throughput multiplexed chromatin ... [https://www.nature.com/articles/s41592-025-02700-8]
- 72. Cryopreservation of human cancers conserves tumour ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00885-z]
- 73. Systematic benchmarking of high-throughput subcellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534522/]
- 74. Optimizing Xenium In Situ data utility by quality assessment ... [https://www.nature.com/articles/s41592-025-02617-2]
- 75. Enhancing RNA Capture Efficiency in Spatial Transcriptomics [https://www.mdpi.com/1422-0067/26/22/11076]
- 76. Single-Cell Sequencing - Center for Genetic Medicine [https://www.cgm.northwestern.edu/cores/nuseq/services/next-generation-sequencing/single-cell-seq.html]
- 77. A comprehensive human embryo reference tool using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11725501/]
- 78. Single-cell RNA sequencing of cells from fresh or frozen ... [https://pubmed.ncbi.nlm.nih.gov/38304946/]
- 79. Optimized nucleus isolation protocol from frozen mouse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10575574/]
- 80. Confronting false discoveries in single-cell differential ... [https://www.nature.com/articles/s41467-021-25960-2]
- 81. Top 10 Bioinformatics Tools for scRNA-seq in 2025 [https://neovarsity.org/blogs/top-bioinformatics-tools-for-scrna-seq]
- 82. Comparison of single-cell RNA-seq methods to enable ... [https://www.sciencedirect.com/science/article/pii/S2667237525002097]
- 83. Comparing Single-Cell Sequencing Platforms [https://www.omicsempower.com/blog/single-cell-sequencing-platforms-comparison/]
- 84. Single Cell Solutions Comparison [https://sgt.uic.edu/single-cell-transcriptomics-comparison/]
- 85. Chapter 1 Quality Control | Basics of Single-Cell Analysis ... [https://bioconductor.org/books/3.23/OSCA.basic/quality-control.html]
- 86. Single-Cell RNA Sequencing of Mutant Whole Mouse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11648987/]
- 87. Navigating single-cell RNA-sequencing: protocols, tools ... [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00044-5]
- 88. Single-cell transcriptomic and genomic changes in the ... [https://www.nature.com/articles/s41586-025-09435-8]