In Situ Hybridization Sensitivity Comparison: A Guide for Researchers and Developers
This article provides a comprehensive comparison of in situ hybridization (ISH) sensitivity for researchers, scientists, and drug development professionals.
In Situ Hybridization Sensitivity Comparison: A Guide for Researchers and Developers
Abstract
This article provides a comprehensive comparison of in situ hybridization (ISH) sensitivity for researchers, scientists, and drug development professionals. It covers foundational principles of ISH and its advantages over immunostaining, explores modern high-sensitivity methodologies like RNAscope and HCR, and offers practical troubleshooting guidance. The content synthesizes validation data and comparative benchmarks to aid in the selection of optimal ISH techniques for specific research and clinical applications, from basic discovery to diagnostic assay development.
Understanding In Situ Hybridization: Principles, Advantages, and Evolution
In situ hybridization (ISH) is a foundational technique in molecular biology that allows for the precise localization of specific DNA or RNA sequences within individual cells, tissue sections, or entire tissues (in whole mount ISH) [1]. The core principle of ISH is the use of a labeled, complementary nucleic acid strand, known as a probe, which binds (hybridizes) to its target sequence within the morphologically preserved biological sample [2] [1]. This technique provides crucial temporal and spatial information about gene expression and genetic loci that cannot be obtained through bulk extraction methods [3].
Within the context of research comparing the sensitivity of different ISH methodologies, understanding the fundamental components and variations of the technique is paramount. This guide details the core aspects of ISH, from its basic principle to its advanced applications and experimental protocols, providing a framework for evaluating its performance.
The Core Principle and Key Variations of ISH
The power of ISH lies in its ability to answer the question of "where" a specific genetic sequence is located. The basic principle involves fixing the target nucleic acids within their cellular context, applying a labeled probe that is complementary to the target sequence under specific hybridization conditions, and then detecting the location of the bound probe [1]. The main variations of the technique are distinguished by the type of target, the method of detection, and the probe design.
Primary ISH Methodologies
| Technique | Target | Detection Method | Primary Advantage | Key Applications |
|---|---|---|---|---|
| CISH (Chromogenic ISH) | DNA or RNA | Chromogenic reaction, visualized with bright-field microscopy [3] | Ability to view signal and tissue morphology simultaneously with common microscopes; permanent slides [3] | Molecular pathology diagnostics [3] |
| FISH (Fluorescent ISH) | DNA or RNA | Fluorophores, visualized with fluorescence microscopy [3] | Multiplexing: ability to visualize multiple targets in the same sample [3] | Gene presence, copy number, location (DNA-FISH); gene expression and RNA localization (RNA-FISH) [3] |
| Branched DNA (bDNA) FISH (e.g., ViewRNA) | RNA (primarily) | Fluorophores with signal amplification, visualized with fluorescence microscopy or flow cytometry [1] [3] | High Sensitivity & Specificity: proprietary signal amplification reduces background and increases signal-to-noise ratio [1] [3] | Detection of low-abundance RNA transcripts; multiplex gene expression analysis [3] |
The following workflow outlines the general stages of a branched DNA FISH assay, a method known for its high sensitivity:
Essential Components: The ISH Toolkit
The successful execution of an ISH experiment relies on a set of key reagents and materials. The table below details these essential components and their functions.
Research Reagent Solutions for ISH
| Item | Function / Description |
|---|---|
| Probes | Labeled, complementary nucleic acids (DNA, RNA, or oligonucleotides) that hybridize to the target sequence. Types include double-stranded DNA, single-stranded DNA, RNA probes (riboprobes), and synthetic oligonucleotides (PNA, LNA) [2]. |
| Labels | Reporter molecules attached to the probe for detection. These can be radioactive isotopes (e.g., 32P, 35S) or non-radioactive labels like biotin, digoxigenin, or fluorescent dyes [2] [1]. |
| Fixatives | Chemicals like formaldehyde used to preserve tissue architecture and cross-link nucleic acids in place, preventing degradation and diffusion [1]. |
| Permeabilization Agents | Substances such as proteinase K that treat cells to open membranes, allowing probe access to the target nucleic acids [1]. |
| Hybridization Buffers | Solutions that control temperature, salt, and/or detergent concentration to ensure specific binding between the probe and its target [1]. |
| Detection Reagents | For non-fluorescent probes, these include enzyme-conjugated antibodies (e.g., anti-digoxigenin) that bind the probe label, followed by a chromogenic substrate to produce a visible precipitate [1]. |
| Signal Amplification System | In branched DNA FISH, a series of oligonucleotides (pre-amplifiers and amplifiers) that bind to the probe to create a tree-like structure, dramatically amplifying the signal [1] [3]. |
Detailed Methodologies: Core Experimental Protocols
Protocol 1: Basic ISH with Digoxigenin-Labeled Probes
This protocol outlines the key steps for a standard chromogenic or fluorescent ISH assay using a hapten-labeled probe [1].
- Sample Preparation: Tissue or cells are fixed, typically with a cross-linking fixative like formaldehyde, to preserve morphology and retain nucleic acids. For some samples, permeabilization with proteinase K is required to allow probe entry [1].
- Probe Hybridization: The labeled complementary DNA or RNA probe is applied to the sample and incubated at an elevated temperature optimized for specific hybridization. This step can take several hours or overnight [1].
- Stringency Washes: After hybridization, non-specifically bound probe is removed through a series of washes. Parameters like temperature, salt concentration, and detergent concentration are manipulated to ensure only perfectly matched sequences remain bound [1].
- Signal Detection:
- For hapten-labeled probes (e.g., digoxigenin), an antibody conjugate (e.g., an antibody linked to alkaline phosphatase) is applied and allowed to bind [1].
- For chromogenic detection, a substrate for the enzyme is added, which produces an insoluble, colored precipitate at the site of the target.
- For fluorescent detection, the antibody is conjugated to a fluorophore, or the probe is directly labeled with a fluorophore.
- Visualization and Analysis: The results are visualized via bright-field microscopy (for chromogenic detection) or fluorescence microscopy. Signal localization within the tissue context is then analyzed.
Protocol 2: Branched DNA (bDNA) Signal Amplification FISH
This method, used in technologies like ViewRNA, employs a multi-step hybridization process to achieve high sensitivity and is particularly suited for detecting low-abundance RNA targets [1] [3].
- Sample Preparation and Fixation: Cells or tissues are fixed to preserve RNA and morphology.
- Target Hybridization: A probe set containing approximately 20 to 40 oligonucleotide pairs is hybridized to the target RNA. These oligo pairs bind side-by-side along the target sequence [1].
- Signal Amplification (Pre-Amplification): A pre-amplifier molecule hybridizes to each pair of the bound oligonucleotides on the target-specific probe [1].
- Signal Amplification (Amplification): Multiple amplifier molecules hybridize to each pre-amplifier [1].
- Label Probe Hybridization: Finally, many label probes, conjugated to alkaline phosphatase (for chromogenic detection) or directly to fluorophores, hybridize to each amplifier molecule. This fully assembled "tree" structure can result in an 8,000-fold signal amplification for a single transcript [1].
- Detection: The signal is detected using fluorescence or bright-field microscopy. The use of independent but compatible signal amplification systems enables the multiplexing of several targets in a single assay [1] [3].
The logical relationship and workflow of the branched DNA signal amplification system is detailed below:
Applications and Quantitative Performance
ISH is a versatile technique with broad applications across biological research and clinical diagnostics. Its utility in sensitivity comparisons becomes clear when examining its use cases and performance metrics.
Key Application Areas of ISH
| Field | Specific Application |
|---|---|
| Microbiology | Morphology and population structure of microorganisms (classic target: 16S rRNA) [2]. |
| Pathology & Diagnostics | Pathogen profiling, assessment of abnormal gene expression, and HER2 testing in breast and gastro-oesophageal carcinoma [2] [4]. |
| Developmental Biology | Gene expression profiling in embryonic tissues [2]. |
| Cytogenetics | Karyotyping and phylogenetic analysis; detection of chromosomal aberrations and fusion genes via unique FISH patterns [2] [1]. |
| Genomic Mapping | Physical mapping of clones to specific chromosomal regions [2]. |
| Gene Expression Studies | Spatial and temporal localization of mRNA, lncRNA, and miRNA within tissues and cells [1] [3]. |
Quantitative Data on ISH Performance
Recent studies, particularly those focusing on automation, provide quantitative data relevant for sensitivity and efficiency comparisons.
Table: Automated vs. Manual FISH Performance Comparison Data from a 2025 validation study of the Leica BOND-III automated staining platform for HER2 FISH testing demonstrates key performance metrics [4].
| Metric | Automated FISH (Leica BOND-III) | Manual FISH (Agilent HER2 IQFISH) |
|---|---|---|
| Sensitivity (Breast Cancer) | 95% | (Baseline) |
| Specificity (Breast Cancer) | 97% | (Baseline) |
| Sensitivity (Gastric Carcinoma) | 100% | (Baseline) |
| Specificity (Gastric Carcinoma) | 100% | (Baseline) |
| Overall Concordance | 98% | (Baseline) |
| Technical Hands-on Time | Significantly decreased | (Baseline) |
| Overall Supply Costs | Reduced | (Baseline) |
This empirical data shows that automated FISH platforms can achieve high diagnostic concordance with manual methods while improving laboratory efficiency and reducing costs, a critical consideration for clinical and research settings [4].
In situ hybridization stands as a critical technique for spatially resolving nucleic acid localization within a cellular context. From its foundational forms (CISH and FISH) to more sensitive, amplified versions (bDNA FISH), the core principle remains the hybridization of a complementary probe to an in-situ target. When framed within sensitivity comparison research, the choice of probe type, labeling method, and detection system—especially the use of sophisticated signal amplification technologies—becomes the defining factor for performance. The ongoing innovation in ISH, including automation and multiplexing, ensures its continued relevance in advancing both basic biological understanding and clinical diagnostic capabilities.
In situ hybridization (ISH) and immunostaining (including immunohistochemistry (IHC) and immunofluorescence) are cornerstone techniques for visualizing molecular targets within tissues and cells. While both methods preserve valuable spatial context lost in bulk analysis techniques, they operate on fundamentally different principles and target distinct molecules. Immunostaining uses antibodies to detect proteins (antigens), providing a snapshot of protein expression and localization [5] [6]. In contrast, ISH uses nucleic acid probes to hybridize to DNA or RNA sequences, revealing information about gene location, transcription, and genomic alterations [7] [6]. Understanding their comparative specificity, reliability, and applications is crucial for selecting the optimal method in research and diagnostics, particularly in sensitive areas like cancer and regenerative biology. This guide provides an in-depth technical comparison of these two powerful techniques, framed within ongoing research aimed at enhancing the sensitivity of in situ hybridization.
Fundamental Principles and Technical Comparison
Core Mechanisms and Targets
The fundamental distinction lies in their detection targets and the chemistry employed. The table below summarizes their core characteristics.
Table 1: Core Principles and Targets of Immunostaining and ISH
| Feature | Immunostaining (IHC/IF) | In Situ Hybridization (ISH) |
|---|---|---|
| Primary Target | Proteins (antigens) [8] | Nucleic Acids (DNA, mRNA, miRNA) [8] [6] |
| Detection Molecule | Antibodies [5] [6] | Nucleic Acid Probes [7] [6] |
| Key Principle | Antigen-antibody binding [6] | Complementary base-pairing hybridization [6] |
| Visualization | Chromogenic (DAB) or Fluorescent tags [5] [6] | Chromogenic (DIG) or Fluorescent (FISH) tags [7] [6] |
| Information Gained | Protein expression, localization, post-translational modifications [5] | Gene expression, genetic aberrations, viral nucleic acids [7] [6] |
Workflow and Signal Detection
The experimental workflows for both techniques share common steps like fixation and permeabilization, but critical differences exist in pre-treatment and detection, which directly impact their specificity and reliability. The following diagram illustrates the core signaling pathways for each method.
Diagram 1: Core signaling pathways for Immunostaining and ISH.
Specificity and Reliability: A Detailed Analysis
Factors Governing Specificity
Immunostaining Specificity: Specificity is almost entirely dependent on the affinity and specificity of the primary antibody used [7]. A well-validated antibody ensures minimal cross-reactivity with non-target proteins. However, antibodies can sometimes recognize proteins with similar epitopes, leading to false-positive results [7]. The method excels at determining the subcellular localization of a target protein (e.g., nuclear, cytoplasmic, membrane) [7].
ISH Specificity: Specificity is determined by the design of the nucleic acid probe, including its length, sequence composition, and hybridization conditions [7]. Probe specificity can be computationally predicted by checking the homology of its sequence with other genes in the relevant database [7] [9]. This reduces the risk of off-target binding and allows for the discrimination between highly homologous genes or different splice variants with careful probe design [7].
Assessing Reliability and Sensitivity
Reliability encompasses sensitivity, reproducibility, and robustness. Key differentiators are highlighted in the table below.
Table 2: Comparison of Reliability and Technical Performance
| Aspect | Immunostaining | In Situ Hybridization | Implications |
|---|---|---|---|
| Sensitivity | Highly dependent on antibody quality and titer [7]. | Can achieve single-molecule sensitivity with advanced methods (e.g., RNAscope) [8]. | ISH is often more reliable for low-abundance targets. |
| Target Validation | Probes can be designed from sequence data for any known gene, independent of species [7]. | Enables protein-level validation of transcriptomic hits [5]. | ISH is superior for novel targets or non-model organisms. |
| Reproducibility | Can be variable between different antibody batches and protocols [7]. | High reproducibility; probe performance is predictable from sequence [7]. | ISH generally offers more consistent results across labs. |
| Sample Preservation | Harsh protease treatments for ISH can destroy protein epitopes, reducing IHC reliability [10] [8]. | Proteinase K digestion can damage tissue morphology [10]. Newer methods (e.g., NAFA) avoid this [10]. | Method choice affects tissue integrity. |
| Clinical Performance | High specificity (e.g., 100% in biliary strictures), but can have low sensitivity (45.8%) [11]. | High sensitivity (84.2% for FISH in biliary strictures), but can have lower specificity (96.0%) [11]. | Combining both can optimize diagnostic accuracy [12] [11]. |
A clinical study on biliary strictures underscores this comparison, where cytology (a morphology-based method similar to IHC) showed 100% specificity but only 45.8% sensitivity, while FISH showed 84.2% sensitivity and 96.0% specificity, demonstrating the trade-offs between these approaches [11].
Experimental Protocols: Key Methodologies
Advanced Immunostaining Protocol for Delicate Tissues
The NAFA (Nitric Acid/Formic Acid) fixation protocol is an advanced method designed for delicate tissues like regenerating planarians, offering superior preservation for both IHC and ISH [10].
- Fixation Solution: 4% formaldehyde in 1x PBS with 0.25% nitric acid and 2.5% formic acid.
- Key Additive: Includes EGTA, a calcium chelator, to inhibit nucleases and preserve RNA integrity during sample preparation [10].
- Critical Modification: Omits Proteinase K digestion, which is standard in many ISH protocols but damages protein epitopes and fragile tissue structures like the epidermis and blastema [10].
- Workflow: After fixation, samples are permeabilized and blocked. For IHC, they are incubated with primary and fluorescently labeled secondary antibodies. For subsequent FISH, the protocol uses formic acid for permeabilization instead of proteases [10].
- Advantage: This protocol robustly preserves fragile external structures (e.g., cilia, epidermis) while allowing antibody and probe penetration, enabling high-quality tandem FISH and immunostaining [10].
High-Sensitivity RNA In Situ Hybridization Workflow
Advanced ISH methods like RNAscope and HCR FISH have significantly improved upon conventional ISH for detecting low-expression genes [7].
- Probe Design (Primary Probe): Use of multiple short, synthetic oligonucleotide probes targeting different regions of the same mRNA transcript. This increases the effective coverage and specificity without compromising tissue penetration [7].
- Signal Amplification (e.g., RNAscope): The primary probes contain tail sequences that serve as binding sites for pre-amplifier molecules. Multiple amplifier molecules then bind to each pre-amplifier, and each amplifier hybridizes with many labeled probes. This branched DNA (bDNA) technology creates a large signal amplification complex at each probe-binding site [8].
- Visualization: The labeled probes are detected using chromogenic (e.g., Fast Red, DAB) or fluorescent substrates. The signal appears as distinct dots, each representing a single mRNA molecule [7] [8].
- Key Advantage: This multi-step amplification allows for single-molecule sensitivity and low background, and the lack of a protease digestion step makes it more compatible with subsequent immunostaining [7] [8].
Application Scenarios and Integration
Distinct and Overlapping Applications
Each technique excels in specific scenarios, which are summarized in the table below.
Table 3: Key Application Scenarios for Immunostaining and ISH
| Application Domain | Immunostaining | In Situ Hybridization |
|---|---|---|
| Cancer Diagnostics & Subtyping | HER2, ER/PR, PD-L1 testing for therapy guidance [5]. | HER2 gene amplification via FISH; detection of gene fusions [4] [6]. |
| Cell Type Identification | Profiling immune cells (CD3, CD20), astrocytes (GFAP) [5]. | Identifying cell types based on specific mRNA expression. |
| Neurobiology | Localizing amyloid-β plaques and tau tangles in Alzheimer's [5]. | Mapping neurotransmitter receptor mRNA expression [8]. |
| Regeneration Research | Visualizing mitotic cells (anti-H3P), muscle fibers (anti-6G10) [10]. | Analyzing gene expression patterns in stem cells (piwi-1) and blastemas [10]. |
| Infectious Disease | - | Detecting viral DNA/RNA (e.g., HPV) [6]. |
| Target Validation | Confirming protein-level expression of transcriptomic hits [5]. | - |
Integrated Workflows: Dual ISH-IHC
Combining ISH and IHC on the same tissue section provides a powerful multi-omics approach, revealing the relationship between gene expression and protein production within the same cell. However, standard protocols for each method are incompatible [8] [9].
- The Fundamental Conflict: The protease treatment required for ISH probe penetration destroys protein epitopes, damaging the IHC signal. Conversely, antibody reagents can introduce RNases that degrade the RNA target for ISH [8].
- Sequential Protocol with Critical Modifications:
- RNase Inhibition: Tissue sections are pretreated with recombinant ribonuclease inhibitors during the IHC antibody incubation to protect RNA integrity [8].
- IHC First: Perform immunostaining first using cross-linked or highly stable antibodies.
- Antibody Crosslinking: After IHC labeling, antibodies are crosslinked to the tissue using a fixative like formaldehyde. Standard fixation is insufficient to withstand the subsequent ISH pretreatments [8].
- ISH Second: Perform the ISH protocol, including its more stringent washes and hybridization steps. The crosslinked antibodies remain bound, preserving the protein signal.
- Outcome: This workflow allows for the robust dual detection of both protein and mRNA targets in the same tissue section, enabling sophisticated spatial biology analysis [8] [9].
The following diagram illustrates the workflow for this powerful integrated approach.
Diagram 2: Sequential workflow for dual ISH-IHC detection.
The Scientist's Toolkit: Essential Reagents and Solutions
Successful implementation of these techniques, especially their integration, relies on key reagents. The following table details essential components for the dual ISH-IHC protocol.
Table 4: Research Reagent Solutions for Dual ISH-IHC
| Reagent / Solution | Function | Key Consideration |
|---|---|---|
| RNase Inhibitor (e.g., RNaseOUT) | Protects RNA targets from degradation during IHC steps [8]. | Critical for preserving RNA integrity when using antibody reagents. |
| Antibody Crosslinker | Covalently binds antibodies to tissue post-IHC, preventing their dissociation during harsh ISH steps [8]. | Standard formaldehyde fixation is often insufficient. |
| Branched DNA (bDNA) ISH Probes (e.g., ViewRNA, RNAscope) | Enable high-sensitivity, multiplexed RNA detection without protease treatment [8] [9]. | Simplify combination with IHC and boost signal for low-expression genes. |
| Formic Acid / NAFA Fixative | Acts as a permeabilization agent for ISH, replacing Proteinase K to better preserve antigen epitopes [10]. | Enhances tissue integrity and compatibility with IHC. |
| Multiplex-Compatible Mountant | Preserves fluorescence and colorimetric signals, prevents photobleaching for archival [8]. | Essential for long-term storage and multi-channel imaging. |
| Spectral Imaging System | Simultaneously resolves multiple fluorescent signals from IHC and FISH with minimal crosstalk [8]. | Enables high-plex data acquisition from a single section. |
Immunostaining and ISH are complementary, not competing, technologies. Immunostaining is the established choice for protein localization and cell phenotyping when high-quality antibodies are available, offering excellent morphological context. ISH provides unparalleled specificity for nucleic acid detection, crucial for genetic aberrations, viral detection, and validating gene expression, especially in non-model organisms or for low-abundance transcripts.
The choice between them hinges on the research question: For "where is the protein?", use immunostaining. For "is the gene active and where?", use ISH. The future of spatial biology lies in their integration. Overcoming technical hurdles with optimized protocols like NAFA fixation and dual ISH-IHC enables true spatial multi-omics, allowing researchers to correlate transcriptional activity with translational output within the complex architecture of intact tissues. This powerful synergy promises to unlock deeper insights into development, disease mechanisms, and regenerative processes.
The journey of in situ hybridization (ISH) from a semi-quantitative morphological technique to a precise single-molecule detection platform represents one of the most significant advancements in molecular pathology and genetic research. This evolution has been driven by fundamental innovations in probe design, signal amplification methodologies, and detection systems that have collectively pushed the sensitivity boundaries of nucleic acid detection in morphologic contexts. This technical review examines the critical developmental pathway from conventional ISH to single-molecule resolution, with particular focus on the quantitative improvements in sensitivity, specificity, and spatial resolution that have transformed research capabilities and diagnostic applications. The progression mirrors a broader thesis in molecular detection: that achieving true single-molecule sensitivity requires not merely incremental improvements but fundamental reimagining of hybridization chemistry, amplification strategies, and visualization technologies.
In situ hybridization has undergone a remarkable transformation since its initial development in 1969, when the first demonstrations used radioactive probes to localize specific DNA sequences [13]. The evolution from these early radioactive methods to contemporary single-molecule detection platforms represents an increase in sensitivity of several orders of magnitude, enabling researchers to visualize and quantify individual nucleic acid molecules within their native cellular and tissue contexts. This enhanced sensitivity has proven particularly valuable in applications requiring detection of low-abundance transcripts, rare genetic variants, and subtle changes in gene expression that were previously undetectable with conventional methodologies.
The drive toward single-molecule sensitivity has been fueled by demands from multiple research domains. In cancer research, detecting rare genetic rearrangements or low-frequency mutations has prognostic and therapeutic implications. In developmental biology, understanding the spatial patterning of gene expression requires precise quantification of transcript localization. In drug development, tracking the distribution of therapeutic oligonucleotides necessitates highly sensitive detection methods [14]. These research needs have catalyzed the technical evolution chronicled in this review, establishing a new paradigm for what is detectable at the molecular level within intact biological systems.
The Historical Context: From Radioactive Probes to Basic Fluorescence
The initial phase of ISH evolution relied heavily on radioactive labeling techniques, which provided the first glimpse into chromosomal organization and gene localization but suffered from significant limitations. These early methods used radiolabeled RNA or DNA probes that required long exposure times (days to weeks) and offered limited spatial resolution due to scatter from radioactive emissions. Despite these constraints, radioactive ISH established the fundamental principle that nucleic acids could be localized within morphological contexts through complementary base pairing [14].
The first major transition in ISH technology came with the development of non-radioactive detection methods using fluorophores in the late 1970s [13]. This fluorescence in situ hybridization (FISH) approach offered several immediate advantages: reduced safety concerns, shorter processing times, and improved spatial resolution. However, early FISH methodologies remained relatively insensitive, capable of detecting only abundant targets such as repetitive DNA sequences or highly expressed genes. The limited brightness of fluorophores and absence of signal amplification strategies restricted applications to targets present in multiple copies per cell.
Table 1: Evolution of ISH Detection Methodologies
| Era | Probe Type | Signal Detection | Sensitivity Limit | Primary Applications |
|---|---|---|---|---|
| 1969-1980s | Radioactive DNA/RNA | Autoradiography | ~10-50 copies/cell | Gene localization, repetitive sequences |
| 1980s-1990s | Biotin/Digoxigenin | Enzyme-based chromogenic | ~5-20 copies/cell | Chromosome analysis, viral detection |
| 1990s-2000s | Directly labeled fluorescence | Epifluorescence microscopy | ~5-10 copies/cell | Karyotyping, gene amplification |
| 2000s-2010s | Oligonucleotide pools | Signal amplification (bDNA, TSA) | ~1-5 copies/cell | mRNA detection, gene fusion analysis |
| 2010s-Present | Multiplexed oligonucleotides | Single-molecule imaging | Single molecules | Spatial transcriptomics, low-abundance transcripts |
The critical limitation of these early fluorescence approaches was their fundamental sensitivity constraint. Without amplification, the signal from a single hybridized probe was insufficient to detect against cellular autofluorescence and background noise. This sensitivity barrier restricted FISH applications to targets present in multiple copies and prevented detection of single transcripts or precise quantification of gene expression at the cellular level.
The Signal Amplification Revolution: Bridging the Sensitivity Gap
The introduction of signal amplification strategies marked a pivotal transition in ISH technology, effectively bridging the gap between conventional detection and single-molecule sensitivity. These methodologies recognized that while the target nucleic acid could not be amplified in situ (as in PCR), the detection signal could be dramatically enhanced through various amplification cascades.
Branched DNA (bDNA) Technology
The branched DNA (bDNA) platform represents one of the most significant innovations in signal amplification for ISH. This approach uses a series of sequential hybridizations to build a complex amplification structure on the primary probe-target hybrid. The methodology typically involves:
- Primary probes that hybridize to the target RNA or DNA
- Preamplifier molecules that bind to the primary probes
- Amplifier molecules that bind to the preamplifiers in a branched configuration
- Labeled probes that hybridize to multiple sites on the amplifiers
This cascade results in an accumulation of thousands of fluorophores at the site of each original probe-target binding event, providing approximately 8,000-fold signal amplification [15]. The commercial implementation of this technology in platforms such as RNAscope and ViewRNA has enabled highly sensitive detection while maintaining specificity through a "double-Z" probe design that requires two independent probe binding events for signal generation [15].
Tyramide Signal Amplification (TSA)
Also known as catalyzed reporter deposition (CARD), tyramide signal amplification utilizes the catalytic activity of horseradish peroxidase to deposit numerous fluorescent or chromogenic tyramide molecules at the probe binding site. The enzyme is conjugated to a detector molecule that binds to the hybridized probe, and when exposed to hydrogen peroxide and tyramide substrates, the enzyme activates the tyramide molecules, causing them to form covalent bonds with nearby tyrosine residues. This results in substantial signal amplification, enabling detection of low-abundance targets [13].
Hybridization Chain Reaction (HCR)
HCR represents a more recent innovation in signal amplification that employs a series of metastable DNA hairpins that undergo a chain reaction of hybridization events initiated by the probe-target binding. This method provides exponential signal amplification while maintaining high specificity and low background, making it particularly suitable for multiplexed applications and whole-mount samples [14].
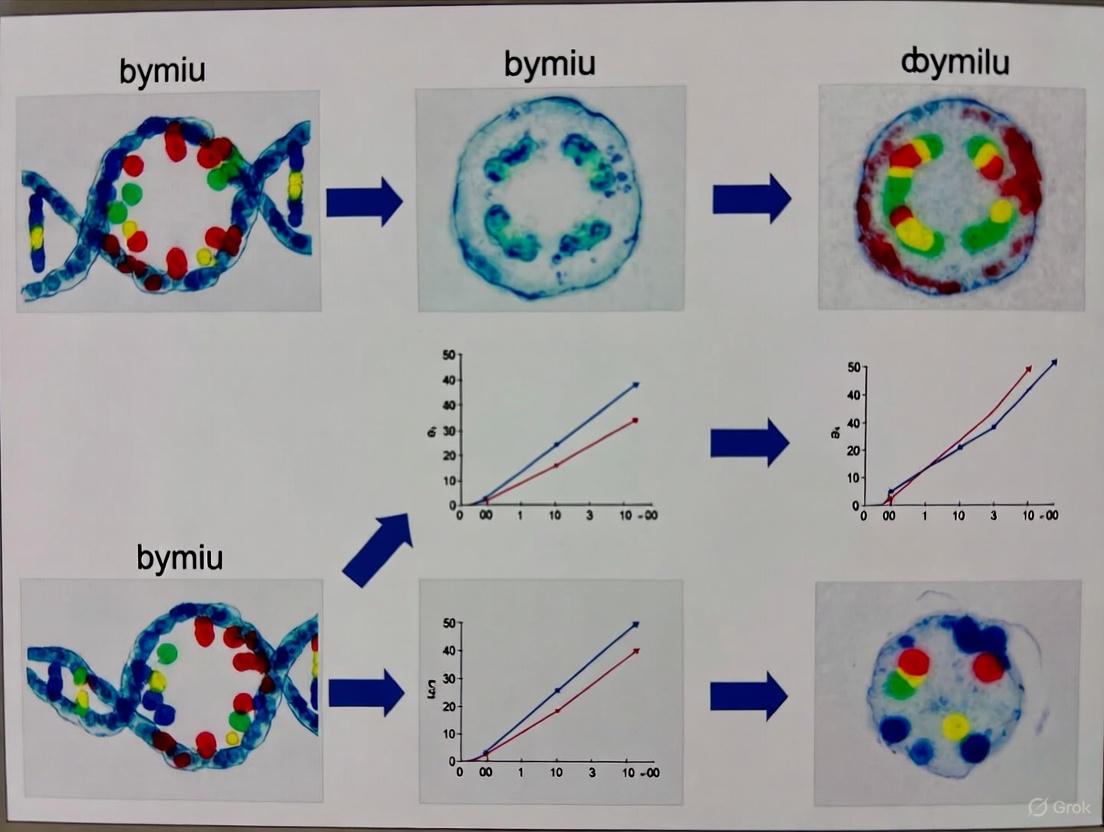
Diagram 1: Evolution of FISH sensitivity through probe design and signal amplification strategies. The transition from conventional FISH to single-molecule detection involved fundamental changes in both probe architecture and amplification methodologies, resulting in progressive improvements in sensitivity.
The Single-Molecule Revolution: smFISH and Beyond
The achievement of true single-molecule sensitivity represents the current apex of ISH evolution, enabled by the convergence of several technological innovations. Single-molecule FISH (smFISH) fundamentally transformed the capacity to detect and quantify RNA expression at the cellular level by providing absolute molecular counts rather than relative expression levels.
Fundamental Principles of smFISH
The core innovation of smFISH lies in the use of multiple short oligonucleotide probes (typically 17-22 base pairs) targeting different regions of the same transcript [16]. Each individual probe carries a single fluorophore, producing insufficient signal for detection against cellular background. However, when multiple probes (typically 20-96) hybridize to a single transcript, their collective signal generates a brightly fluorescent spot that can be easily detected by standard fluorescence microscopy [16]. Each bright spot corresponds to a single RNA molecule, enabling direct quantification of transcript abundance without amplification bias.
The statistical nature of this approach ensures high specificity, as false-positive signals from individual mis-bound probes are unlikely to co-localize. Raj et al. (2008) first demonstrated that this method could achieve true single-molecule resolution, fundamentally changing the landscape of gene expression analysis in situ [16].
Advanced smFISH Methodologies
Recent innovations have further enhanced the basic smFISH approach:
High-throughput multiplexed variations such as MERFISH (Multiplexed Error-Robust FISH) and seqFISH (sequential FISH) enable simultaneous detection of hundreds to thousands of different RNA species through combinatorial barcoding and sequential hybridization schemes [17]. These approaches maintain single-molecule sensitivity while dramatically expanding multiplexing capability.
Locked Nucleic Acid (LNA) and Peptide Nucleic Acid (PNA) probes incorporate modified nucleic acid analogs that increase binding affinity and specificity, allowing for shorter probes and improved discrimination of single-nucleotide variants [18].
CRISPR-integrated systems such as the CRISPR FISHer platform have recently emerged, combining CRISPR-mediated target recognition with signal amplification for live-cell imaging of genomic loci [13].
Table 2: Quantitative Comparison of smFISH Methodologies
| Method | Probe Type | Probes per Transcript | Multiplexing Capacity | Sensitivity (Detection Limit) | Key Applications |
|---|---|---|---|---|---|
| Conventional smFISH | DNA oligonucleotides | 30-48 | 1-3 colors | Single molecules | Quantification of abundant transcripts |
| Branched DNA smFISH | Z-probe pairs | 10-20 pairs | 3-4 colors | Single molecules | Low-abundance transcripts, clinical samples |
| MERFISH/seqFISH | Barcoded oligonucleotides | 30-96 | 100-10,000 genes | Single molecules | Spatial transcriptomics, cell atlas |
| HCR-FISH | DNA initiators | 1-4 initiators | 5-10 colors | Single molecules | Whole-mount samples, thick tissues |
| LNA/PNA FISH | Modified nucleotides | 15-30 | 1-3 colors | Single molecules | Short transcripts, miRNA, SNP detection |
The Researcher's Toolkit: Essential Reagents and Methodologies
Implementing single-molecule FISH requires careful selection of reagents and methodologies optimized for specific applications. The following table summarizes key solutions and their functions in contemporary smFISH workflows.
Table 3: Research Reagent Solutions for Single-Molecule FISH
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| Probe Design Platforms | Stellaris Designer, TrueProbes, MERFISH Designer | Computational probe selection and optimization | TrueProbes integrates genome-wide BLAST and thermodynamic modeling for enhanced specificity [19] |
| Probe Synthesis | Biosearch Technologies, IDT, GeneCopoeia | Custom oligonucleotide production | 5 nmol scale sufficient for hundreds of experiments; 3' amine modification for fluorophore coupling [16] |
| Fluorophores | Cy5, Alexa 594, Quasar 670, TMR | Signal generation | Cy5 and Alexa 594 provide optimal signal-to-noise; avoid shorter wavelengths (e.g., Alexa 488) due to autofluorescence [16] |
| Signal Amplification Systems | RNAscope, ViewRNA, HCR | Signal enhancement | RNAscope provides 8000-fold amplification via branched DNA [15] |
| Tissue Preparation | Neutral buffered formalin, paraformaldehyde | Tissue fixation and morphology preservation | 24±12 hours fixation optimal; under-fixation and over-fixation both detrimental to RNA integrity [14] |
| Permeabilization | Proteinase K, Triton X-100, Tween-20 | Tissue and cell permeability | Concentration and timing critical for probe accessibility while maintaining morphology [14] |
| Image Analysis Software | Spotiflow, Localize, FISH-quant | Automated spot detection and quantification | Spotiflow uses deep learning for subpixel-accurate detection in diverse image types [17] |
Experimental Protocol: smFISH for Single-Molecule Detection in Oocytes and Embryos
The following detailed protocol adapts smFISH for challenging samples, based on the optimized methodology described by [15] for use in murine oocytes and embryos. This protocol highlights the critical modifications required to achieve single-molecule sensitivity in delicate cellular contexts.
Probe Design and Synthesis (Days 1-2)
Design Parameters: Generate 17-22 base pair oligonucleotide probes targeting different regions of the transcript of interest. Ideal parameters include:
- GC content approximately 45% for uniform binding efficiency
- Minimum of 2 base pair spacing between adjacent probes
- 30-96 probes per transcript target for optimal signal-to-noise
- Include 3' amine group for fluorophore conjugation if synthesizing independently
Probe Synthesis and Coupling:
- Combine 1 nmol of each oligonucleotide in a single microcentrifuge tube
- Add 0.11 volumes of 1M sodium bicarbonate (pH 8.0) to achieve 0.1M final concentration
- Dissolve fluorophore (Cy5, Alexa 594, or TMR) in 50μL of 0.1M sodium bicarbonate
- For TMR, first dissolve in <5μL DMSO before adding bicarbonate
- Mix fluorophore solution with oligonucleotides and incubate overnight at room temperature with gentle rocking protected from light
Purification:
- Precipitate oligonucleotides by adding 10% volume 3M sodium acetate and 2.5 volumes 100% ethanol
- Store at -70°C for at least 1 hour (or overnight)
- Centrifuge at 16,000 RCF for 15 minutes at 4°C
- Remove supernatant and purify coupled probes via HPLC using C18 column with triethylammonium acetate (Buffer A) and acetonitrile (Buffer B) gradient
Sample Preparation and Fixation (Day 3)
Fixation:
- Transfer oocytes or embryos to drops of 4% paraformaldehyde in PBS
- Fix for 30-60 minutes at room temperature
- Note: Commercial permeabilization buffers often cause lysis of delicate cells; replace with PBS-based buffers
Permeabilization:
- Wash samples 3×5 minutes in PBS with 0.1% Tween-20
- Permeabilize in PBS with 0.5% Triton X-100 for 30 minutes
- Transfer to Agtech 6-well plates for subsequent steps to minimize sample loss
Hybridization and Signal Detection (Days 3-4)
Hybridization:
- Resuspend probe set in proprietary hybridization buffer (commercial buffers essential for preventing aggregation)
- Hybridize with transcript-specific probes overnight at 37°C
- Perform sequential hybridizations with pre-amplifier and amplifier molecules per manufacturer protocols
Washing and Imaging:
- Wash 3×10 minutes in PBS-based wash buffers
- Mount samples with anti-fade mounting medium
- Image using confocal microscopy with appropriate filter sets
- Acquire z-stacks through entire cell volume to ensure complete transcript capture
Data Analysis and Quantification
Image Processing:
- Use software such as Localize or Spotiflow for automated spot detection
- Apply background subtraction and flat-field correction
- Implement 3D spot detection for accurate counting throughout cell volume
Quantification:
- Each punctate fluorescent spot represents a single mRNA molecule
- Compare signal patterns across channels to exclude cross-talk
- Validate using negative controls (bacterial DapB gene) and positive controls (housekeeping genes)
Diagram 2: Workflow for single-molecule FISH experiments. The process involves four major phases: probe design and synthesis, sample preparation, hybridization and detection, and quantitative data analysis, with specific critical steps at each phase.
Technological Frontiers and Future Directions
The evolution of ISH sensitivity continues with several emerging technologies pushing the boundaries of what is detectable. Recent innovations focus on enhancing multiplexing capabilities, improving quantitative accuracy, and expanding applications to new sample types.
Automated platforms such as the Leica BOND-III have demonstrated significant improvements in reproducibility and efficiency while maintaining sensitivity equivalent to manual methods. Studies show 98% concordance between automated and manual FISH with significant reduction in hands-on time and supply costs [4].
Digital PCR integration with single-molecule detection principles has enabled ultra-sensitive quantification of nucleic acids in solution. BEAMing (Bead, Emulsion, Amplification and Magnetics) technology achieves a limit of detection of 0.01%, an order of magnitude improvement over conventional digital PCR [20].
Spatial transcriptomics platforms represent the current cutting edge, combining single-molecule sensitivity with genome-scale multiplexing. Techniques such as Xenium In Situ and MERFISH enable mapping of hundreds to thousands of genes simultaneously while maintaining subcellular resolution [17].
Computational probe design advances through platforms like TrueProbes integrate genome-wide BLAST analysis with thermodynamic modeling to optimize probe specificity and sensitivity. These tools address limitations in conventional design algorithms that struggle with short genes, low-abundance transcripts, and sequences with shared motifs [19].
The continued evolution of ISH sensitivity promises to further transform biological research and clinical diagnostics, enabling increasingly precise spatial and quantitative analysis of gene expression in the context of intact biological systems.
In situ hybridization (ISH) technologies have become indispensable tools in biomedical research and clinical diagnostics, enabling the precise localization of nucleic acids within their native cellular and tissue contexts. The efficacy of these methods hinges on two fundamental metrics: signal-to-noise ratio (SNR), which determines assay clarity and specificity, and detection limits, which define the minimum number of target molecules that can be reliably identified. This technical guide explores the core principles and experimental methodologies for quantifying these critical parameters across various ISH platforms, including RNAscope, HCR, Yn-situ, and U-FISH. By examining recent advancements in probe design, signal amplification, and computational analysis, we provide a framework for standardized sensitivity assessment that supports accurate comparison between methodologies and informs appropriate technique selection for specific research or diagnostic applications.
The sensitivity of any in situ hybridization assay is fundamentally governed by its ability to distinguish true signal from background noise across a range of target abundances. The signal-to-noise ratio (SNR) provides a quantitative measure of this distinction, calculated as the intensity of specific staining divided by non-specific background fluorescence or chromogenic precipitation. A higher SNR directly correlates with improved assay reliability, enabling more accurate target identification and quantification. Closely related is the concept of detection limits, which defines the lowest concentration of a target nucleic acid that can be reliably detected with statistical confidence. These parameters are influenced by multiple interconnected factors including probe design, signal amplification efficiency, tissue preservation, hybridization conditions, and detection methodology.
Recent innovations in ISH technologies have progressively pushed these sensitivity boundaries. Traditional single-molecule FISH (smFISH) methods typically required 20-50 probes targeting different regions of the same transcript to achieve robust detection. However, emerging methodologies like Yn-situ have demonstrated that sophisticated probe architecture can reduce this requirement to just 3-5 probes while maintaining or even improving sensitivity [21]. Similarly, the integration of deep learning approaches such as U-FISH has enabled significant enhancement of SNR in complex image data by transforming raw images with variable characteristics into enhanced images with uniform signal spots and improved signal-to-noise ratios [22]. Understanding how to quantify and compare these advancements requires standardized methodological approaches and metrics, which form the core focus of this technical guide.
Quantitative Performance Benchmarks
Table 1: Comparative Performance of ISH Methods and Analysis Tools
| Method / Tool | Key Innovation | Signal-to-Noise Ratio | Detection Limit | Probe Requirements |
|---|---|---|---|---|
| Yn-situ [21] | Y-branched preamplifier with 20 initiator repeats | Higher SNR than HCR with 20-probe sets; smaller puncta | Detects RNA with just 3 probe pairs; works for short transcripts | 3-5 probe pairs (vs. 20 for standard HCR) |
| U-FISH [22] | Deep learning-based image enhancement | Superior signal enhancement; median F1 score: 0.924; distance error: 0.290 pixels | Enables detection in diverse imaging conditions & 3D data | Not applicable (post-imaging analysis) |
| RNAscope [23] | Proprietary probe design and amplification | 97.3% concordance with FISH in unequivocal cases; superior in heterogeneous samples | Single-molecule sensitivity at cellular level | Not specified in detail |
| QuantISH [24] | Computational framework for CISH analysis | Enables quantification in chromogenic ISH with single-channel data | Cell-type specific quantification in carcinoma, immune, and stromal cells | Not applicable (image analysis pipeline) |
The quantitative comparison of ISH methodologies reveals distinct approaches to optimizing sensitivity parameters. The Yn-situ method achieves its performance through a novel preamplifier design that creates a Y-shaped structure upon hybridization, with each preamplifier carrying 20 initiator repeats that simultaneously trigger 20 hybridization chain reactions [21]. This architecture produces a significant amplification cascade from minimal probe input, fundamentally altering the relationship between probe count and detection sensitivity. Experimental data demonstrates that while one probe pair produces detectable but weak signals unsuitable for quantitative studies, three probe pairs generate strong, quantifiable signals, and five probe pairs achieve optimal performance with high specificity and intensity [21].
Computational approaches offer a different pathway to sensitivity enhancement by operating on the output of various ISH methods rather than modifying the hybridization chemistry itself. U-FISH employs a U-Net model trained on a comprehensive dataset of over 4000 images and 1.6 million verified targets from seven diverse sources [22]. This approach transforms raw FISH images with variable characteristics into enhanced images with uniform signal spot characteristics, achieving a median F1 score of 0.924 and a distance error of just 0.290 pixels in benchmark analyses [22]. The method's compact architecture of only 163k parameters enables efficient processing while maintaining high accuracy, demonstrating particularly strong noise resistance capabilities in tests with simulated noise [22].
For chromogenic ISH (CISH) methods, where fluorescent multi-channel advantages are absent, the QuantISH pipeline addresses the significant analytical challenge of segmenting nuclei and quantifying signal from a single channel containing both marker and counterstain [24]. Through color deconvolution to separate brown marker RNA stain from blue nucleus stain, followed by sophisticated computational processing including void-filling using textural synthesis algorithms, this framework enables cell-type specific classification and expression quantification based on nuclear morphology [24]. This approach maintains sensitivity despite the constraints of chromogenic detection systems.
Experimental Protocols for Sensitivity Quantification
Yn-situ Assay Optimization and Sensitivity Determination
The Yn-situ protocol employs a systematic approach to sensitivity optimization beginning with probe design and validation. The core innovation centers on a single-strand DNA preamplifier approximately 1 kb long containing repetitive sequences that serve as initiation sites. To generate this probe, a plasmid containing the double-stranded preamplifier sequence is amplified using LongAmp Taq DNA polymerase (identified as optimal through comparative testing of five commercial polymerases), with optimal results achieved at 0.5 μM primer concentration and 0.05 ng/μL template concentration [21]. Following amplification, single-stranded preamplifier is produced through strandase digestion of the phosphorylated antisense strand, with completeness of digestion verified empirically through testing of reverse primers at varying positions.
Critical to the assay's sensitivity is an improved fixation protocol that incorporates 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) following standard formaldehyde fixation. This chemical modification crosslinks the phosphate groups of cellular RNA with amine groups from proteins, dramatically reducing RNA degradation and improving signal detection, particularly in suboptimal samples such as 6-month-old tissue stored at -80°C [21]. Hybridization conditions follow established HCR parameters but with significantly fewer probe pairs. Experimental determination of optimal preamplifier concentration identified 0.2 ng/μL as ideal, though signals can be detected at concentrations as low as 0.002 ng/μL [21]. To reduce potential background from PCR handle sequences in the preamplifier, short oligos corresponding to these sequences are used at 10 times the preamplifier concentration as blocking reagents.
Sensitivity limits were quantitatively determined through systematic reduction of probe pairs. Experiments demonstrated that strong, quantifiable signals are achieved with three probe pairs, while one pair produces detectable but weak signals unsuitable for rigorous quantification [21]. This establishes the practical detection limit for the method while highlighting the relationship between probe number and signal intensity. The high sensitivity and wide dynamic range of Yn-situ further enables quantification of genes expressed at different levels, as demonstrated in olfactory sensory neurons [21].
U-FISH Image Enhancement and Analysis Protocol
The U-FISH methodology employs a deep learning approach to sensitivity enhancement that begins with comprehensive dataset preparation. The model was trained on a meticulously curated collection comprising over 4000 images with more than 1.6 million verified targets from seven diverse sources, ensuring broad generalizability across different experimental conditions and biological samples [22]. This dataset diversity is a cornerstone feature that enables the development of a universal spot detection model capable of consistent performance without dataset-specific parameter adjustments.
The U-Net architecture underlying U-FISH contains only 163k parameters, making it computationally efficient while maintaining high accuracy [22]. The network transforms raw FISH images with variable characteristics into enhanced images with uniform signal spot characteristics and improved signal-to-noise ratio. This enhanced output enables reliable spot detection with fixed parameters, eliminating the need for time-consuming manual adjustment for each image. Benchmark testing against both deep learning-based (deepBlink, DetNet, SpotLearn) and rule-based (Big-FISH, RS-FISH, Starfish, TrackMate) methods demonstrated U-FISH's superior performance across diverse datasets, with particular advantages in processing 3D FISH data through an innovative approach that enables a 2D network to effectively handle 3D data [22].
For experimental validation, the sensitivity and specificity of U-FISH were quantified using F1 scores and distance errors on test datasets. The method achieved a median F1 score of approximately 0.924, surpassing deepBlink (F1: 0.901), DetNet (F1: 0.886), SpotLearn (F1: 0.910), and rule-based methods [22]. The distance error of 0.290 pixels further confirmed its high precision in spot localization. Additional tests on datasets with simulated noise revealed strong noise resistance capabilities, highlighting its suitability for analyzing FISH images under various experimental conditions [22].
Signal-to-Noise Optimization in Chromogenic ISH
For chromogenic ISH methods, the QuantISH pipeline employs specific preprocessing steps to maximize sensitivity despite the limitations of single-channel detection. The process begins with extraction of contiguous images from tiled microscope scans, typically in MIRAX (MRXS) format from digital slide scanners with 40x magnification [24]. Individual tissue microarray spots are cropped using a module based on the HistoCrop method, followed by color deconvolution using the ImageJ software to separate brown marker RNA stain from blue nucleus stain [24].
A critical sensitivity-enhancing step involves cleaning demultiplexing artifacts through void-filling using a resynthesizer textural synthesis plug-in for GIMP software based on an algorithm that performs best-fit texture synthesis on user-specified regions of interest [24]. This process mitigates the effect of voids in the demultiplexed nucleus staining caused by overlapping signals in RNA-CISH images, enabling more accurate cell segmentation. The void-filled nucleus signal and demultiplexed marker RNA signal are then used in subsequent analysis steps.
Cell segmentation employs CellProfiler software with RescaleIntensity and IdentifyPrimaryObjects components, using Otsu's method with adaptive thresholding [24]. Non-default parameters were determined experimentally through multiple iterations: object diameter 25 to 170 pixels, and threshold smoothing scale of 1.3488. Cell type classification follows, based on nuclear morphology features extracted from the filtered nucleus channel and its segmentation, enabling cell-type specific expression quantification even without separate fluorescent channels for different cell types [24].
Visualization of Signaling Pathways and Workflows
Yn-situ Probe Hybridization and Amplification
Yn-situ Signal Amplification Pathway - This diagram illustrates the cascade of molecular interactions in the Yn-situ method that enable high-sensitivity RNA detection with minimal probes, culminating in significantly improved signal-to-noise ratio.
U-FISH Computational Enhancement Workflow
U-FISH Computational Enhancement Workflow - This workflow diagrams the transformation of variable-quality raw images into standardized enhanced outputs through deep learning, enabling consistent spot detection without manual parameter adjustment.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Their Functions in Sensitive ISH Detection
| Reagent Category | Specific Examples | Function in Sensitivity Optimization |
|---|---|---|
| Polymerases | LongAmp Taq DNA Polymerase, KAPA HiFi DNA Polymerase, Q5 High-Fidelity DNA Polymerase | Amplification of preamplifier probes with high yield and specificity; critical for Yn-situ probe production [21] |
| Fixation Reagents | Formaldehyde, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), PBS | Tissue preservation and RNA crosslinking; EDC significantly reduces RNA degradation, improving signal in stored samples [21] |
| Detection Systems | DAB, AEC, NBT/BCIP, Fast Red, Tyramide Signal Amplification (TSA) | Chromogenic or fluorescent signal generation and amplification; TSA enables detection of low-abundance targets [25] |
| Hybridization Buffers | SSC buffer, PBST with Tween 20, Prehybridization buffers | Control of stringency conditions; precise temperature and buffer composition critical for signal-to-noise optimization [25] |
| Enzymes | Pepsin, Strandase, Restriction enzymes (SfiI), HRP, Alkaline phosphatase | Tissue permeabilization, probe generation, and signal detection; pepsin digestion time must be optimized for each tissue type [21] [25] |
| Computational Tools | U-FISH, QuantISH, CellProfiler, ImageJ | Image analysis, spot detection, and quantification; deep learning methods enhance signal-to-noise post-acquisition [22] [24] |
The selection and optimization of research reagents directly impacts the achievable sensitivity of ISH experiments. For probe-based amplification methods like Yn-situ, the choice of DNA polymerase significantly affects preamplifier production. Comparative testing of five commercial polymerases identified LongAmp Taq DNA Polymerase as providing the highest yield of the desired 1 kb product, a critical factor in assay consistency [21]. For detection systems, the matching of enzyme conjugates with appropriate substrates is essential - HRP should be used with DAB and AEC, while alkaline phosphatase pairs with NBT/BCIP and Fast Red [25]. Mismatched combinations result in failed experiments regardless of target abundance.
Fixation protocols represent another crucial sensitivity determinant. While standard formaldehyde fixation preserves tissue architecture, the addition of EDC crosslinking dramatically improves signal detection in challenging samples by covalently linking RNA phosphate groups to protein amines, effectively immobilizing targets and reducing degradation [21]. This is particularly valuable for archival tissues or samples with partial RNA degradation. Enzymatic permeabilization using pepsin requires careful optimization, with typical digestion times of 3-10 minutes at 37°C, but must be adjusted for specific tissue types - over-digestion weakens or eliminates signal, while under-digestion decreases hybridization efficiency [25].
Computational reagents in the form of software tools and algorithms have emerged as essential components in modern ISH sensitivity optimization. The U-FISH platform, with its compact 163k parameter architecture, demonstrates how specialized computational tools can enhance signal-to-noise ratio post-acquisition, achieving a median F1 score of 0.924 in benchmark tests [22]. Similarly, the QuantISH pipeline enables sensitivity extraction from chromogenic ISH images through sophisticated color deconvolution and segmentation algorithms that would be impossible through visual assessment alone [24]. These tools effectively extend the detection limits of established laboratory methods through computational means.
The continuous refinement of in situ hybridization technologies has progressively lowered detection limits while improving signal-to-noise ratios through innovations in probe design, amplification strategies, and computational analysis. The quantitative framework presented here establishes standardized metrics and methodologies for sensitivity assessment that enable direct comparison across platforms and experimental conditions. As ISH applications expand into increasingly challenging domains including low-abundance targets, spatially resolved transcriptomics, and clinical diagnostics, rigorous attention to these fundamental sensitivity parameters becomes essential for methodological validation and appropriate technique selection. The integration of computational enhancement with biochemical amplification represents a particularly promising direction, offering complementary pathways to overcome the inherent limitations of each approach individually. By establishing clear metrics and standardized assessment protocols, the field can advance more rapidly toward the ultimate goal of comprehensive, quantitative spatial mapping of nucleic acids with single-molecule sensitivity across diverse biological contexts.
High-Sensitivity ISH Methods: Protocols, Costs, and Practical Applications
In molecular pathology, diagnostics, and research, the ability to evaluate gene expression within its native tissue context is invaluable. Traditional RNA in situ hybridization (ISH) techniques have often been hampered by technical complexity, insufficient sensitivity, and high background noise, limiting their clinical and research utility [26]. Similarly, while immunohistochemistry (IHC) is a popular alternative, it depends on the availability of specific antibodies, which can be expensive, time-consuming to develop, and sometimes impossible to create for species other than human, rat, and mouse [27]. Grind-and-bind methods like PCR, though useful, sacrifice all spatial and morphological context [27]. To solve these persistent problems, Advanced Cell Diagnostics (ACD) developed the RNAscope platform—an advanced ISH technology that combines single-molecule sensitivity with high specificity in a format amenable to automation, thereby enabling simplified and robust workflows for researchers and drug development professionals [27] [26].
Core Technology: The RNAscope Platform and Its Mechanism
The RNAscope technology represents a significant evolution in RNA ISH. Its foundational innovation is a proprietary double Z (ZZ) probe design that enables simultaneous signal amplification and background suppression, allowing for single-molecule visualization while preserving tissue morphology [26].
The Double Z Probe Design and Signal Amplification
The high performance of RNAscope stems from its unique probe architecture and amplification cascade, detailed below and illustrated in Figure 1.
dot code block
Figure 1. RNAscope Signal Amplification Mechanism. The diagram illustrates the proprietary double Z probe design and subsequent hybridization steps that enable specific signal amplification, resulting in a detectable dot for each target RNA molecule.
The process functions as follows:
- Probe Hybridization: Each target RNA molecule is hybridized by 20 double Z probe pairs on average [28]. A minimum of 7 Z pairs is required to generate a signal [29]. The double Z probe consists of three regions: a lower region that hybridizes to the target RNA, a spacer sequence, and a tail that binds to the pre-amplifier [28].
- Signal Amplification Cascade: The binding of the pre-amplifier to the Z probe tails initiates a multi-stage amplification process. Each pre-amplifier subsequently binds to multiple amplifier molecules, which in turn are conjugated with numerous labeled probes (either chromogenic or fluorescent) [28]. This cascade can result in up to 8,000-fold signal amplification, as approximately 400 labeled probes attach to each dimer [28].
- Detection: The final output is visualized as a distinct punctate dot, where each dot corresponds to a single RNA transcript [30]. The intensity or size of a dot can vary based on the number of ZZ probes bound to the target molecule, but the critical quantitative factor is the number of dots, not their size [30].
Assay Portfolio: RNAscope, BaseScope, and miRNAscope
ACD's technology suite includes three complementary assays tailored for different RNA targets, as summarized in Table 1.
Table 1: Comparison of ACD's RNAscope Technology Assays
| Feature | RNAscope Assay | BaseScope Assay | miRNAscope Assay |
|---|---|---|---|
| ZZ Pairs per Target | 20 (minimum of 7) [29] | 1 to 3 [29] | N/A (Unique probe design) [29] |
| Target Molecules | mRNA & lncRNA >300 bases [29] | Short targets (50-300 bases), exon junctions, splice variants, point mutations [29] | Small RNAs (17-50 bases) including miRNAs, ASOs, siRNAs [29] |
| Multiplex Capability | Single-plex up to 12-plex [29] | Single-plex to Duplex [29] | Single-plex [29] |
| Primary Application | Detection of long RNA transcripts [29] | Detection of short sequences, splice variants, and genetic mutations [29] | Detection of small non-coding RNAs [29] |
Implementing Simplified and Automated Workflows
A key advantage of the RNAscope platform is its compatibility with automated instrumentation, which standardizes the staining process, reduces manual hands-on time, and improves reproducibility.
Standardized Workflow Steps
The RNAscope procedure involves a series of standardized steps, whether performed manually or on an automated system. The entire workflow, from sample preparation to analysis, is depicted in Figure 2.
dot code block
Figure 2. RNAscope Automated Workflow. The process flows from sample preparation through pretreatment, hybridization, and finally to visualization and analysis, and is compatible with automated platforms like the Leica Bond Rx.
- Sample Preparation: The assay is compatible with formalin-fixed paraffin-embedded (FFPE) tissues, fresh frozen tissues, and cultured cells [28]. For FFPE samples, tissue fixation and processing are critical first steps.
- Pretreatment and Permeabilization: Slides are subjected to a universal pretreatment to expose target RNA sequences and permeabilize the tissue, allowing probe access [29] [28].
- Hybridization and Amplification: Target-specific probes are hybridized, followed by the sequential amplification steps. These key steps can be fully automated on instruments like the Leica Bond Rx and Roche Discovery ULTRA [28].
- Visualization and Imaging: Results can be visualized using a standard bright-field microscope for chromogenic dyes or an epifluorescent/confocal microscope for fluorescent assays [30].
- Analysis: Quantification is performed by counting the punctate dots, each representing an individual RNA transcript. This can be done manually or using image analysis software like HALO, QuPath, or Aperio [30] [28].
Performance and Validation Data
Independent studies and systematic reviews have validated the performance of RNAscope against established gold standard methods, confirming its place in modern research and diagnostic development.
Sensitivity and Specificity Compared to Gold Standard Methods
A 2021 systematic review comparing RNAscope to techniques like IHC, qPCR, and DNA ISH in human samples confirmed that RNAscope is a highly sensitive and specific method [28]. The review, which included 27 retrospective studies, found a high concordance rate (CR) between RNAscope and PCR-based methods (qPCR and qRT-PCR) and DNA ISH, ranging from 81.8% to 100% [28]. Its concordance with IHC was lower (58.7% to 95.3%), which is expected given that the two techniques measure different biomolecules (RNA vs. protein) that may not always correlate perfectly due to post-transcriptional regulation [28].
Performance in Challenging Sample Types
The robustness of RNAscope is demonstrated by its application on demanding sample types. A 2025 study successfully employed the miRNAscope assay on FFPE human brain samples stored for up to 76 years [31]. The combination of Nanostring nCounter profiling for candidate selection followed by miRNAscope ISH enabled the spatial detection of specific miRNAs like miR-124-3p in these decades-old samples, opening avenues for investigating epigenetic mechanisms in historical collections [31].
Table 2: Key Performance Metrics of RNAscope Technology
| Metric | Performance Data | Context / Comparison |
|---|---|---|
| Sensitivity | Single-molecule sensitivity [32] [26] | Capable of detecting individual RNA transcripts |
| Specificity | High specificity due to double-Z probe design [28] | Suppresses background noise; off-target binding is unlikely |
| Concordance with qPCR/qRT-PCR | 81.8% - 100% [28] | High agreement with quantitative PCR methods |
| Detection Efficiency | Similar to other sensitive ISH-based commercial SRT platforms [33] | Independent analysis shows performance matches other top platforms |
| Application in Old Archives | Successful detection in 76-year-old FFPE samples [31] | Validates use in long-term stored, challenging samples |
A Practical Guide to Data Analysis and Interpretation
A significant strength of the RNAscope assay is that its output—discrete dots corresponding to individual RNA molecules—lends itself to both semi-quantitative and quantitative analysis [34]. The analysis approach should be tailored to the biological question and expression pattern.
Controls and Sample Qualification
Before analyzing target probe data, it is crucial to review control slides. ACD recommends running a minimum of three slides per sample: the target marker panel, a positive control, and a negative control probe [30].
- Positive Control (e.g., PPIB, Polr2A, UBC): Validates tissue RNA integrity and successful assay execution. Failure indicates RNA degradation or technical issues [28] [33].
- Negative Control (bacterial dapB gene): Confirms the absence of background noise and non-specific signal [30] [28].
Analysis Methodologies for Different Expression Scenarios
The RNAscope analysis guide outlines several common expression scenarios and appropriate methodologies for each [34]:
- Homogeneous Target Expression: When a cell population shows uniform staining, the overall expression level can be reported as the average number of dots per cell using semi-quantitative scoring or image analysis software [34].
- Heterogeneous Target Expression: When cells within a population display different expression levels, the dynamic range should be quantified. Cell-by-cell expression profiles can be binned, and data can be presented as a histogram or calculated as a Histo score (H-score). The H-score is calculated as: H-score = Σ (ACD score x percentage of cells per bin), providing a value from 0 to 400 [34].
- Target Expression in Multiple Cell Types or Subpopulations: In this case, each cell type or region of interest should be analyzed independently according to the methodologies above [34].
- Co-expression of Two Targets: To determine the percentage of cells co-expressing two genes, quantitative software analysis is used to count the number of cells positive for both Target 1 and Target 2 divided by the total number of cells [34].
Essential Research Reagent Solutions
Implementing the RNAscope workflow effectively requires a set of core reagents and tools. The following toolkit is essential for researchers.
Table 3: Research Reagent Solutions for RNAscope Workflows
| Item | Function | Examples & Notes |
|---|---|---|
| RNAscope Probe Sets | Target-specific detection; hybridize to RNA of interest | Probes for >20,000 targets in multiple species; available in chromogenic or fluorescent formats [27] |
| RNAscope Reagent Kit | Contains reagents for pretreatment, hybridization, amplification, and detection | Kit components are specific to each assay type (e.g., RNAscope, BaseScope) and cannot be interchanged [29] |
| Positive Control Probes | Verify tissue RNA integrity and assay performance | PPIB (moderate expression), Polr2A (low expression), UBC (high expression) [28] [33] |
| Negative Control Probe | Assess background noise and non-specific binding | Bacterial dapB gene; should not produce signal in animal tissues [30] [28] |
| Automation Platform | Standardize staining, improve reproducibility, enable high-throughput | Compatible with Leica Bond Rx and Roche Discovery ULTRA automated stainers [28] |
| Image Analysis Software | Quantify dot counts and perform cell-based or region-based analysis | HALO (Indica Labs), QuPath, Aperio; enables quantitative, high-throughput data extraction [30] [28] |
RNAscope technology, with its innovative double Z probe design, has established a new standard for sensitive and specific in situ RNA analysis. Its compatibility with automated platforms translates this powerful detection capability into a simplified, reproducible workflow that is accessible for both research and clinical diagnostic development. As the field moves towards greater integration of AI-driven image analysis and expanded multiplexing capabilities, RNAscope and related commercial ISH platforms are poised to remain at the forefront of spatial biology, enabling deeper insights into gene expression within the native tissue architecture [35].
Spatial transcriptomics has revolutionized biological research by enabling precise mapping of gene expression within intact tissues and cells. The sensitivity and specificity of in situ hybridization (ISH) techniques are paramount for detecting low-abundance RNAs, short transcripts, and achieving single-molecule resolution. This whitepaper provides an in-depth technical comparison of three powerful ISH methods—Hybridization Chain Reaction (HCR), Signal Amplification By Exchange Reaction (SABER FISH), and click-amplifying FISH (clampFISH)—focusing on their customization potential for diverse research applications. We evaluate their performance characteristics, detailed experimental protocols, and reagent requirements to guide researchers in selecting and optimizing these methods for enhanced spatial RNA detection.
Advanced in situ hybridization techniques employ enzyme-free amplification to achieve high sensitivity and multiplexing capabilities while preserving tissue morphology. The core principle involves using DNA probes that bind to target RNAs and initiate localized signal amplification cascades.
- Hybridization Chain Reaction (HCR) utilizes metastable DNA hairpins that undergo a triggered, self-assembly process upon binding to an initiator sequence attached to the target probe. This results in the formation of a long, nicked double helix that tethers numerous fluorophores, providing linear signal amplification [36] [37].
- SABER FISH (Signal Amplification By Exchange Reaction) employs Primer Exchange Reaction (PER) to synthesize long, single-stranded DNA concatemers in vitro, which are appended to the primary target probes. These concatemers serve as scaffolds for hybridizing multiple fluorescent "imager" strands, significantly boosting the signal per probe [38] [39].
- clampFISH uses "C"-shaped probes that hybridize adjacently on the target RNA. These probes are then ligated via click chemistry, forming a stable circularized complex. This complex can withstand stringent washes and serves as a template for exponential signal amplification through successive hybridization of fluorescent secondary and tertiary probes [40].
Table 1: Quantitative Performance Comparison of HCR, SABER FISH, and clampFISH
| Performance Metric | HCR v3.0 | HCR-Cat (Next-Gen) | SABER FISH | clampFISH |
|---|---|---|---|---|
| Signal Amplification Type | Linear | Catalytic (Linear + Enzymatic) | Linear (concatemer-based) | Exponential (multi-round) |
| Typical Signal Gain | Baseline | ~240-fold (vs. HCR v3.0) [36] | Customizable (via concatemer length) [38] | ~1.74-fold per amplification round [40] |
| Probe Requirement for Detection | Multiple (e.g., 8-20) [36] | Can work with a single probe pair [36] | 15-30 short oligonucleotides [38] | Not specified |
| Short RNA Detection Capability | Limited for very short targets | Enabled via catalytic deposition [36] | Enabled via concatemer amplification [38] | Not specified |
| Multiplexing Capability | High (orthogonal hairpins) [36] | High (combined with immuno-detection) [36] | High (orthogonal concatemers + DNA exchange imaging) [39] | High (sequential rounds) [40] |
| Nuclear RNA/Transcription Site Detection | Challenging, lower contrast [40] | Not specifically reported | Not specifically reported | Excellent with nuclampFISH variant [40] |
The following diagram illustrates the fundamental mechanism and customization workflow for each method.
Experimental Protocols for Key Applications
Next-Generation HCR for Challenging Targets in Thick Tissues
Application: Robust detection of short or low-abundance RNA targets in whole-mount zebrafish, mouse brain, and Drosophila tissues with high autofluorescence [36].
Detailed Protocol:
- Fixation and Permeabilization: Fix samples (e.g., 5 dpf larval zebrafish) with 4% PFA. For thick tissues, perform permeabilization with 0.5% Triton X-100 for 24-48 hours.
- Hybridization: Hybridize split-initiator probe sets (e.g., 8 probes for hcrt mRNA) in a standard hybridization buffer (e.g., 30% formamide, 5x SSC) at 37°C for 12-36 hours.
- HCR v3.0 Amplification (Baseline): Wash off excess probes and amplify with fluorophore-labeled HCR hairpins (e.g., Alexa Fluor dyes) in amplification buffer at room temperature for 12-24 hours.
- HCR-Cat Enhancement: a. Instead of fluorophores, use HCR hairpins conjugated to haptens like FITC or Digoxigenin (DIG). b. After HCR amplification, incubate samples with an HRP-conjugated anti-FITC or anti-DIG antibody. c. Catalytically deposit fluorescent tyramide (e.g., Cy3- or Cy5-tyramide) for 10-30 minutes to achieve ~240-fold signal increase.
- Imaging: Clear samples using PACT or iDISCO+ methods for deep tissue imaging. Image using confocal or light-sheet microscopy.
SABER FISH for a Unified and Multiplexed Platform
Application: A "one probe fits all" approach for multiplexed RNA detection in whole-mount flatworms (Macrostomum lignano), planarians, and FFPE mouse intestinal sections [38].
Detailed Protocol:
- Probe Design and Synthesis: a. Design a pool of 15-30 short ssDNA oligonucleotides (~35-45 nt) complementary to the target RNA. Each probe should contain a 9-nt 3' initiator sequence. b. Synthesize long concatemeric probes via Primer Exchange Reaction (PER). Combine probes, PER primer, catalytic hairpin, and strand-displacing polymerase (e.g., Bst DNA Polymerase) in 1x Isothermal Amplification Buffer. Incubate at 37°C for 2-4 hours. Control concatemer length by varying reaction time.
- Sample Preparation: Fix and permeabilize samples (e.g., whole-mount M. lignano) according to standard protocols. For FFPE sections, perform deparaffinization and rehydration.
- Hybridization: Hybridize the PER-synthesized concatemerized probes to the sample in a standard FISH hybridization buffer. Use a hybridization temperature of 37°C for 12-48 hours.
- Signal Readout and Multiplexing: a. Wash and hybridize fluorescent 20-nt "imager" strands to the concatemers for 30-60 minutes. b. For multiplexing, use orthogonal concatemer sequences for different targets. Perform iterative imaging: strip imager strands gently (e.g., using 65% formamide) and re-hybridize with new imagers for the next target set.
- Imaging: Image using a standard epifluorescence or confocal microscope.
NuclampFISH for Nuclear RNA and Transcription Site Detection
Application: Specifically designed to amplify nuclear RNA signals and enable cell sorting based on transcriptional activity, compatible with downstream chromatin analysis [40].
Detailed Protocol:
- Reversible Crosslinking and Nuclear Isolation: Fix cells with a reversible crosslinker (e.g., DSG) instead of formaldehyde. Isolate nuclei by lysing the cell membrane and cytoplasm with a detergent-based lysis buffer (e.g., 0.5% Triton X-100).
- Probe Hybridization with Optimization: Hybridize clampFISH "C"-shaped primary probes targeting exons or introns in a high-salt hybridization buffer (e.g., 5x SSC) containing 0.1% Triton X-100 to enhance nuclear accessibility. Incubate at 37°C overnight.
- Click Ligation: Perform click chemistry ligation to circularize the probes bound to the target RNA.
- Exponential Signal Amplification: a. Hybridize fluorescent secondary probes complementary to the clampFISH backbone. b. Wash stringently. For higher signal, perform multiple rounds of amplification (tertiary, quaternary probes). Each round typically provides a ~1.74-fold signal increase. c. For nuclampFISH, 4-8 rounds of amplification are feasible to achieve bright, quantifiable nuclear spots.
- Flow Sorting and Downstream Analysis: Sort cells based on transcription site signal intensity using a flow cytometer. After sorting, reverse the crosslinks to perform downstream biochemical assays like ATAC-seq or ChIP on the sorted populations.
Research Reagent Solutions and Material Essentials
Table 2: Essential Research Reagents and Materials
| Item Name | Function / Description | Example Application / Note |
|---|---|---|
| Split-Initiator DNA Probes | Binds target mRNA and provides initiator sequence for HCR. Typically 36-39 nt [37]. | Core component for HCR v3.0 to minimize background. |
| HCR Hairpin Amplifiers | Metastable DNA hairpins (H1, H2) that self-assemble into polymer. Can be conjugated to fluorophores or haptens (FITC, DIG) [36]. | For HCR-Immuno/Cat, use hapten-labeled hairpins. |
| Primer Exchange Reaction (PER) Mix | Enzymatic system (Bst Polymerase, catalytic hairpin) to synthesize long concatemers on primary probes [38] [39]. | Core of SABER for in vitro signal scaffold synthesis. |
| Fluorescent Imager Strands | Short (~20 nt) oligonucleotides conjugated to fluorophores; hybridize to SABER concatemers [39]. | Readout for SABER FISH; easily exchanged for multiplexing. |
| ClampFISH "C" Probes | Circularizable probes that hybridize adjacently on target RNA [40]. | Form stable complex via click chemistry ligation. |
| Click Chemistry Ligation Kit | Reagents for bioorthogonal ligation of clampFISH probes [40]. | Creates stable, wash-resistant template for amplification. |
| Tyramide Reagents (e.g., Cy3-tyramide) | Fluorescent tyramide for HRP-catalyzed deposition; provides massive signal gain [36]. | Used in HCR-Cat for extreme sensitivity. |
| Reversible Crosslinker (e.g., DSG) | Crosslinks biomolecules while allowing reversal for downstream analysis [40]. | Critical for nuclampFISH compatibility with chromatin assays. |
HCR, SABER FISH, and clampFISH each offer distinct paths to customizing and enhancing in situ hybridization. HCR excels with its simple, enzyme-free protocol and new catalytic enhancements for the most challenging targets. SABER FISH provides a unified, highly multiplexable platform with predictable signal tuning via concatemer length. clampFISH, particularly nuclampFISH, offers unique capabilities for nuclear transcript detection and compatibility with flow sorting and downstream omics assays. The choice of method depends critically on the experimental priorities: utmost sensitivity for low-abundance targets, maximum multiplexing capability, or analysis of nuclear transcription and chromatin state. Understanding the principles, protocols, and reagent ecosystems of these methods empowers researchers to tailor spatial transcriptomics to their specific research needs.
The analysis of gene expression within its native spatial context is fundamental to deciphering complex biological systems, from embryonic development to disease pathogenesis. In situ hybridization (ISH) has served as a cornerstone technique for visualizing RNA distribution within tissues and cells. However, conventional ISH methods have historically faced a significant limitation: limited multiplexing capacity. The ability to simultaneously detect multiple RNA species is crucial for understanding coordinated gene expression networks, cellular heterogeneity, and complex tissue organization. While single-molecule fluorescence in situ hybridization (smFISH) achieves high sensitivity for detecting individual transcripts, its scalability is constrained by the spectral overlap of available fluorophores, typically allowing only a handful of targets to be visualized simultaneously [41] [42].
Recent technological advances have produced powerful new methods that overcome these limitations through innovative molecular and computational strategies. This technical guide focuses on two emerging platforms—DART-FISH (Decoding Amplified taRgeted Transcripts with Fluorescence in Situ Hybridization) and Yn-situ—that enhance multiplexing capabilities while addressing critical challenges in sensitivity, cost, and application to challenging samples like human tissues. These technologies represent significant progress in spatial transcriptomics, enabling researchers to map dozens to hundreds of genes within their anatomical contexts with cellular resolution.
Core Principles and Workflow
DART-FISH is a padlock probe-based technology capable of profiling hundreds to thousands of genes in centimeter-sized human tissue sections. The method combines padlock probe hybridization, rolling circle amplification (RCA), and a combinatorial barcoding system to achieve highly multiplexed RNA detection [43] [42].
The following diagram illustrates the core workflow of DART-FISH:
Key Methodological Innovations
Padlock Probe Design and In Situ Amplification
DART-FISH utilizes padlock probes—linear DNA oligonucleotides that hybridize to cDNA targets and are subsequently circularized through ligation. These probes contain:
- Target-complementary sequences at both ends that flank the cDNA region of interest
- A universal amplification sequence for subsequent rolling circle amplification
- Gene-specific barcode regions composed of concatenated decoder sequences [42]
Following circularization, the probes undergo rolling circle amplification (RCA), generating repetitive DNA nanoballs known as RCA colonies (rolonies). Each rolony contains hundreds of copies of the barcode sequence, significantly amplifying the signal from individual transcripts and enabling robust detection [43].
Combinatorial Barcoding and Isothermal Decoding
A distinctive feature of DART-FISH is its implementation of a combinatorial barcoding system adapted from Illumina BeadArray technology. In this scheme:
- Each gene is assigned a unique barcode that is "on" in exactly k out of n imaging rounds
- When "on," the barcode fluoresces in one of three color channels
- With n=6 rounds and k=3, the system can generate 540 unique barcodes—sufficient for profiling hundreds of genes [42]
The decoding process is enzyme-free and isothermal, relying solely on sequential hybridization of fluorescent decoding probes at room temperature. This simplifies the experimental procedure and reduces between-cycle preparation time to approximately 10 hours for imaging 121 genes in large tissue sections [42].
RiboSoma Cytoplasmic Staining
A significant challenge in spatial transcriptomics of human tissues is accurate cell segmentation. DART-FISH introduces RiboSoma, an omni-cell type cytoplasmic stain that substantially improves the identification of cell bodies. RiboSoma is implemented by adding a 5' handle to reverse-transcription primers, enabling collective visualization of all cDNA molecules with fluorescent oligos. When crosslinked to a polyacrylamide gel, this method enhances cDNA retention and provides a quality control metric for in situ reactions [43] [42].
Cost-Effective Probe Production
To address the prohibitive costs of synthesizing individual padlock probes for hundreds of genes, DART-FISH employs an enzymatic protocol for in-house probe production from microarray-synthesized oligo pools. This approach reduces costs to less than 25% of direct synthesis methods and becomes increasingly economical with larger probe sets [42].
Core Principles and Workflow
Yn-situ was developed to address limitations in conventional smFISH methods, particularly their high cost when using proprietary reagents and limited application to long transcripts. This method enhances the robustness, sensitivity, and ease of use while reducing the cost of single-molecule RNA FISH approaches [44].
While the search results provide limited technical details about Yn-situ, the methodology appears to focus on optimizing probe design and hybridization conditions to improve detection efficiency. Compared to traditional smFISH that requires many short probes tiling the target transcript, Yn-situ potentially reduces the number of probes needed while maintaining high sensitivity, making it more accessible for research laboratories [44].
Comparative Technical Specifications
Performance Metrics and Applications
Table 1: Technical Specifications and Performance Comparison of DART-FISH and Yn-situ
| Parameter | DART-FISH | Yn-situ |
|---|---|---|
| Multiplexing Capacity | 100-1000+ genes [43] [42] | Limited data available, improved over standard smFISH [44] |
| Detection Sensitivity | Detects transcripts as short as 461 nucleotides (e.g., neuropeptides) [43] | Designed for robust single-molecule detection [44] |
| Signal Amplification Method | Rolling circle amplification (RCA) [42] | Not specified in available literature |
| Barcoding Strategy | Combinatorial barcoding with Illumina-derived system [42] | Not specified |
| Cell Segmentation Aid | RiboSoma cytoplasmic stain [43] | Not specified |
| Typical Applications | Large human tissue sections (e.g., brain cortex, kidney) [42] | Cellular RNA detection with improved cost efficiency [44] |
| Protocol Duration | <10 hours for 121 genes [42] | Not specified |
| Key Innovation | Enzyme-free isothermal decoding and computational deconvolution [42] | Improved robustness and reduced cost [44] |
Table 2: Key Experimental Procedures for DART-FISH and Yn-situ
| Protocol Step | DART-FISH Methodology | Yn-situ Methodology |
|---|---|---|
| Sample Preparation | Fresh-frozen tissue sections fixed with PFA, permeabilized [42] | Standard FISH sample preparation (inferred) |
| cDNA Synthesis | Reverse transcription with random/dT primers containing 5' handle [42] | Not specified |
| Target Hybridization | Padlock probe hybridization and circularization at high temperature [42] | Optimized oligonucleotide probe hybridization |
| Signal Amplification | Rolling circle amplification to generate rolonies [43] | Not specified |
| Detection | Sequential fluorescent decoding probes with 6 rounds of imaging [42] | Fluorescence detection |
| Image Analysis | Computational decoding-by-deconvolution for crowded regions [43] | Standard smFISH analysis (inferred) |
Research Reagent Solutions
Table 3: Essential Research Reagents for Advanced Multiplexed FISH Technologies
| Reagent/Category | Function | Technology Application |
|---|---|---|
| Padlock Probes | Target-specific probes for cDNA hybridization and circularization | DART-FISH [42] |
| RCA Enzymes | Enzymes for rolling circle amplification to generate DNA nanoballs | DART-FISH [42] |
| Decoder Probes | Fluorescently-labeled oligonucleotides for barcode readout | DART-FISH [42] |
| Polyacrylamide Gel | Matrix for cDNA cross-linking and retention | DART-FISH [42] |
| RiboSoma Oligos | Fluorescent probes against 5' handle for cytoplasmic staining | DART-FISH [43] |
| Optimized smFISH Probes | Short oligonucleotide probes for efficient target binding | Yn-situ [44] |
Technical Considerations and Implementation Challenges
Addressing Optical Crowding and Tissue Autofluorescence
A significant challenge in highly multiplexed FISH, particularly in human tissues, is optical overcrowding of signals and high autofluorescence background. DART-FISH employs both computational and molecular strategies to address these issues:
- Computational deconvolution: A pixel-based decoding method resolves densely packed rolonies without requiring physical tissue expansion, maintaining imaging throughput [43]
- Signal amplification: RCA-generated rolonies provide intense, localized signals that overcome tissue autofluorescence from lipofuscin, collagen, and mitochondria [42]
- Robust barcoding: The Illumina-derived barcoding system with limited cross-hybridization ensures specific decoding even in suboptimal signal-to-noise conditions [42]
Application to Challenging Tissue Types
Both technologies show particular utility for complex tissue environments:
- DART-FISH has been successfully applied to human neocortex and diseased kidney tissues, demonstrating robustness in tissues with inherent autofluorescence and structural complexity [42]
- The enhanced sensitivity of Yn-situ makes it suitable for detecting lower abundance transcripts that may be challenging with conventional smFISH [44]
DART-FISH and Yn-situ represent significant advancements in multiplexed RNA in situ hybridization technologies, each addressing critical limitations in conventional approaches. DART-FISH stands out for its exceptional multiplexing capacity, innovative combinatorial barcoding system, and application to challenging human tissue samples. Its enzyme-free isothermal decoding and cost-effective probe production make it particularly suitable for large-scale spatial transcriptomics studies. Yn-situ offers improved robustness and cost efficiency for single-molecule RNA detection, potentially broadening access to highly sensitive in situ hybridization.
For researchers designing studies requiring highly multiplexed spatial gene expression analysis, particularly in complex human tissues, DART-FISH provides a powerful and scalable solution. For applications requiring enhanced sensitivity over conventional FISH with potentially lower complexity, Yn-situ represents a promising alternative. As these technologies continue to evolve, they will undoubtedly expand our ability to map complex gene expression patterns with unprecedented resolution and scale, advancing our understanding of tissue organization in health and disease.
In situ hybridization (ISH), a foundational technique for visualizing nucleic acids within tissues, has undergone significant evolution to overcome challenges related to sensitivity, complexity, and cost [7]. For researchers and drug development professionals engaged in sensitivity comparison studies, selecting an appropriate ISH method involves a critical trade-off between monetary cost, time investment, and technical ease of use. While conventional ISH methods offer lower costs, newer, high-sensitivity variants provide simplified workflows and robust multiplexing capabilities at a higher price point [7]. This comparative analysis examines established and emerging ISH technologies, providing a structured framework for evaluating these critical parameters within a broader thesis on ISH sensitivity. The decision extends beyond mere technical performance to encompass practical considerations of budget, personnel expertise, and project scope, all of which directly impact research efficiency and scalability in both academic and industrial settings.
Method Comparison: Quantitative Analysis of ISH Variants
The selection of an ISH method requires a balanced consideration of financial and temporal resources. The table below summarizes the key cost and time characteristics of major ISH variants, providing a direct comparison for researchers [7].
Table 1: Comparative Analysis of ISH Methods: Cost, Time, and Usability
| Method | Monetary Cost (Total) | Monetary Cost (Per Sample) | Staining Time | Difficulty of Experimental Procedures | Probe Design & Synthesis |
|---|---|---|---|---|---|
| DIG-RNA ISH | Low | Low | 2–3 days | Difficult | Done by user (can be outsourced) |
| RNAscope | High | High | 1 day | Easy | Provided by manufacturer only |
| HCR ISH | Moderate | Decreases with increasing sample size | 1–3 days | Moderate | Done by user (can be outsourced) |
| clampFISH | Moderate | Decreases with increasing sample size | 1–3 days | Moderate | Done by user |
| SABER FISH | Moderate | Decreases with increasing sample size | 2–3 days | Moderate | Done by user |
The data reveals a clear inverse relationship between user effort and monetary cost. Conventional digoxigenin (DIG)-labeled RNA ISH offers the lowest monetary cost but demands the highest level of expertise and the longest experimental time, typically requiring 2–3 days to complete and involving difficult procedures [7]. In contrast, the commercialized RNAscope platform significantly reduces the hands-on time and expertise barrier, completing staining in 1 day with easy procedures, but this convenience comes at a high monetary cost [7]. Other high-sensitivity variants like HCR ISH, clampFISH, and SABER FISH occupy a middle ground in terms of total monetary cost. A key feature of these methods is that the cost per sample becomes more economical with larger sample sizes, making them particularly suitable for larger-scale studies [7]. However, they require a moderate level of experimental skill and a time investment for protocol optimization [7].
Detailed Experimental Protocols for Key ISH Methods
A deeper understanding of the time investment and ease of use is rooted in the specific procedural requirements of each method. The protocols below outline the core workflows for conventional and a high-sensitivity method.
Conventional ISH Protocol
The conventional ISH protocol, often using DIG-labeled RNA probes, is a multi-day process requiring careful optimization at each stage [7] [45]. The following workflow details the critical steps.
Fixation and Permeabilization: Tissue samples are fixed with paraformaldehyde (e.g., 4% in PBS) to preserve structure and nucleic acid integrity [45]. This is followed by permeabilization, often using proteinase K digestion and detergents like Triton X-100 or SDS, to allow probe access [7] [45]. An optional acetylation step can be included here to reduce background by blocking positively charged amines [45].
Pre-hybridization Blocking: Samples are incubated with a pre-hybridization buffer containing blocking agents (e.g., Denhardt's solution, salmon sperm DNA, heparin) for 30-60 minutes at 37-45°C to minimize non-specific probe binding [45]. A typical buffer recipe includes 50% formamide, 1X SSC, and various blocking components [45].
Hybridization and Washes: Denatured, labeled probes are applied to the sample and incubated overnight (16-18 hours) in a humidified chamber at a optimized temperature (37-45°C) [45]. Subsequent stringency washes with buffers like SSC, performed at controlled temperatures, remove unbound and weakly bound probes to ensure specificity [45].
Signal Detection and Imaging: For chromogenic detection, an enzyme-conjugated antibody (e.g., anti-DIG-AP) is applied, followed by a substrate (e.g., NBT/BCIP) which develops until the signal appears [45]. For fluorescence (FISH), samples are counterstained with DAPI and mounted in an antifade medium before imaging with a fluorescence microscope [45].
RNAscope Protocol
RNAscope represents a major shift towards standardized, user-friendly assays. Its "ready-to-use" probe design and simplified workflow drastically reduce hands-on time and the need for optimization.
The RNAscope technology is based on a proprietary signal amplification system using a novel type of "ZZ" probe [7]. The key differentiator is its simplified, standardized workflow enabled by commercial kits. The procedure uses reagents supplied in drop bottles to facilitate easy application [7]. The protocol involves a series of sequential hybridizations: first with the target-specific ZZ probes, followed by pre-amplifier and amplifier molecules that create a branching structure for significant signal amplification [7]. This entire process, from sample pretreatment to final signal development, can be completed in a single day, a significant reduction from the multi-day conventional protocol [7]. Furthermore, the method is compatible with automated staining systems, enhancing reproducibility and throughput for clinical and high-volume research applications [7].
The Scientist's Toolkit: Essential Research Reagent Solutions
The execution of any ISH protocol relies on a suite of critical reagents and equipment. The following table details the essential components of an ISH workflow and their functions, providing a checklist for researchers.
Table 2: Key Research Reagent Solutions for In Situ Hybridization
| Item Category | Specific Examples | Function/Purpose |
|---|---|---|
| Fixatives | Paraformaldehyde (4%, PBS), Formalin (10%) | Preserves tissue architecture and nucleic acid integrity by cross-linking. |
| Permeabilizers & Detergents | Proteinase K, Triton X-100, Tween-20, SDS, NP-40 | Disrupts membranes to allow probe entry into cells and subcellular compartments. |
| Blocking Agents | Bovine Serum Albumin (BSA), Casein, Salmon Sperm DNA, Heparin | Reduces nonspecific binding of probes and detection antibodies, lowering background. |
| Hybridization Buffers & Salts | Saline Sodium Citrate (SSC), Denhardt's Solution, Formamide | Creates optimal ionic and chemical environment for specific probe-target hybridization. |
| Probes | DIG-labeled RNA probes, RNAscope ZZ Probes, HCR hairpin DNA | The labeled nucleic acid sequence that binds specifically to the target of interest. |
| Detection Systems | Anti-DIG-AP, NBT/BCIP (Chromogenic), Fluorescently labeled tyramide (TSA) | Visualizes the bound probe through enzymatic color reaction or fluorescence. |
| Key Equipment | Humidified hybridization chamber, incubator/oven, water bath, fluorescence microscope | Provides controlled conditions for hybridization, washes, and signal visualization. |
The landscape of in situ hybridization offers multiple paths, each with distinct advantages in cost, time, and usability. The choice for a research or drug development project is not one of identifying a universally "best" method, but of aligning technical capabilities with project constraints and goals. For large-scale, cost-sensitive projects where technical expertise is available, user-dependent methods like HCR ISH or optimized conventional ISH provide excellent value. Conversely, for targeted studies prioritizing rapid results, reproducibility, and minimal protocol development, the high monetary cost of commercial solutions like RNAscope is justified by significant savings in time and labor. As the field advances, driven by trends in automation, multiplexing, and AI-integration, the balance of these factors will continue to evolve, offering scientists an ever more powerful and accessible toolkit for spatial genomics.
In situ hybridization (ISH) stands as a cornerstone technique in molecular pathology and research, enabling the localization of specific nucleic acid sequences within cells and tissues. Since its inception in 1969, ISH has evolved from radioactive methodologies to sophisticated non-radioactive systems that offer unprecedented sensitivity and specificity [14] [46]. This evolution has proven particularly valuable for drug development professionals who require precise spatial information about gene expression, therapeutic oligonucleotide distribution, and biomarker localization [14]. The technique's unique capacity to provide spatial context at the single-cell level makes it indispensable for understanding disease heterogeneity, validating therapeutic targets, and elucidating mechanisms of action for novel therapeutics.
Within toxicologic pathology and drug research, ISH has gained increasing prominence as a complement to immunohistochemistry, especially for targets where specific antibodies are unavailable or for detecting nucleic acid-based therapeutics [14]. The ability to visualize low-abundance transcripts, short microRNAs (miRNAs), and therapeutic oligonucleotides within their morphological context provides insights that bulk molecular methods cannot achieve. This guide systematically compares ISH methodologies across different nucleic acid targets, providing evidence-based recommendations for researchers navigating the complex landscape of available technologies.
Core Principles and Technical Fundamentals
Essential ISH Workflow and Critical Decision Points
The fundamental ISH procedure consists of several interconnected steps, each requiring careful optimization to ensure specific detection while preserving tissue morphology and nucleic acid integrity. The workflow begins with appropriate tissue preparation, followed by probe selection and hybridization, and concludes with signal detection and visualization [14]. Understanding these core principles is essential for selecting the appropriate method for specific experimental goals.
Tissue preparation represents perhaps the most critical pre-analytical variable influencing ISH success. Proper fixation preserves both morphological detail and nucleic acid integrity. 10% neutral buffered formalin (NBF) has emerged as the standard fixative for ISH, with fixation time optimally spanning 24±12 hours at room temperature using a 10:1 fixative-to-tissue ratio [14]. Inadequate fixation risks under-preservation and subsequent nucleic acid degradation, while over-fixation can mask target sequences through excessive cross-linking, reducing probe accessibility [14]. For miRNA targets specifically, extended fixation times (up to 144 hours) have demonstrated improved signal intensity when combined with appropriate retrieval methods [47].
Following fixation, permeabilization becomes necessary to allow probe access to intracellular targets. This typically involves proteinase treatments (e.g., proteinase K) or heat-induced retrieval methods [14] [47]. The intensity and duration of permeabilization must be carefully titrated, as insufficient treatment limits probe access while excessive digestion compromises tissue morphology [14]. Recent evidence indicates that heat-induced retrieval using high-pH buffers (pH 9) outperforms traditional proteinase K digestion for miRNA detection, providing superior signal intensity while better preserving morphological detail [47].
Table 1: Critical Factors in Tissue Preparation for Different Targets
| Parameter | RNA/mRNA Targets | microRNA Targets | DNA Targets |
|---|---|---|---|
| Optimal Fixation | 10% NBF, 24±12 hours [14] | 10% NBF, up to 144 hours [47] | 10% NBF, 24±12 hours [14] |
| Permeabilization Methods | Proteinase K or heat-induced retrieval [14] | Heat-induced retrieval (TRS pH 9) preferred [47] | Proteinase K or heat-induced retrieval [14] |
| Storage Conditions | -20°C or -80°C for long-term; use within 3 months at room temperature [14] [48] | Similar to RNA; cool, dry environment essential [48] | Less critical; room temperature often acceptable |
| Key Challenges | RNase degradation; over-fixation reducing accessibility [14] [48] | Small size; association with protein complexes [49] [47] | Target accessibility in chromatin |
Probe Design and Hybridization Chemistry
Probe selection constitutes another fundamental decision point significantly influencing sensitivity, specificity, and signal-to-noise ratio. Probes can be broadly categorized into DNA probes, RNA probes (riboprobes), and synthetic oligonucleotides incorporating various backbone modifications [14] [46]. For conventional RNA detection, digoxigenin (DIG)-labeled RNA probes between 200-1000 bases offer high sensitivity and have been widely adopted [48] [7]. These probes typically employ antisense strands complementary to target mRNAs, with sense strands serving as negative controls [48].
For challenging targets like miRNAs, synthetic oligonucleotides incorporating specialized modifications provide enhanced performance. Locked nucleic acid (LNA) nucleotides contain a methylene bridge that locks the ribose in a C3'-endo conformation, increasing duplex stability and melting temperature [49]. Similarly, 2'-O-methyl RNA (2OMe) modifications demonstrate increased probe:RNA duplex stability, faster hybridization kinetics, and improved capability to bind structured targets [49]. Optimal designs often combine these technologies, such as 2OMeRNA+LNA probes at a 2:1 ratio, which have demonstrated superior sensitivity for low-copy-number miRNAs like miR-130a in mouse brain compared to DNA+LNA probes [49].
Hybridization conditions must be carefully optimized according to probe characteristics. Key parameters include temperature (typically 18-24°C below melting temperature for DNA+LNA probes), time, and buffer composition [49] [48]. Recent innovations include substituting toxic formamide with 4M urea in hybridization buffers, achieving equally specific but nontoxic assays [49]. Additionally, exclusion of yeast RNA from hybridization buffers when using high-affinity 2OMeRNA+LNA probes can improve sensitivity by reducing nonspecific probe retention [49].
Target-Specific Method Selection and Optimization
microRNA Detection Strategies
The short length of miRNAs (18-25 nucleotides) presents unique challenges for detection, requiring specialized approaches to achieve sufficient sensitivity and specificity. Traditional ISH methods often struggle with these low-copy-number targets, necessitating either signal amplification or probe modification strategies.
LNA-modified DNA probes represent a well-established approach for miRNA ISH, significantly enhancing duplex stability and discrimination between perfectly matched and mismatched targets [49] [47]. These probes have enabled the specific detection of miRNAs in FFPE tissues across various research and diagnostic applications. However, detection of low-copy-number miRNAs may still require additional enhancements, such as the use of 2OMeRNA+LNA probes which provide superior performance in terms of sensitivity and signal-to-noise ratio compared to DNA+LNA probes [49].
For the most challenging miRNA targets, highly sensitive commercial systems like miRNAscope offer robust solutions. This technology employs a proprietary Z-probe design combined with branched DNA (bDNA) signal amplification to achieve single-molecule sensitivity [31]. The method has successfully detected miRNAs including miR-124-3p, miR-9-5p, and miR-145-5p in human brain samples stored for decades as FFPE blocks, demonstrating remarkable robustness for archival specimens [31]. The automated staining protocol available for platforms like the Leica Bond RX further enhances reproducibility, making it particularly valuable for standardized drug research applications [31].
Table 2: Comparison of High-Sensitivity ISH Methods for Different Targets
| Method | Principle | Best Applications | Sensitivity | Multiplexing Capability |
|---|---|---|---|---|
| Conventional DIG-RNA ISH | Enzyme-labeled RNA probes; chromogenic/fluorescent detection [7] | mRNA, lncRNAs; low-cost applications [7] | Moderate | Difficult [7] |
| RNAscope/ miRNAscope | Proprietary Z-probes with branched DNA amplification [7] [31] | Low-abundance mRNAs, miRNAs; standardized workflows [7] [31] | Very high (single-molecule) | Easy (up to 12-plex reported) [7] |
| HCR ISH | Hybridization chain reaction; self-assembling fluorescent DNA nanostructures [7] | mRNA, miRNAs; customizable amplification [7] | High | Easy [7] |
| clampFISH | Padlock probes with click chemistry fixation [7] | mRNA; repeated imaging studies [7] | High | Easy [7] |
| SABER FISH | Primer exchange reaction to generate concatemers [7] | mRNA; customizable signal amplification [7] | High (adjustable) | Easy [7] |
mRNA and Long RNA Detection
For mRNA targets, the selection criteria shift toward balancing sensitivity with morphological preservation. Conventional DIG-labeled RNA probes remain a robust choice for many applications, particularly when targeting moderately to highly expressed transcripts [48] [7]. These probes typically range from 250-1500 bases, with approximately 800 bases offering optimal sensitivity and specificity [48]. A key advantage of RNA probes is their high affinity for complementary RNA sequences, enabling strong hybridization signals.
Recent innovations have significantly expanded options for mRNA detection. RNAscope technology provides exceptional sensitivity for low-abundance transcripts while offering simplified workflows compatible with automation [7]. The method's design incorporates multiple proprietary Z-probes that each bind adjacent target sequences, followed by signal amplification through branched DNA structures. This approach visualizes individual mRNA molecules as distinct dots, enabling both qualitative localization and quantitative analysis [7].
For researchers requiring custom probe designs or wishing to avoid commercial systems, HCR ISH (hybridization chain reaction) presents an attractive alternative. This method uses initiator probes that trigger self-assembly of fluorescent DNA hairpins, resulting in amplified signal that can be tuned by varying reaction time [7]. The open-source nature of HCR allows for greater customization and cost-effectiveness, particularly for large-scale studies.
DNA Target Localization
DNA ISH primarily focuses on detecting genomic alterations, chromosomal abnormalities, and viral DNA integration events. While early DNA ISH methods relied heavily on radioactive probes, contemporary approaches predominantly use fluorescent in situ hybridization (FISH) with locus-specific probes [46]. These techniques enable visualization of specific genes, chromosomal regions, or entire chromosomes, providing crucial information for cancer diagnostics, prenatal testing, and basic research.
Chromogenic ISH (CISH) offers an alternative to FISH for DNA detection, utilizing peroxidase- or alkaline phosphatase-labeled reporter antibodies that generate permanent chromogenic signals visible by bright-field microscopy [46]. CISH provides the advantage of signal permanence and compatibility with standard histopathology workflows, making it particularly valuable for clinical diagnostics and retrospective analyses.
Advanced Applications and Integrated Workflows
Multiplexing and Combination Techniques
The ability to detect multiple nucleic acid targets simultaneously within a single sample provides powerful insights into complex biological systems and disease processes. Modern ISH methods have made significant strides in multiplexing capability, with several platforms now supporting simultaneous detection of 3-12 targets [7]. Approaches like MERFISH (multiplexed error-robust FISH) enable highly multiplexed imaging of numerous RNA species in their native cellular environment, mapping cellular diversity at subcellular resolution [46].
Combining ISH with protein detection via immunohistochemistry (IHC) creates particularly valuable experimental paradigms for correlating transcriptional and translational events within individual cells. Traditional ISH methods often presented challenges for combination with IHC due to protein-denaturing hybridization conditions and protease-based permeabilization [7]. However, newer high-sensitivity ISH variants typically employ lower hybridization temperatures that better preserve antigen integrity, facilitating robust simultaneous detection of RNA and protein targets [7].
Quantitative Analysis and Validation
As ISH methodologies achieve single-molecule sensitivity, quantitative analysis becomes increasingly feasible and valuable. Techniques like smFISH (single-molecule FISH) enable precise counting of individual RNA molecules within single cells, revealing natural cell-to-cell variations in gene expression that bulk analyses obscure [46]. For drug development applications, this capability supports precise assessment of target engagement, pharmacodynamic effects, and heterogeneity in treatment response.
Validation remains essential when implementing any ISH method, particularly for novel targets or applications. Recommended practices include:
- Using sense strand probes or scramble sequences as negative controls [48] [47]
- Including tissues with known expression patterns as positive controls [47]
- Comparing results with orthogonal methods like RT-qPCR or sequencing when feasible [31]
- Performing titration experiments for probe concentration and permeabilization conditions [48]
- Assessing specificity through mismatch controls or target-specific blockers [49]
Research Reagent Solutions Toolkit
Table 3: Essential Reagents and Their Applications in Modern ISH
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Probe Technologies | LNA-modified DNA [49] [47], 2OMeRNA+LNA [49], DIG-labeled RNA [48] | LNA enhances duplex stability; 2OMeRNA improves kinetics; DIG enables enzymatic detection |
| Labeling Systems | Digoxigenin (DIG) [48], Fluorescein (FITC) [49], Biotin [46], Fluorescent dyes (Cy3, Cy5, Alexa dyes) [46] | DIG offers high sensitivity for chromogenic detection; fluorophores enable multiplexing |
| Fixation Agents | 10% Neutral Buffered Formalin [14], Paraformaldehyde [48] | NBF is standard; PFA often used for frozen sections and whole-mount ISH |
| Permeabilization Methods | Proteinase K [14] [48], Pepsin [47], Heat-induced retrieval (TRS pH 6/9) [47] | Enzymatic methods require titration; heat-induced retrieval preferred for miRNAs [47] |
| Hybridization Buffers | Formamide-based [48], Urea-based [49] | Urea provides non-toxic alternative to formamide with equal specificity [49] |
| Signal Amplification | Tyramide Signal Amplification (TSA) [7], Branched DNA [7], Hybridization Chain Reaction [7] | TSA enhances sensitivity; bDNA enables single-molecule detection; HCR is customizable |
Experimental Workflow and Visualization
The diagram below illustrates the key decision points and corresponding methodological recommendations for selecting optimal ISH strategies based on experimental goals:
Selecting the appropriate ISH method requires careful consideration of multiple factors, including target type (DNA, mRNA, miRNA), abundance, required sensitivity, multiplexing needs, and available resources. For DNA targets, FISH and CISH provide robust detection of genomic alterations. mRNA detection spans from conventional DIG-RNA ISH for moderate-abundance transcripts to RNAscope and HCR ISH for low-copy targets. miRNA detection benefits most from specialized approaches including LNA-modified probes, 2OMeRNA+LNA chemistry, and miRNAscope technology, particularly when combined with heat-induced retrieval methods.
Emerging trends in ISH technology continue to expand application possibilities. Microfluidic approaches are addressing hybridization efficiency challenges by actively delivering probes to targets, significantly reducing assay times [46]. Multiplexing capabilities are rapidly advancing, with methods like MERFISH enabling simultaneous visualization of hundreds to thousands of RNA species [46]. Integration with immunohistochemistry is becoming more robust, facilitated by gentler hybridization conditions that preserve protein epitopes [7]. These developments promise to further establish ISH as an indispensable tool for drug development, diagnostic applications, and basic research requiring spatial context for nucleic acid localization.
For researchers navigating this complex landscape, the key lies in aligning methodological choices with specific experimental questions while implementing appropriate validation controls. The continued refinement of ISH technologies ensures this decades-old technique will remain at the forefront of spatial molecular analysis for the foreseeable future.
Optimizing ISH Experiments: Troubleshooting Common Pitfalls and Enhancing Signal
In-situ hybridization (ISH) is a cornerstone technique in molecular pathology, diagnostics, and research, enabling the visualization of specific nucleic acid sequences within intact cells and tissues. However, its diagnostic sensitivity and analytical performance are frequently compromised by three common experimental failures: absence of signal, weak signal, and high background fluorescence. These challenges are particularly critical in comparative sensitivity studies, where consistent, high-quality results are paramount for accurate evaluation of different ISH methodologies. Technological advances, including the development of branched DNA (bDNA) amplification systems like RNAscope, have demonstrated superior sensitivity against traditional ISH methods in validation pilots [35]. Nevertheless, optimizing fundamental parameters across all ISH platforms remains essential for reliable data interpretation. This guide provides researchers with a systematic troubleshooting framework and detailed protocols to address these core failure modes, thereby enhancing the reliability of ISH sensitivity comparisons.
Troubleshooting Common ISH Failures
The following sections provide a detailed analysis of the most common ISH failures, their root causes, and evidence-based solutions.
No Signal or Weak Signal
A absent or weak signal is often the result of inadequate probe hybridization or target accessibility. The key is to systematically examine each step of the protocol, from sample preparation to final detection.
Table 1: Troubleshooting No or Weak Signal
| Problem Cause | Underlying Principle | Recommended Solution | Supporting Data/Example |
|---|---|---|---|
| Poor Fixation [50] [51] | Under-fixation compromises cellular architecture, leading to nucleic acid degradation and loss of target. | Use freshly prepared fixative (e.g., formaldehyde, Carnoy's solution) and adhere strictly to recommended fixation times. | For FFPE tissues, aim for sections of 3-4μm thickness to ensure optimal probe penetration and interpretation [50]. |
| Inefficient Permeabilization [51] | Cellular membranes and proteins block probe access to the target nucleic acids. | Optimize concentration, time, and temperature for permeabilizing agents (e.g., Triton X-100, Tween-20, proteinase K). | Over-digestion with protease can damage tissue morphology, while under-digestion prevents probe access [25] [51]. |
| Suboptimal Denaturation [50] | Incomplete denaturation of target DNA prevents single-stranded probe binding. | Ensure denaturation is performed at 95 ± 5°C for 5-10 minutes. Use a calibrated hot plate and a moist environment to prevent slide drying [25]. | For FFPE samples with extensive cross-linking, confirm the denaturation temperature and time are sufficient [50]. |
| Low Probe Quality or Concentration [50] [51] | Inefficiently labeled probes or insufficient probe volume result in weak hybridization. | Check probe design and labeling efficiency. Optimize probe concentration; too little leads to weak signal, while too much increases background [50]. | Ensure probes are specific and sensitive. For low-abundance targets, consider signal amplification methods like Tyramide Signal Amplification (TSA) [25]. |
| Target Degradation [25] | Long intervals between tissue acquisition and fixation, or RNase contamination for RNA targets, degrade the nucleic acid. | Minimize the time between tissue collection and fixation. Use RNase-free conditions for RNA ISH. | Always run positive control slides to distinguish between protocol failure and true negative results [25]. |
The flowchart below outlines a systematic diagnostic approach for resolving no or weak signal issues.
High Background
High background fluorescence obscures specific signal and complicates data interpretation, often stemming from non-specific probe binding or inadequate washing.
Table 2: Troubleshooting High Background
| Problem Cause | Underlying Principle | Recommended Solution | Supporting Data/Example |
|---|---|---|---|
| Insufficient Stringency Washes [25] [50] | Failure to remove unbound or non-specifically bound probes leads to widespread fluorescence. | Optimize wash stringency (pH, temperature, salt concentration). Use freshly prepared SSC buffer (75-80°C) for stringent washes [25]. | For ≥2 slides, increase SSC temperature by 1°C per slide, but do not exceed 80°C [25]. Always use buffers with detergents like Tween 20 (PBST) [25]. |
| Over-fixation [50] | Excessive formalin cross-linking masks target sequences, increasing non-specific probe binding. | Use freshly prepared fixative and adhere closely to recommended fixation times. | Over-fixation reduces signal intensity and elevates background by promoting non-specific binding [50]. |
| Inadequate Enzymatic Digestion [25] | Under-digestion leaves cellular debris that causes autofluorescence and non-specific binding sites. | Titrate enzyme (e.g., pepsin) digestion time (3-10 min at 37°C for most tissues). Prevent evaporation during digestion [25]. | Over-digestion can also eliminate signal and prevent counterstaining, while under-digestion decreases specific signal [25]. |
| Probe Mismatch or Contamination [25] [51] | Using biotin-labeled probes with anti-digoxigenin conjugate (or vice versa) causes detection failure and background. | Verify probe-conjugate matches (biotin/anti-biotin, digoxigenin/anti-digoxigenin). Ensure HRP is used with DAB/AEC, and AP with NBT/BCIP/Fast Red [25]. | Pipette carefully to avoid cross-contamination between probe vials [51]. |
| Dark Counterstaining [25] | Intense counterstain can mask the specific signal, particularly with DAB or NBT/BCIP. | Use a light counterstain (e.g., Mayer’s hematoxylin for 5 sec - 1 min) [25]. | For chromogens like AEC or Fast Red, note that the precipitate is solvent-soluble; DAB is solvent-insoluble [25]. |
| Damaged Optical Filters [50] | Worn or damaged microscope filters produce a mottled appearance and weak signal, increasing perceived background. | Regularly inspect filters for damage and replace according to manufacturer guidelines (typically every 2-4 years). | Close the microscope shutter when not in use to extend filter life [50]. |
The following diagram illustrates the primary pathways leading to high background and the corresponding corrective actions.
Advanced Protocols for Sensitivity Optimization
High-Sensitivity ViewRNA ISH for Viral Detection
The development of a high-sensitivity BK virus (BKV) detection system using ViewRNA ISH exemplifies how advanced probe design can overcome limitations of conventional methods. This protocol leverages branched DNA (bDNA) signal amplification to achieve superior specificity and sensitivity.
Background: Conventional immunohistochemistry (IHC) for BKV large T antigen (LT) demonstrates suboptimal diagnostic sensitivity, failing in ~30% of biopsy-proven BKV nephropathy cases. Traditional DNA-targeted ISH shows even lower sensitivity [52].
Key Methodology:
- Probe Design: A set of 40 oligonucleotide probes targeting the BKV-LT DNA and mRNA sequences was designed. This multi-probe strategy enhances signal amplification.
- Technology Platform: The ViewRNA ISH system utilizes bDNA technology. Each target-specific probe pair hybridizes to the nucleic acid, and then multiple amplification molecules bind, each carrying many label probes. This creates a substantial signal amplification cascade without the need for enzymatic reactions from the sample.
- Specificity Validation: The BKV-LT ViewRNA-ISH system demonstrated stringent species specificity, differentiating BKV from the closely related JCV and SV40 polyomaviruses. This is a critical improvement over cross-reactive SV40-LT antibodies used in conventional IHC [52].
- Versatility: The system is adaptable for both chemical and fluorescent ISH detection, making it suitable for various laboratory settings and applications [52].
Outcome: The BKV-LT ViewRNA-ISH system provided excellent sensitivity and specificity at the cellular and tissue level, proving to be a versatile and powerful tool for the accurate clinical diagnosis of challenging infections like BKV nephropathy [52].
Reflex FISH Testing for Unsuccessful Cultures
A large-scale retrospective study demonstrated the utility of a reflex FISH assay in salvaging genetic information from products of conception (POC) samples that failed conventional karyotyping.
Background: Approximately 20% of POC cultures are unsuccessful due to microbial contamination or lack of viable dividing cells, preventing a genetic diagnosis [53].
Key Methodology:
- Experimental Design: 5555 POC samples were simultaneously processed for both conventional chromosome analysis and a reflex interphase FISH assay.
- FISH Protocol: The reflex FISH assay targeted chromosomes 13, 16, 18, 21, 22, X, and Y to detect common aneuploidies. If the chromosome analysis failed, the FISH assay was automatically performed ("reflexed") on the same sample [53].
- Data Analysis: A 3-year retrospective comparative analysis of FISH results versus chromosome results was performed to validate the assay's performance [53].
Outcome and Quantitative Data:
- Of 762 successful FISH studies on chorionic villi/fetal tissue that failed karyotyping, 181 (25%) detected an abnormality.
- The targeted FISH panel detected approximately 70% of the chromosome abnormalities in POC that are detectable by a full karyotype.
- When FISH and chromosome analysis results were combined for the 4189 chorionic villi/fetal tissue specimens, the overall abnormality detection rate was 47% [53].
This protocol highlights how a validated, targeted FISH approach can effectively complement broader cytogenetic methods, ensuring critical data is recovered from suboptimal samples.
Table 3: Key Research Reagent Solutions for ISH Troubleshooting
| Reagent/Kit | Primary Function | Application Note |
|---|---|---|
| CytoCell LPS 100 Tissue Pretreatment Kit [50] | Heat and enzyme-based pretreatment of FFPE tissues to break cross-links and expose target nucleic acids. | Critical for reducing background. Heat Tissue Pretreatment Solution to 98–100°C before enzyme treatment. Refresh solution between slide batches. |
| UroVysion Probe Set [54] | Multi-color FISH probe for detecting aneuploidy in chromosomes 3, 7, 17, and 9p21 loss. | Used in meta-analysis of biliary strictures. Defining "polysomy only" as positive provided 96.2% specificity for malignancy [54]. |
| Rembrandt CISH Kit [25] | Complete solution for chromogenic ISH, including all necessary buffers, conjugates, and substrates. | Ensure the correct PanWash solution is used based on the probe (e.g., different solutions for HPV vs. CMV/EBV probes). |
| ViewRNA ISH System [52] | Branched DNA (bDNA) technology for high-sensitivity detection of RNA/DNA targets with single-molecule sensitivity. | Utilizes a set of ~40 probes for specific signal amplification. Demonstrated superior specificity vs. immunofluorescence for BKV detection. |
| Mayer’s Hematoxylin [25] | Nuclear counterstain for chromogenic ISH. Provides a light blue counterstain to visualize tissue architecture. | A light counterstain (5 sec - 1 min) is recommended to avoid masking the brown DAB or blue NBT/BCIP signal. |
| Histomount [25] | Aqueous mounting medium for preserving stained slides under a coverslip for long-term storage and imaging. | Apply to wet sections, avoiding bubbles, to preserve signal and morphology. |
The sensitivity of Fluorescence In Situ Hybridization (FISH) is fundamentally determined by the quality of its initial sample preparation steps. Within the context of comparative sensitivity research, inconsistent fixation, permeabilization, or suboptimal probe design introduce significant variability that can compromise the validity of findings. This technical guide details the critical protocols for these foundational steps, providing a standardized framework for researchers aiming to generate reliable, reproducible, and high-quality FISH data. The ultimate goal is to minimize technical artifacts, thereby ensuring that observed signal variations genuinely reflect biological differences rather than preparation inconsistencies. As the field advances with techniques like single-molecule FISH (smFISH) and highly multiplexed error-robust FISH (MERFISH), the precision of these preparatory phases becomes even more critical for accurate signal quantification and sensitivity comparisons [55] [56].
Sample Fixation: Preserving Morphology and Nucleic Acid Accessibility
Fixation is the first and perhaps most crucial step, as it arrests cellular metabolism and preserves morphological structure while rendering the cell permeable to nucleic acid probes. The choice of fixative and protocol directly impacts the trade-off between structural preservation and target accessibility.
Aldehyde-Based Fixation Protocol
Cross-linking fixatives, such as formaldehyde, are the most common for FISH as they preserve cellular structure with minimal distortion.
- Reagent Preparation: Prepare a 4% formaldehyde solution in a suitable buffer, typically phosphate-buffered saline (PBS) or saline-sodium citrate (SSC). The solution should be prepared fresh from paraformaldehyde or from high-quality commercial formaldehyde stocks to avoid formic acid degradation products that can increase autofluorescence.
- Procedure:
- Gently wash cells or tissue sections with PBS to remove culture media or debris.
- Immerse the sample in an adequate volume of 4% formaldehyde for 10–15 minutes at room temperature. Note: Over-fixation can lead to excessive cross-linking, reducing probe accessibility and thus sensitivity.
- Terminate fixation by washing the sample three times with PBS.
- Sensitivity Consideration: Consistent fixation time and temperature are paramount for sensitivity comparisons. Variations can alter the degree of cross-linking, leading to batch-to-batch differences in hybridization efficiency [55].
Permeabilization: Enabling Probe Delivery
Following fixation, permeabilization creates pores in the cellular membranes and, for cross-linking fixatives, reverses some protein-nucleic acid cross-links to allow probe entry.
Detergent-Based Permeabilization Protocol
This method is standard for most cell culture and tissue section preparations.
- Reagent Preparation: Prepare a permeabilization solution containing 0.1% to 0.5% Triton X-100 or Tween-20 in PBS.
- Procedure:
- Incubate the fixed sample in the permeabilization solution for 5–10 minutes at room temperature.
- Wash the sample three times with PBS to remove the detergent.
- Alternative Protease-Based Treatment: For tissues or cells with dense matrices, a protease digestion step (e.g., using proteinase K) may be necessary. However, this requires careful optimization, as over-digestion can destroy tissue architecture and leak nucleic acids [55].
The following workflow diagram summarizes the key stages of sample preparation and their impact on the final result, which is crucial for sensitivity.
Probe Design: The Key to Specificity and Signal Strength
Probe design is arguably the most active area of innovation in enhancing FISH sensitivity. The shift from single, long probes to multiple, short oligonucleotide probes has been fundamental to the development of smFISH, allowing for the precise localization and quantification of individual mRNA molecules [55] [19].
Modern smFISH Probe Design Principles
Contemporary probe design focuses on creating sets of short, singly-labeled oligonucleotides that collectively span a target RNA.
- Probe Length and Labeling: Typical probes are 20-mer to 30-mer oligonucleotides, each tagged with a single fluorophore at one terminus. Using multiple such probes (e.g., 30-50) per transcript ensures that the combined fluorescent signal from a single mRNA molecule is bright enough to be distinguished from background noise [55] [19].
- Computational Design and Specificity: Advanced software tools are now essential for designing high-performance probe sets. They evaluate factors such as:
- GC Content: Maintained within a narrow window (e.g., 40-60%) to ensure uniform hybridization thermodynamics.
- Melting Temperature (Tm): Probes are designed to have a consistent Tm for uniform behavior during hybridization.
- Specificity and Off-Target Binding: This is a critical factor for sensitivity. Tools like TrueProbes perform genome-wide BLAST analyses to minimize cross-hybridization to off-target transcripts, directly reducing background fluorescence and improving the signal-to-noise ratio [19]. This is especially vital for detecting low-abundance transcripts.
The table below summarizes the key design parameters and their impact on assay performance.
Table 1: Key Parameters in smFISH Probe Design for Optimal Sensitivity
| Design Parameter | Typical Optimal Range | Impact on Sensitivity and Specificity |
|---|---|---|
| Probe Length | 20 - 30 nucleotides | Shorter probes improve penetration but require more for a bright signal; longer probes may have higher non-specific binding [55] [19]. |
| Number of Probes per Transcript | 30 - 50 probes | A higher number of probes increases the signal per transcript, directly enhancing detection sensitivity for low-abundance RNAs [19]. |
| GC Content | 40% - 60% | Content outside this range can lead to non-specific binding (low GC) or overly stable secondary structures (high GC), reducing effective hybridization [19]. |
| Specificity Screening | Genome-wide BLAST | Critically minimizes off-target binding, which is a primary source of background noise, thereby improving the signal-to-noise ratio [19]. |
Probe Design Workflow
The process of designing a sensitive and specific probe set involves multiple filtering and selection steps, as illustrated in the following logic diagram.
The Researcher's Toolkit: Essential Reagents for FISH
A successful FISH experiment relies on a suite of specific reagents, each serving a distinct function in the preparation and hybridization process.
Table 2: Essential Research Reagents for FISH Experiments
| Reagent / Solution | Primary Function | Key Considerations |
|---|---|---|
| Formaldehyde (4%) | Cross-linking fixative that preserves cellular architecture and immobilizes nucleic acids. | Freshness is critical; degraded formaldehyde increases autofluorescence. Incubation time must be standardized [55]. |
| Triton X-100 (0.1-0.5%) | Detergent that permeabilizes lipid membranes to allow fluorescent probes to enter the cell. | Concentration and time must be optimized; over-permeabilization can damage cellular structures [55]. |
| Ethanol (70-100%) | Used for dehydration and storage of samples post-fixation, helping to preserve them. | A common step for storing samples before hybridization, preventing degradation. |
| Hybridization Buffer | A solution that creates optimal chemical conditions (pH, ionic strength) for probe-target annealing. | Contains salts (e.g., in SSC buffer) to control stringency and formamide to lower the melting temperature [55] [56]. |
| Formamide | A key component of hybridization buffers that destabilizes hydrogen bonds, allowing hybridization at lower temperatures. | Enables the use of more stable, specific probes without damaging the sample with high heat [55]. |
| Saline-Sodium Citrate (SSC) Buffer | Provides the ionic strength (via sodium ions) necessary for the annealing of the probe to its target sequence. | Higher SSC concentration increases stringency; used in both hybridization and post-hybridization washes [55]. |
| Fluorescently-Labelled DNA Oligos | Synthesized oligonucleotide probes that bind complementary mRNA sequences. | The core detection reagent. Design, labeling efficiency, and purity are paramount for sensitivity [19]. |
Mastering sample preparation is a prerequisite for any rigorous investigation into FISH sensitivity. By standardizing and optimizing the protocols for fixation, permeabilization, and—most critically—probe design, researchers can establish a robust foundation for their experiments. The adoption of computationally designed, highly specific probe sets, coupled with careful attention to pre-hybridization conditions, directly addresses the major sources of background noise and variability. This disciplined approach ensures that the high sensitivity offered by modern FISH platforms is fully realized, enabling accurate and reliable detection of nucleic acids in their native cellular context.
In situ hybridization (ISH) is a cornerstone technique for visualizing nucleic acid targets within their native morphological context. The sensitivity and specificity of any ISH experiment are profoundly dependent on the fine-tuning of its core steps: the hybridization itself and the subsequent post-hybridization washes. Temperature, duration, and chemical stringency are not merely procedural details; they are powerful levers that control the thermodynamic equilibrium of probe-target binding, directly impacting the signal-to-noise ratio. In the context of sensitivity comparison research, a deep understanding and precise control of these parameters are what enable the reliable detection of low-abundance transcripts and the discrimination of highly similar sequences, such as single-nucleotide variants (SNVs) [57] [58]. This guide provides a detailed examination of these critical factors, equipping researchers with the knowledge to systematically optimize ISH protocols for maximum performance.
The Thermodynamics of Hybridization and Wash Stringency
The underlying principle of ISH is the hybridization of a labeled probe to a complementary nucleic acid target, forming a stable duplex. The stability of this duplex is governed by its melting temperature (Tm), the temperature at which half of the duplexes dissociate. The goal of optimization is to find conditions that allow the specific probe-target duplex (which has a higher Tm) to form stably, while preventing the formation of imperfect, non-specific duplexes (with lower Tm) with off-target sequences.
- Probe-Target Hybridization: During this phase, probes diffuse through the sample and bind to their targets. A temperature too far below the optimal Tm will promote non-specific binding and cross-hybridization, increasing background noise. A temperature too high will impede even specific binding, reducing the desired signal [59] [58].
- Post-Hybridization Washes: These steps are designed to remove poorly bound and non-specifically bound probes. Washes performed at a stringency (a combination of temperature and chemical environment) that is slightly below the Tm of the specific duplex will denature and wash away imperfect duplexes while leaving the perfect match intact [58].
The following diagram illustrates this core optimization workflow and the relationship between key parameters.
Optimizing Key Parameters for Sensitivity and Specificity
Temperature
Temperature is the most critical parameter for controlling hybridization specificity. Even minor deviations from the optimum can have dramatic consequences.
Table 1: Impact of Hybridization Temperature on Experimental Outcomes
| Temperature Condition | Impact on Sensitivity & Specificity | Experimental Evidence |
|---|---|---|
| Too Low (Below Optimum) | Increased cross-hybridization and reduced specificity due to stable binding of probes to non-targets with similar sequences [59]. | A study on oligonucleotide microarrays found that suboptimal temperatures lead to a loss of differentially expressed genes, with transcription factors and other low-copy-number regulators being disproportionately affected [59]. |
| Optimal | Best compromise: High specificity binding is achieved while maintaining strong signal intensity [59]. | The same study established that calibrated protocols are essential for the sensitive profiling of low-copy-number molecules, which is a key challenge in ISH sensitivity [59]. |
| Too High (Above Optimum) | Reduced sensitivity and signal intensity due to impaired probe-target binding, leading to a degraded signal-to-noise ratio [59]. | A temperature deviation of just 1°C was shown to cause a loss of up to 44% of differentially expressed genes that could otherwise be identified [59]. |
Stringency
Stringency determines the "strictness" of the binding conditions and is controlled by both temperature and the chemical composition of the hybridization and wash buffers.
Table 2: Methods for Controlling Stringency
| Method | Principle & Role | Application Notes |
|---|---|---|
| Formamide | Denaturing agent that reduces the effective Tm of duplexes, allowing high-stringency washes to be performed at lower, more practical temperatures (e.g., 37-50°C instead of >65°C) [58]. | Commonly used at concentrations of 0-50% in hybridization buffers. Higher concentrations increase stringency. |
| Saline-Sodium Citrate (SSC) | The ionic strength of the buffer (e.g., 2x SSC, 0.2x SSC) affects duplex stability. Higher salt concentration (e.g., 5x SSC) stabilizes duplexes (lower stringency), while lower salt (e.g., 0.1x SSC) destabilizes them (higher stringency) [58]. | Post-hybridization washes often progress from higher to lower salt concentrations to gradually increase stringency. |
| Detergents (e.g., SDS, Tween-20) | Reduce non-specific binding of probes to the tissue or slide surface, thereby lowering background noise without directly affecting Tm [58]. | SDS (e.g., 0.1%) is often included in hybridization and wash buffers. |
Duration
The duration of hybridization and washes must be balanced to achieve complete reaction without introducing unnecessary background or damaging the sample.
- Hybridization Duration: Must be sufficient for the probes to diffuse into the tissue and reach their targets. This can range from 2-4 hours for thin cultured cells to over 16 hours (overnight) for thick tissue sections or whole-mounts [58]. Excessively long hybridization can increase background.
- Wash Duration: Must be long enough to allow the diffusion of unbound and non-specifically bound probes out of the sample. Typically, multiple washes of 15-30 minutes each are performed [58]. The most stringent wash is often given more time to ensure effective removal of off-target probes.
Practical Optimization and Experimental Protocols
An Objective Workflow for Empirical Temperature Optimization
While theoretical calculations provide a starting point, empirical calibration is essential. The following workflow, adapted from microarray optimization studies, can be applied to ISH protocol development [59].
Step-by-Step Protocol:
- Sample Selection: Choose two biologically distinct samples expected to show differential expression for a set of target genes, alongside a large number of non-differentially expressed "housekeeping" genes.
- Hybridization Series: Process the sample pairs in parallel using identical protocols, varying only the hybridization temperature across a sensible range (e.g., 45°C, 50°C, 55°C, 60°C).
- Data Analysis: For each temperature condition, quantify the results. The measure of success is the amount of information about sample differences that can be extracted. This translates to:
- The number of target genes reliably identified as differentially expressed.
- The statistical significance (p-values) of these differences.
- The intensity and clarity of signal for low-copy-number transcripts.
- Determine Optimum: The temperature that yields the greatest power in detection (most reliable differentially expressed genes, smallest p-values, best signal for low-abundance targets) is the generally optimal hybridization temperature for that probe set and sample type [59].
Protocol Snippets: Establishing a Baseline
The following protocols provide a starting point for optimizing a branched DNA (bDNA) ISH assay and a standard RNA FISH assay.
Protocol 1: High-Sensitivity bDNA-Based ViewRNA ISH This protocol is adapted from a study that developed a highly sensitive BK virus detection system, which is relevant for detecting low-abundance targets [52].
- Probe Design: Use a set of ~40 oligonucleotide probes targeting the same mRNA transcript to enable signal amplification via the bDNA system.
- Hybridization: Hybridize probes to samples according to the manufacturer's instructions. The specific temperature and duration should be empirically determined but often fall within 37°C - 50°C for several hours.
- Signal Amplification: Perform a series of amplifier hybridizations per the bDNA protocol to build the signal cascade.
- Washes: Between each hybridization and amplification step, wash the samples with a standardized wash buffer to remove unbound reagents. The final post-hybridization wash should be performed at a pre-optimized stringency (controlled by temperature and salt concentration) to minimize background.
Protocol 2: Standard RNA FISH with Formamide-Based Stringency This is a generalized protocol for fluorescent oligonucleotide probes [58].
- Hybridization Buffer: 2x SSC, 0-50% Formamide, 0.1% SDS, and a blocking agent (e.g., 0.1 mg/mL tRNA or salmon sperm DNA).
- Hybridization: Apply probe in hybridization buffer and incubate in a humidified chamber at 37-55°C for 4-16 hours.
- Post-Hybridization Washes:
- Wash 1: 2x SSC / 0.1% SDS at room temperature for 15-30 minutes.
- Wash 2: 1x SSC / 0.1% SDS at the hybridization temperature or slightly below (e.g., 37°C) for 15-30 minutes. This is a medium-stringency wash.
- Wash 3 (High-Stringency): 0.1x SSC / 0.1% SDS at 37-50°C for 15-30 minutes. This is the most critical wash for removing cross-hybridized probes.
- Final Rinse: A brief rinse in a low-salt buffer (e.g., 0.1x SSC) at room temperature before mounting.
The Scientist's Toolkit: Essential Reagents for Hybridization and Washes
Table 3: Key Research Reagent Solutions for ISH
| Reagent | Function in Hybridization/Washes |
|---|---|
| Formamide | Denaturing agent used to lower the effective melting temperature of nucleic acid duplexes, enabling high-stringency washes at biologically compatible temperatures [58]. |
| Saline-Sodium Citrate (SSC) | A buffer providing the ionic strength necessary for nucleic acid hybridization. The concentration (e.g., 2x vs 0.2x) is a primary factor in controlling stringency [58]. |
| Sodium Dodecyl Sulfate (SDS) | An ionic detergent used in wash buffers to reduce non-specific hydrophobic interactions between probes and cellular components, thereby lowering background [58]. |
| Tween-20 | A non-ionic detergent used to minimize non-specific adsorption of probes and antibodies to surfaces, further reducing background noise. |
| Blocking Agents (tRNA, DNA) | Unrelated nucleic acids (e.g., yeast tRNA, salmon sperm DNA) are used to pre-emptively bind to non-specific sites on the sample, blocking probe attachment and reducing background [58]. |
| Padlock Probes & RCA Reagents | Padlock probes are circularizable oligonucleotides that, upon correct target binding, can be amplified via Rolling Circle Amplification (RCA) to generate a massive, localized signal for ultra-sensitive detection [42] [60]. |
| Branched DNA (bDNA) Amplifiers | Synthetic, multi-arm DNA structures that hybridize to target-bound probes and provide numerous binding sites for labeled probes, enabling significant signal amplification without the use of enzymes during the detection phase [52]. |
The journey to a robust and sensitive ISH protocol is one of meticulous optimization. There is no universal "one-size-fits-all" condition for hybridization and washes. The optimal temperature, stringency, and duration are a function of the specific probe sequence, the sample type, and the target abundance. By understanding the thermodynamics underlying hybridization, employing a systematic empirical approach to optimization as outlined in this guide, and leveraging modern signal amplification technologies, researchers can push the boundaries of detection. This enables highly sensitive and specific spatial gene expression analysis, which is fundamental for advanced research in development, disease, and drug development.
The simultaneous detection of nucleic acids and proteins within their native tissue context through the combination of in situ hybridization (ISH) and immunostaining provides a powerful tool for understanding spatial relationships in gene expression and protein localization. This integrated approach is particularly valuable for investigating complex biological processes such as cellular differentiation, signal transduction, and disease pathogenesis [61]. Despite its considerable potential, technical challenges, particularly signal impairment and tissue degradation, have historically limited its widespread application [62] [10].
This technical guide outlines current methodologies and optimized protocols for successfully combining ISH with immunostaining. We focus on practical solutions for overcoming key hurdles, framed within the context of advancing sensitivity and reliability in molecular localization studies. The techniques described herein are designed to provide researchers with robust frameworks adaptable to various tissue types and experimental requirements.
Core Technical Challenges and Innovative Solutions
The integration of ISH with immunostaining presents several interconnected technical challenges that can compromise experimental outcomes if not properly addressed.
Primary Technical Hurdles
- Signal Reduction Post-ISH: A common obstacle is the significant weakening of immunostaining signals following ISH procedures. This is frequently attributed to harsh permeabilization treatments, including proteinase K digestion, which can damage protein epitopes recognized by antibodies [10].
- Tissue Integrity Compromise: Delicate tissues, particularly fragile structures like regeneration blastemas in planarians or embryonic tissues, are susceptible to degradation from aggressive permeabilization steps required for probe penetration [10].
- Antigen Masking: Fixation methods, particularly over-fixation with aldehydes, can mask antigenic sites through excessive cross-linking, thereby reducing antibody binding capacity [63].
- Protocol Compatibility: Sequential staining requires careful optimization to ensure that reagents from the first staining round do not interfere with the second, particularly when using enzymatic detection systems [64].
Emerging Solutions and Protocol Innovations
Recent methodological developments have yielded promising strategies to overcome these challenges:
- Alternative Fixation Protocols: The Nitric Acid/Formic Acid (NAFA) protocol represents a significant advancement by eliminating proteinase K digestion. This approach preserves both tissue architecture and antigen integrity while maintaining effective probe penetration for ISH [10].
- Sequential Fixation Strategies: Consecutive fixation with 4% paraformaldehyde (PFA) followed by cold methanol has proven effective for preserving immunoreactivity in cultured neurons after ISH, offering a simple yet robust alternative [62].
- Optimized Permeabilization: The use of acid-based treatments instead of enzymatic digestion enhances tissue permeability while minimizing damage to epitopes and delicate cellular structures [10].
- Dual Assay Design: Meticulous optimization of hybridization temperatures, antibody concentrations, and detection systems enables successful simultaneous visualization of both targets [64].
Optimized Experimental Protocols
This section provides detailed methodologies for implementing successful combined ISH and immunostaining protocols, drawing from recent technical advances.
The NAFA Fixation Protocol for Delicate Tissues
The NAFA protocol is particularly suited for fragile tissues and whole-mount preparations, demonstrating excellent performance in planarian and killifish fin regeneration studies [10].
Workflow Diagram: NAFA Protocol
Step-by-Step Procedure:
- Sample Preparation: Collect and rinse tissues in appropriate physiological buffer.
- NAFA Fixation: Fix tissues in Nitric Acid/Formic Acid solution.
- EGTA Treatment: Incubate samples with the calcium chelator EGTA to inhibit nucleases and preserve RNA integrity.
- ISH Protocol:
- Pre-hybridize with ISH buffer for 30 minutes.
- Hybridize with labeled probes at appropriate temperature (2 hours).
- Perform stringent washes with saline-sodium citrate buffer.
- Immunostaining:
- Block with 2% serum and 1% BSA.
- Incubate with primary antibody overnight at room temperature.
- Incubate with fluorochrome-conjugated secondary antibody.
- Detection: Visualize signals using chromogenic or fluorescent detection.
Key Advantages: This protocol preserves delicate external structures like cilia and epidermis while permitting effective probe penetration into internal tissues, enabling accurate co-localization studies [10].
Combined ISH-IHC on Archival Tissues
For formalin-fixed, paraffin-embedded tissues, particularly in clinical samples, the following protocol has demonstrated efficacy for simultaneous miRNA and protein detection [64].
Workflow Diagram: Sequential ISH-IHC
Step-by-Step Procedure:
- Tissue Preparation: Cut 3-4µm sections from FFPE blocks and mount on charged slides.
- Deparaffinization: Heat slides at 60°C for 30-120 minutes, followed by xylene and graded ethanol series.
- Antigen Retrieval:
- Proteinase K treatment (15 µg/mL, 37°C, 10 minutes) or
- Heat-induced epitope retrieval with Tris-EDTA (pH 9) in microwave for 15 minutes.
- ISH Procedure:
- Pre-hybridize with ISH buffer for 30 minutes.
- Hybridize with DIG-labeled LNA probes at 48-53°C for 2 hours.
- Stringent washes with SSC buffer.
- Incubate with alkaline phosphatase-conjugated anti-DIG antibody overnight.
- Develop with NBT/BCIP substrate (2 hours at 30°C or overnight at RT).
- IHC Staining:
- Block with appropriate serum.
- Incubate with primary antibody (e.g., pan-cytokeratin, 1:800) for 1 hour at RT.
- Incubate with HRP-conjugated secondary antibody for 30 minutes.
- Develop with DAB substrate.
- Counterstaining and Mounting: Counterstain with nuclear fast red or hematoxylin, dehydrate, and mount.
Critical Optimization Points: This method allows precise delimitation of tumor compartments through simultaneous epithelial marker staining and miRNA detection, enabling accurate stromal expression analysis [64].
Research Reagent Solutions
Successful combined ISH and immunostaining requires careful selection of reagents and tools. The following table outlines essential materials and their functions in the experimental workflow.
Table 1: Essential Reagents for Combined ISH-Immunostaining
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Fixatives | 4% PFA, NAFA solution, Methanol | Tissue preservation while maintaining RNA integrity and antigenicity [62] [10] |
| Permeabilization Agents | Proteinase K, Triton X-100, Acids | Enable probe/antibody penetration; concentration and timing critical [64] |
| Probes | DIG-labeled LNA probes, Fluorescent RNA probes | Target detection; LNA probes offer enhanced binding affinity [64] |
| Antibodies | Primary: Target-specific; Secondary: ALP/HRP-conjugated | Protein detection; species-specific secondary antibodies minimize cross-reactivity [63] |
| Detection Substrates | NBT/BCIP (ALP), DAB (HRP), Fluorophores | Signal generation; enzymatic for permanent record, fluorescent for multiplexing [64] |
| Blocking Agents | BSA, Sheep Serum, Levamisole | Reduce non-specific binding; levamisole inhibits endogenous ALP [64] |
Quantitative Performance Assessment
Rigorous validation of combined ISH-immunostaining results requires quantitative assessment and comparison with established methods.
Method Performance Metrics
Table 2: Performance Comparison of ISH-Immunostaining Methods
| Method | Tissue Preservation | RNA Detection | Protein Detection | Best Application |
|---|---|---|---|---|
| NAFA Protocol [10] | Excellent (95% integrity) | High (comparable to NAC) | High (bright signals) | Delicate tissues, whole mounts |
| PFA+Methanol [62] | Good | High | High | Cultured neurons, cell lines |
| Traditional NAC [10] | Poor (epidermal damage) | High | Reduced (epitope damage) | Robust tissues, RNA-focused |
| Proteinase K-based [64] | Moderate | High | Variable | Archival FFPE tissues |
AI-Enhanced Quantification Approaches
Advanced computational methods significantly enhance the accuracy and reproducibility of signal quantification in combined ISH-immunostaining:
- Digital Image Analysis: Software platforms including ImageJ and Aperio ImageScope enable precise quantification of signal intensity and distribution, particularly valuable for stromal-epithelial differentiation in cancer samples [64].
- AI-Powered Algorithms: Machine learning approaches facilitate optimal nuclear segmentation and signal quantification, reducing subjectivity in interpretation [61].
- Automated Scoring Systems: For clinical applications, AI-based systems demonstrate high accuracy in classifying expression levels, with one meta-analysis showing pooled sensitivity of 0.97 and specificity of 0.82 for HER2 status classification, highlighting their potential for standardized assessment [65].
The technical hurdles associated with combining ISH and immunostaining are being successfully addressed through innovative fixation strategies, optimized permeabilization methods, and careful protocol sequencing. The development of techniques such as the NAFA protocol and sequential PFA-methanol fixation enables researchers to simultaneously visualize nucleic acids and proteins while preserving tissue integrity and antigenicity.
As the field advances, the integration of automated staining platforms, AI-powered quantification tools, and highly sensitive detection systems will further enhance the reproducibility and accessibility of these powerful multimodal approaches. These methodologies provide researchers with increasingly sophisticated tools to explore spatial relationships in gene expression and protein localization, ultimately advancing our understanding of complex biological systems in both health and disease.
Benchmarking ISH Technologies: Validation, Specificity, and Clinical Translation
The advent of highly multiplexed, spatially-resolved gene expression profiling technologies represents a powerful advance for understanding complex biological systems, enabling researchers to study cellular and sub-cellular localization patterns that are lost using single-cell RNA-seq technologies [66]. However, the burgeoning diversity of academic and commercial profiling technologies has created formidable challenges for cross-platform comparison and interpretation. While standard sensitivity metrics—such as the number of unique molecules detected per cell—have been widely adopted, these measures provide an incomplete picture of data quality because they do not account for off-target molecular artifacts that compromise specificity [66]. This technical guide examines the critical role of specificity metrics, with a focused analysis of the novel Mutually Exclusive Co-expression Rate (MECR) metric, in standardizing the evaluation of in situ hybridization technologies within the broader context of sensitivity comparison research.
The fundamental challenge in cross-technology benchmarking stems from substantial differences in probe design, signal amplification methods, detection approaches, and computational processing pipelines [66]. These technical variations manifest as differing levels of non-specific background signal and off-target binding that confound accurate gene expression quantification. Without standardized metrics to control for these artifacts, comparisons of sensitivity across platforms remain fundamentally flawed, potentially leading to incorrect biological interpretations in both basic research and drug development applications.
Understanding Specificity inIn SituTechnologies
The Specificity Problem
In multiplexed in situ gene expression profiling, specificity refers to a technology's ability to correctly identify and measure target transcripts without detecting non-target signals. The molecular artifacts that reduce specificity can originate from multiple sources:
- Non-specific hybridization: Probes binding to unintended RNA sequences with partial complementarity
- Incomplete probe removal: Residual probe from previous hybridization cycles in sequential detection methods
- Background autofluorescence: Native tissue fluorescence that interferes with signal detection
- Enzymatic amplification artifacts: Non-specific signal amplification during tyramide or other amplification steps [67]
These artifacts manifest biologically as implausible gene expression patterns, including the co-expression of marker genes in cell types where they are known to be mutually exclusive [66]. For example, in mouse brain studies, the neuronal marker Slc17a7 and astrocyte marker Gfap exhibit nearly mutually exclusive expression patterns in validated single-cell RNA-seq data, yet some in situ technologies detect widespread co-expression of these markers, indicating substantial off-target effects [66].
Limitations of Traditional Sensitivity Metrics
Traditional quality metrics for gene expression technologies have emphasized sensitivity—the ability to detect genuine low-abundance transcripts—often measured as the number of unique molecules detected per cell [66]. However, when technologies exhibit different levels of specificity, sensitivity metrics become misleading. A technology with high rates of non-specific binding may appear highly sensitive because it detects many molecules per cell, but a component of this detected signal represents background noise rather than true biological signal [66].
This limitation is particularly problematic when comparing technologies with different target panel sizes and composition. Technologies targeting different genes cannot be fairly compared using raw molecular counts, as the expression levels of targeted genes vary substantially across the transcriptome. Furthermore, the common practice of benchmarking using well-characterized tissues like mouse brain, while providing conserved spatial organization, does not inherently control for platform-specific artifacts [66].
MECR: A Novel Metric for Quantifying Specificity
Conceptual Framework
The Mutually Exclusive Co-expression Rate (MECR) represents a novel approach to quantifying specificity in multiplexed in situ gene expression technologies [66]. This metric was specifically designed to address the absence of multi-species "barnyard" experiments—the gold standard for specificity assessment in single-cell RNA-seq—in spatial transcriptomics datasets.
The core principle underlying MECR leverages the well-established biological knowledge that certain genes exhibit mutually exclusive expression patterns in specific cell types. By analyzing pairs of marker genes known to be restricted to distinct, non-overlapping cell populations in validated reference data, MECR quantifies the rate at which a technology incorrectly detects co-expression of these markers within individual cells [66].
Calculation Methodology
The MECR calculation incorporates several key steps and validation components:
Table: Key Components for MECR Calculation
| Component | Description | Validation Requirement |
|---|---|---|
| Marker Gene Pairs | Selection of gene pairs with mutually exclusive expression patterns | Must be verified using scRNA-seq reference data |
| Cell Type Annotations | Assignment of cell identities using reference datasets | Based on well-established marker genes |
| Co-expression Detection | Measurement of cells expressing both markers simultaneously | Normalized for abundance of each gene |
| Normalization | Adjustment for varying expression levels across genes | Ensures fair comparison across technologies |
The specific workflow for implementing MECR analysis involves:
- Reference Dataset Selection: Obtain a high-quality scRNA-seq dataset with well-annotated cell types for the tissue of interest (e.g., mouse brain) [66].
- Marker Gene Identification: Identify pairs of genes that exhibit mutually exclusive expression patterns across cell types in the reference data [66].
- Spatial Data Processing: Process the in situ hybridization dataset(s) to obtain gene expression matrices with cells as rows and genes as columns [66].
- Co-expression Quantification: For each marker pair, calculate the percentage of cells that express both genes simultaneously in the in situ data [66].
- Normalization and Aggregation: Normalize co-expression rates for the abundance of each gene and aggregate across multiple marker pairs to compute the final MECR value [66].
The following diagram illustrates the logical workflow for MECR calculation:
Experimental Validation Protocols
Implementation of MECR analysis requires careful experimental design and validation:
Reference Data Requirements:
- Use well-established scRNA-seq references with comprehensive cell type annotation [66]
- Select references with sufficient cellular resolution to distinguish target cell populations
- Verify marker gene specificity through literature review and data re-analysis
Marker Gene Selection Criteria:
- Genes must show highly specific expression in one cell population (>95% specificity)
- Genes must show minimal expression (<5% detection) in mutually exclusive populations
- Marker pairs should represent biologically distinct cell lineages
- Include multiple pairs across different cell types to ensure robust metric calculation
Benchmarking Experimental Design:
- Process identical tissue sections with different in situ technologies when possible
- Ensure consistent cell segmentation parameters across compared datasets
- Include technical replicates to assess metric reproducibility [66]
- Analyze sufficient cell numbers (>10,000 cells per dataset) for statistical power
Comparative Analysis of In Situ Technologies Using MECR
Technology Performance Variation
Application of MECR across six prominent in situ technologies revealed substantial variation in specificity performance. In a landmark comparative benchmark analysis using publicly available mouse brain datasets, technologies exhibited widely differing MECR values, with the highest MECR (indicating lower specificity) being observed in the Xenium dataset [66].
This finding was particularly significant because Xenium also demonstrated the highest number of average molecular counts per cell based on traditional sensitivity metrics [66]. The elevated MECR suggested that a component of Xenium's high molecular counts might be attributable to non-specific signals rather than genuinely higher sensitivity.
Table: Performance Comparison of In Situ Technologies
| Technology | Type | Panel Size | Avg. Molecules/Cell | MECR Performance | Key Specificity Finding |
|---|---|---|---|---|---|
| Xenium | Commercial | Not specified | 297 | Highest MECR | High apparent sensitivity correlated with reduced specificity [66] |
| MERFISH | Academic | 1,147 genes | Not specified | Lower MECR | Better specificity control despite large panel [66] |
| MERSCOPE | Commercial | Receptor-focused | Not specified | Intermediate MECR | Target list enrichment for lowly expressed genes [66] |
| Molecular Cartography | Commercial | 99 genes | Not specified | Higher MECR | Specificity challenges despite small panel [66] |
| STARmap PLUS | Academic | Not specified | Not specified | Lower MECR | Favorable specificity profile [66] |
| EEL FISH | Academic | Not specified | 42 | Lower MECR | Low molecular count but favorable specificity [66] |
Impact on Biological Interpretation
The practical consequences of varying specificity levels extend beyond technical metrics to significantly impact biological interpretation. Technologies with elevated MECR values (indicating lower specificity) can produce seriously confounded results in downstream analyses:
Spatially-Aware Differential Expression:
- Reduced specificity inflates apparent expression levels, particularly for lowly expressed genes [66]
- Artificial spatial patterns may emerge from platform-specific artifacts rather than biological variation
- False positive findings in neighborhood analysis and cell-cell communication inference
Cell Type Identification:
- Contaminated marker expression profiles reduce accuracy of cell type annotation
- Erosion of distinct cell population boundaries in clustering analysis
- Incorrect assignment of cellular identities in spatially resolved data
The confounding effect is particularly pronounced for lowly expressed genes, where the relative contribution of non-specific background is proportionally greater [66]. This creates systematic biases in comparative expression analysis that cannot be corrected through normalization alone.
Standardization Framework for Cross-Technology Comparison
Integrated Quality Assessment
Moving beyond single-metric evaluation, comprehensive technology assessment requires an integrated framework that incorporates both sensitivity and specificity measures:
Multi-dimensional Quality Metrics:
- Specificity: Quantified using MECR and related metrics
- Sensitivity: Measured through detection limits for low-abundance transcripts
- Reproducibility: Assessed via technical replicate correlation [66]
- Accuracy: Evaluated through comparison with orthogonal validation methods
Experimental Design Requirements:
- Include common marker genes across technology panels to enable direct comparison [66]
- Standardize reference tissues and cell type annotations
- Implement consistent cell segmentation methodologies [66]
- Share raw data to permit re-analysis with standardized pipelines
Experimental and Computational Best Practices
Wet-Lab Protocol Considerations:
- Optimize probe design to minimize cross-hybridization potential
- Include negative control probes to estimate background signal levels
- Titrate amplification reagents to avoid signal saturation [67]
- Implement rigorous wash conditions to reduce non-specific binding [48]
Computational Analysis Recommendations:
- Apply background subtraction methods informed by negative controls
- Implement cell segmentation approaches that account for tissue morphology [66]
- Utilize expression thresholds that maximize signal-to-noise ratio
- Develop platform-specific normalization approaches that address artifact patterns
Research Reagent Solutions for Specificity Optimization
Table: Essential Reagents for Specificity-Enhanced In Situ Hybridization
| Reagent Category | Specific Examples | Function in Specificity Control |
|---|---|---|
| Labeled Probes | Digoxigenin (DIG)-labeled RNA probes [48] | Hapten incorporation for highly specific antibody detection |
| Detection Systems | Tyramide signal amplification [67] | Controlled amplification for enhanced signal-to-noise ratio |
| Hybridization Solutions | Formamide-based hybridization buffer [48] | Stringency control for specific probe-target binding |
| Stringency Washes | SSC buffers at controlled temperatures [48] | Removal of non-specifically bound probes |
| Blocking Reagents | BSA, milk, or serum in blocking buffers [48] | Reduction of non-specific antibody binding |
| Permeabilization Agents | Proteinase K [48] | Controlled tissue permeabilization for probe access |
The development and implementation of specificity metrics like MECR represents a critical advancement in the standardization of in situ hybridization technologies. By providing a quantitative framework for assessing off-target artifacts, these metrics enable more meaningful comparison across platforms and more accurate interpretation of biological results. The consistent finding that traditional sensitivity metrics can be misleading when specificity varies highlights the necessity of integrated quality assessment in technology selection and experimental design.
For the research community, widespread adoption of specificity metrics like MECR will require:
- Development of standardized reference datasets with established marker genes
- Creation of open-source computational tools for metric calculation
- Consensus building around reporting standards for technology performance
- Integration of specificity metrics into technology development cycles
As the field of spatially resolved transcriptomics continues to evolve, with technologies expanding in multiplexing capacity and spatial resolution, maintaining rigorous attention to specificity will be essential for ensuring that biological discoveries reflect genuine phenomena rather than technological artifacts. The MECR metric provides a foundation for this rigorous approach, enabling researchers to make informed decisions about technology selection and application in both basic research and drug development contexts.
Within the broader scope of in situ hybridization (ISH) sensitivity comparison research, a critical pillar is the rigorous validation of findings against other transcriptional analysis methods. Validation studies are not merely a supplementary step; they are fundamental for confirming the accuracy, sensitivity, and quantitative potential of ISH assays. Correlating ISH data with orthogonal techniques like single-cell RNA sequencing (scRNA-seq) and quantitative PCR (qPCR) provides a multi-faceted view of gene expression, strengthening the reliability of biological conclusions. This guide details the methodologies and quantitative frameworks for performing these essential correlations, providing researchers and drug development professionals with a robust toolkit for method verification.
Quantitative Data Comparison: scRNA-seq and qPCR
A foundational study provides a direct quantitative comparison of scRNA-seq methods against the gold standard, multiplexed qPCR, offering a benchmark for how sequencing-based data can be validated [68].
Table 1: Quantitative Performance of Single-Cell RNA-seq Methods Against qPCR Benchmark
| Method | Reaction Volume | Correlation with qPCR (r) | Linear Regression Slope | Key Advantages |
|---|---|---|---|---|
| C1 (SMARTer on Fluidigm) | Nanoliter | > 0.84 | ~1.0 | Highest accuracy, fewer false positives, reduced amplification bias [68] |
| SMARTer Ultra Low | Microliter | > 0.84 | < 1.0 | Good correlation with established protocol [68] |
| TransPlex | Microliter | > 0.84 | < 1.0 | Good correlation, tube-based method [68] |
Key insights from this comparative analysis include:
- qPCR as a Gold Standard: The high correlation (r > 0.84) across methods confirms that scRNA-seq can perform quantitative transcriptome measurements consistent with qPCR [68].
- Volume-Dependent Accuracy: The near-ideal slope of 1.0 for the nanoliter-volume system highlights the impact of reaction volume. The reduced volume increases reactant concentration and minimizes amplification bias, leading to superior accuracy [68].
- Methodological Trade-offs: While the Ovation method with Nextera was noted for high reproducibility, it demonstrated low sensitivity, underscoring that reproducibility and sensitivity are not always linked [68].
Experimental Protocols for Key Validation Experiments
Protocol: Validating scRNA-seq with Multiplexed qPCR
This protocol is designed to benchmark the accuracy of scRNA-seq data against qPCR measurements in a matched cell type [68].
Detailed Methodology:
- Cell Preparation: Use a homogeneous cell line (e.g., HCT116) to minimize biological variability. A total of 457 single cells were analyzed in the benchmark study [68].
- Single-Cell Isolation: Isolate single cells either by:
- Fluorescence-activated cell sorting (FACS), sorting individual cells into well plates.
- Using an integrated microfluidic system (e.g., Fluidigm C1) for capture and processing [68].
- cDNA Synthesis & Amplification:
- Perform cDNA synthesis and amplification using the scRNA-seq method being validated (e.g., SMARTer Ultra Low RNA Kit) and, in parallel, for qPCR.
- For scRNA-seq, include External RNA Controls Consortium (ERCC) spike-in RNAs before cell lysis to monitor technical reproducibility [68].
- Library Construction and Sequencing: Construct libraries (e.g., using Illumina Nextera) and sequence. For the scRNA-seq validation, an average of 2 million raw reads per cell was sufficient for quantitative comparison [68].
- Multiplexed qPCR: Perform multiplexed qPCR on the same cell type to measure the expression of a curated set of genes (e.g., 40 genes including high, low, and non-expressed genes) [68].
- Data Analysis for Correlation:
- For both qPCR and scRNA-seq data, calculate gene expression values relative to the median expression across all transcripts for each cell. This creates a dimensionless number for direct comparison [68].
- Calculate the Pearson correlation coefficient (r) and perform linear regression between the relative expression values from qPCR and scRNA-seq to assess accuracy and any systematic bias (slope) [68].
Protocol: Correlating ISH with scRNA-seq
This protocol outlines a general approach for validating ISH findings using scRNA-seq data, which can reveal cellular heterogeneity.
Detailed Methodology:
- Sample Preparation: Apply the ISH assay (e.g., RNAscope) and scRNA-seq to serial sections of the same tissue sample or to genetically identical cell populations under the same treatment conditions.
- ISH Analysis: Quantify the signal intensity and the proportion of positive cells for the target genes using microscopy and image analysis software.
- scRNA-seq Analysis: Process the tissue or cell population with scRNA-seq. A dataset of 400 cells per condition/timepoint, with an average of 6,415 genes detected per cell, provides a robust profile [69]. Data processing includes normalization and clustering to identify cell populations.
- Differential Expression Analysis: Identify differentially expressed genes (DEGs) in the scRNA-seq data using tests like MAST, with a fold-change cutoff (e.g., 1.25) and adjusted p-value (e.g., 0.01), while excluding genes detected in fewer than 10% of cells [69].
- Data Correlation:
- Expression Level: Correlate the average expression level of a gene from scRNA-seq with the quantitative ISH signal intensity across multiple samples or conditions.
- Cellular Detection: Correlate the proportion of cells expressing a particular transcript (as determined by ISH) with the proportion of cells in which the transcript is detected above a defined threshold in the scRNA-seq data. scRNA-seq is biased towards highly expressed genes, so this correlation is strongest for genes in the top expression quintiles [69].
Figure 1: Workflow for correlating ISH, scRNA-seq, and qPCR data in a validation study.
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential materials and kits used in the featured validation experiments.
Table 2: Key Research Reagents and Kits for Validation Studies
| Item | Function / Application | Example Use Case |
|---|---|---|
| SMARTer Ultra Low RNA Kit (Clontech) | cDNA synthesis and amplification from low-input RNA samples. | Used for single-cell RNA-seq library preparation in tube-based and C1 microfluidic methods [68]. |
| TransPlex Kit (Sigma-Aldrich) | Whole-transcriptome amplification from single cells. | Utilized for tube-based single-cell RNA-seq library construction [68]. |
| C1 System (Fluidigm) | Integrated microfluidic circuit for automated single-cell capture, lysis, and cDNA synthesis. | Enabled high-throughput single-cell RNA-seq in nanoliter volumes, reducing bias and false positives [68]. |
| Nextera Kit (Illumina) | Library preparation for next-generation sequencing. | Used for sequencing library construction from amplified cDNA in the referenced scRNA-seq study [68]. |
| ERCC Spike-In Controls | Exogenous RNA controls to monitor technical variation and reproducibility. | Added to single-cell lysates on the C1 platform to assess sample-to-sample reproducibility [68]. |
| Leica BOND-III | Automated staining platform for FISH/ISH assays. | Validated for HER2 FISH testing, demonstrating 98% concordance with manual methods, reducing hands-on time and costs [4]. |
| RNAscope Assays (ACD) | Highly sensitive ISH for RNA detection in tissue. | Used in pilot studies against traditional ISH, demonstrating superior sensitivity and faster turnaround [35]. |
scRNA-seq as a Discovery Tool for ISH Targets
Beyond validation, scRNA-seq can guide ISH by identifying novel targets within heterogeneous populations. A study on glucocorticoid response in breast cancer used scRNA-seq to reveal over 100 differentially expressed genes (DEGs) in response to hormone treatment that were not detected by bulk RNA-seq [69]. This highlights the power of scRNA-seq to uncover regulatory events masked in population-averaged measurements. These newly discovered DEGs from scRNA-seq become prime candidates for further spatial validation using ISH assays to confirm their expression in specific tissue contexts and cell types.
Figure 2: scRNA-seq complements bulk sequencing to identify novel ISH targets.
The accurate detection of Human Epidermal Growth Factor Receptor 2 (HER2) status in breast cancer has transitioned from a binary classification to a spectrum-based paradigm, driven by the clinical efficacy of novel antibody-drug conjugates (ADCs) against tumors with low HER2 expression. This evolution presents significant challenges for pathological assessment, requiring enhanced sensitivity and reproducibility across detection platforms. Historically, HER2 testing aimed to identify overexpression (IHC 3+) or gene amplification for trastuzumab eligibility, with IHC 1+ considered clinically negative [70]. The landmark DESTINY-Breast04 trial demonstrated that trastuzumab deruxtecan (T-DXd) improved survival in patients with metastatic breast cancer showing HER2-low expression (IHC 1+ or 2+/ISH-negative), necessitating precise distinction between HER2-negative and HER2-low categories [71]. Subsequent findings further extended this spectrum to include HER2-ultralow (IHC 0 with faint membrane staining in ≤10% of tumor cells) [71]. This paradigm shift exposes critical limitations in conventional immunohistochemistry (IHC) assays originally validated for detecting HER2 overexpression rather than low-level expression, highlighting the urgent need for standardized methodologies with improved analytical sensitivity and dynamic range within the context of in situ hybridization sensitivity comparison research [70].
Technical Foundations of HER2 Detection Methodologies
Immunohistochemistry (IHC): Protein Expression Analysis
IHC detects HER2 protein expression on the cell membrane through antibody-mediated staining visualized via chromogenic reactions. The American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines establish a semi-quantitative scoring system:
- Score 0 (Negative): No staining or membrane staining in <10% of tumor cells
- Score 1+ (Negative/Low): Faint/barely perceptible partial membrane staining in >10% of tumor cells
- Score 2+ (Equivocal): Weak to moderate complete membrane staining in >10% of tumor cells
- Score 3+ (Positive): Strong complete membrane staining in >10% of tumor cells [72]
The clinical interpretation of these scores has evolved significantly. While scores 0 and 1+ were historically grouped as "HER2-negative," they now represent distinct therapeutic categories, with IHC 1+ (HER2-low) potentially eligible for T-DXd therapy [71]. This refined classification demands exceptional consistency in staining and interpretation, particularly at the critical 0 versus 1+ boundary.
In Situ Hybridization (ISH): Gene Amplification Analysis
ISH methodologies detect HER2 gene amplification at the DNA level, providing a quantitative assessment of gene copy number. The principal ISH techniques include:
Fluorescence ISH (FISH): Uses fluorescently labeled DNA probes to target HER2 and chromosome 17 centromere (CEP17) sequences. Amplification is defined by HER2:CEP17 ratio ≥2.0 with an average HER2 copy number ≥4.0 signals/cell, or HER2:CEP17 ratio <2.0 with an average HER2 copy number ≥6.0 signals/cell [73]. Despite being considered the gold standard, FISH requires fluorescence microscopy and suffers from signal fading over time.
Chromogenic ISH (CISH) and Silver-Enhanced ISH (SISH): Utilize enzyme-mediated chromogenic or silver precipitation reactions visible under standard bright-field microscopy. SISH offers permanent slide preservation and clearer morphological correlation, with processing time reduced to approximately 6 hours compared to 12-16 hours for FISH [74].
Emerging Methodologies: Digital PCR and Computational Approaches
Digital PCR (dPCR) provides absolute quantification of HER2 gene copy number relative to reference genes without requiring tissue morphology assessment. Studies demonstrate 82.8% sensitivity and 97.3% specificity compared to standard assays, with excellent inter-platform reproducibility (97% concordance) [75]. dPCR shows particular utility for resolving equivocal cases, accurately classifying 75% of IHC 2+ amplified cases and 95% of non-amplified cases [75].
Artificial intelligence (AI) platforms employing deep learning algorithms have demonstrated remarkable proficiency in HER2 scoring, achieving pooled sensitivity of 0.97 and specificity of 0.82 for distinguishing HER2-low/positive (1+/2+/3+) from true negative (0) cases [65]. Performance improves with higher expression levels, reaching near-perfect classification for IHC 3+ (sensitivity 0.97, specificity 0.99) [65].
Comparative Analytical Performance of HER2 Detection Methods
Table 1: Performance Characteristics of HER2 Detection Methodologies
| Method | Target | Sensitivity | Specificity | Concordance with FISH | Key Limitations |
|---|---|---|---|---|---|
| IHC (Conventional) | Protein | 85.7% (for 3+) [70] | 100% (for 3+) [70] | 93.8% [73] | Poor dynamic range for HER2-low; inter-observer variability |
| FISH | Gene | - | - | Gold standard | Fluorescence fading; specialized equipment required |
| SISH | Gene | Comparable to FISH [74] | Comparable to FISH [74] | >95% [74] | - |
| Digital PCR | Gene | 82.8% [75] | 97.3% [75] | 97% (inter-platform) [75] | Loses tissue morphology context |
| AI-Assisted IHC | Protein | 97% (1+/2+/3+ vs 0) [65] | 82% (1+/2+/3+ vs 0) [65] | - | Dependent on training data quality |
Table 2: AI Performance Across HER2 IHC Scores Based on Meta-Analysis
| HER2 Score | Sensitivity | Specificity | Area Under Curve (AUC) | Concordance with Pathologists |
|---|---|---|---|---|
| 1+ | 0.69 [95% CI 0.57-0.79] [65] | 0.94 [95% CI 0.90-0.96] [65] | 0.92 [95% CI 0.90-0.94] [65] | 88% [95% CI 86-90%] [65] |
| 2+ | 0.89 [95% CI 0.84-0.93] [65] | 0.96 [95% CI 0.93-0.97] [65] | 0.98 [95% CI 0.96-0.99] [65] | - |
| 3+ | 0.97 [95% CI 0.96-0.99] [65] | 0.99 [95% CI 0.97-0.99] [65] | 1.00 [95% CI 0.99-1.00] [65] | 97% [95% CI 96-98%] [65] |
Critical Research Findings and Case Studies
The CASI-01 Study: Revealing Fundamental Assay Limitations
The Consortium for Analytic Standardization in Immunohistochemistry (CASI-01) study, involving 54 IHC laboratories across Europe and the U.S., provided pivotal insights into HER2 assay performance across the expression spectrum. This investigation revealed that conventional FDA-cleared HER2 assays exhibit detection thresholds ranging from 30,000-60,000 HER2 molecules per cell, rendering them adequate for identifying HER2 overexpression (3+) with 85.7% sensitivity and 100% specificity, but fundamentally limited for detecting low-expression categories due to insufficient dynamic range [70].
The study demonstrated that enhanced analytical sensitivity achieved through optimized IHC assays combined with image analysis yielded a six-fold improvement in dynamic range for HER2-low scores (p=0.0017) [70]. This finding underscores the necessity of reporting IHC analytical sensitivity and dynamic range as standard performance metrics, particularly for HER2-low detection. The CASI-01 study further established that image analysis can surpass pathologist readout accuracy in specific clinical contexts, marking a pivotal transition in IHC companion diagnostics from a qualitative "stain" approach to a quantitative "assay" model incorporating calibration, reference standards, and statistical process control [70].
Inter-laboratory Variability: A Nationwide Assessment
A comprehensive Dutch nationwide study analyzing 103,505 breast tumors across 34 pathology laboratories revealed substantial variability in HER2-low frequency (33.4%-94.5% among non-amplified cases) [76]. This variability persisted even among laboratories using identical antibody clones and detection systems, indicating that factors beyond reagent selection contribute to staining differences.
Critical findings included antibody-specific detection patterns, with clone A0485 identifying the highest proportion of HER2-low cases (71.5%), followed by DG44 (66.7%), SP3 (60.1%), and 4B5 (59.1% with Ultraview, 57.0% with Optiview) [76]. The study additionally documented a gradual increase in HER2-low frequency since 2022, potentially reflecting evolving diagnostic thresholds and increased pathologist awareness following T-DXd approval [76].
Resolving Equivocal Cases: The Role of Advanced Methodologies
The challenge of HER2 equivocal (IHC 2+) cases necessitates reflexive testing, typically by ISH. A study of 44 breast cancer specimens demonstrated significant discrepancy between IHC and FISH (p=0.019), with 81.8% (36/44) of cases classified as IHC 2+ but only 47.7% (21/44) showing amplification by FISH [77]. Digital PCR has emerged as a valuable adjunctive method for these challenging cases, demonstrating 75% sensitivity and 95.3% specificity in classifying IHC 2+ cases in an independent validation cohort [75]. Notably, discordant cases frequently exhibited lower percentages of HER2-positive cells, highlighting the impact of tumor heterogeneity on assay performance [75].
Standardized Experimental Protocols
Silver-Enhanced In Situ Hybridization (SISH) Protocol
The SISH protocol enables simultaneous detection of HER2 gene and chromosome 17 centromere (CEP17) using bright-field microscopy:
Sample Preparation: Tissue sections (4μm) are baked at 60°C for 20 minutes to ensure adhesion.
Pretreatment: Slides undergo cell conditioning using EDTA-based buffer (pH 8.0) for 8-32 minutes, followed by protease digestion ( protease 3 for 16 minutes) to expose target sequences [74].
Hybridization: HER2 and CEP17 probes are denatured at 95°C for 12 minutes, then hybridized at 52°C for 6 hours. The HER2 probe is labeled with dinitrophenyl and detected with ultraView SISH DNP Detection Kit, while the CEP17 probe is labeled with digoxigenin and detected with ultraView Red ISH DIG Detection Kit [74].
Signal Detection: Silver precipitation is initiated for HER2 (black nuclear signal) followed by red chromogenic development for CEP17 (red nuclear signal).
Counterstaining: Hematoxylin II is applied for 8 minutes followed by bluing reagent for 4 minutes [74].
Interpretation: HER2 amplification is defined as HER2:CEP17 ratio ≥2.0 or average HER2 signals/cell ≥6.0. A minimum of 20 tumor cells within morphologically representative areas are counted.
Artificial Intelligence-Assisted HER2 Scoring Workflow
AI-based HER2 scoring integrates computational pathology with traditional assessment:
Whole Slide Imaging: IHC-stained slides are digitized using high-resolution scanners (40x magnification recommended).
Image Preprocessing: Whole slide images are partitioned into smaller patches (typically 256x256 or 512x512 pixels), with color normalization to minimize staining variability [65].
Region of Interest Selection: Convolutional neural networks automatically identify and segment invasive tumor regions, excluding DCIS, normal epithelium, and stromal elements.
Patch-Level Classification: Deep learning algorithms classify each patch based on membrane staining intensity and completeness, generating probability scores for HER2 categories (0, 1+, 2+, 3+).
Whole Slide Scoring: Patch-level predictions are aggregated using attention-based multiple instance learning frameworks to generate final HER2 scores, incorporating spatial relationships and tumor heterogeneity [65].
Pathologist Review: Cases with uncertain predictions or technical artifacts are flagged for pathologist review, maintaining human oversight in the diagnostic process.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagents and Platforms for HER2 Detection
| Reagent/Platform | Manufacturer | Function | Application Notes |
|---|---|---|---|
| HercepTest | Dako/Agilent | IHC detection of HER2 protein | Uses clone A0485; demonstrates higher HER2-low detection rates (71.5%) [76] |
| PATHWAY anti-HER2/neu (4B5) | Ventana/Roche | IHC detection of HER2 protein | Used in DESTINY-Breast04 trial; multiple detection systems (Ultraview, Optiview) with varying sensitivity [76] |
| INFORM HER2 Dual ISH | Ventana/Roche | Simultaneous detection of HER2 and CEP17 | SISH methodology; 6-hour processing time; bright-field compatible [74] |
| PathVysion HER2 FISH | Abbott Molecular | FISH detection of HER2 amplification | Gold standard; requires fluorescence microscopy and darkroom conditions [72] |
| QX200 Droplet Digital PCR | Bio-Rad | Absolute quantification of HER2 copy number | 97.3% specificity; useful for equivocal cases [75] |
| Ventana Benchmark Series | Ventana/Roche | Automated staining platform | Standardized IHC and ISH processing; reduces inter-laboratory variability [72] |
The evolving taxonomy of HER2 expression from binary positive/negative classification to a spectrum encompassing ultralow, low, and positive categories demands fundamental advances in detection technologies. The integration of standardized reference materials, automated image analysis, and artificial intelligence represents the most promising path toward reproducible HER2 scoring across the expression continuum. The CASI-01 study establishes that conventional IHC assays, while adequate for identifying HER2 overexpression, lack the dynamic range necessary for reliable HER2-low detection [70]. Emerging solutions include optimized high-sensitivity IHC assays, objective image analysis systems, and AI platforms that demonstrate exceptional performance particularly for distinguishing 2+ and 3+ scores [65].
The profound inter-laboratory variability in HER2-low classification [76] underscores the urgent need for standardized validation protocols incorporating quantitative metrics of analytical sensitivity and dynamic range. Future HER2 testing paradigms will likely integrate multiple complementary methodologies—combining traditional IHC/ISH with digital PCR and computational pathology—to resolve challenging cases and account for tumor heterogeneity. As therapeutic options continue to expand across the HER2 expression spectrum, precision pathology must evolve accordingly, embracing standardized controls, quantitative platforms, and computational tools to ensure optimal patient selection for targeted therapies.
In situ hybridization (ISH) continues to be a cornerstone technique in molecular pathology, diagnostics, and research, enabling the precise detection and localization of specific nucleic acid sequences within cells or tissue sections [35]. The selection of an appropriate platform—whether a commercial automated system or an academically developed manual protocol—profoundly impacts the sensitivity, specificity, and overall reliability of experimental outcomes. This review provides an in-depth technical comparison of the performance characteristics of various ISH platforms, framed within the broader context of advancing sensitivity comparison research. For researchers, scientists, and drug development professionals, understanding these nuances is critical for selecting the optimal technology for applications in cancer diagnostics, spatial biology, and drug discovery, where accuracy and reproducibility are paramount [78].
The market for ISH technologies is evolving rapidly, driven by technological innovations and increasing integration into precision medicine and laboratory-developed tests (LDTs) [78]. This review synthesizes current quantitative performance data, details core experimental methodologies, and provides a practical toolkit to inform platform selection and implementation strategies.
Quantitative Performance Data Comparison
The performance of an ISH platform is primarily quantified by its sensitivity and specificity. These metrics were analyzed across several prominent platforms and methodologies, with key quantitative findings summarized in the table below.
Table 1: Comparative Performance Metrics of ISH Platforms and Methodologies
| Platform / Methodology | Application Context | Reported Sensitivity | Reported Specificity | Key Performance Notes |
|---|---|---|---|---|
| Leica BOND-III Automated FISH [4] | HER2 FISH Testing (Breast Cancer) | 0.95 | 0.97 | High concordance (98%) with manual methods; significantly reduces hands-on time. |
| Leica BOND-III Automated FISH [4] | HER2 FISH Testing (Gastric Cancer) | 1.0 | 1.0 | Perfect sensitivity and specificity in validated cohort. |
| RNAscope (ACD Bio) [35] | Pilot vs. Traditional ISH | Superior Sensitivity (Not Quantified) | N/A | Noted for superior sensitivity and faster turnaround time. |
| NanoString Spatial Profiling [35] | Multiplexed Spatial Profiling | Enhanced Resolution (Not Quantified) | N/A | Demonstrated enhanced spatial resolution and data richness. |
Beyond individual platform studies, the overall ISH market demonstrates strong growth, projected to reach USD 2.35 billion by 2030, with a compound annual growth rate (CAGR) of 7.4% [78]. This growth is a indirect indicator of the technology's increasing reliability and adoption in critical diagnostic and research applications. Fluorescence ISH (FISH), particularly DNA FISH, currently holds the largest market share within the segment due to its well-established superior sensitivity and accuracy in detecting genetic anomalies like gene amplifications and chromosomal rearrangements [78].
Detailed Experimental Protocols
A critical factor influencing the sensitivity and specificity of any ISH experiment, regardless of the platform, is the underlying protocol. The following details a core methodology for a sensitive, DIG-labeled RNA probe ISH protocol, which can be adapted for both commercial kits and academic laboratory-developed tests [48].
Core Workflow: DIG-Labeled RNA In Situ Hybridization
The entire experimental process, from sample preparation to final detection, is visualized in the following workflow diagram. This diagram outlines the logical sequence of steps and key decision points.
Stage 1: Sample Storage and Tissue Preparation
Proper sample handling is the most critical first step for preserving nucleic acid integrity and ensuring reliable results [48].
- Sample Storage: Tissue samples must be handled to prevent RNA degradation. Immediate flash-freezing in liquid nitrogen or fixation in formalin followed by paraffin embedding (FFPE) are standard approaches. FFPE tissues can be stored long-term in a cool, dry environment. For prepared slides, avoid dry storage at room temperature; instead, store in 100% ethanol at -20°C or in a covered box at -80°C to preserve them for years [48].
- Tissue Preparation and Sectioning: Fixation using agents like paraformaldehyde or formalin preserves tissue structure and nucleic acid integrity. Fixed tissues are embedded in paraffin and sectioned thinly with a microtome. RNase contamination is a major threat; it is present on skin, glassware, and reagents. Use sterile techniques, gloves, and RNase-free solutions throughout [48].
Stage 2: Probe Design and Selection
The choice of probe is a key determinant of assay sensitivity and specificity [48].
- Probe Type: RNA probes (riboprobes), particularly antisense RNA probes, are preferred for high sensitivity and specificity for target RNA sequences. DNA probes offer high sensitivity but hybridize less strongly to mRNA than RNA probes.
- Probe Design: RNA probes should ideally be 250–1,500 bases long, with approximately 800 bases often providing the highest sensitivity and specificity. Transcription templates should allow for both antisense (probe) and sense strand (negative control) RNA synthesis. Probe specificity is critical; a probe with >5% non-complementary base pairs may hybridize loosely and be washed away during subsequent steps [48].
Stage 3: Detailed Hybridization and Detection Protocol
This section details the key wet-bench procedures following the workflow diagram.
- Deparaffinization and Rehydration: For FFPE sections, immerse slides in a rack and wash sequentially: Xylene (2x3 min), Xylene:100% ethanol 1:1 (3 min), 100% ethanol (2x3 min), 95% ethanol (3 min), 70% ethanol (3 min), 50% ethanol (3 min), followed by a rinse with cold tap water. From this point onward, slides must not dry out, as this causes non-specific antibody binding and high background [48].
- Antigen Retrieval and Permeabilization: Digest tissue with 20 µg/mL proteinase K in pre-warmed 50 mM Tris for 10–20 min at 37°C. This step is highly variable; a proteinase K titration experiment is recommended to optimize for each tissue type. Insufficient digestion reduces signal, while over-digestion damages tissue morphology. Follow with a 20-second immersion in ice-cold 20% (v/v) acetic acid to further permeabilize cells [48].
- Hybridization: Apply hybridization solution to the slides and pre-hybridize for 1 hour in a humidified chamber at the desired temperature (typically 55–62°C). Meanwhile, dilute the denatured probe (heated to 95°C for 2 min, then chilled on ice) in hybridization solution. Apply 50–100 µL of probe per section, cover with a coverslip, and incubate overnight at 65°C in a humidified chamber [48].
- Stringency Washes: These washes remove non-specifically bound probe and are critical for specificity.
- Wash 1: 50% formamide in 2x SSC, 3x5 min at 37–45°C. This removes excess probe.
- Wash 2: 0.1-2x SSC, 3x5 min at 25-75°C. This removes non-specific/repetitive hybridization. The temperature and stringency (SSC concentration) here are crucial. Use lower temperature (up to 45°C) and lower stringency (1–2x SSC) for short or complex probes. Use higher temperature (~65°C) and higher stringency (below 0.5x SSC) for single-locus, large, or repetitive probes [48].
- Blocking and Detection: Wash slides twice in MABT (Maleic Acid Buffer with Tween) for 30 min. Transfer to a humidified chamber and block with MABT + 2% blocking reagent (BSA, milk, or serum) for 1–2 hours. Drain the buffer and incubate with anti-DIG antibody (concentration per datasheet) in blocking buffer for 1–2 hours at room temperature. Perform final washes (5x10 min with MABT) before proceeding to the colorimetric or fluorescent visualization substrate [48].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful execution of an ISH experiment relies on a suite of essential reagents and solutions, each serving a specific function to ensure optimal performance. The following table details these key components.
Table 2: Essential Reagents and Solutions for In Situ Hybridization
| Reagent/Solution | Function / Purpose | Key Considerations & Examples |
|---|---|---|
| Probes | Hybridizes to the target nucleic acid sequence for detection. | RNA probes offer high sensitivity. DIG-labeled probes allow for antibody-based detection. Probe length (optimal ~800 bases) and specificity are critical [48]. |
| Fixatives | Preserves tissue morphology and nucleic acid integrity. | Formalin and paraformaldehyde are standard for FFPE samples [48]. |
| Permeabilization Agents | Disrupts cell membranes to allow probe and antibody access. | Proteinase K digests proteins; concentration and time require optimization [48]. |
| Hybridization Buffer | Creates the chemical environment for specific probe-target binding. | Contains formamide, salts (SSC), Denhardt's solution, dextran sulfate, and blocking agents to control stringency and reduce background [48]. |
| Stringency Wash Buffers | Removes non-specifically bound probe post-hybridization. | SSC (Saline Sodium Citrate) solutions at varying concentrations and temperatures are used to fine-tune specificity [48]. |
| Blocking Buffer | Prevents non-specific binding of the detection antibody. | Typically contains a protein source (BSA, milk, or serum) in a buffer like MABT [48]. |
| Detection Antibody | Binds to the probe label (e.g., DIG) for visualization. | e.g., Anti-DIG antibody conjugated to an enzyme (for colorimetric detection) or a fluorophore [48]. |
Discussion & Technical Considerations
The quantitative data indicates that automated platforms like the Leica BOND-III can achieve performance on par with, or even exceeding, manual methods while significantly improving operational efficiency [4]. This addresses key market challenges such as technical complexity and inter-operator variability [78]. However, the choice between commercial and academic platforms involves trade-offs. Commercial kits offer standardization, validation support, and ease of use, which is vital for clinical diagnostics. In contrast, academically developed ("home-brew") protocols provide unparalleled flexibility for probe design and assay customization, which is often necessary for novel research targets.
A significant driver of adoption in commercial platforms is their integration into precision medicine and companion diagnostics. For instance, collaborations between diagnostic companies and pharmaceutical firms to develop validated ISH-based companion diagnostics are becoming more common, enhancing the precision of targeted therapies [78]. Furthermore, technological advancements such as multiplexing capabilities, automated image analysis, and the integration of AI are expected to be key strategic focuses for vendors by 2025, further pushing the boundaries of sensitivity and quantitative analysis [35].
This performance review underscores that both commercial automated systems and optimized academic protocols are capable of achieving high sensitivity and specificity in ISH. The decision matrix for platform selection must extend beyond raw performance metrics to include factors such as throughput requirements, available expertise, assay customization needs, and regulatory considerations. The ongoing innovation in probe chemistry, automation, and digital analysis promises to further elevate the performance standards of ISH platforms, solidifying their indispensable role in spatial genomics, molecular pathology, and translational research.
In situ hybridization (ISH) continues to be a cornerstone technique in molecular pathology, diagnostics, and research, yet its path to clinical diagnostics has been hampered by challenges in reproducibility and analytical variability. Technological advances are accelerating, with a growing number of vendors offering innovative solutions tailored to diverse needs—from clinical labs to biotech research [35]. The reproducibility of molecular diagnostics represents a fundamental requirement for clinical implementation, affecting everything from basic research validation to patient management decisions. For techniques like ISH, which provide essential spatial context for nucleic acid detection, ensuring that results are consistent both within and between laboratories is particularly challenging due to the multi-step, complex nature of these assays. The growing integration of automated analysis and artificial intelligence is poised to address these challenges, but requires rigorous validation to ensure reliable performance across diverse clinical settings.
Reproducibility Challenges in Conventional ISH Technologies
Limitations of Traditional Detection Methods
Traditional ISH methods and immunohistochemistry (IHC) demonstrate significant limitations in both sensitivity and specificity that directly impact reproducibility. Conventional IHC shows suboptimal diagnostic sensitivity, failing to detect viral antigens in approximately 30% of biopsy-proven BK virus-associated nephropathy (BKVAN) cases despite concurrent viremia exceeding clinical alert thresholds [52]. Such variability in detection can result in inappropriate patient management, including erroneous immunosuppressive treatment that exacerbates disease progression. Similarly, standard DNA-targeted ISH demonstrates inferior sensitivity for detecting low amounts of viral DNA, creating substantial inter-laboratory variability and compromising the reliability of clinical diagnoses [52].
The specificity challenges are equally problematic. Conventional immunofluorescence staining demonstrates broad-spectrum cross-reactivity when detecting polyomaviruses, with SV40 large T antigen targeting antibodies detecting BK virus, JC virus, and SV40 variants simultaneously due to 60-70% amino acid sequence homology [52]. This cross-reactivity introduces significant interpretation variability between laboratories and technologists, particularly for low-abundance targets where subjective assessment plays a larger role.
Quantitative Assessment of Diagnostic Variability
Table 1: Diagnostic Performance of FISH for Biliary Strictures Based on Different Positivity Criteria
| FISH Positivity Criteria | Sensitivity (%) | Specificity (%) | Clinical Application |
|---|---|---|---|
| Polysomy only | 49.4 (43.2-55.5) | 96.2 (92.7-98.1) | Highest specificity for conclusive positive results |
| Polysomy + Tetrasomy/Trisomy | 64.3 (55.4-72.2) | 78.9 (64.4-88.5) | Increased sensitivity with reduced specificity |
| Polysomy + 9p deletion | 54.7 (42.4-66.5) | 95.1 (84.0-98.6) | Balanced approach maintaining high specificity |
A systematic review and meta-analysis of fluorescence in situ hybridization (FISH) for biliary strictures reveals how definitional variability directly impacts diagnostic reproducibility. The analysis of 18 studies comprising 2516 FISH specimens demonstrated that the threshold for a "positive" result significantly affects test characteristics [54]. When using thresholds as defined in individual studies, the overall sensitivity of FISH was 57.6% (95% CI: 49.4–65.4%) with specificity of 87.8% (95% CI: 79.2–93.2%). However, standardized criteria substantially improved performance, with polysomy-only thresholds providing near-perfect specificity (96.2%) albeit with reduced sensitivity (49.4%) [54]. This variability in diagnostic performance based solely on interpretation criteria underscores the profound reproducibility challenges in conventional ISH approaches.
Advanced ISH Platforms: Enhancing Sensitivity and Reproducibility
Signal Amplification Technologies
Novel ISH platforms employing advanced signal amplification strategies have demonstrated marked improvements in both sensitivity and reproducibility. The ViewRNA ISH system, based on branched DNA (bDNA) amplification technology, represents a novel ISH strategy with enhanced sensitivity [52]. This system utilizes a set of 40 probes targeting both DNA and mRNA of the large T antigen coding region, providing stringent species specificity that eliminates the cross-reactivity problems of conventional immunofluorescence. In comparative studies, the fluorescent BKV-LT ViewRNA-ISH system produced intense nucleic acid signals exclusively in BK virus-infected cells, whereas conventional immunofluorescence demonstrated broad cross-reactivity across multiple polyomaviruses [52].
The reproducibility advantages of these advanced systems extend beyond specificity. The bDNA-based approach provides consistent signal amplification that reduces operator-to-operator variability and enables more standardized quantification. This technological advancement addresses a fundamental limitation of traditional ISH: the inconsistent signal-to-noise ratio that plagues low-abundance target detection and introduces interpretation variability between laboratories.
Automated Analysis and Integration
Automation represents a critical pathway to addressing reproducibility challenges in ISH diagnostics. By 2025, vendors are expected to focus on integrating AI-driven image analysis and expanding multiplexing capabilities [35]. The reduction of manual steps through automation directly decreases error rates and improves inter-laboratory consistency. This trend is reflected across molecular diagnostics, where automation of even well-established techniques like ELISA has demonstrated measurable improvements in reproducibility [79].
Table 2: Reproducibility Metrics in Automated Diagnostic Systems
| System Type | Metric | Performance | Impact on Reproducibility |
|---|---|---|---|
| Automated ELISA | Coefficient of Variation | Reduction from 2.8% to 2.4% for amyloid-beta (1-40) [79] | Improved precision across multiple runs |
| Deep Learning System for CBCT | Symmetric Mean Curve Distance | Within-subject coefficient of repeatability: 0.969 mm [80] | High temporal reproducibility in anatomical localization |
| Deep Learning System for CBCT | Likert Score Repeatability | Repeatability measure: 0.877 for DLS vs 0.923 for radiologist [80] | Comparable to human expert consistency |
The implementation of automated deep learning systems for anatomical localization demonstrates how computational approaches can enhance reproducibility. In mandibular canal identification on CBCT scans, a deep learning system achieved a within-subject coefficient of repeatability of 0.969 mm for symmetric mean curve distance, with a repeatability measure of 0.877 for Likert scoring by experts [80]. This high level of reproducibility is particularly notable given the system's performance across heterogeneous datasets with varying scanning parameters and clinical conditions.
Experimental Protocols for Reproducible ISH Analysis
High-Sensitivity BKV Detection Using ViewRNA ISH
Cell Culture and Preparation
- Maintain human bladder cancer cell lines (TCCSUP and 5637) and 293T cells in appropriate media supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin [52]
- Culture TCCSUP cells in MEM/EBSS medium, 5637 cells in RPMI-1640 medium, and 293T cells in DMEM medium [52]
- For BKV detection, establish infection models and transient transfection systems for comparative specificity analysis
ViewRNA ISH Procedure
- Design a probe set targeting both BKV DNA and mRNA of the large T antigen coding region (40 probes) [52]
- Follow manufacturer's protocol for branched DNA signal amplification
- Perform both chemical and fluorescent ISH adaptations depending on detection requirements
- Include appropriate controls: BKV-infected cells, uninfected cells, and cells transfected with related viruses (JCV, SV40) for specificity validation [52]
Image Analysis and Interpretation
- Acquire images using standardized exposure settings across samples
- Implement automated counting algorithms for signal quantification
- Establish threshold values based on negative control samples
- Perform blinded analysis by multiple independent reviewers to assess inter-observer variability
Diagnostic FISH for Biliary Strictures
Sample Preparation
- Obtain biliary stricture specimens through endoscopic retrograde cholangiopancreatography (ERCP)-based brushing [54]
- Process samples immediately or preserve using appropriate fixation methods
- Prepare slides using standardized cytospin protocols to ensure consistent cell distribution
FISH Hybridization
- Utilize commercial UroVysion probe set (Abbott Molecular Inc.) containing labeled DNA probes for chromosomes 3, 7, 17, and 9p21 [54]
- Follow manufacturer's hybridization protocol with strict temperature and time controls
- Include control samples with known chromosomal abnormalities in each run
Microscopy and Interpretation
- Score a minimum of 50 cells per sample by trained technologists
- Define abnormality categories: polysomy (≥3 copies of ≥2 probes), trisomy (≥3 copies of 1 probe), tetrasomy (4 copies of each probe), and 9p21 deletion [54]
- Establish positivity thresholds based on clinical requirements (see Table 1)
- Implement digital imaging systems for archival and second opinion reviews
The Researcher's Toolkit: Essential Reagents and Platforms
Table 3: Research Reagent Solutions for Reproducible ISH
| Reagent/Platform | Function | Key Features |
|---|---|---|
| ViewRNA ISH System | Signal amplification for RNA/DNA detection | Branched DNA technology with 40-probe sets; higher sensitivity and specificity [52] |
| UroVysion FISH Probe Set | Chromosomal abnormality detection | Probes for chromosomes 3, 7, 17, and 9p21; standardized commercial kit [54] |
| Automated Liquid Handling Systems | Reagent dispensing and washing | Fixed steel tips reduce analyte loss (16-19% improvement for hydrophobic peptides) [79] |
| AI-Driven Image Analysis Platforms | Quantitative assessment of ISH signals | Reduced inter-observer variability; consistent threshold application [35] |
| Multiplexed ISH Platforms | Simultaneous detection of multiple targets | Spatial profiling with enhanced resolution and data richness [35] |
Workflow Visualization: Automated ISH Analysis Pathway
Automated ISH Analysis Workflow: This diagram illustrates the integrated pathway from sample preparation to quantitative results, highlighting critical steps where standardization and automation enhance reproducibility.
Implementation Framework: Validation and Quality Assurance
Pilot Program Design
Validation is crucial before full adoption of any ISH platform in clinical diagnostics. For example, a leading hospital conducted a pilot comparing ACD's RNAscope with traditional ISH methods, noting superior sensitivity and faster turnaround [35]. Similarly, a biotech firm tested Leica's automated system, achieving consistent results across multiple operators, which facilitated regulatory approval processes [35]. These pilot programs should include:
- Comparison with established reference methods using well-characterized samples
- Assessment of inter-operator and inter-instrument variability
- Determination of analytical sensitivity and specificity using dilution series
- Evaluation of clinical sensitivity and specificity in relevant patient cohorts
Quality Control Metrics
Implementing robust quality control measures ensures ongoing reproducibility:
- Establish batch-specific control samples with predetermined acceptable ranges
- Monitor assay performance through statistical process control methods
- Participate in external proficiency testing programs when available
- Document all procedural deviations and their impact on results
- Regular calibration and maintenance schedules for automated equipment
Future Directions: AI Integration and Standardization
By 2025, expect vendors to focus on integrating AI-driven image analysis and expanding multiplexing capabilities [35]. Mergers and acquisitions are likely as companies seek to broaden their portfolios—e.g., larger firms acquiring niche spatial genomics startups [35]. Pricing strategies will evolve, with some vendors offering tiered models to accommodate different lab sizes and budgets.
Innovation will also target ease of use and automation, reducing manual steps and error rates [35]. Regulatory pathways may tighten, prompting vendors to prioritize compliance and validation support. The convergence of highly multiplexed ISH technologies with computational pathology platforms will enable comprehensive spatial biology analysis while maintaining the reproducibility required for clinical diagnostics.
The critical importance of reproducibility in clinical diagnostics ensures that technologies demonstrating consistent performance across multiple sites and operators will see the most rapid adoption. As ISH technologies continue to evolve, the integration of automated workflows, standardized validation protocols, and AI-enhanced analysis will ultimately bridge the gap between research applications and routine clinical implementation.
Conclusion
The landscape of in situ hybridization is defined by a trade-off between sensitivity, specificity, cost, and workflow complexity. While commercial kits like RNAscope offer user-friendly, standardized solutions, academic methods provide flexibility and lower per-sample costs for large-scale studies. Future directions point toward increased multiplexing capabilities, enhanced sensitivity for short transcripts, and greater integration with computational analysis and automation for robust clinical diagnostics. The choice of ISH method must be strategically aligned with the project's specific goals, whether for high-content discovery research or validated clinical assay development.
References
- 1. In situ hybridization [https://en.wikipedia.org/wiki/In_situ_hybridization]
- 2. In Situ Hybridization (ISH) - NCBI - NIH [https://www.ncbi.nlm.nih.gov/probe/docs/techish/]
- 3. In Situ Hybridization (ISH), CISH, and FISH Reagents [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cellular-imaging/in-situ-hybridization-ish.html]
- 4. Automation of fluorescent in situ hybridization (FISH) ... [https://pubmed.ncbi.nlm.nih.gov/40962484/]
- 5. Top applications of immunohistochemistry [https://www.abcam.com/en-us/stories/articles/top-applications-of-immunohistochemistry]
- 6. What are the differences between IHC and ISH and how do they... - Enzo [https://www.enzo.com/note/what-are-the-differences-between-ihc-and-ish-and-how-do-they-help-in-the-fight-against-cancer/]
- 7. Recent Advances in High-sensitivity In Situ Hybridization and Costs... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10323200/]
- 8. Spatial Omics x Neuroscience: Dual ISH-IHC Brain Mapping [https://www.thermofisher.com/blog/life-in-the-lab/spatial-omics-dual-ish-ihc-brain-mapping/]
- 9. Dual ISH and IHC [https://acdbio.com/science/applications/research-solutions/sequential-rna-protein-detection]
- 10. A powerful and versatile new fixation protocol for immunostaining and... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-024-02052-3]
- 11. Comparison of fluorescence in situ hybridization and ... [https://pubmed.ncbi.nlm.nih.gov/40345927/]
- 12. Comparison of fluorescence in situ hybridization and ... [https://www.sciencedirect.com/science/article/abs/pii/S2213294525000456]
- 13. Technical innovations of signal amplification in ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925004366]
- 14. Review of In Situ Hybridization Techniques for Drug Research ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10467011/]
- 15. Using Single Molecule mRNA Fluorescent in Situ ... [https://www.nature.com/articles/s41598-018-26345-0]
- 16. Single molecule fluorescent in situ hybridization (smFISH) of C ... [https://www.ncbi.nlm.nih.gov/books/NBK126649/]
- 17. Fluorescence in situ hybridization - Latest research and news [https://www.nature.com/subjects/fluorescence-in-situ-hybridization]
- 18. Selected In Situ Hybridization Methods: Principles and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8270300/]
- 19. TrueProbes: Quantitative Single-Molecule RNA-FISH ... [https://elifesciences.org/reviewed-preprints/108599]
- 20. Analytical techniques for nucleic acid and protein detection ... [https://www.nature.com/articles/s12276-025-01453-w]
- 21. Robust and sensitive in situ RNA detection using Yn-situ [https://pmc.ncbi.nlm.nih.gov/articles/PMC9046451/]
- 22. U-FISH: a fluorescent spot detector for imaging-based spatial ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03736-x]
- 23. Automated Quantitative RNA in Situ Hybridization for ... [https://www.sciencedirect.com/science/article/pii/S1525157812003157]
- 24. QuantISH: RNA in situ hybridization image analysis ... [https://www.nature.com/articles/s41374-022-00743-5]
- 25. In Situ Hybridization Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/gene-expression-analysis-genotyping-support-center/in-situ-hybridization-support/ish-support-troubleshooting.html]
- 26. RNAscope: A Novel in Situ RNA Analysis Platform for ... [https://www.sciencedirect.com/science/article/pii/S1525157811002571]
- 27. RNAscope, In Situ Hybridization Technique | ACD [https://acdbio.com/referenceguide]
- 28. Evaluation of the Suitability of RNAscope as a Technique ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8710359/]
- 29. RNAscope, BaseScope and miRNAscope Assays [https://acdbio.com/rnascope-basescope-and-mirnascope-assays]
- 30. RNAscope Assay Visualization and Interpretation [https://acdbio.com/support-solution-category/rnascope-assay-visualization-and-interpretation]
- 31. Application of microRNA In Situ Hybridization on Long-term ... [https://link.springer.com/article/10.1007/s12035-025-05077-z]
- 32. RNAscope® technology | In Situ Hybridization, RNA-ISH [https://acdbio.com/dataanalysisguide]
- 33. Optimizing Xenium In Situ data utility by quality assessment ... [https://www.nature.com/articles/s41592-025-02617-2]
- 34. A Guide for RNAscope™ Data Analysis [https://www.bio-techne.com/reagents/rnascope-ish-technology/rnascope/guide-rnascope-data-analysis]
- 35. Top In Situ Hybridization Companies & How to Compare ... [https://www.linkedin.com/pulse/top-situ-hybridization-companies-how-compare-them-2025-cathe/]
- 36. Next-generation hybridization chain reaction tools with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12440025/]
- 37. Modified in situ Hybridization Chain Reaction Using Short ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2020.00075/full]
- 38. One probe fits all: a highly customizable modular RNA in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12148028/]
- 39. SABER-FISH – Signal amplification for multiplexed ... [https://www.protocols.io/view/saber-fish-signal-amplification-for-multiplexed-fl-dzp975r6.html]
- 40. NuclampFISH enables cell sorting based on nuclear RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11818-0]
- 41. Multiplexed RNA imaging and in situ profiling in living cells [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06220a?page=search]
- 42. Mapping human tissues with highly multiplexed RNA in situ ... [https://www.nature.com/articles/s41467-024-46437-y]
- 43. Mapping Human Tissues with Highly Multiplexed RNA in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10462101/]
- 44. Robust and sensitive in situ RNA detection using Yn-situ [https://www.sciencedirect.com/science/article/pii/S2667237522000595]
- 45. ISH Protocol [https://www.bostonbioproducts.com/resources/ish-protocol/]
- 46. In Situ Hybridization: Tips, Techniques, and Expert Insights [https://www.the-scientist.com/tips-and-tricks-for-in-situ-hybridization-71928]
- 47. A novel approach for microRNA in situ hybridization using ... [https://www.nature.com/articles/s41598-021-83888-5]
- 48. ISH: in situ hybridization protocol [https://www.abcam.com/en-us/technical-resources/protocols/in-situ-hybridization]
- 49. A Sensitive Alternative for MicroRNA In Situ Hybridizations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3201160/]
- 50. How do I reduce high background in my FISH assay? [https://www.ogt.com/us/resources/fish-resources-and-support/fish-resources/optimize-your-fish-assay-simple-fixes-for-reducing-high-background-signal/]
- 51. FISH Tips and Troubleshooting [https://www.creative-bioarray.com/support/fish-tips-and-troubleshooting.htm]
- 52. High-sensitivity BK virus detection system using viewRNA ... [https://www.sciencedirect.com/science/article/abs/pii/S0732889325001130]
- 53. Reflex fluorescent in situ hybridization testing for ... [https://pubmed.ncbi.nlm.nih.gov/21415758/]
- 54. Diagnostic Accuracy Performance of Fluorescence In Situ ... [https://www.mdpi.com/2077-0383/13/21/6457]
- 55. A technical review and guide to RNA fluorescence in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7085896/]
- 56. Protocol optimization improves the performance of multiplexed ... [https://pubmed.ncbi.nlm.nih.gov/40887478/]
- 57. Chemical tools for discriminating single nucleotide variants [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d5cs01006c?page=search]
- 58. A technical review and guide to RNA fluorescence in situ ... [https://peerj.com/articles/8806/]
- 59. The impact of quantitative optimization of hybridization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3065421/]
- 60. A review of reaction enhancement strategies for isothermal ... [https://www.sciencedirect.com/science/article/pii/S2666053921000096]
- 61. Optimizing your in situ hybridization for success [https://cellcarta.com/science-hub/optimizing-your-in-situ-hybridization-for-success/]
- 62. article A Simple Method for Combined Fluorescence In Situ ... [https://www.sciencedirect.com/science/article/pii/S1016847823107588]
- 63. Ten Approaches That Improve Immunostaining: A Review of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8836139/]
- 64. Combined In Situ Hybridization and Immunohistochemistry ... [https://www.mdpi.com/2072-6694/13/6/1307]
- 65. Systematic review and meta-analysis of artificial ... [https://www.nature.com/articles/s41746-025-01483-8]
- 66. Comparative analysis of multiplexed in situ gene ... [https://elifesciences.org/reviewed-preprints/96949]
- 67. Quantitative methods for genome-scale analysis of in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2395252/]
- 68. Quantitative assessment of single-cell RNA-sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4022966/]
- 69. Single-cell RNA sequencing reveals a heterogeneous ... [https://www.nature.com/articles/s42003-020-0837-0]
- 70. New standards in HER2-low testing: the CASI-01 comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571579/]
- 71. International Expert Consensus Recommendations for ... [https://www.sciencedirect.com/science/article/pii/S0893395225002236]
- 72. Comparison of the IHC, FISH, SISH and qPCR methods ... [https://www.spandidos-publications.com/10.3892/mmr.2012.919]
- 73. Concordance of HER2 Immunohistochemistry and ... [https://ar.iiarjournals.org/content/37/6/3323]
- 74. Review of In Situ Hybridization (ISH) Stain Images Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11430898/]
- 75. Robust and accurate digital measurement for HER2 ... [https://www.nature.com/articles/s41598-017-07176-x]
- 76. HER2-low breast cancer in routine practice: a nationwide ... [https://link.springer.com/article/10.1007/s00428-025-04266-4]
- 77. Comparison of Immunohistochemical Methods (IHC) and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9985809/]
- 78. In Situ Hybridization Market: Growth, Size, Share, and Trends [https://www.marketsandmarkets.com/Market-Reports/in-situ-hybridization-market-212028829.html]
- 79. For data reproducibility: automation of ELISA test kit ... [https://www.tecan.com/blog/data-reproducibility-automation-of-elisa-test-kit-protocols]
- 80. Reproducibility analysis of automated deep learning based ... [https://www.nature.com/articles/s41598-023-40516-8]