Optimizing Blocking Solutions for FISH: A Comprehensive Guide to Enhance Signal and Reduce Background
This article provides a systematic guide for researchers and scientists on optimizing blocking solutions in Fluorescence in Situ Hybridization (FISH), with a specific focus on challenging Formalin-Fixed Paraffin-Embedded (FFPE) tissues.
Optimizing Blocking Solutions for FISH: A Comprehensive Guide to Enhance Signal and Reduce Background
Abstract
This article provides a systematic guide for researchers and scientists on optimizing blocking solutions in Fluorescence in Situ Hybridization (FISH), with a specific focus on challenging Formalin-Fixed Paraffin-Embedded (FFPE) tissues. It covers the foundational role of blocking in preventing non-specific probe binding, details practical methodological protocols, offers extensive troubleshooting for common issues like high background and weak signals, and outlines rigorous validation strategies. By integrating established practices with emerging optimization techniques, this guide aims to enhance the reliability, reproducibility, and accuracy of FISH assays in both research and clinical diagnostics.
The Critical Role of Blocking Solutions in FISH Assay Success
Fluorescence in situ hybridization (FISH) has revolutionized molecular cytogenetics, enabling precise localization of specific DNA and RNA sequences within cells and tissues. However, a significant challenge that frequently compromises data quality and reliability is non-specific binding, which manifests as high background fluorescence. This obscures critical data, complicates interpretation, and may lead to erroneous conclusions [1]. This technical guide addresses the root causes of non-specific binding and provides actionable solutions, with a particular focus on the critical role of blocking solution optimization and rigorous protocol refinement to suppress off-target probe interactions.
Frequently Asked Questions (FAQs) on Non-Specific Binding
1. What are the primary causes of high background in FISH assays? High background, or non-specific signal, arises from multiple factors. These include inadequate wash stringency, which fails to remove loosely bound probes; over- or under-fixation of samples, which can either mask targets or fail to preserve cellular structure; insufficient pre-treatment, leaving cellular debris that causes autofluorescence; non-optimized denaturation conditions (temperature and time); and the use of degraded or contaminated buffers. Even worn-out microscope filters can contribute to signal noise [2] [3] [4].
2. How can I optimize my blocking strategy to reduce non-specific binding? While blocking agents are a cornerstone for reducing background, their optimization is sample-specific. For probes containing repetitive sequences (like Alu or LINE elements), adding COT-1 DNA to the hybridization mixture is essential to block probe binding to these non-target sites [4]. Furthermore, ensuring proper pre-treatment with enzymes like pepsin or proteinase K is a form of indirect blocking, as it removes proteins that may non-specifically trap probes. The digestion must be carefully optimized, as both under- and over-digestion can increase background [4] [1].
3. My signals are weak and my background is high. What should I check first? Begin by verifying your denaturation conditions. Use a calibrated hotplate to ensure the denaturation temperature is exactly 75°C for 2 minutes, as recommended for many protocols [3]. Sub-optimal denaturation leads to poor probe access and weak true signals, while excessive heat can increase non-specific binding. Next, check the freshness and pH of your wash buffers (SSC solutions) and ensure the stringent wash is performed at the correct temperature (75-80°C) [3] [4].
4. How does sample fixation contribute to non-specific binding? Fixation is a critical balancing act. Under-fixation results in incomplete preservation of cellular structure, leading to DNA degradation and increased non-specific probe binding. Over-fixation, particularly with formalin, causes excessive protein-nucleic acid cross-linking, which can mask target sequences and paradoxically increase background by forcing probes to bind to non-target sites. Always use freshly prepared fixative and adhere strictly to recommended fixation times [1].
Troubleshooting Guide: Common Issues and Solutions
The table below summarizes common problems related to non-specific binding and their targeted solutions.
Table 1: Troubleshooting Guide for Non-Specific Binding in FISH
| Problem & Symptoms | Primary Causes | Recommended Solutions |
|---|---|---|
| High Background Levels [2] [1] | Inadequate stringent washes; Over-/under-fixation; Insufficient pre-treatment; Old buffers. | Optimize wash stringency (temperature, pH, time); Use fresh wash buffers; Standardize fixation protocol; Optimize enzymatic pre-treatment. |
| Weak/Faded Signal with High Background [2] [3] | Poor denaturation; Worn microscope filters; Over-fixation. | Calibrate denaturation equipment (75°C for 2 mins); Replace optical filters per manufacturer's guidelines (typically every 2-4 years). |
| Autofluorescence & Non-Specific Probe Binding [3] [1] | Cellular debris; Probe exposure to light; Inappropriate slide type. | Minimize light exposure to probes and slides; Aliquot probes for single use; Use non-adhesive/charge-neutral slides for cell samples. |
| Uneven or Patchy Signal [2] | Uneven probe distribution; Air bubbles during mounting; Inconsistent permeabilization. | Apply probes carefully to avoid squeezing from under coverslip; Use a template for consistent probe application; Ensure even pre-treatment. |
Experimental Protocols for Key Optimization Steps
Protocol 1: Optimized Pre-treatment and Hybridization for FFPE Tissues
This protocol is designed to maximize target accessibility while minimizing background in challenging formalin-fixed paraffin-embedded (FFPE) samples [4] [1].
- Deparaffinization and Rehydration: Follow standard protocols using xylene and ethanol series.
- Heat-Induced Epitope Retrieval: Immerse slides in pre-heated Tissue Pretreatment Solution (e.g., Citrate buffer). Maintain at 98–100°C for 30 minutes in a water bath. The duration may require extension based on tissue type and fixation.
- Enzymatic Digestion: Cool slides and treat with a pre-warmed enzyme solution (e.g., Pepsin at 37°C for 3-10 minutes). Microscopic monitoring is crucial to prevent under- or over-digestion.
- Denaturation: Perform on a calibrated hotplate. Denature slides with applied probe at 75°C for 2-5 minutes. Ensure slides are cover-slipped and in a humidified environment to prevent drying.
- Hybridization: Hybridize overnight (16 hours) at 37°C in a sealed, humidified chamber.
Protocol 2: High-Stringency Post-Hybridization Washes
This protocol is critical for removing non-specifically bound probes without disrupting true hybrids [4].
- Remove Coverslips: Soak slides in the appropriate buffer (e.g., PBST or SSC) to gently remove coverslips.
- Initial Rinse: Rinse slides briefly at room temperature with SSC buffer.
- Stringent Wash: Immerse slides in a coplin jar containing SSC buffer (concentration as per probe protocol). Place the jar in a water bath set at 75°C for 5 minutes. Increase the temperature by 1°C per slide if processing more than two, but do not exceed 80°C.
- Final Rinses: Rinse slides with TBST or a similar buffer to prepare for counterstaining. Avoid using water or PBS without detergent at this stage.
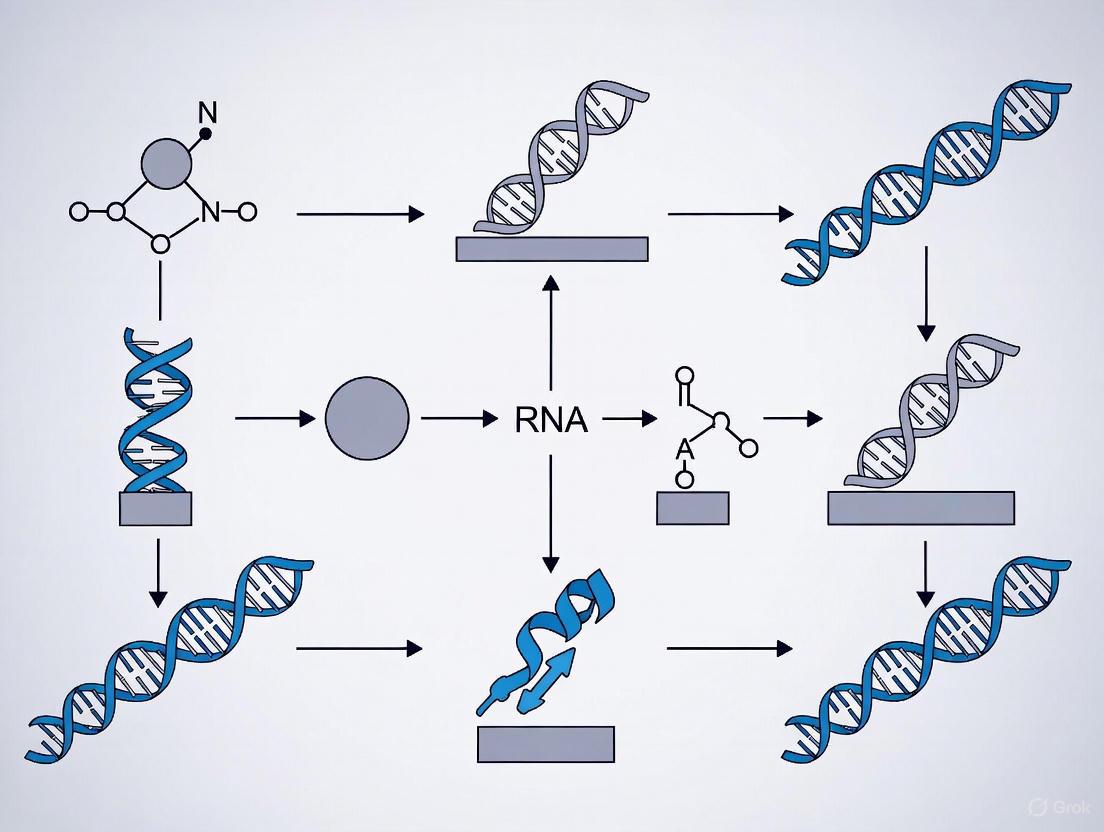
The following diagram illustrates the logical workflow for diagnosing and addressing the root causes of non-specific binding in a FISH experiment.
Research Reagent Solutions
The table below lists key reagents and materials essential for minimizing non-specific binding and achieving high-quality FISH results.
Table 2: Essential Reagents for Mitigating Non-Specific Binding in FISH
| Reagent/Material | Function & Role in Reducing Background |
|---|---|
| COT-1 DNA [4] | Blocks repetitive genomic sequences (e.g., Alu, LINE) to prevent non-specific binding of probes to these regions. |
| Formamide [2] [5] | A chemical denaturant used in hybridization buffers. Its concentration is key to controlling stringency and specificity. |
| Proteinase K / Pepsin [2] [3] [4] | Enzymes that digest proteins, removing cellular debris that causes autofluorescence and unmasking target nucleic acids. |
| SSC Buffer (Saline-Sodium Citrate) [3] [4] | The primary buffer for post-hybridization washes. Its concentration, temperature, and pH are critical for stringency. |
| Blocking Agents | Various proprietary formulations are included in commercial FISH kits to competitively inhibit non-probe binding to cellular components. |
| Methanol/Acetic Acid Fixative [3] | A freshly prepared Carnoy's solution (3:1 methanol:glacial acetic acid) is preferred for metaphase spreads to preserve morphology. |
| Hypotonic Solution (e.g., KCl) [3] [1] | Used during cell harvest to swell cells, improving chromosome spreading and reducing cytoplasmic background. |
Core Components of an Effective Blocking Solution
Frequently Asked Questions (FAQs)
1. What is the primary purpose of a blocking solution in FISH assays? The primary purpose is to reduce non-specific binding, which improves the signal-to-noise ratio by blocking non-target interactions, such as those with Fc receptors or other off-target binders. This enhances the specificity and sensitivity of the assay, allowing for more accurate detection of the authentic signal [6].
2. What are the consequences of inadequate blocking in FFPE-FISH? Inadequate blocking can lead to high background noise, non-specific staining, and reduced hybridization efficiency. This compromises the accuracy of the results and can lead to false positives or false negatives, making data interpretation difficult [7].
3. Which components are critical for an effective blocking solution? Critical components include normal sera from the host species of your primary antibodies (e.g., mouse, rat), tandem dye stabilizers to prevent fluorophore degradation, and, for some protocols, additives like sodium azide as a preservative [6].
4. How do I select the appropriate serum for my blocking solution? You should select normal sera from the same host species as the primary antibodies used in your panel. For example, if you are using primarily rat-derived antibodies, you should use rat serum. Avoid using serum from the same species as the cells being stained if you are detecting immunoglobulins [6].
5. What are the main technical challenges for FISH in FFPE tissues? Key challenges include issues related to sample fixation (such as inadequate fixation), contamination, the age of the tissue blocks and slides, inadequate pretreatment, and the FISH technique itself. These can all negatively impact signal quality [7].
Troubleshooting Common Blocking Issues
| Issue | Potential Cause | Recommended Solution |
|---|---|---|
| High Background Noise | Inadequate blocking of Fc receptors or other non-specific interactions. | Optimize blocking solution with appropriate normal sera; ensure complete coverage of all potential non-specific binding sites [6] [8]. |
| Poor Hybridization Efficiency | Inadequate pretreatment or sample over-fixation leading to masked targets. | Implement optimized pretreatment protocols; monitor and control fixation time carefully [7]. |
| Weak or Absent Signal | Over-blocking, which may prevent antibody access to the target epitope. | Titrate blocking reagents to find the optimal concentration that reduces noise without compromising the specific signal [6]. |
| Inconsistent Results Between Runs | Variability in blocking solution preparation or incubation conditions. | Standardize the blocking protocol, including reagent concentrations, incubation time (e.g., 15 min at room temperature), and temperature [6]. |
| Signal Degradation Over Time | Breakdown of tandem fluorophores; insufficient stabilizer in solution. | Incorporate a tandem stabilizer (at a 1:1000 dilution) in the blocking and resuspension buffers to preserve dye integrity [6]. |
Quantitative Effects of Blocking on Assay Performance
The following table summarizes key quantitative data related to blocking optimization.
| Parameter | Without Optimized Blocking | With Optimized Blocking | Notes & Context |
|---|---|---|---|
| Non-specific Binding | High (Baseline) | Reduced, as shown by lower fluorescence in blocked samples [8] | Demonstrated in flow cytometry using Fc receptor blocking. |
| Assay Sensitivity | Limited | Enhanced signal-to-noise ratio [6] | Blocking improves detection of authentic signals above background noise. |
| Data Accuracy | Prone to false positives/negatives [7] | Improved specificity and reliable results [7] | Critical for clinical diagnosis and research using FFPE-FISH. |
| Signal Preservation | Tandem dye breakdown possible | Dye integrity maintained with stabilizer [6] | Tandem stabilizer used at 1:1000 dilution. |
Detailed Experimental Protocol for Blocking Solution Preparation
This protocol provides an optimized, general-use approach for preparing a blocking solution for high-parameter assays, adapted from best practices in flow cytometry which are applicable to FISH methodology [6].
Materials
- Mouse serum (e.g., Thermo Fisher, cat. no. 10410)
- Rat serum (e.g., Thermo Fisher, cat. no. 10710C)
- Tandem stabilizer (e.g., BioLegend, cat. no. 421802)
- Sodium azide (10%) [Precaution: Highly toxic; handle with appropriate care.]
- FACS buffer or appropriate molecular biology buffer
Methodology
- Prepare the Blocking Solution: Combine the following reagents to create a 1 mL mixture [6]:
- Mouse serum: 300 µl
- Rat serum: 300 µl
- Tandem stabilizer: 1 µl
- Sodium azide (10%): 10 µl (Can be omitted for short-term use)
- FACS buffer: 389 µl (Add to reach the final volume)
- Application: Dispense cells or tissue sections into your staining vessel (e.g., a 96-well V-bottom plate for cells).
- Centrifuge: For cells, centrifuge for 5 minutes at 300 × g and carefully remove the supernatant.
- Blocking Step: Resuspend the pellet or cover the tissue section with the prepared blocking solution (e.g., 20 µl for a cell pellet in a well).
- Incubation: Incubate for 15 minutes at room temperature, protected from light.
- Proceed with Staining: After incubation, proceed directly with your primary antibody or probe staining protocol without a wash step.
Research Reagent Solutions
Essential materials and their functions for effective blocking in FISH and related assays.
| Reagent | Function | Example & Context |
|---|---|---|
| Normal Sera | Blocks non-specific binding to Fc receptors and other cellular structures by providing excess inert immunoglobulins. | Mouse and rat serum; chosen to match the host species of the primary antibodies [6]. |
| Fc Receptor Blocking Solution | Specifically blocks Fc receptors on immune cells to prevent antibody binding independent of variable domain specificity, reducing false positives [8]. | Human Fc Receptor Blocking Solution; critical for live cell assays on human immune cells [8]. |
| Tandem Stabilizer | Prevents the breakdown of tandem fluorophores, which can lead to erroneous signals and misassignment of fluorescence [6]. | Added to blocking and resuspension buffers at a 1:1000 dilution to preserve signal integrity [6]. |
| BSA or Protein Albumin | A common protein used in blocking buffers to coat non-specific binding sites on tissues and cells. | Often used at 1-5% concentration in various blocking buffer recipes. |
| Sodium Azide | A preservative that inhibits microbial growth in reagent stocks and buffers for long-term storage [6]. | Used at a 0.09-0.1% final concentration; handle with extreme care due to high toxicity [6]. |
Workflow for Blocking Solution Optimization
This diagram outlines the key decision points and steps in developing and troubleshooting an effective blocking strategy.
Blocking Solution Efficacy Decision Pathway
This flowchart helps troubleshoot results after the initial blocking step to guide optimization efforts.
The choice of sample type—Formalin-Fixed Paraffin-Embedded (FFPE) tissues or single-cell suspensions—is a critical first step in Fluorescence In Situ Hybridization (FISH) that directly influences experimental design, protocol optimization, and the reliability of your results. Each sample type presents unique advantages and technical challenges, particularly concerning sample preservation, macromolecule accessibility, and the optimization of blocking solutions to reduce background noise. Understanding these differences is essential for designing robust and reproducible FISH experiments within a research thesis focused on blocking solution optimization.
Frequently Asked Questions
Q1: What is the single most critical step in preparing FFPE tissues for FISH? A: The most critical step is achieving a balance during tissue pretreatment, which includes deparaffinization, antigen retrieval, and permeabilization. Insufficient pretreatment leads to weak or absent probe signals due to poor probe penetration, while over-treatment causes tissue fragmentation and loss of morphology [9]. The optimal pretreatment conditions, especially enzyme digestion time, must be empirically determined for each tissue type (e.g., 10-40 minutes for breast tissue, 15-20 minutes for lung) [9].
Q2: Why does the fixation protocol differ between cell suspensions and FFPE tissues? A: Cell suspensions and tissue blocks have different physical properties and integrity requirements.
- Cell Suspensions: Are typically fixed with a low concentration of paraformaldehyde (e.g., 3%) for a short duration (e.g., 1 hour). This is sufficient to preserve cell structure without excessively cross-linking proteins or compromising DNA accessibility for FISH probes [10].
- FFPE Tissues: Require more robust fixation and processing (including embedding in paraffin wax) to maintain complex tissue architecture over long-term storage. The extensive cross-linking from formalin fixation necessitates the rigorous pretreatment steps mentioned above.
Q3: My FFPE FISH results show high background autofluorescence. What could be the cause? A: High background in FFPE samples can stem from several factors related to suboptimal blocking or pretreatment:
- Incomplete deparaffinization: Residual paraffin can bind nonspecifically to fluorescent dyes [9].
- Over-fixation: Samples fixed in formalin for too long can exhibit increased autofluorescence [7].
- Insufficient blocking: A blocking solution that is not optimized for the specific endogenous components of the FFPE tissue may fail to quench autofluorescence.
- Old or suboptimal reagents: Ensure pretreatment solutions and enzymes are stored correctly and refreshed regularly [9].
Q4: How does sample age affect FISH quality? A: Sample age impacts both sample types differently.
- Cell Suspensions: Fixed cells stored in phosphate-buffered saline (PBS) show a significant deterioration in FISH signal quality after 10 days of storage [10].
- FFPE Tissue Blocks: While paraffin embedding allows for room-temperature storage for decades, the age of the cut slides is critical. Aged slides can lead to decreased RNA integrity and increased background, affecting the performance of even the latest commercial spatial transcriptomics platforms [11] [7].
Troubleshooting Guides
Common Issues with Cell Suspensions
| Issue | Possible Cause | Solution |
|---|---|---|
| Poor or No Signal | Over-fixation with PFA compromising DNA accessibility [10]. | Optimize fixation: use 3% PFA for 1 hour [10]. |
| Inadequate permeabilization. | Optimize permeabilization conditions (concentration, time, temperature) using agents like Triton X-100 [2]. | |
| Morphological Distortion | Over-fixation or over-permeabilization [2]. | Standardize fixation and permeabilization times; use gentler methods for cell dissociation [2]. |
| Weak/Faded Signal | Fluorophore degradation or sample over-exposure to light [2]. | Use fresh, sensitive fluorophores; minimize light exposure during imaging; include antifade reagents in mounting medium [2]. |
Common Issues with FFPE Tissues
| Issue | Possible Cause | Solution |
|---|---|---|
| Tissue Fragmentation | Over-digestion during enzyme pretreatment [9]. | Decrease enzyme digestion time. After digestion, check morphology with DAPI; over-digested cells should be <15% [9]. |
| Weak Probe Signal | Insufficient digestion or denaturation [9]. | Increase enzyme digestion time; ensure denaturation temperature is correctly calibrated (75°C for 5 mins, up to 85°C for difficult specimens) [9]. |
| High Background / Autofluorescence | Incomplete deparaffinization or over-fixation [7] [9]. | Ensure complete paraffin clearing with extended xylene washes; use optimized blocking solutions; refresh pretreatment solutions regularly [9]. |
| Uneven or Patchy Signal | Non-uniform application of probes or uneven pretreatment [2]. | Check for uniform distribution of probes; avoid air bubbles during mounting [2]. |
Comparative Experimental Data
Table 1: Performance of Commercial iST Platforms on FFPE Tissues
A systematic benchmark of imaging-based Spatial Transcriptomics (iST) platforms on FFPE tissues revealed key performance differences [11].
| Platform | Signal Amplification Method | Relative Transcript Counts (on matched genes) | Concordance with scRNA-seq | Spatially Resolved Cell Typing |
|---|---|---|---|---|
| 10X Xenium | Padlock probes with rolling circle amplification | Consistently higher | Yes | Slightly more clusters found, with varying false discovery rates [11]. |
| Nanostring CosMx | Branch chain hybridization | High, in concordance with scRNA-seq | Yes | Slightly more clusters found, with varying false discovery rates [11]. |
| Vizgen MERSCOPE | Direct hybridization with transcript tiling | Lower than Xenium and CosMx | Information Missing | Fewer clusters found [11]. |
Table 2: Impact of Sample Type on FISH Experimental Parameters
| Parameter | Cell Suspensions | FFPE Tissues |
|---|---|---|
| Optimal Fixation | 3% PFA for 1 hour [10]. | Standard formalin fixation followed by paraffin embedding [11] [7]. |
| Key Challenge | Maintaining cell integrity and DNA accessibility after fixation [10]. | Breaking protein cross-links and retrieving antigens without destroying tissue morphology [7] [9]. |
| Storage Stability | Deterioration after 10 days in PBS [10]. | Years to decades at room temperature as blocks, but cut slides degrade faster [11] [7]. |
| Primary Signal Issue | Weak or no signal from over-fixation [2] [10]. | High background autofluorescence and weak signal from incomplete pretreatment [9]. |
Detailed Experimental Protocols
Protocol 1: Optimized FISH for Cell Suspensions
Methodology:
- Fixation: Prepare healthy, actively growing cells. Fix cells using a 3% paraformaldehyde (PFA) solution for 1 hour at room temperature [10].
- Permeabilization: Permeabilize the fixed cells using a buffer containing 0.5% Triton X-100 for 10 minutes on ice to allow probe access [2].
- Denaturation: Denature target nucleic acids using heat or alkaline treatment to make them single-stranded. Optimize conditions to maintain sample integrity [2].
- Hybridization: Incubate with labeled FISH probes in a humidified chamber at the appropriate hybridization temperature (e.g., 37°C) overnight.
- Stringent Washes: Perform post-hybridization washes with saline-sodium citrate (SSC) buffer at a defined stringency to remove unbound probes [2].
- Counterstaining and Mounting: Counterstain nuclei with DAPI (0.5 µg/mL) for 10 minutes, mount with an antifade mounting medium, and image [2].
Protocol 2: Optimized FISH for FFPE Tissue Sections
Methodology:
- Sectioning: Cut 4-6 µm thick sections. Place the tissue block on ice for 20 minutes before sectioning for smoother cutting. Mount on positively charged slides and dry in an oven at 50°C overnight or 60°C for 1 hour to adhere [9].
- Deparaffinization and Hydration: Clear paraffin by immersing slides in xylene (3 changes, 5 minutes each), followed by rehydration through a graded ethanol series (100%, 95%, 70%) to water [9].
- Antigen Retrieval and Pretreatment: Immerse slides in pre-treatment solution (e.g., citrate buffer, pH 6.0) and heat in a 98-100°C water bath for 30 minutes [9]. Cool slides to room temperature.
- Enzyme Digestion: Treat sections with a pre-warmed enzyme (e.g., pepsin) at 37°C. The duration must be optimized for tissue type (e.g., 10-40 min for breast, 15-20 min for lung) [9].
- Denaturation and Hybridization: Co-denature sample and probe at 75°C for 5 minutes using a calibrated hotplate or hybridizer, then incubate at 37°C in a humid chamber overnight [9].
- Post-Hybridization Washes and Counterstaining: Perform stringent washes. Counterstain with DAPI, mount with antifade medium, and image.
Signaling Pathways and Workflows
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in FISH |
|---|---|
| Paraformaldehyde (PFA) | A cross-linking fixative that preserves cellular and tissue morphology by creating covalent bonds between proteins, maintaining structural integrity for probing [2] [10]. |
| Permeabilization Agents (Triton X-100) | A detergent that dissolves lipids in cell and nuclear membranes, creating pores that allow FISH probes to access the interior of the cell and hybridize to target nucleic acids [2]. |
| Proteolytic Enzymes (e.g., Pepsin) | Critical for FFPE tissue pretreatment. These enzymes digest proteins cross-linked by formalin fixation, thereby uncovering target sequences and enabling probe penetration [9]. |
| Blocking Solution | A key reagent for optimization. Typically contains proteins (e.g., BSA) and other agents that adsorb to nonspecific binding sites on the sample, reducing background signal and improving the signal-to-noise ratio. |
| Formamide | A denaturing agent included in hybridization buffers. It lowers the melting temperature of double-stranded nucleic acids, allowing hybridization to occur at lower, more manageable temperatures that preserve tissue morphology. |
| Saline-Sodium Citrate (SSC) Buffer | A key component in post-hybridization washes. The salt concentration and temperature determine the stringency, washing away imperfectly matched or unbound probes to ensure signal specificity [2]. |
FAQs: Core Concepts and Problem Solving
How does fixation impact the effectiveness of my blocking step? Fixation preserves tissue architecture but can mask epitopes or introduce autofluorescence, which blocking must subsequently overcome. Inconsistent fixation directly undermines blocking efficacy. Over-fixation, in particular, reduces target accessibility, making it difficult for blocking reagents to prevent non-specific probe binding, leading to high background [7] [12].
Why is permeabilization critical even when using a blocking solution? Blocking reagents reduce non-specific binding to cellular components, but they cannot access targets sealed within membranes. Permeabilization physically disrupts these membranes, allowing blocking agents and probes to reach their intracellular targets. Without adequate permeabilization, your blocking step will be ineffective for intracellular targets, resulting in poor or no signal [2].
My blocking solution doesn't seem to be reducing background. What pretreatment issues should I investigate? The problem likely stems from your pretreatment workflow. Key issues to check include:
- Incomplete paraffin clearing: Excess paraffin preferentially binds to certain dyes (e.g., FITC) and creates a physical barrier, preventing proper blocking and probe penetration [12].
- Inadequate or excessive enzyme digestion: Under-digestion leaves proteins that cause non-specific binding, while over-digestion destroys tissue morphology and creates "ghost" nuclei, both of which blocking cannot fix [12].
- Sample age: Older FFPE blocks can have increased autofluorescence, which a standard blocking step may not be sufficient to quench [7].
Troubleshooting Guide: Common Issues and Solutions
The following table outlines common problems, their potential causes related to the interaction between pretreatment and blocking, and targeted solutions.
| Issue Observed | Potential Root Cause (Linked to Pretreatment) | Recommended Solution |
|---|---|---|
| High background or non-specific signal [2] [12] | - Incomplete paraffin clearing.- Under-digestion during enzymatic pretreatment.- Over-fixation masking epitopes. | - Ensure complete paraffin removal with fresh xylene washes [12].- Optimize enzyme digestion time and temperature; validate by checking DAPI staining post-digestion [12]. |
| Weak or absent FISH signal [2] [12] | - Over-fixation or over-permeabilization, damaging target.- Inadequate permeabilization, blocking probe access.- Incorrect denaturation temperature. | - Optimize fixation time and permeabilization agent concentration [2].- Calibrate denaturation equipment; increase temperature up to 85°C if needed [12]. |
| Poor tissue morphology or cell damage [2] [13] | - Over-permeabilization, destroying cellular structure.- Over-digestion with enzyme. | - Titrate permeabilization agents (e.g., Triton X-100) and use gentler methods [2].- Reduce enzyme digestion time to prevent tissue loss [12]. |
| Uneven or patchy hybridization [2] | - Non-uniform permeabilization across sample.- Air bubbles during probe application. | - Ensure even application of permeabilization reagents and avoid sample drying [2].- Use a template for consistent probe application [2]. |
Optimized Experimental Protocols
Protocol 1: Integrated Pretreatment and Blocking for FFPE FISH
This protocol is adapted from standard cytogenetic practices for challenging FFPE samples [12].
Materials:
- Reagents: Xylene (or substitute), Ethanol series (100%, 95%, 70%), Pretreatment Solution (e.g., Citrate-based), Proteolytic Enzyme (e.g., Pepsin), Blocking Solution (e.g., containing serum), FISH Probes, DAPI.
- Equipment: Ceramic jars, Heated water bath (98-100°C), Hotplate or hybridizer, Humidified chamber, Fluorescence microscope.
Methodology:
- Dewaxing: Devax slides in fresh xylene (2 changes, 10 minutes each) to ensure complete paraffin removal. Hydrate through a graded ethanol series (100%, 95%, 70%) and rinse in distilled water [12].
- Pretreatment: Immerse slides in pre-warmed pretreatment solution (98-100°C) for 30 minutes in ceramic jars to maintain temperature. Rinse in distilled water [12].
- Enzymatic Digestion: Treat slides with a pre-optimized concentration of proteolytic enzyme (e.g., Pepsin) at 37°C. Critical Step: Time must be validated per tissue type; typically 5-20 minutes. Stop reaction in water. Check digestion quality by staining a test slide with DAPI; over-digested cells should be <15% [12].
- Denaturation: Co-denature slides and probe at 75°C for 5 minutes on a calibrated hotplate. For difficult specimens, temperature may be increased to 85°C [12].
- Hybridization: Hybridize in a humidified chamber at 37°C for 4-16 hours. Ensure humidity strips are pre-soaked to prevent signal drop-out [12].
- Post-Hybridization Washes & Counterstaining: Perform stringent washes per probe manufacturer's instructions. Counterstain with DAPI and mount with an anti-fade reagent [2].
Protocol 2: Blocking for High-Parameter Assays (Flow Cytometry Context)
This protocol highlights blocking principles that are analogous to FISH, focusing on reducing non-specific interactions [6] [14].
Materials:
- Blocking Solution: Normal sera (e.g., Rat serum, Mouse serum), Tandem stabilizer, Sodium azide (optional), FACS Buffer.
- Staining Buffer: Brilliant Stain Buffer (for polymer dye panels), Tandem stabilizer, FACS Buffer.
Methodology:
- Prepare Blocking Solution: Create a solution containing 30% (v/v) mouse serum, 30% (v/v) rat serum, and tandem stabilizer at a 1:1000 dilution in an appropriate buffer (e.g., FACS buffer) [6].
- Apply Blocking: Resuspend prepared and permeabilized (if needed) cells in the blocking solution. Incubate for 15 minutes at room temperature in the dark [6].
- Staining: Without washing, add the staining master mix (containing antibodies and Brilliant Stain Buffer) directly to the blocked cells. Incubate for 1 hour at room temperature [6].
- Wash and Analyze: Wash cells twice with buffer, resuspend in buffer with tandem stabilizer, and acquire data [6].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents used in the featured protocols and their specific functions in the context of pretreatment and blocking.
| Research Reagent | Function in Pretreatment & Blocking |
|---|---|
| Normal Sera (e.g., Rat, Mouse) | Contains antibodies and other proteins that bind to non-specific sites (e.g., Fc receptors), preventing off-target binding of your primary detection reagents [6]. |
| Proteolytic Enzyme (e.g., Pepsin) | Digests proteins that cross-link during fixation, thereby unmasking target epitopes and allowing probe access. Requires precise optimization [12]. |
| Tandem Stabilizer | Prevents the degradation of tandem fluorophores, which can cause erroneous signal spillover and increased background, thereby preserving signal-to-noise ratio [6]. |
| Brilliant Stain Buffer | Contains agents that disrupt dye-dye interactions between polymer-based fluorophores, reducing non-specific background signal in highly multiplexed panels [6]. |
| Triton X-100 / Tween-20 | Detergents used for permeabilization. They create pores in lipid membranes, enabling blocking reagents and probes to access intracellular targets [2]. |
Workflow Visualization
The diagram below illustrates the logical workflow for integrating pretreatment with blocking, highlighting critical decision points to ensure optimal outcomes.
Integrated Pretreatment and Blocking Workflow
The diagram below outlines a troubleshooting decision tree to systematically address high background, a common issue often stemming from the interaction of pretreatment and blocking.
Troubleshooting High Background
Protocol Development: Implementing Effective Blocking Strategies
Step-by-Step Guide to Blocking Solution Preparation and Application
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents used in the preparation and application of blocking solutions for FISH (Fluorescence In Situ Hybridization) experiments.
Table: Essential Reagents for FISH Blocking Solutions
| Reagent | Function & Explanation |
|---|---|
| BSA (Bovine Serum Albumin) [15] | A primary blocking agent used at 1-3% concentration to coat the slide and minimize non-specific binding of detection antibodies. |
| Fish Gelatin [16] | A blocking agent purified from cold-water fish skin. It contains no IgG or serum proteins, making it ideal for avoiding cross-reactivity with mammalian antibodies. |
| Fish Serum [17] | A ready-to-use blocking buffer optimized to reduce non-specific background binding in immunodetection procedures. |
| Normal Serum [17] | Serum (e.g., from goat) that matches the host species of the secondary antibody. Used at 5-10% to bind non-specific sites. |
| Tween 20 [15] [4] | A detergent (0.1% concentration) added to buffers to reduce surface tension and wash away unbound reagents, thereby lowering background. |
| SSC (Saline-Sodium Citrate) [15] | A buffer salt solution (e.g., 4x SSC) that provides the optimal ionic strength and pH for hybridization and washing steps. |
| COT-1 DNA [15] [4] | Used to block repetitive DNA sequences (like Alu and LINE elements) in the genome, which prevents non-specific binding of the probe and reduces background. |
Standard Protocol: Preparing and Applying a BSA-Based Blocking Solution
This is a standard methodology for a blocking solution commonly used in FISH protocols for tissue sections [15].
Detailed Methodology
Recipe for 100 mL of 3% BSA Blocking Solution:
- BSA: 3 grams
- 20x SSC: 20 mL
- Tween 20: 100 µL
- Deionized Water: Add up to a final volume of 100 mL [15]
Application Protocol:
- Preparation: Prepare the blocking solution and pre-warm it to 37°C before use.
- Application: After the post-hybridization stringency washes, incubate the slides in the blocking solution.
- Incubation: Incubate the slides at 37°C for 30 minutes in a moist chamber to prevent evaporation [15].
- Proceed to Detection: After blocking, proceed directly to the application of the detection antibody (e.g., anti-DIG-rhodamine diluted in a detection buffer containing 1% BSA) without a post-blocking wash [15].
Diagram: Workflow for Blocking Solution Preparation and Application.
Frequently Asked Questions (FAQs) & Troubleshooting
Q1: How can I reduce high background staining in my FISH experiment? A: High background can have several causes. Systematically check the following:
- Insufficient Blocking: Ensure the blocking step is performed for the full recommended time and temperature. Consider testing alternative blocking agents like fish gelatin or fish serum [16] [17].
- Inadequate Stringency Washes: The post-hybridization stringent wash is critical. Use an SSC buffer at the correct temperature (75-80°C) [4].
- Probe Specificity: Probes containing repetitive sequences can cause background. Add COT-1 DNA to your hybridization mix to block these sequences [4].
- Detection Issues: Titrate your primary and secondary antibodies to the lowest effective concentration. Over-incubation with the substrate can also cause background to develop; monitor the reaction under a microscope and stop it as soon as the desired signal appears [4] [17].
Q2: What is the advantage of using fish gelatin over BSA for blocking? A: Fish gelatin is purified from cold-water fish skin and contains no IgG or serum proteins. This makes it an excellent choice for minimizing cross-reactivity when using antibodies raised in mammals, as there is no risk of interaction with contaminating mammalian immunoglobulins that might be present in other blocking agents [16].
Q3: I am seeing low or no specific signal. Could my blocking solution be the problem? A: While the blocking solution itself is unlikely to abolish a specific signal, issues in sample preparation and processing upstream of blocking are common culprits. These include improper tissue fixation, over-digestion during enzymatic pretreatment, or inadequate denaturation of the target and probe [4] [7]. Always run positive and negative control slides to validate your entire assay workflow [4] [18].
Q4: Which blocker should I use for my specific application? A: The optimal blocker can be target and tissue-dependent. Common options include:
- 2-5% BSA: A standard, reliable choice for many applications [17].
- 5-10% Normal Serum: Use serum from the species in which your secondary antibody was raised [17].
- Fish Serum or Gelatin: Ideal for reducing cross-reactivity and for use in specialized kits or with mammalian antibodies [16] [17].
Table: Troubleshooting Common Blocking and FISH Issues
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High Background Staining | Inadequate stringent washes [4]. | Ensure stringent wash with SSC buffer is performed at 75-80°C. |
| Probe binds repetitive sequences [4]. | Add COT-1 DNA to the hybridization mixture to block non-specific binding. | |
| Detection antibody concentration too high [17]. | Titrate the antibody to find the optimal, lowest concentration. | |
| Low or No Signal | Sample degradation from improper fixation [4] [7]. | Ensure tissues are fixed promptly in fresh neutral-buffered formalin. |
| Over-digestion during enzymatic pretreatment [4]. | Optimize pepsin or protease digestion time (e.g., 3-10 minutes at 37°C). | |
| Non-specific Antibody Binding | Insufficient blocking [17]. | Use an alternative blocking agent (e.g., Fish Serum [17]) or increase blocking time. |
| Secondary antibody cross-reactivity [17]. | Ensure the secondary antibody host species is different from your sample species. |
Diagram: Logical troubleshooting path for high background in FISH experiments.
Frequently Asked Questions (FAQs)
1. What are the primary causes of high background fluorescence, and how can they be fixed? High background, or noise, often stems from suboptimal sample preparation, denaturation, or washing steps. Key fixes include:
- Sample Fixation: Avoid under-fixation (causes non-specific probe binding) and over-fixation (masks target sequences) by using freshly prepared fixatives and adhering strictly to recommended fixation times [19].
- Denaturation: Optimize denaturation temperature and time. Temperatures that are too low prevent probe binding, while those that are too high cause non-specific binding. Similarly, prolonged denaturation times can increase background [19].
- Washing Stringency: Perform stringent post-hybridization washes. Adjust the pH, temperature, and salt concentration of wash buffers to remove non-specifically bound probes. Always use freshly prepared wash solutions [19] [4].
- Probe Volume: Using an excessive probe volume can elevate background. Ensure the probe concentration is optimized for your specific assay [19].
2. Why might my FISH assay produce weak or absent signals? Weak or absent signals are typically related to poor probe hybridization or detection.
- Hybridization Conditions: Ensure the denaturation temperature is correctly calibrated. For hematology FISH, 75°C for 2 minutes is recommended [3].
- Probe Integrity: Check probe design and labeling efficiency. Protect probes from light and aliquot them for single use to prevent degradation [3] [4].
- Tissue Permeabilization: Inadequate permeabilization or enzymatic digestion (e.g., with pepsin) can prevent probe access to the target. Optimize digestion time and temperature; over-digestion can damage the sample, while under-digestion reduces signal [19] [4].
- Optical Filters: Worn or damaged microscope filters can weaken detected signals. Check and replace filters according to the manufacturer's guidelines, typically every 2-4 years [19] [3].
3. How can I improve the reproducibility of my FISH experiments? Consistency is key to reproducibility.
- Protocol Standardization: Standardize all steps, from sample preparation and fixation to hybridization and washing [2] [4].
- Control Samples: Always run appropriate positive and negative controls to validate your results and technique [4].
- Reagent Quality: Use consistent, high-quality reagents. Prepare fixative and wash buffers fresh for each experiment [19] [3].
Parameter Optimization Data
The following tables summarize critical parameters and their optimized ranges based on experimental data.
Table 1: Optimized Parameters for Key FISH Protocol Steps
| Protocol Step | Parameter | Optimal Range / Condition | Impact of Deviation |
|---|---|---|---|
| Sample Fixation | Fixative [2] [3] | Fresh 3:1 methanol/glacial acetic acid or formaldehyde | Under-fixation: High background [19].Over-fixation: Reduced signal, high background [19]. |
| Tissue Pre-treatment | Enzyme Digestion [4] | Pepsin, 37°C for 3-10 min (tissue-dependent) | Under-digestion: High background, weak signal [19] [4].Over-digestion: Cell damage, weak signal [19]. |
| Denaturation | Temperature [3] | 75°C (for hematology) / 95±5°C (for CISH) [4] | Too Low: Weak/absent signal [19].Too High: High background [19]. |
| Time [4] | 2-10 minutes | Too Short: Weak signal [19].Too Long: High background [19]. | |
| Hybridization | Time [20] [4] | 4 hours (rapid protocol) to 16 hours (overnight) | Too Short: Weak signal [19].Too Long: May increase background. |
| Temperature [4] | 37°C | Deviation can reduce hybridization efficiency and signal strength [4]. | |
| Stringent Wash | Temperature [4] | 75-80°C in SSC buffer | Too Low: High background [4].Too High: Signal loss [4]. |
Table 2: Quantitative Data from an Optimized Digital FISH Workflow [20]
| Profiled Parameter | "Low Profile" Setting | "High Profile" Setting | Application Context |
|---|---|---|---|
| Denaturation/Hybridization | 4 hours (with IntelliFISH buffer) | 18 hours (conventional) | Faster turnaround with strong signals [20]. |
| Scanning Exposure Time | 150 ms | 2000 ms | Routine use vs. weak signal/high background cases [20]. |
| Mean Scanning Time | 15 minutes | 159 minutes | LP ideal for routine; HP for challenging samples [20]. |
| Mean Digital File Size | 458 MB | 1129 MB | LP reduces data storage needs [20]. |
Detailed Experimental Protocols
Protocol 1: Optimized FISH for FFPE Tissue Sections with Rapid Hybridization
This protocol is adapted from a study that implemented a digital FISH workflow, significantly reducing hybridization time while maintaining high signal quality [20].
Slide Pre-treatment:
- Deparaffinize and rehydrate FFPE tissue sections according to standard laboratory protocols.
- Perform heat-induced epitope retrieval by incubating slides in a preheated pretreatment buffer at 98–100°C for 30 minutes [19].
- Digest tissues with pepsin at 37°C. The duration (e.g., 15-30 minutes) must be optimized for your specific tissue type and fixation [20] [4].
- Dehydrate the slides in an ethanol series and air-dry.
Denaturation and Hybridization:
- Apply the probe mixture to the target area on the slide and add a coverslip.
- Denature the probe and target DNA simultaneously on a preheated hotplate. For FFPE tissues, denature at 82°C for 5-10 minutes [4].
- Immediately transfer the slides to a humidified chamber and hybridize at 37°C for 4 hours using the IntelliFISH Hybridization buffer [20].
Post-Hybridization Washes and Detection:
- Remove coverslips by soaking in a mild buffer like PBST [4].
- Perform a stringent wash in 1X SSC buffer at 75°C for 5 minutes [4].
- Rinse slides in TBST or PBST at room temperature.
- Apply DAPI-containing mounting medium (e.g., VECTASHIELD HardSet) and allow it to harden for at least 30 minutes before imaging [20].
Protocol 2: Troubleshooting and Optimization of the Blocking Step
While a specific "blocking solution" is not always explicitly named in FISH protocols, the principles of blocking non-specific sites are achieved through several key steps. Optimizing these is crucial for reducing background.
Post-Hybridization Stringent Washes: This is the most critical step for "blocking" non-specific signal.
- Solution: Use a solution of 1X SSC with 0.1% Tween-20.
- Method: After hybridization, wash slides in this solution at a stringently controlled temperature of 75-80°C for 5 minutes. The temperature is the key parameter; increase it by 1°C per additional slide but do not exceed 80°C [4].
- Optimization: If background persists, incrementally increase the wash temperature within the 75-80°C range or the wash duration by 1-2 minutes.
Enzymatic Pre-treatment: This step unmasks target nucleic acids and reduces background from cellular debris.
- Solution: Pepsin or Proteinase K solution.
- Method: Incubate slides with the enzyme at 37°C for 3-10 minutes. The exact time must be empirically determined for each tissue type [4].
- Optimization: If background is high, slightly increase digestion time. If signal is lost or morphology is damaged, reduce digestion time [19] [4].
Probe Design and Concentration: The probe itself can be a source of background.
- Optimization: Ensure probes do not contain repetitive sequences (e.g., Alu elements). If they do, add unlabeled COT-1 DNA to the hybridization mix to block these non-specific binding sites [4].
- Probe Volume: Using the correct probe volume is crucial. Excess probe leads to high background, while insufficient volume causes weak signals [19].
Experimental Workflow and Parameter Relationships
The following diagram illustrates the interconnected workflow of a FISH experiment and how optimizing key parameters at each stage influences the final outcome.
Figure 1: A sequential workflow diagram for FISH experiments, highlighting the key parameters (Concentration, Time, Temperature) to optimize at each step to ensure high signal-to-noise ratio.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Optimized FISH Assays
| Reagent / Solution | Function | Optimization Tip |
|---|---|---|
| Methanol/Acetic Acid Fixative | Preserves cellular architecture and nucleic acids. | Always prepare fresh and use pre-chilled (-20°C). For blood smears, use with a hypotonic solution like potassium chloride to reduce background [3]. |
| Pepsin / Proteinase K | Enzymatically digests proteins to unmask target nucleic acids. | Concentration and incubation time (e.g., 37°C for 3-10 min) must be titrated for each tissue type to avoid over- or under-digestion [4]. |
| Formaldehyde/PFA Fixative | Cross-links proteins to preserve tissue morphology (common for FFPE). | Avoid over-fixation, which causes excessive cross-linking and masks targets, leading to high background [19]. |
| IntelliFISH Hybridization Buffer | A specialized buffer that facilitates probe binding. | Enables a significant reduction in hybridization time (from 18 hrs to 4 hrs) while maintaining strong signals [20]. |
| SSC Wash Buffer | A saline-sodium citrate buffer used for post-hybridization stringent washes. | The stringency is controlled by temperature (75-80°C) and salt concentration. Use freshly prepared [19] [4]. |
| DAPI Mounting Medium | Counterstains nuclei and preserves fluorescence during imaging. | Use a hardening medium (e.g., VECTASHIELD HardSet) for stability and to prevent drying [20]. |
Synergy with Probe Design and Hybridization Conditions
Frequently Asked Questions (FAQs)
1. How does probe design influence the required hybridization conditions? Probe design is intrinsically linked to hybridization conditions. Key probe characteristics like length, sequence composition (GC content), and type (DNA, RNA, or oligonucleotide) directly determine the optimal hybridization temperature and stringency [21] [22]. For instance, RNA probes (riboprobes) form more stable RNA-RNA hybrids, allowing for potentially higher stringency conditions compared to DNA-DNA hybrids [21] [23]. The probe's melting temperature (Tm), which is influenced by its length and GC content, serves as the primary guide for setting the hybridization temperature [22]. Furthermore, complex probes containing repetitive sequences require the addition of unlabeled DNA, like COT-1 DNA, to the hybridization buffer to block non-specific binding [4] [23].
2. What are the primary causes of high background noise in FISH, and how can they be addressed? High background can arise from several sources, each with specific solutions:
- Insufficient Stringency: Washes that are not stringent enough fail to remove imperfectly matched probes. This can be corrected by increasing the temperature or decreasing the salt concentration in the post-hybridization wash buffers [21] [4] [2].
- Over-digestion with Proteases: Excessive proteinase K or pepsin treatment damages tissue morphology, leading to probe trapping and high background. This requires careful titration of the protease concentration and digestion time [21] [4].
- Endogenous Biotin: When using biotin-labeled probes, endogenous biotin in tissues can cause non-specific staining. This can be blocked with an avidin/biotin blocking step or avoided by using digoxigenin-labeled probes [21].
- Probe Binding to Repetitive Sequences: If probes contain repetitive elements, they must be pre-blocked with unlabeled COT-1 DNA during hybridization to prevent genome-wide binding [4].
- Inadequate Washes: Using the wrong wash buffer, such as PBS without detergent, can elevate background. Always use the specified buffers, like PBST (PBS with Tween 20) [4].
3. My FISH signal is weak or absent. What steps should I take? A weak or absent signal often stems from issues with target accessibility, probe quality, or denaturation.
- Optimize Permeabilization and Digestion: Insufficient protease (e.g., proteinase K, pepsin) digestion prevents probe access to the target. A titration experiment is recommended to find the optimal balance between signal intensity and tissue preservation [21] [4].
- Verify Probe Quality and Denaturation: Check probe labeling efficiency, fragment size, and concentration [23]. Ensure that both the probe and target DNA are completely denatured. For DNA probes, a denaturation step of 5-10 minutes at 95±5°C is typical [4].
- Confirm Detection Reagent Activity: If using an indirect detection system, verify that enzyme conjugates (e.g., HRP or AP) are active by testing them with their substrate [4].
- Consider Signal Amplification: For low-abundance targets, consider switching to an amplification method like HCR-FISH (Hybridization Chain Reaction) or CARD-FISH to enhance the signal [24] [25].
Troubleshooting Guide
This guide summarizes common problems, their potential causes, and recommended solutions.
| Problem | Potential Causes | Troubleshooting Strategies |
|---|---|---|
| High Background | Insufficient post-hybridization washes [4] [2]Inadequate stringency [21]Endogenous biotin (for biotin probes) [21]Over-digestion with protease [21] [4] | Increase temperature and/or decrease salt concentration in wash buffers [21] [2].Block endogenous biotin or use digoxigenin probes [21].Titrate protease concentration and incubation time [21]. |
| Weak or No Signal | Insufficient permeabilization/digestion [21] [4]Poor probe quality or low concentration [2] [23]Incomplete denaturation [4] [2]Low target abundance [4] | Optimize proteinase K (1-5 µg/mL) or pepsin digestion [21] [4].Check probe labeling, fragment size (100-250 bp for DNA), and use adequate concentration [23].Ensure denaturation at 95±5°C for 5-10 mins [4].Use signal amplification (e.g., HCR-FISH) [24]. |
| Poor Morphology | Over-digestion with protease [21]Over-fixation of sample [4] [23] | Titrate protease to find optimal concentration [21].Limit formalin fixation to less than 24 hours [23]. |
| Uneven or Patchy Signal | Non-uniform probe distribution [2]Air bubbles during hybridization [2]Sample drying out during procedure [4] | Ensure even application of probe and avoid hard pressure on coverslips [2].Use a humidified chamber and prevent drying at all steps [4]. |
Experimental Protocols for Key Optimizations
Protocol: Proteinase K Digestion Titration
Purpose: To optimize sample pretreatment for maximum signal while preserving tissue morphology [21]. Materials: Proteinase K, TE Buffer (pH 7.5), PBS. Method:
- Deparaffinize and rehydrate FFPE tissue sections.
- Prepare a series of Proteinase K solutions in TE buffer (e.g., 0, 1, 2, 5, 10 µg/mL).
- Apply each concentration to replicate sections and incubate at room temperature for 10 minutes.
- Stop the reaction by washing slides in PBS.
- Proceed with the standard FISH protocol, including hybridization with your target probe.
- Evaluate slides for hybridization signal intensity and tissue integrity. The optimal concentration produces the strongest signal with the least disruption to morphology [21].
Protocol: Optimization of Hybridization Stringency
Purpose: To determine the optimal hybridization and wash stringency for a specific probe [21] [23]. Materials: Hybridization buffer with formamide, Saline-Sodium Citrate (SSC) buffer, Water bath. Method:
- Hybridization Temperature: Perform hybridizations at a range of temperatures (e.g., 37°C to 65°C) while keeping other factors constant. The typical starting range is 55-62°C [23]. Formamide in the buffer allows for lower temperatures while maintaining stringency [21] [23].
- Post-Hybridization Washes: After hybridization, perform stringent washes with SSC buffer at varying temperatures. A common stringent wash uses 0.1X to 1X SSC at 75-80°C for 5 minutes [4]. Increase the temperature by 1°C per slide when processing multiple slides, but do not exceed 80°C [4].
- Analyze results to find the conditions that give the strongest specific signal with the lowest background.
Research Reagent Solutions
This table details key reagents used in FISH to optimize blocking, hybridization, and detection.
| Reagent | Function in FISH | Key Considerations |
|---|---|---|
| COT-1 DNA | Blocks repetitive DNA sequences to prevent non-specific probe binding, reducing background [4] [23]. | Added directly to the hybridization mixture [23]. |
| Formamide | A denaturing agent included in hybridization buffer. It lowers the melting temperature (Tm) of nucleic acid hybrids, allowing hybridization to occur at lower temperatures that better preserve tissue morphology [21] [23]. | Concentration must be optimized for each probe-target pair. |
| Proteinase K / Pepsin | Proteolytic enzymes used for sample pretreatment. They digest proteins to unmask target nucleic acids and permit probe access, crucial for signal intensity [21] [4]. | Requires careful titration; over-digestion destroys morphology, under-digestion reduces signal [21]. |
| Dextran Sulfate | A volume excluder added to hybridization buffer. It increases the effective probe concentration, accelerating hybridization kinetics [25]. | Helps to drive probe-target hybridization. |
| Blocking Reagents | Used to prevent non-specific binding of detection reagents (e.g., antibodies). Common blockers include BSA or proprietary formulations in detection kits [21]. | Essential for reducing background in indirect detection methods. |
Workflow and Pathway Diagrams
FISH Optimization Workflow
Probe Hybridization Mechanisms
COT-1 DNA is a critical biochemical reagent extensively used in fluorescence in situ hybridization (FISH) and microarray assays to block non-specific hybridization of repetitive DNA sequences. The human genome consists of approximately 50% repetitive sequences, including Short Interspersed Nuclear Elements (SINEs, such as Alu elements) and Long Interspersed Nuclear Elements (LINEs, such as L1 elements) [26]. When using labeled probes in FISH, these repetitive elements can bind to multiple genomic locations, creating substantial background noise that obscures specific signals from target sequences. COT-1 DNA addresses this problem by preemptively binding to these repetitive sequences, thereby significantly reducing cross-hybridization and improving the signal-to-noise ratio for accurate interpretation of FISH results [27] [28].
The reagent is derived from human placental DNA through a process of shearing, denaturation, and reannealing under conditions that preferentially enrich for repetitive elements [28]. This process, known as Cot filtration, results in DNA fragments predominantly 50-300 base pairs in size that are highly enriched for the repetitive sequences that cause non-specific hybridization in molecular assays [27]. Commercial COT-1 DNA preparations typically demonstrate a 3-4 fold enrichment of major repetitive elements compared to the normal genome, providing an effective competitive inhibitor for non-specific hybridization [29].
Troubleshooting Guide: COT-1 DNA in FISH Experiments
Common Technical Issues and Solutions
Problem: High Background Staining
- Potential Cause: Probes containing abundant repetitive sequences (Alu or LINE elements) binding non-specifically throughout the genome.
- Solution: Increase the amount of COT-1 DNA in the hybridization mixture. Ensure COT-1 DNA is thoroughly mixed with the probe before application [4].
- Additional Measures: Verify that stringent wash steps use the correct temperature (75-80°C) and buffer (SSC) conditions. Wash slides with PBST (PBS with 0.025% Tween 20) rather than water or plain PBS [4].
Problem: Weak or No Specific Signal
- Potential Cause: Excessive COT-1 DNA concentration may compete with target-specific probe binding.
- Solution: Titrate COT-1 DNA concentration to find the optimal level for your specific probe [29].
- Additional Measures: Check probe integrity and target accessibility. For formalin-fixed paraffin-embedded (FFPE) tissues, optimize pretreatment protocols to ensure adequate target exposure without over-digestion [7].
Problem: Inconsistent Results Between Experiments
- Potential Cause: Variability in COT-1 DNA composition or concentration between different manufacturers or lots.
- Solution: Use COT-1 DNA from the same manufacturer and lot for comparable experiments. Validate concentration using fluorometry rather than spectrophotometry for more accurate quantification [29] [27].
- Additional Measures: Include appropriate positive and negative controls in each experiment to monitor hybridization efficiency [4].
Optimization Parameters for COT-1 DNA Blocking
Table 1: Key Optimization Parameters for COT-1 DNA in FISH
| Parameter | Optimal Range | Effect of Deviation | Validation Method |
|---|---|---|---|
| Concentration | 50-150 μg/reaction | Too low: high background; Too high: reduced specific signal | Titration with control probes |
| Fragment Size | 50-300 bp | Smaller fragments may not block effectively; Larger fragments may reduce hybridization kinetics | Gel electrophoresis |
| Purity | A260/A280 ≈ 1.8 | Impurities may inhibit hybridization or increase background | Spectrophotometry |
| Hybridization Time | 16 hours (overnight) | Shorter times: incomplete blocking; Longer times: no significant improvement | Time-course experiment |
Frequently Asked Questions (FAQs)
Q1: What exactly is COT-1 DNA and how does it work? COT-1 DNA is human genomic DNA that has been processed to enrich for repetitive sequences. It works through competitive inhibition - when added to FISH hybridization mixtures, it binds to repetitive elements throughout the genome, preventing labeled probes from binding non-specifically to these sequences. This mechanism significantly reduces background noise and improves specific signal detection [27] [28].
Q2: When should I use COT-1 DNA in my FISH experiments? COT-1 DNA is essential when your probe contains repetitive elements that are also present elsewhere in the genome. This is particularly important for:
- Whole chromosome painting probes [30]
- Probes derived from genomic regions rich in SINEs/LINEs [26]
- Probes larger than 10 kb that likely contain repetitive elements [4]
- Any FISH application where you observe high background staining [4]
Q3: How much COT-1 DNA should I use in my experiments? The optimal concentration varies by application but typically ranges from 50-150 μg per reaction. For initial experiments, a dilution series is recommended to determine the ideal concentration for your specific probe and tissue system. Quantitative studies have shown that hybridization intensity increases with COT-1 DNA concentration up to a point, after which specific signal may diminish [29].
Q4: Can COT-1 DNA ever enhance non-specific hybridization? Surprisingly, yes. Some studies have reported that COT-1 DNA can enhance non-specific hybridization between probes and genomic targets containing conserved repetitive elements, potentially increasing background rather than decreasing it. This effect is particularly notable for probes mapping to genomic regions with conserved repetitive sequences [29]. This underscores the importance of empirical optimization for each experimental system.
Q5: Are there alternatives to COT-1 DNA for blocking repetitive sequences? Yes, alternatives include:
- Computational design of single-copy probes that avoid repetitive elements altogether [29]
- Synthetic repetitive elements complementary to specific repeats in your probes [29]
- For specialized applications, species-specific COT-1 DNA (e.g., mouse COT-1 DNA for mouse studies) [27]
Research Reagent Solutions
Table 2: Essential Reagents for COT-1 DNA-Based FISH Experiments
| Reagent | Function | Specifications |
|---|---|---|
| Human COT-1 DNA | Blocks repetitive sequences | 50-300 bp fragments, enriched for SINEs/LINEs [27] |
| Species-Specific COT-1 DNA | Blocks repetitive sequences in non-human studies | Mouse COT-1 available for murine systems [27] |
| Stringent Wash Buffer (SSC) | Removes non-specifically bound probes | 1X SSC, 75-80°C for optimal stringency [4] |
| PBST (PBS with Tween 20) | Washing without increasing background | 0.025% Tween 20 concentration [4] |
| Proteolytic Enzymes (Pepsin) | Tissue pretreatment for target accessibility | 3-10 minutes at 37°C, optimized per tissue type [7] |
Workflow: Integrating COT-1 DNA in FISH Protocols
Advanced Considerations and Limitations
While COT-1 DNA significantly improves FISH specificity, researchers should be aware of its limitations. Quantitative studies have demonstrated that the composition of COT-1 DNA is not purely repetitive sequences; it also contains linked single-copy sequences that can adventitiously associate with probes, potentially distorting quantitative measurements [29]. This effect is more pronounced for probes mapping to genomic regions containing conserved repetitive elements.
For formalin-fixed paraffin-embedded (FFPE) tissues, additional challenges exist. The fixation process can alter DNA accessibility, requiring optimized pretreatment protocols. COT-1 DNA concentration may need adjustment for FFPE samples compared to cell preparations [7]. Monitoring signal quality and background through rigorous control experiments is essential when adapting COT-1 DNA blocking to different sample types.
Emerging technologies, including artificial intelligence and digital pathology, offer new opportunities to standardize and improve the assessment of COT-1 DNA blocking efficiency in FISH experiments [7]. These approaches may help quantify background reduction and specific signal preservation more objectively than visual inspection alone.
Diagnosing and Solving Common Blocking-Related FISH Issues
Solving High Background and Non-Specific Signal
High background and non-specific signal are common challenges in Fluorescence In Situ Hybridization (FISH) assays that can obscure critical data, complicate interpretation, and potentially lead to erroneous conclusions. These issues can stem from multiple aspects of the FISH procedure, from sample preparation through final imaging. This guide provides a systematic troubleshooting framework to help researchers identify and correct the root causes of high background, ensuring the accuracy and reliability of their FISH analyses.
FAQs and Troubleshooting Guides
Q1: How can sample preparation affect background signal?
Improper sample preparation is a primary contributor to high background. The fixation process requires a delicate balance to preserve cellular architecture while maintaining target DNA accessibility [19].
- Under-fixation: Leads to incomplete preservation of cellular structure, increasing the risk of DNA degradation and non-specific probe binding, which elevates background fluorescence [19].
- Over-fixation: Especially with formalin, causes excessive cross-linking of proteins and nucleic acids. This can mask target sequences, reduce overall signal intensity, and paradoxically increase background through non-specific binding [19].
- Section Thickness: For FFPE tissue, sections that are too thick can cause issues with probe penetration and interpretation. Aim for sections of 3–4μm for optimal results [19].
Q2: What is the role of pre-treatment, and how should it be optimized?
Pre-treatment steps, such as enzyme digestion or heat treatment, are designed to break down proteins, lipids, and other cellular components that may mask target DNA sequences. Both insufficient and excessive pre-treatment can cause problems [19].
Table: Troubleshooting Pre-Treatment Steps
| Issue | Consequence | Solution |
|---|---|---|
| Insufficient Pre-treatment | Leaves autofluorescent cellular debris and creates nonspecific binding sites, increasing background [19]. | Optimize pre-treatment time and temperature. Use a dedicated pretreatment kit and refresh solutions between slide batches [19]. |
| Over-digestion | Damages the sample and target sequence, resulting in a weak or lost specific signal [19]. | Titrate enzyme concentration and duration. Adhere closely to protocols specific to your tissue type and fixation [19]. |
Q3: How do probe volume and denaturation conditions influence signal clarity?
Using an optimal probe volume is key to maximizing specific binding and minimizing background. Denaturation conditions are particularly critical for FFPE samples due to their high cross-linking [19].
- Probe Volume: Too low a volume results in weak specific signals, while too much can increase background [19].
- Denaturation Temperature: Too low a temperature prevents effective probe binding; too high a temperature promotes non-specific binding [19].
- Denaturation Time: Short times may reduce probe binding, yielding weak signals. Prolonged times can unmask non-specific binding sites, increasing off-target probe binding and background [19].
Q4: Why are washing steps critical, and how can they be optimized?
Effective washing removes excess, unbound, or non-specifically bound probes, which is a critical step for reducing background fluorescence [19] [4].
- Stringency Washes: The stringency (pH, temperature, and time) must be carefully optimized. High stringency helps remove probes bound to non-target sequences, but too much can strip specific signals. Begin with the protocol's recommended settings and make incremental adjustments [19].
- Wash Buffer Quality: Always use freshly prepared wash buffers to prevent contamination or degradation, which can fail to remove background or introduce new fluorescence [19].
- Correct Buffers: Using the wrong buffer (e.g., PBS without Tween 20 instead of PBST) can lead to elevated background. For post-hybridization stringent washes, use SSC buffer at 75–80°C [4].
Q5: Can my equipment cause high background?
Yes, the optical components of your microscope can be a source of problems. Worn or damaged optical filters will exhibit a mottled appearance and can significantly weaken signals and increase background noise. Protect filters from the light source by closing the microscope shutter when not in use, and replace them according to the manufacturer's guidelines, typically every 2–4 years [19].
Q6: What blocking buffers are recommended for FISH assays?
Blocking buffers are essential for occupying non-specific binding sites on the membrane or tissue before probe application. The choice of blocker can significantly impact your signal-to-noise ratio [31] [32].
Table: Comparison of Blocking Buffers for FISH
| Blocking Buffer / Agent | Benefits | Best Used When |
|---|---|---|
| Non-fat Milk (2-5%) | Inexpensive; contains multiple protein types [31]. | Working with robust, high-abundance targets on a budget. Not suitable for biotin-streptavidin systems or phosphoprotein detection [31]. |
| Bovine Serum Albumin (BSA) (2-3%) | Good for biotin-streptavidin systems and phosphoprotein detection; can increase sensitivity [31]. | Targeting phosphoproteins or low-abundance targets. Can be a weaker blocker, potentially allowing more non-specific binding [31]. |
| Purified Proteins (e.g., Casein) | Single-protein buffer reduces chances of cross-reaction; ideal when milk blocks antigen-antibody binding [31]. | Standard blockers like milk cause high background or mask your specific signal [31]. |
| Fish Serum Blocking Buffer | Non-mammalian source minimizes immunological interactions with mammalian samples; reduces chances of false positives [32]. | Working with human or other mammalian samples, especially in multiplex fluorescence imaging [32]. |
| Specialty Blocking Buffers (e.g., SuperBlock, StartingBlock) | Serum- and biotin-free; designed to be compatible in situations where traditional agents fail; often block quickly (<15 min) [31]. | Troubleshooting persistent background issues or developing a new assay for maximum reliability [31]. |
Experimental Protocols & Workflows
Detailed Protocol: Optimized Digital FISH Workflow
An optimized diagnostic workflow incorporating rapid hybridization and digital imaging has been shown to reduce turnaround time while maintaining high quality [20].
- Tissue Marking: A pathologist encircles a representative tumor area on an H&E slide. The corresponding area is marked on the back of the slide to be hybridized using a diamond pen to narrow the scanning area [20].
- FISH Technique:
- Tissue: Use formalin-fixed, paraffin-embedded (FFPE) specimens, sectioned at 3–4μm [19] [20].
- Pre-treatment: Vary pretreatment time from 30 to 40 minutes depending on tissue type. For example, use a Tissue Pretreatment Solution at 98–100°C for 30+ minutes, followed by enzyme treatment at 37°C [19] [20].
- Hybridization: Apply specific probes in IntelliFISH Hybridization Buffer and hybridize at 37°C for 4 hours (a substantial reduction from traditional 18-hour protocols) [20].
- Mounting: Use DAPI VECTASHIELD HardSet as a nuclear stain and mounting medium, allowing a minimum hardening time of 30 minutes [20].
- Slide Imaging:
- Scan with a 40x objective. Two scanning profiles can be used [20]:
- Low Profile (LP): Exposure time 150 ms, digital gain 3–4. Ideal for routine use, with shorter scan times and smaller file sizes.
- High Profile (HP): Exposure time 2000 ms, digital gain 0–2. Provides a better signal-to-noise ratio for samples with weak signals or high inherent background.
- Activate the Z-stack function (5-7 layers with 0.4μm distance) to account for tissue fluctuations [20].
- Scan with a 40x objective. Two scanning profiles can be used [20]:
- Analysis: Use digital imaging software for manual or automated signal counting. Note that automated counting may require manual editing in densely packed tissues [20].
Workflow Diagram: FISH Assay Optimization Pathway
The following diagram outlines a logical pathway for troubleshooting high background in your FISH assay, from start to finish.
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting the right reagents is fundamental to a successful FISH assay with low background.
Table: Essential Reagents for FISH Optimization
| Reagent / Kit | Function | Key Feature / Benefit |
|---|---|---|
| CytoCell LPS 100 Tissue Pretreatment Kit [19] | Breaks down proteins and lipids that mask target DNA. | Pre-optimized for effective pretreatment of FFPE tissue, reducing a key variable. |
| IntelliFISH Hybridization Buffer [20] | Medium for probe hybridization. | Enables rapid hybridization (4 hours vs. 18 hours), shortening assay time and potentially reducing background. |
| UltraBlock-FISH Blocking Buffer [32] | Blocks non-specific binding sites on the membrane/tissue. | Non-mammalian fish proteins minimize interactions with mammalian samples, reducing false positives. |
| VECTASHIELD HardSet with DAPI [20] | Mounting medium with nuclear counterstain. | Fast hardening time provides stable preparation for imaging. |
| Freshly Prepared Wash Buffers [19] | Removes unbound and non-specifically bound probes. | Critical for reducing background; contaminated or degraded buffers are a common failure point. |
| Optical Filters [19] | Microscope component for isolating fluorescence signals. | Worn filters degrade image quality; regular replacement (every 2-4 years) is essential for signal clarity. |
Addressing Weak or Absent Probe Signals
Troubleshooting Guide
Q1: What are the primary causes of weak or absent FISH signals?
Weak or absent signals in FISH experiments can stem from issues across multiple procedural stages. The table below summarizes the common causes and their direct solutions.
| Problem Cause | Specific Issue | Recommended Solution |
|---|---|---|
| Probe Quality | Inefficient dye incorporation, unexpected fragment length, or low yield [23]. | Verify probe yield, dye incorporation, and fragment length (100-250 bp for DNA probes) [23]. |
| Sample Preparation | Over-fixation (>24 hours) reducing target accessibility [2] [23]; insufficient permeabilization [2]. | Optimize fixation time [2] [23]; use enzymatic digestion (e.g., Pepsin, 3-10 min at 37°C) to remove cytoplasm [4] [3]. |
| Denaturation | Incomplete denaturation of target DNA/probe [2]; incorrect temperature [3]. | Calibrate hotplate; ensure denaturation at 75°C for 2 minutes or 95±5°C for 5-10 minutes [4] [3]. |
| Hybridization | Low probe concentration or short hybridization time [2]. | Increase probe concentration or hybridization time (e.g., overnight for 16 hours) [2] [4]. |
| Signal Detection | Use of a fluorophore with low sensitivity [2]. | Use a more sensitive fluorophore or employ signal amplification methods like tyramide signal amplification (TSA) [2] [4]. |
Q2: How can I optimize the blocking and pretreatment steps to improve signal clarity?
Effective blocking and sample pretreatment are crucial for reducing background noise and enhancing specific signal detection. The following workflow and table detail the key optimization steps.
Key Experimental Protocol: Enzymatic Digestion For tissue sections, a critical pretreatment step is enzymatic digestion to remove proteins that obscure the target nucleic acids [4].
- Reagent Preparation: Prepare a fresh pepsin solution in Tris-EDTA buffer or a 3.0 μg/mL solution of proteinase K [4] [33].
- Application: Apply the digestion solution to the sample and incubate at 37°C [4].
- Optimization: The incubation time typically ranges from 3 to 10 minutes for pepsin and about 15 minutes for proteinase K [4] [33]. Caution: Over-digestion can weaken the FISH signal and damage morphology, while under-digestion may also decrease signal [4].
Q3: What advanced probe technologies can enhance signal detection?
For challenging targets, such as low-abundance sequences, standard DNA probes may be insufficient. Advanced nucleic acid analogs provide higher binding affinity and specificity.
| Technology | Description | Application Benefit |
|---|---|---|
| Peptide Nucleic Acid (PNA) Clamps | Synthetic nucleic acids with a peptide backbone that form exceptionally stable hybrids with DNA/RNA, effectively suppressing non-target amplification [34]. | Completely suppresses predator DNA amplification in metabarcoding (99.3-99.9% efficiency), allowing for the detection of prey organisms in herbivorous fish diets [34]. |
| Locked Nucleic Acid (LNA) Probes | Nucleic acid analogs containing LNA nucleotides that significantly increase the melting temperature (Tm) of the probe-target duplex [33]. | Enhances sensitivity and specificity for detecting small, low-abundance RNA targets, such as bacterial small non-coding RNA [33]. |
Research Reagent Solutions
The following table lists key reagents and their roles in optimizing FISH experiments to address weak signals.
| Reagent | Function | Consideration |
|---|---|---|
| Pepsin / Proteinase K | Enzymatic digestion to remove cytoplasmic proteins and improve probe accessibility [4] [3]. | Concentration and time must be optimized for each sample type to prevent over- or under-digestion [4]. |
| FISH-Blocker | A blocking agent using non-mammalian fish proteins to minimize non-specific antibody binding [35]. | Ideal for immuno-detection steps, offering an alternative to mammalian protein-based blockers like BSA [35]. |
| Formamide | A component of hybridization buffer that lowers the melting temperature of nucleic acid duplexes [33]. | Allows for hybridization at lower temperatures, which helps preserve sample morphology [33] [23]. |
| Dextran Sulfate | A volume-excluding polymer used in the hybridization buffer [33]. | Concentrates the probe and increases the hybridization rate [33]. |
| Cot-1 DNA | Genomic DNA enriched for repetitive sequences [4]. | Added during hybridization to block non-specific binding of probes to repetitive DNA sequences, reducing background [4]. |
Correcting for Autofluorescence and Morphological Distortion
Troubleshooting Guides
Frequently Asked Questions
Q1: What are the primary causes of autofluorescence in FFPE tissue FISH samples, and how can they be mitigated? Autofluorescence in FFPE tissues primarily stems from inadequate fixation, endogenous fluorophores in the tissue, and reagent interactions. Mitigation strategies include optimized pretreatment protocols to clear endogenous pigments, using probes with bright, specific fluorophores to outcompete background signals, and employing spectral imaging systems capable of autofluorescence unmixing. Implementing rigorous quality control during tissue processing and fixation is also crucial to prevent its introduction [7].
Q2: How does suboptimal blocking solution contribute to high background noise and morphological distortions? An ineffective blocking solution fails to prevent nonspecific binding of probes and antibodies, leading to high background fluorescence that obscures true signals. This can distort morphological assessment by creating a diffuse, hazy appearance, making precise signal localization within tissue architecture difficult. Optimized blocking is fundamental for preserving tissue morphology and ensuring that only specific hybridization events are detected [7].
Q3: What are the best practices for validating blocking solution efficiency in a FISH protocol? Validation should include a no-probe control to assess inherent tissue autofluorescence and a negative control with a non-targeting probe to measure nonspecific binding. The use of internal positive controls is also recommended. Comparing signal-to-noise ratios between different blocking formulations quantitatively measures efficiency. Implementing these controls as part of standard laboratory quality control is essential for reliable results [7].
Q4: Can automated platforms improve consistency in FISH results for FFPE tissues? Yes, automation significantly improves consistency. A recent validation of the Leica BOND-III automated staining platform for HER2 FISH testing demonstrated a 98% concordance with manual methods. Automation standardizes critical steps like pretreatment, denaturation, and hybridization, reducing inter-run and inter-operator variability. It also significantly decreases technical hands-on time and overall supply costs [36].
Q5: How does spectral flow cytometry's approach to autofluorescence differ from conventional methods? Conventional flow cytometry cannot separate autofluorescence from specific fluorophore signals, often requiring compensation that may subtract real signal. In contrast, spectral flow cytometry collects the full emission spectrum and uses unmixing algorithms to digitally separate the unique spectral signature of autofluorescence from that of specific fluorochromes. This allows for the autofluorescence to be identified and subtracted, improving resolution and accuracy [37].
Troubleshooting Common Experimental Issues
Problem: High, diffuse background autofluorescence obscuring specific FISH signals.
- Potential Cause #1: Inadequate blocking or nonspecific probe binding.
- Solution: Re-optimize the blocking solution by testing different concentrations of blocking agents (e.g., serum, BSA, or commercial blockers) and increasing the blocking incubation time. Ensure probe sequences are specific and of high purity.
- Potential Cause #2: Endogenous tissue fluorophores or fixative-induced fluorescence.
- Solution: Incorporate a photobleaching step with strong light exposure before hybridization. Use spectral imaging and unmixing to digitally separate and remove the autofluorescence signal [37] [7].
Problem: Morphological distortion of tissue or cellular architecture.
- Potential Cause #1: Over-fixation or improper tissue processing.
- Solution: Standardize fixation time using neutral buffered formalin and do not exceed 24-48 hours. Ensure proper dehydration and paraffin embedding protocols are followed.
- Potential Cause #2: Overly aggressive pretreatment (proteinase K or heat).
- Solution: Titrate the pretreatment conditions (e.g., enzyme concentration, time, temperature) using a test slide to find the optimal balance between antigen retrieval and tissue preservation [7].
Problem: Weak or absent specific FISH signal.
- Potential Cause #1: Insufficient probe penetration due to cross-linking.
- Solution: Optimize the pretreatment protocol to adequately break down cross-links without destroying morphology. Increase probe concentration or use probes with brighter fluorophores.
- Potential Cause #2: Denaturation or hybridization steps are suboptimal.
- Solution: Calibrate the temperature of the thermal cycler or hot plate for denaturation. Ensure hybridization buffer is correctly formulated and the hybridization chamber is sealed to prevent evaporation [7].
Problem: Inconsistent results between experiment runs.
- Potential Cause #1: Manual protocol leading to operator variability.
- Solution: Transition to an automated staining platform to ensure precise and reproducible timing, temperature, and reagent application for every step [36].
- Potential Cause #2: Degradation of reagents or aged FFPE blocks/slides.
- Solution: Implement strict reagent quality control, aliquot reagents to avoid freeze-thaw cycles, and use freshly cut sections from well-preserved FFPE blocks whenever possible [7].
Experimental Data & Protocols
Quantitative Performance of Automated FISH
The following table summarizes key performance metrics from a validation study of the automated Leica BOND-III platform for HER2 FISH testing, demonstrating its reliability compared to manual methods [36].
Table 1: Performance Metrics of Automated vs. Manual FISH
| Metric | Automated FISH (Leica BOND-III) | Manual FISH |
|---|---|---|
| Sensitivity (Breast Cancer) | 95% | - |
| Specificity (Breast Cancer) | 97% | - |
| Sensitivity (Gastric Cancer) | 100% | - |
| Specificity (Gastric Cancer) | 100% | - |
| Overall Concordance | 98% | (Baseline) |
| Technical Hands-on Time | Significantly Decreased | (Baseline) |
Key Reagent Solutions for FISH Optimization
The following table details essential reagents and their optimized functions for addressing autofluorescence and morphological issues in FISH.
Table 2: Research Reagent Solutions for FISH Optimization
| Reagent / Solution | Function & Optimization Purpose |
|---|---|
| Optimized Blocking Solution | Prevents nonspecific binding of probes/antibodies to minimize background. Optimization is thesis-central for clean signal-to-noise [7]. |
| Proteinase K / Pretreatment Solution | Digests proteins to enable probe access to targets. Concentration and time must be titrated to balance signal with tissue integrity [7]. |
| Spectral Fluorophores | Fluorochromes with distinct spectral signatures for use with spectral imaging/cytometry, enabling autofluorescence unmixing [37]. |
| Automated Staining Platform Reagents | Formulated specifically for consistent performance on automated systems, reducing variability and improving reproducibility [36]. |
| High-Quality, Specific FISH Probes | DNA probes with high binding specificity and brightness to outcompete background autofluorescence [7]. |
Detailed Protocol: Blocking Solution Optimization for FISH
Objective: To systematically evaluate and optimize blocking solutions for reducing background and improving signal clarity in FISH on FFPE tissue sections.
Materials:
- FFPE tissue sections known to exhibit autofluorescence.
- Candidate blocking solutions: e.g., 1-5% BSA in PBS, 5-10% normal serum, commercial blocking reagents.
- Proteinase K solution.
- Target-specific FISH probe and detection system.
- Humidified hybridization chamber.
Methodology:
- Section Pretreatment: Deparaffinize and rehydrate FFPE sections. Perform a standardized proteinase K digestion.
- Blocking: Apply different blocking solutions to serial sections. Include a negative control with no blocking. Incubate for 30-60 minutes at room temperature.
- Hybridization: Apply the FISH probe according to the standard protocol (denaturation, hybridization, post-hybridization washes).
- Detection and Imaging: Complete the detection steps and image all sections using identical microscope and camera settings.
- Analysis: Quantify the signal-to-noise ratio by measuring the mean fluorescence intensity of the specific signal and the background in a cell-free area for each section.
Experimental Workflow and Signaling Pathways
FISH Optimization Workflow
Autofluorescence Unmixing Pathway
Ensuring Reproducibility and Standardization Across Experiments
Frequently Asked Questions (FAQs)
Q: What are the most critical factors for achieving reproducible FISH results? A: The most critical factors include standardized sample fixation, optimized permeabilization, precise denaturation and hybridization conditions, and consistent post-hybridization washes. Always run appropriate positive and negative controls with every experiment to monitor performance [2].
Q: My FISH signal is weak or absent. What should I check first? A: First, verify probe design and labeling efficiency. Then, optimize denaturation and hybridization conditions (time and temperature). Ensure adequate sample permeabilization and check fluorescence microscope settings and filters. Increasing probe concentration or hybridization time may also help [2].
Q: How can I reduce high background noise in my FISH assays? A: Optimize post-hybridization wash conditions by increasing stringency (temperature, salt concentration, and duration). Ensure complete denaturation of target DNA/RNA and check for probe cross-reactivity with non-target sequences. Using blocking solutions effectively and optimizing probe concentration are also key [2].
Q: Why do I get uneven or patchy signals across my sample? A: This is often due to non-uniform distribution of the probe during hybridization or uneven permeabilization and denaturation of the sample. Avoid air bubbles and ensure the sample does not dry out during any step of the procedure. Using a template for consistent probe application can improve uniformity [2].
Q: How does sample fixation impact FISH reproducibility? A: Fixation is crucial. Over-fixation can reduce target accessibility, leading to weak signals, while under-fixation can degrade morphology and nucleic acids. Use appropriate fixatives like formaldehyde or paraformaldehyde and optimize fixation time and concentration for your specific sample type [2].
Troubleshooting Guide
The following table summarizes common FISH issues, their potential causes, and recommended solutions to ensure reproducible and high-quality results.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Poor or No Signal [2] | Inadequate permeabilization, insufficient denaturation/hybridization, inactive probe. | Optimize permeabilization agents (Triton X-100, proteinase K) and conditions; verify denaturation (95±5°C for 5-10 min [4]); check probe activity and increase concentration. |
| High Background [4] [2] | Inadequate stringent washes, non-specific probe binding, sample drying. | Increase stringency of SSC washes (75-80°C [4]); optimize probe design; use blocking agents (e.g., COT-1 DNA [4]); ensure slides remain humidified. |
| Weak/Faded Signal [2] | Fluorophore quenching, over-fixed sample, signal degradation. | Use antifade mounting medium; minimize light exposure; optimize fixation time; consider signal amplification methods (e.g., Tyramide Signal Amplification [4]). |
| Morphological Distortion [2] | Over-fixation, over-permeabilization, harsh tissue handling. | Optimize fixation and permeabilization conditions; use gentler dissociation methods for cells/tissues. |
| Autofluorescence [7] | Inadequate fixation, endogenous fluorophores in FFPE tissues. | Implement optimized pretreatment protocols; use light and reagent controls to identify and mitigate specific causes [7]. |
Experimental Protocol: Blocking Solution Optimization for FISH
Background
Blocking is a critical step to reduce non-specific probe binding and lower background noise, which is essential for achieving a high signal-to-noise ratio. This protocol outlines a method to systematically optimize blocking conditions for challenging samples, such as Formalin-Fixed Paraffin-Embedded (FFPE) tissues, which are prone to high background [7].
Materials
- Standard saline citrate (SSC) buffer
- Formamide
- Dextran sulfate
- Denatured salmon sperm DNA
- COT-1 DNA (human or species-specific)
- tRNA
- BSA (Bovine Serum Albumin)
- Detergent (e.g., Tween 20)
- Humidified hybridization chamber
Procedure
- Prepare Base Hybridization Buffer: Create a standard hybridization buffer containing SSC, formamide, dextran sulfate, and a baseline concentration of denatured salmon sperm DNA.
- Design Blocking Experiment: Prepare several aliquots of the base hybridization buffer, supplementing them with different blocking agents as outlined in the table below.
- Apply Probe Mixture: For each experimental condition, mix your FISH probe with the corresponding blocking buffer. Denature the probe mixture according to your standard protocol (e.g., 85°C for 5 minutes for some Rembrandt kit probes [4]).
- Hybridize: Apply the probe-blocking mixture to your pre-treated and denatured tissue sections. Perform hybridization overnight (~16 hours) in a humidified chamber at 37°C [4].
- Wash and Detect: The following day, perform stringent washes (e.g., in SSC at 75-80°C for 5 minutes [4]) to remove unbound probe. Proceed with your standard detection protocol.
- Evaluate: Image all slides under identical microscope settings. Compare the signal intensity and background levels across the different blocking conditions to identify the optimal formulation.
The table below provides a structured framework for testing different blocking agent combinations to systematically optimize your FISH protocol.
| Condition | Base Buffer | Additional Blocking Agents | Expected Outcome |
|---|---|---|---|
| A (Control) | Standard Hybridization Buffer | Denatured salmon sperm DNA only | Baseline signal and background. |
| B (Protein Block) | Standard Hybridization Buffer | Denatured salmon sperm DNA + 1-5% BSA | Reduction in background from non-specific protein binding. |
| C (RNA Block) | Standard Hybridization Buffer | Denatured salmon sperm DNA + tRNA (50-100 µg/mL) | Reduction in background from non-specific RNA binding. |
| D (Repeat Block) | Standard Hybridization Buffer | Denatured salmon sperm DNA + COT-1 DNA (1-10 µg/mL) | Significant reduction in background from repetitive sequences (e.g., Alu, LINE) [4]. |
| E (Combo Block) | Standard Hybridization Buffer | Denatured salmon sperm DNA + BSA + COT-1 DNA | Greatest reduction in complex, high background; may slightly reduce specific signal. |
Workflow and Signaling Diagrams
FISH Optimization Workflow
Signal-to-Noise Optimization Logic
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents used in FISH experiments, along with their primary functions.
| Reagent | Function in FISH |
|---|---|
| Formaldehyde/PFA | Fixative that preserves cell/tissue morphology and maintains the integrity of target nucleic acids [2]. |
| Proteinase K | Permeabilization enzyme that digests proteins, allowing probe access to the target nucleic acids [2]. |
| Triton X-100/Tween-20 | Detergents used for permeabilization of cell and tissue membranes [2]. |
| Formamide | A denaturing agent included in hybridization buffers to lower the melting temperature (Tm), allowing hybridization to occur at lower, less destructive temperatures [4]. |
| Dextran Sulfate | A polymer added to hybridization buffers to increase viscosity and probe concentration, enhancing the hybridization kinetics and signal intensity. |
| COT-1 DNA | Blocking agent used to suppress non-specific hybridization of probe to repetitive DNA sequences (e.g., Alu, LINE elements), reducing background [4]. |
| Salmon Sperm DNA | A non-specific DNA blocker used to bind to and saturate areas of non-specific charge interaction on the sample, reducing background noise. |
| BSA (Bovine Serum Albumin) | A protein blocker used to reduce non-specific binding of probes or detection reagents to proteins in the sample [2]. |
| SSC Buffer (Saline-Sodium Citrate) | A salt solution used during hybridization and stringent washes; its concentration and temperature determine the stringency of the washing process [4]. |
| DAPI | A DNA-binding fluorescent dye used as a counterstain to visualize the nuclei and overall cell architecture [2]. |
Assaying Performance: Validation and Quality Control for Blocking Solutions
Establishing Effective Positive and Negative Controls
Frequently Asked Questions
Q1: What constitutes an effective positive control for a FISH experiment? An effective positive control is a sample with a known, verified genetic abnormality or expression pattern that your FISH probe targets. This control should be processed identically to your test samples. A successful result in the positive control, where the expected fluorescent signal is clear and specific, confirms that every step of your FISH protocol—from sample preparation and denaturation to hybridization and washing—was performed correctly. It validates the functionality of your probes and the entire assay.
Q2: Why is my positive control showing a weak or absent signal? A weak or absent signal in your positive control indicates a potential failure in one or more steps of the FISH procedure [2] [23]. Consider the following troubleshooting steps:
- Probe Integrity: Verify the quality of your probe. Check for efficient dye incorporation and ensure the probe fragments are of the correct length (typically a smear between 100-250 bp for DNA probes) [23].
- Sample Permeabilization: Inadequate permeabilization prevents the probe from reaching its target. Optimize the concentration, time, and temperature of your permeabilization agent (e.g., Triton X-100, proteinase K) [2].
- Denaturation Conditions: Ensure the target DNA and probe are completely denatured. Suboptimal denaturation temperature or time will hinder hybridization [2] [23].
- Hybridization Stringency: The hybridization temperature or salt concentration in the buffer may be incorrect. Typically, hybridization occurs between 55°C and 62°C; carefully tune these parameters for specificity [23].
Q3: What is the purpose of a negative control, and what types are available? A negative control is used to identify non-specific binding and false-positive signals. A successful negative control, which shows no specific fluorescence, confirms the specificity of your probe and the stringency of your wash conditions [2]. Common types include:
- No-Probe Control: A sample processed without any FISH probe applied. This detects background autofluorescence from the sample or mounting medium.
- Sense Probe Control: A sample hybridized with a probe that is complementary to the non-coding strand, which should not hybridize to the target mRNA in an RNA-FISH experiment.
- Cell/Locus Control: A sample from a cell line or tissue known not to contain the genetic target you are probing for.
Q4: My negative control shows high background fluorescence. How can I resolve this? High background in the negative control is caused by non-specific probe binding or insufficient washing [2]. To resolve this:
- Increase Wash Stringency: Perform more stringent post-hybridization washes by adjusting the temperature, salt concentration (SSC), or duration. Higher temperatures and lower salt concentrations increase stringency [2] [23].
- Optimize Probe Concentration: A too-high probe concentration can lead to non-specific binding. Titrate your probe to find the optimal concentration that gives a strong specific signal with low background [23].
- Use Cot-1 DNA: Include unlabeled Cot-1 DNA in your hybridization mix to block repetitive DNA sequences and reduce non-specific hybridization [23].
- Check Fixation: Avoid over-fixation, as this can trap biomolecules and increase autofluorescence. Ensure fixation times and concentrations are optimized [2] [23].
Experimental Protocols for Control Validation
The following table outlines a standard FISH protocol with key steps for control validation [2] [23].
Table 1: Standardized FISH Protocol with Critical Control Checkpoints
| Step | Protocol Description | Control Validation Focus |
|---|---|---|
| Sample Preparation | Use healthy, actively growing cells or fresh tissues. Fix with formaldehyde or paraformaldehyde (avoid over-fixation beyond 24 hours). For cells, use a 3:1 methanol/acetic acid solution. | Preserve nucleic acid integrity and cell morphology. Ensure samples are comparable across tests and controls. |
| Permeabilization | Treat with agents like Triton X-100, Tween-20, or proteinase K. | Balance between allowing probe access and maintaining morphology. Critical for signal strength in positive control. |
| Denaturation | Denature target DNA and probe using heat or alkaline treatment. | Ensure complete denaturation to make nucleic acids single-stranded. Failure here affects both controls and tests. |
| Hybridization | Apply labeled probe in appropriate buffer. Incubate at 55-62°C in a humidified chamber for 4-16 hours. | Prevents evaporation and patchy hybridization. Temperature is key for specificity (negative control). |
| Post-Hybridization Washes | Perform stringent washes with SSC buffer. | Remove unbound/non-specifically bound probes. Primary step for reducing background (negative control). |
| Counterstaining & Mounting | Apply DAPI or propidium iodide. Use antifade mounting medium. | Visualize nuclei. Prevent photobleaching. Check for even signal distribution. |
Quantitative Data for Control Assessment
Establishing expected signal metrics is crucial for objectively assessing your controls and experimental results.
Table 2: Quantitative Signal Assessment Guide for FISH Controls
| Metric | Positive Control Expectation | Negative Control Expectation | Troubleshooting Guidance |
|---|---|---|---|
| Signal-to-Noise Ratio | High (Clear, distinct signals over minimal background). | Low (No specific signals; background should be minimal). | Low ratio in positive control: Check probe quality and denaturation. High ratio in negative: Increase wash stringency. |
| Signal Intensity | Bright, easily detectable fluorescence. | No specific fluorescent foci. | Weak signal: Optimize permeabilization, increase probe concentration/hybridization time. |
| Background Level | Low, uniform background across the sample. | Low, uniform background across the sample. | High, uneven background: Increase wash stringency, optimize probe concentration, check for over-fixation. |
| Specificity | >95% of target cells show the expected signal pattern. | >95% of cells show no specific signal. | Non-specific signals: Verify probe specificity, include Cot-1 DNA, re-optimize hybridization temperature. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for FISH Control Experiments
| Reagent / Solution | Function in FISH Protocol |
|---|---|
| Formaldehyde/Paraformaldehyde | Crosslinking fixative that preserves cell and tissue morphology and maintains the integrity of target nucleic acids. |
| Protease (e.g., Proteinase K) | Enzyme used for permeabilization; digests proteins to allow probe access to the intracellular target. |
| Formamide | Component of hybridization buffer; allows hybridization to occur at lower temperatures, preserving sample morphology. |
| Cot-1 DNA | Unlabeled DNA enriched for repetitive sequences; used as a blocking agent to suppress non-specific hybridization. |
| Saline-Sodium Citrate (SSC) Buffer | A salt buffer used during hybridization and post-hybridization washes; concentration and temperature determine stringency. |
| DAPI (4',6-diamidino-2-phenylindole) | A fluorescent DNA-binding dye used as a counterstain to visualize the nucleus and overall chromosome morphology. |
| Antifade Mounting Medium | Preserves fluorescence by reducing photobleaching during microscopy and storage. |
Experimental Workflow and Troubleshooting Diagrams
FISH Control Validation Workflow
FISH Control Troubleshooting Guide
Comparative Analysis of Different Blocking Agents and Formulations
In molecular biology techniques such as Fluorescence In Situ Hybridization (FISH) and flow cytometry, blocking is a critical preparatory step that determines the specificity and quality of experimental results. Effective blocking prevents non-specific binding of probes or antibodies by occupying reactive sites on samples and membranes, thereby reducing background noise and improving the signal-to-noise ratio. This technical guide provides a comparative analysis of various blocking agents and formulations, offering researchers troubleshooting guidance and optimized protocols to address common experimental challenges. Proper blocking strategy implementation is essential for generating reliable, reproducible data in both diagnostic and research applications.
Key Blocking Agent Types and Their Applications
The selection of an appropriate blocking agent is highly dependent on the specific application, detection system, and sample type. No single blocking agent is ideal for every situation, as each has distinct advantages and limitations.
Table 1: Characteristics of Common Blocking Agents
| Blocking Agent | Optimal Concentration | Primary Applications | Advantages | Limitations |
|---|---|---|---|---|
| Normal Serum | 1-5% [6] | Flow Cytometry (Fc receptor blocking) [38] | Inexpensive; effective for Fc-mediated binding [38] | Lot-to-lot variation; may contain activating compounds [38] |
| Bovine Serum Albumin (BSA) | 2-3% [31] | Western Blot (phosphoprotein detection) [31] | Low biotin content; compatible with streptavidin systems [31] | Weaker blocking can lead to non-specific binding [31] |
| Non-Fat Dry Milk | 2-5% [31] | General Western Blotting [31] | Inexpensive; contains multiple protein types [31] | Contains biotin & phosphoproteins; may mask antigens [31] |
| Purified Proteins (Casein, etc.) | Varies by product [31] | High-sensitivity applications [31] | Fewer cross-reactions; defined composition [31] | More expensive than traditional options [31] |
| Specialized Commercial Blockers | As per manufacturer | Fluorescent detection, challenging systems [31] | Optimized for specific techniques; consistent performance [31] | Higher cost; proprietary formulations [31] |
Experimental Protocols for Optimal Blocking
Standardized Blocking Protocol for High-Parameter Flow Cytometry
This protocol provides an optimized, general-use approach for reducing non-specific interactions in high-parameter flow cytometry, incorporating strategies to address Fc receptor binding, dye-dye interactions, and tandem dye breakdown [6].
Materials Required:
- Mouse serum (Thermo Fisher, cat. no. 10410 or equivalent)
- Rat serum (Thermo Fisher, cat. no. 10710C or equivalent)
- Tandem stabilizer (BioLegend, cat. no. 421802)
- Brilliant Stain Buffer (Thermo Fisher, cat. no. 00-4409-75) or BD Horizon Brilliant Stain Buffer Plus (BD Biosciences, cat. no. 566385)
- FACS buffer
- Sterilin clear microtiter plates, 96-well V-bottom (Fisher Scientific, cat. no. 1189740)
- Centrifuge
- 20- and 200-µl multichannel pipettes and tips
- Flow cytometer
Step-by-Step Procedure:
Prepare Blocking Solution: Create a blocking solution comprised of rat serum, mouse serum, tandem stabilizer, and serum from any other host species present in your antibody panel according to the following formulation [6]:
- Mouse serum: 300 μL (for 1-mL total mix)
- Rat serum: 300 μL
- Tandem stabilizer: 1 μL (1:1000 dilution)
- Sodium azide (10%): 10 μL (optional for short-term use)
- FACS buffer: 389 μL (remaining volume)
Cell Preparation: Dispense cells into V-bottom, 96-well plates for staining. Cell numbers should be standardized to reduce batch effects [6].
Initial Blocking: Centrifuge plates at 300 × g for 5 minutes at 4°C or room temperature and remove supernatant. Resuspend cells in 20 μL blocking solution and incubate for 15 minutes at room temperature in the dark [6].
Staining Master Mix Preparation: While blocking, prepare surface staining master mix [6]:
- Tandem stabilizer: 1 μL (1:1000 dilution)
- Brilliant Stain Buffer: 300 μL (up to 30% v/v)
- Antibodies: As appropriate for your panel
- FACS buffer: Remaining volume to 1 mL
Staining: Add 100 μL surface staining mix to each sample and mix by pipetting. Incubate for 1 hour at room temperature in the dark [6].
Washing: Wash with 120 μL FACS buffer, centrifuge for 5 minutes at 300 × g, and discard supernatant. Repeat wash with 200 μL FACS buffer [6].
Sample Acquisition: Resuspend samples in FACS buffer containing tandem stabilizer at 1:1000 dilution and acquire on your cytometer [6].
FISH Blocking and Pre-Treatment Protocol
Effective FISH requires careful sample preparation and pre-treatment to ensure probe accessibility while maintaining sample morphology [2] [23].
Sample Fixation and Preparation:
- For tissue samples: Fix with formalin or paraformaldehyde. Avoid precipitating fixatives like alcohols. Do not exceed 24 hours of fixation to prevent increased autofluorescence and reduced probe penetration [23].
- For cell samples: Use healthy, actively growing cells. Treat with a hypotonic solution (e.g., sodium citrate and BSA) and then fix with a freshly prepared 3:1 methanol/acetic acid solution [23].
- Section thickness: Optimal section thickness is 3-4μm. Thicker sections impede probe penetration, while thinner sections may truncate signals [23].
Slide Preparation and Pre-Treatment:
- Pre-clean glass slides with 70% ethanol before use. For enhanced adhesion, treat with poly-lysine [23].
- Apply appropriate pre-treatment based on sample type:
- For FFPE sections: Ensure proper dewaxing
- For all sample types: Optimize permeabilization using proteases, detergents, or alcohols
- Denature target sequences using heat or pH adjustment
- Age cell spreads if necessary [23]
Hybridization and Post-Hybridization Washes:
- Hybridization temperature typically ranges between 55°C and 62°C - this is the most critical variable for specificity [23].
- Include formamide in hybridization buffer to allow lower hybridization temperatures and preserve sample morphology [23].
- Include Cot DNA sequences to reduce nonspecific hybridization to repetitive DNA sequences [23].
- Control humidity to prevent drying or overconcentration of the solution [23].
- Perform stringent post-hybridization washes with gradually increasing stringency to remove weak, non-specific interactions [23].
Frequently Asked Questions (FAQs)
Q1: What is the most effective blocking strategy for flow cytometry with mouse immune cells? For mouse immune cells stained primarily with rat antibodies, a combination of normal rat serum (for Fc receptor blocking) and Brilliant Stain Buffer (for dye-dye interactions) is highly effective [6] [38]. Normal serum from the same species as your primary antibodies provides species-matched immunoglobulins that effectively compete for Fc receptor binding sites [38].
Q2: Why might my Western blot show high background even after blocking? High background can result from insufficient blocker concentration, inadequate blocking time, or using an inappropriate blocking agent for your detection system [31]. For chemiluminescent detection with biotin-streptavidin systems, avoid milk-based blockers as they contain biotin that can interfere [31]. Instead, use BSA or specialized commercial blockers. Also ensure your blot doesn't dry out during the blocking or antibody incubation steps [31].
Q3: How can I reduce non-specific binding in FISH experiments? Several strategies can reduce FISH background: (1) Ensure proper sample fixation without exceeding 24 hours; (2) Optimize permeabilization to balance accessibility and morphology preservation; (3) Use the correct hybridization temperature (55-62°C); (4) Include Cot DNA in hybridization buffer to block repetitive sequences; (5) Perform stringent post-hybridization washes [2] [23].
Q4: What are the benefits of specialized commercial blocking buffers versus traditional options? Specialized commercial blockers offer consistent, optimized formulations with defined composition, eliminating the lot-to-lot variability seen with normal sera [31]. They are often designed for specific applications (e.g., fluorescent detection) and are frequently biotin-free, making them ideal for streptavidin-based detection systems [31]. The main trade-off is the higher cost compared to traditional options like BSA or milk.
Q5: How do I handle tandem dye breakdown in flow cytometry panels? Tandem dye breakdown can be minimized by: (1) Using tandem stabilizer in your staining buffer; (2) Staining with tandem dyes after fixation (for intracellular targets); (3) Strategic panel design that places tandem dyes on post-fix T cell markers rather than monocytes; (4) Proper storage and handling of tandem-conjugated antibodies protected from light [6] [39].
Troubleshooting Guide
Table 2: Common Blocking Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Background Staining | Inadequate Fc receptor blocking [38]Insufficient blocking concentration/time [31]Wrong blocking agent for detection system [31] | Use species-appropriate normal serum or purified IgG [38]Increase blocker concentration or duration [31]Switch to BSA for biotin-streptavidin systems [31] |
| Weak or No Signal | Over-blocking masking antigens [31]Incompatible blocking agent [31]Fluorochrome fading [40] | Reduce blocking concentration/time [31]Test alternative blockers (e.g., casein vs. BSA) [31]Use fresh antibodies protected from light [40] |
| Non-Specific Signal in FISH | Inadequate permeabilization [2]Suboptimal hybridization stringency [23]Sample over-fixation [23] | Optimize permeabilization conditions [2]Adjust hybridization temperature/stringency [23]Limit fixation to ≤24 hours [23] |
| Uneven Staining | Uneven blocker distribution [2]Air bubbles during mounting [2]Inconsistent sample preparation [2] | Ensure uniform application of solutions [2]Avoid bubbles during mounting [2]Standardize sample prep protocols [2] |
Visual Guide: Blocking Optimization Workflow
The following diagram illustrates a systematic approach to troubleshooting and optimizing blocking conditions in experimental workflows:
Systematic Troubleshooting for Blocking Issues
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Blocking Optimization
| Reagent Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Normal Sera | Rat serum, Mouse serum [6] | Fc receptor blocking | Use serum from same species as primary antibodies [38] |
| Specialized Buffers | Brilliant Stain Buffer, Tandem Stabilizer [6] | Prevent dye-dye interactions, tandem breakdown | Essential for polymer dyes (Brilliant, Super Bright) [6] |
| Purified Proteins | BSA, Casein, purified IgG [31] | Membrane blocking, non-specific site occupation | BSA ideal for phosphoprotein detection & biotin-free systems [31] |
| Commercial Blockers | StartingBlock, Blocker Casein, SuperBlock [31] | Ready-to-use optimized blocking | Consistent performance; application-specific formulations [31] |
| Detergents | Tween-20, Triton X-100 [2] [31] | Reduce hydrophobic interactions | Add to wash buffers (0.05-0.2%); weak antibodies may be affected [31] |
Effective blocking is a foundational element of successful experimental outcomes in techniques ranging from FISH to flow cytometry. The optimal blocking strategy depends on multiple factors including sample type, detection method, and specific reagents employed. By understanding the mechanisms of non-specific binding and implementing the systematic troubleshooting approaches outlined in this guide, researchers can significantly improve their assay sensitivity, specificity, and reproducibility. Continual optimization and validation of blocking protocols remain essential as new reagents and detection technologies emerge in the field of molecular biology.
Troubleshooting Guides
What are the primary causes of high background staining in FISH, and how can blocking optimize it?
High background staining, or non-specific signal, is a common issue that effective blocking and optimized washing can resolve.
| Cause | Troubleshooting Strategy | Role of Blocking Optimization |
|---|---|---|
| Insufficient blocking | Increase blocker concentration; extend blocking time; ensure blocker is fresh and properly prepared. | Blocking agents like BSA or casein saturate non-specific binding sites on the sample and membrane. |
| Inadequate post-hybridization washes | Perform stringent washes with appropriate salt concentration (e.g., SSC buffer) and temperature (75–80°C); ensure wash buffers are used correctly [41]. | Optimal blocking reduces the initial non-specific probe attachment, making it easier to wash away any remaining unbound probes. |
| Over-digestion during pretreatment | Optimize enzyme (e.g., pepsin) concentration and incubation time (typically 3-10 minutes at 37°C) [41]. | Over-digestion can damage tissue morphology, creating more non-specific sites that the blocker must cover. |
| Sample drying during procedure | Ensure slides remain covered and in a humidified chamber during all incubation steps, especially hybridization [2]. | Drying can concentrate salts and probes, overwhelming the blocking agent's capacity. |
How do I troubleshoot a weak or absent FISH signal?
A weak or absent signal can result from issues with probe accessibility or integrity, often related to pre-hybridization steps.
| Cause | Troubleshooting Strategy | Role of Blocking Optimization |
|---|---|---|
| Over-fixation of sample | Optimize fixation time and concentration (e.g., 24 hours in 10% buffered formalin) [42]. | Over-fixation excessively cross-links proteins, hindering probe access to the target; blocking cannot overcome this physical barrier. |
| Inadequate permeabilization | Optimize permeabilization conditions (e.g., concentration of Triton X-100, time, temperature) [2]. | Insufficient permeabilization prevents the blocker and probe from reaching all intracellular targets. |
| Under-digestion during pretreatment | Titrate proteinase K or pepsin digestion time to increase target accessibility without damaging morphology [2]. | Like over-fixation, under-digestion leaves targets masked. |
| Denaturation issues | Ensure denaturation is performed at 95±5°C for 5-10 minutes on a calibrated hot plate [41]. | Incomplete denaturation of target DNA prevents probe hybridization. |
What factors contribute to uneven or patchy hybridization signals?
Uneven signals often stem from inconsistencies in sample preparation or reagent application.
| Cause | Troubleshooting Strategy | Role of Blocking Optimization |
|---|---|---|
| Non-uniform reagent application | Apply probes and blocking buffers carefully; use a template to ensure consistent placement; avoid air bubbles and squeezing of coverslips [2]. | Ensures the blocking agent uniformly covers the entire sample. |
| Inconsistent permeabilization or denaturation | Ensure samples are treated evenly across slides; use calibrated equipment for temperature-critical steps. | Creates a uniform landscape for the blocker to act upon. |
| Uneven sample thickness or adhesion | Use standardized section thickness and appropriate charged or silanized slides to ensure good adhesion [42]. | A uniform sample allows for even binding of the blocking agent. |
Frequently Asked Questions (FAQs)
What is the fundamental mechanism of blocking in FISH?
Blocking works by saturating non-specific binding sites on the tissue sample and the surrounding matrix with inert proteins or polymers. This prevents the fluorescently labeled probes from attaching to these sites, thereby reducing background noise and enhancing the specificity of the true signal from the target nucleic acid sequence [2].
How does the choice of blocking agent impact FISH results?
The optimal blocking agent depends on the specific experiment. Here is a comparison of common blockers:
| Blocking Agent | Best For | Considerations |
|---|---|---|
| Bovine Serum Albumin (BSA) | General use; often a good starting point; phosphoprotein detection. | Lacks phosphoproteins that can cause interference; highly purified [43]. |
| Non-Fat Dry Milk | General use when cost is a factor. | May contain phosphoproteins and biotin, which can cause high background with certain targets or detection systems [43]. |
| Casein | High-sensitivity applications; reducing non-specific binding. | Effective at minimizing background [43]. |
| Fish Gelatin | Reducing background in problematic assays. | Can be effective where other protein-based blockers fail. |
| Commercial Blocking Buffers | Standardized, ready-to-use formulations for consistency. | Often optimized for specific applications or detection methods. |
Should Tween 20 be included in the blocking buffer for fluorescent detection?
The inclusion of Tween 20, a non-ionic detergent, requires careful consideration. It helps reduce non-specific hydrophobic interactions and prevents excessive binding of blockers to the membrane. However, if not thoroughly washed out before the slides are dried, Tween 20 can itself autofluoresce and create high background [44]. A best practice is to include Tween 20 in the blocking and washing buffers but ensure a final wash in TBS or SSC without detergent before drying and imaging.
How long should the blocking incubation be, and at what temperature?
A typical blocking incubation is 30 minutes to 1 hour at room temperature with gentle agitation. For more challenging samples with high background, blocking can be extended overnight at 4°C to enhance efficiency. The optimal condition should be determined empirically [2] [43].
How does sample fixation affect blocking efficiency?
Fixation preserves tissue morphology but can mask target sequences. Over-fixation (using too high a concentration or too long a time) creates excessive cross-links, making it difficult for both the blocking agent and the probe to access their sites. This can lead to high background and a weak specific signal. Adhering to standardized fixation protocols (e.g., 24 hours in 10% neutral buffered formalin) is crucial for reproducible blocking and hybridization [42].
Experimental Protocol: Standardization of a FISH Blocking Procedure
This protocol is adapted from a study on HER-2 FISH standardization, which achieved a 98.6% hybridization success rate [42].
Methodology
Sample Preparation:
- Use formalin-fixed, paraffin-embedded (FFPE) tissue sections cut at 4µm thickness.
- Mount on positively charged or silanized slides to ensure adhesion.
- Bake slides at 56°C overnight or at 70°C for 35 minutes to melt paraffin and adhere tissue.
Deparaffinization and Hydration:
- Deparaffinize slides in xylene (or substitute) followed by a graded ethanol series (100%, 95%, 70%) and finally rehydrate in deionized water.
Pretreatment and Digestion (Critical for Accessibility):
- Heat-Induced Epitope Retrieval: Immerse slides in preheated citrate or EDTA-based buffer (pH 6.0-9.0) and heat for 15-40 minutes at 95-100°C. Cool slides to room temperature.
- Enzymatic Digestion: Treat slides with pepsin (0.5-4 mg/mL) or proteinase K at 37°C for 3-20 minutes. This step must be optimized for each tissue type. Over-digestion damages morphology, while under-digestion reduces signal.
Blocking (The Key Step):
- Prepare a blocking solution of 2-5% BSA in TBST (Tris-Buffered Saline with 0.1% Tween 20).
- Apply enough solution to cover the tissue section completely.
- Incubate in a humidified chamber for 1 hour at room temperature (or overnight at 4°C for difficult samples).
Denaturation and Hybridization:
- Denature target DNA and probe according to kit manufacturer's instructions (typically 5-10 minutes at 95±5°C).
- Apply the denatured probe to the tissue, add a coverslip, and seal with rubber cement.
- Hybridize in a dark, humidified chamber overnight (16 hours) at 37°C.
Post-Hybridization Washes and Detection:
- Remove coverslips carefully and perform a stringent wash in 0.5X SSC or 2X SSC / 0.1% NP-40 buffer at 75-80°C for 5-10 minutes [41].
- Wash slides in TBST at room temperature.
- Apply a DAPI counterstain (e.g., 100-200 µL of a 125 ng/mL solution) for 10-20 minutes in the dark.
Mounting and Visualization:
- Mount slides with an anti-fade mounting medium.
- Visualize signals using a fluorescence microscope with appropriate filters.
Workflow Diagram
Troubleshooting Decision Pathway
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in FISH (Especially Blocking) |
|---|---|
| Bovine Serum Albumin (BSA) | A purified protein used as a primary blocking agent to saturate non-specific binding sites, effectively reducing background. |
| Non-Fat Dry Milk | A cost-effective protein-based blocking agent. Avoid for phospho-targets or biotin-streptavidin systems due to potential interference. |
| Casein | A milk-derived protein known for providing a "clean" background with low non-specific binding in many applications. |
| Fish Gelatin | A protein blocker useful for reducing background in assays where mammalian proteins might cause cross-reactivity. |
| Tween 20 | A non-ionic detergent added to blocking and wash buffers to reduce hydrophobic interactions and prevent non-specific adhesion. |
| Tris-Buffered Saline (TBS) | A common buffer used to prepare blocking solutions and for washing steps. Preferred over PBS for fluorescent detection to minimize autofluorescence. |
| Proteinase K / Pepsin | Enzymes used for tissue digestion to unmask target nucleic acid sequences, making them accessible to probes and blocking agents. |
| Formalin/Paraformaldehyde | Cross-linking fixatives used to preserve tissue morphology and immobilize nucleic acids in situ. |
Correlating FISH Results with Orthogonal Techniques like qPCR or Sequencing
Technical Support Center
Fluorescence in situ hybridization (FISH) serves as a powerful cytogenetic technique for visualizing specific DNA or RNA sequences within cells and tissues. However, as with any analytical method, FISH results can sometimes yield unexpected or ambiguous findings. In these situations, correlation with orthogonal techniques like quantitative PCR (qPCR) or sequencing becomes essential for validation and accurate interpretation. This technical guide addresses common challenges researchers face when integrating these methodologies, providing troubleshooting advice and protocols to ensure data reliability within the context of blocking solution and general assay optimization.
Troubleshooting Guides & FAQs
FAQ: What are the primary reasons for discordant results between FISH and qPCR?
Answer: Discordant results often stem from the fundamental differences in what each technique measures. The table below summarizes common causes and their resolutions.
Table 1: Resolving Discordant Results Between FISH and qPCR
| Cause of Discordance | FISH Result | qPCR Result | Recommended Action |
|---|---|---|---|
| Spatial vs. Bulk Analysis | Detects a genetic abnormality in a subset of cells. | Averages nucleic acid content across the entire sample, diluting the signal from a small abnormal population. | Correlate with patient history; consider if mosaicism or minimal residual disease is plausible [45]. |
| Sequence Variation | Probe fails to hybridize due to a sequence variant in the target region. | Amplification may still occur with lower efficiency, yielding an unexpected Cq value. | Redesign the FISH probe or verify the assay's performance against carefully quantified controls [46]. |
| Sample Quality | Poor sample morphology or over-fixation can reduce signal. | PCR inhibitors in the sample can lead to poor amplification efficiency and higher-than-expected Cq values. | Use a different sample region; dilute the sample to reduce inhibitors; treat RNA samples with DNAse [2] [46]. |
| Technical Artifacts | High background or poor probe penetration. | Primer-dimer formation, contaminated reagents, or inaccurate baseline adjustment in amplification plots. | Optimize wash stringency and permeabilization; run a No Template Control (NTC); check primer specificity and pipetting accuracy [2] [46]. |
FAQ: How should we handle ambiguous FISH findings?
Answer: Ambiguous FISH results, such as unexpected signal patterns or weak signals, require a systematic approach to rule out technical artifacts and confirm the genetic finding.
- Verify Probe Specificity: Check for potential cross-reactivity of the probe with non-target sequences or repetitive regions [2].
- Check for Sequence Variations: Use sequencing to identify potential single nucleotide polymorphisms (SNPs) or other sequence changes in the probe-binding region that could prevent hybridization [2].
- Confirm with an Alternative FISH Probe: If available, use a different FISH probe targeting another region of the same gene or locus.
- Correlate with Karyotype or Microarray: Ambiguous structural rearrangements or marker chromosomes identified by FISH can be further clarified using karyotyping or microarray analysis to provide a genome-wide view [45].
FAQ: Our qPCR validation for a FISH-identified deletion shows a later Cq but not the expected magnitude. Why?
Answer: This is a common issue. A standard qPCR assay might not be sensitive enough to detect a heterozygous deletion, especially if the sample is a mixture of normal and abnormal cells (e.g., contaminated with normal tissue). The FISH result provides spatial information, showing that only a proportion of cells carry the deletion, while the qPCR result reflects the average gene dosage across all cells.
- Troubleshooting Steps:
- Redesign Primers/Probe: Ensure your qPCR assay is highly efficient. The slope of the standard curve should be between -3.1 and -3.6, with an R² value >0.98 [46].
- Use Digital PCR (dPCR): For absolute quantification of copy number variations, dPCR is a superior orthogonal method to qPCR as it is less influenced by amplification efficiency and can provide a more precise measure of gene dosage [45].
- Check Sample Purity: Ensure the analyzed sample for qPCR is representative of the area analyzed by FISH.
Experimental Protocols for Validation
Protocol 1: Validating a FISH-Identified Gene Amplification using qPCR
This protocol is designed to confirm a gene amplification (e.g., HER2 in breast cancer) suspected from FISH analysis.
1. Sample and Reagent Preparation
Table 2: Research Reagent Solutions for Validation Experiments
| Item | Function |
|---|---|
| FISH Probes (Locus-specific) | Binds to the target genomic region to visualize its copy number and location [47]. |
| DNA Extraction Kit | Iserts high-quality, high-molecular-weight DNA from the same sample used for FISH. |
| qPCR Master Mix | Contains DNA polymerase, dNTPs, and optimized buffers for efficient amplification. |
| Hydrolysis Probes (e.g., TaqMan) | Fluorescently-labeled probes that provide sequence-specific detection and high specificity in qPCR. |
| Primers (Target & Reference) | Amplify the target gene of interest and a stable reference gene (e.g., on a non-amplified chromosome). |
2. Workflow Diagram
3. Procedure
- Step 1: Parallel Sample Processing. Section the sample (e.g., FFPE tissue block). Use consecutive sections for FISH and DNA extraction to ensure analysis of the same cell population [45].
- Step 2: FISH Analysis. Perform FISH according to standard protocols, using a locus-specific probe for the target gene. Count amplification signals across multiple cells to confirm the initial finding [2] [47].
- Step 3: DNA Extraction and qPCR.
- Extract DNA from the dedicated section, quantifying it with a spectrophotometer.
- Designate a confirmed normal sample as a calibrator.
- Run the qPCR assay in triplicate for both the target gene and a reference gene on all test samples and the calibrator. Use a negative control (no template) to check for contamination.
- Step 4: Data Analysis.
- Calculate the ΔCq for each sample (Cqtarget - Cqreference).
- Calculate the ΔΔCq (ΔCqsample - ΔCqcalibrator).
- The fold-change is given by 2^(-ΔΔCq). A fold-change significantly greater than 2.0 supports the presence of gene amplification.
Protocol 2: Using Sequencing to Resolve Unexpected FISH Signals
This protocol is used when a FISH probe shows no signal, a weak signal, or an atypical binding pattern, suggesting a possible sequence variant.
1. Workflow Diagram
2. Procedure
- Step 1: Generate Hypothesis. Based on weak or absent FISH signal, hypothesize a sequence variant preventing probe binding [2].
- Step 2: PCR Amplification. Design primers to amplify the genomic region targeted by the FISH probe. Use DNA from the same sample.
- Step 3: Sequencing. Purify the PCR product and submit it for Sanger sequencing.
- Step 4: Analysis. Align the sequenced DNA against the reference human genome. Look for single nucleotide polymorphisms (SNPs), insertions, or deletions (indels) within the probe's target sequence.
- Step 5: Validation. The definitive validation involves designing a new FISH probe that accounts for the identified sequence variant. Successful hybridization with the new probe confirms the hypothesis.
Conclusion
Optimizing the blocking solution is not an isolated step but a foundational element that permeates every phase of a successful FISH protocol, directly impacting signal-to-noise ratio, assay reproducibility, and diagnostic accuracy. As highlighted throughout this guide, effective blocking requires a meticulous, integrated approach that considers sample preparation, probe design, and hybridization stringency. Future directions point towards the development of more robust, standardized commercial blocking reagents and the integration of artificial intelligence and digital pathology to provide quantitative, automated assessment of FISH signal quality. Embracing these optimized and validated blocking strategies will be crucial for advancing the application of FISH in precision medicine, ensuring reliable results in both research and clinical diagnostics.
References
- 1. How do I reduce high background in my FISH assay? [https://www.ogt.com/ca/resources/fish-resources-and-support/fish-resources/optimize-your-fish-assay-simple-fixes-for-reducing-high-background-signal/]
- 2. FISH Tips and Troubleshooting [https://www.creative-bioarray.com/support/fish-tips-and-troubleshooting.htm]
- 3. What's wrong with my hematology FISH? [https://www.ogt.com/us/resources/fish-resources-and-support/fish-support/whats-wrong-with-my-hematology-fish/]
- 4. In Situ Hybridization Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/gene-expression-analysis-genotyping-support-center/in-situ-hybridization-support/ish-support-troubleshooting.html]
- 5. Protocol optimization improves the performance of ... [https://www.nature.com/articles/s41598-025-17477-1]
- 6. Optimization of the Blocking and Signal Preservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462741/]
- 7. Challenges and solutions in FISH for formalin-fixed paraffin ... [https://pubmed.ncbi.nlm.nih.gov/39315587/]
- 8. What support reagents do I need for flow cytometry? - CST Blog [https://blog.cellsignal.com/cell-preparation-for-flow-cytometry]
- 9. What's wrong with my FFPE FISH? [https://www.ogt.com/resources/fish-resources-and-support/fish-support/whats-wrong-with-my-ffpe-fish/]
- 10. Optimized fixation and storage conditions for FISH analysis of ... [https://pubmed.ncbi.nlm.nih.gov/9671447/]
- 11. Systematic benchmarking of imaging spatial ... [https://www.nature.com/articles/s41467-025-64990-y]
- 12. What's wrong with my FFPE FISH? [https://www.ogt.com/us/resources/fish-resources-and-support/fish-support/whats-wrong-with-my-ffpe-fish/]
- 13. Enhancing Histological Techniques for Small Crustaceans [https://www.mdpi.com/2076-2615/15/12/1715]
- 14. Optimization of the Blocking and Signal Preservation ... [https://pubmed.ncbi.nlm.nih.gov/40996492/]
- 15. Protocols < Krause Lab - Yale School of Medicine [https://medicine.yale.edu/lab/krause/protocols/]
- 16. 10X Fish Gelatin Blocking Agent [https://biotium.com/product/fish-gelatin-blocking-buffer-10x/]
- 17. Fish Serum Blocking Buffer, 500 mL - FAQs [https://www.thermofisher.com/store/v3/products/faqs/37527]
- 18. RNAscope Troubleshooting Guide and FAQ [https://acdbio.com/technical-support/solutions/faq]
- 19. How do I reduce high background in my FISH assay? [https://www.ogt.com/us/resources/fish-resources-and-support/fish-resources/optimize-your-fish-assay-simple-fixes-for-reducing-high-background-signal/]
- 20. Optimized workflow for digitalized FISH analysis in pathology [https://pmc.ncbi.nlm.nih.gov/articles/PMC8114497/]
- 21. 10 Tips for Optimizing In Situ Hybridization (ISH) [https://www.enzo.com/note/10-tips-for-optimizing-in-situ-hybridization-ish/]
- 22. Guidelines for the Design of FISH Probes [https://www.creative-bioarray.com/support/guidelines-for-the-design-of-fish-probes.htm]
- 23. FISH Tips and Troubleshooting [https://www.clinicallab.com/fish-tips-and-troubleshooting-22053]
- 24. Optimizing the hybridization chain reaction-fluorescence in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10077247/]
- 25. A technical review and guide to RNA fluorescence in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7085896/]
- 26. Stable CoT-1 repeat RNA is abundant and associated with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4023122/]
- 27. Cot-1 DNA® Blocking Reagent [https://www.thermofisher.com/us/en/home/life-science/gene-expression-analysis-genotyping/genotyping-genomic-profiling/genomic-profilling-acgh/cot-1-dna.html]
- 28. Human Cot DNA [https://www.enzo.com/product/human-cot-dna/]
- 29. Distortion of quantitative genomic and expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1316118/]
- 30. Fluorescence In Situ Hybridization (FISH) [http://www.nature.com/scitable/topicpage/fluorescence-in-situ-hybridization-fish-327]
- 31. Blocking Buffers for Western Blot and ELISA [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-buffers-western-blot-elisa.html]
- 32. UltraBlock-FISH™ Blocking Buffer 'Ready to Use' [https://www.leinco.com/p/ultrablock-fish-blocking-buffer-ready-to-use/]
- 33. Locked Nucleic Acid Flow Cytometry-fluorescence in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3369778/]
- 34. Effectiveness of blocking primers and a peptide nucleic acid ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0266268]
- 35. FISH-Blocker| A non-mammalian protein blocking agent [https://www.gbiosciences.com/Buffers-Reagents-Chemicals/FISH-Blocker?srsltid=AfmBOorXF_cKBSDF1b1pdpl_c0YApZ5IrSwrp9OG_YcNvH6cjceeuEut]
- 36. Automation of fluorescent in situ hybridization (FISH) ... [https://pubmed.ncbi.nlm.nih.gov/40962484/]
- 37. Spectral Flow Cytometry [https://www.bdbiosciences.com/en-us/learn/applications/multicolor-flow-cytometry/spectral-flow-cytometry]
- 38. Optimizing Flow Cytometry Experiments-Part 2 How To Block ... [https://expertcytometry.com/optimizing-flow-cytometry-experiments-part2-how-to-block-samples-sample-blocking/]
- 39. Optimisation of blocking during flow cytometry [https://www.listonlab.uk/blog/2025/9/25/optimisation-of-blocking-during-flow-cytometry.html]
- 40. Flow cytometry troubleshooting [https://www.abcam.com/en-us/technical-resources/applications/flow-cytometry/troubleshooting]
- 41. In Situ Hybridization Support—Troubleshooting [https://www.thermofisher.com/id/en/home/technical-resources/technical-reference-library/gene-expression-analysis-genotyping-support-center/in-situ-hybridization-support/ish-support-troubleshooting.html]
- 42. Standardization and optimization of fluorescence in situ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5988532/]
- 43. Western blot blocking: Best practices [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-blocking-methods]
- 44. Optimization of blocking conditions for fluorescent Western ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269719307389]
- 45. Fluorescent in situ hybridisation (FISH) — Knowledge Hub [https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/fluorescent-in-situ-hybridisation-fish/]
- 46. Troubleshooting qPCR: Interpreting Amplification Curves [https://blog.biosearchtech.com/thebiosearchtechblog/bid/63622/qpcr-troubleshooting]
- 47. FISH testing: How it works, conditions, and what to expect [https://www.medicalnewstoday.com/articles/fish-testing]