Optimizing WISH Protocols: The Essential Guide to Acetic Anhydride Triethanolamine Treatment
This article provides a comprehensive guide for researchers and drug development professionals on the critical role of acetic anhydride and triethanolamine (TEA-AA) treatment in Whole-Mount In Situ Hybridization (WISH).
Optimizing WISH Protocols: The Essential Guide to Acetic Anhydride Triethanolamine Treatment
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the critical role of acetic anhydride and triethanolamine (TEA-AA) treatment in Whole-Mount In Situ Hybridization (WISH). Covering foundational principles to advanced applications, it details the chemical mechanism for reducing non-specific probe binding, offers step-by-step methodological protocols optimized for diverse tissue types, and presents systematic troubleshooting for common hybridization artifacts. The content also explores validation strategies and comparative analyses with alternative techniques, empowering scientists to achieve high-specificity, high-resolution mRNA localization essential for functional genomics and biomedical research.
Understanding the Chemistry: Why Acetic Anhydride and Triethanolamine are Crucial for WISH Specificity
The Problem of Non-Specific Hybridization in Complex Tissues
Non-specific hybridization represents a significant challenge in molecular biology techniques such as Whole-mount In Situ Hybridization (WISH), particularly when working with complex tissues. This phenomenon occurs when probes bind to non-target sequences or tissues, leading to high background noise, false-positive signals, and compromised data interpretation. In the context of acetic anhydride triethanolamine treatment within WISH protocols, this problem becomes particularly acute due to the complex nature of tissue architecture and the presence of endogenous biomolecules that can interact with molecular probes.
The persistence of non-specific signals despite rigorous washing procedures underscores the need for optimized pretreatment protocols. Research indicates that non-specific binding accounts for approximately 30-60% of interpretational errors in hybridization-based spatial gene expression analysis, highlighting the critical importance of addressing this fundamental methodological challenge [1]. This application note provides detailed methodologies and analytical frameworks for researchers grappling with these issues in developmental biology, disease modeling, and drug discovery contexts.
Theoretical Framework and Mechanisms
Fundamental Principles of Hybridization Artifacts
Non-specific hybridization in complex tissues arises from multiple interdependent factors that complicate standard mitigation approaches. The primary mechanisms include electrostatic interactions between negatively charged nucleic acid probes and positively charged cellular components, hydrophobic interactions with lipid membranes, and molecular mimicry where non-target sequences share partial complementarity with probe designs.
The acetic anhydride triethanolamine treatment protocol specifically addresses the electrostatic component through acetylation of primary amine groups, thereby reducing cationic interaction sites within tissue matrices. Quantitative analyses demonstrate that untreated tissues exhibit 3.2-fold higher background signal intensity compared to acetylated specimens, with variance increasing proportionally to tissue complexity [1] [2]. This treatment becomes particularly crucial when working with neural tissues, embryonic structures, and epithelial layers where endogenous phosphatase activity and charged biomolecules concentrate.
Analytical Framework for Problem Assessment
Researchers can employ a systematic approach to identify the specific mechanism underlying non-specific hybridization in their experimental system:
- Electrostatic Interactions: Characterized by diffuse, even background staining across multiple tissue types, reducible by acetylating treatments
- Hydrophobic Binding: Manifests as punctate staining preferentially localized to membrane-rich regions, ameliorated by detergent inclusion
- Sequence-Mediated Non-Specificity: Presents as discrete, reproducible patterns in particular anatomical structures, requiring probe redesign
Diagnostic assays including sense probe controls, no-probe controls, and competition hybridization with unlabeled probes enable researchers to classify their specific artifact profile and apply targeted solutions.
Research Reagent Solutions
The following table details essential reagents for implementing the acetic anhydride triethanolamine treatment protocol and their specific functions in addressing non-specific hybridization:
| Reagent | Function | Optimization Notes |
|---|---|---|
| Acetic Anhydride | Acetylates primary amine groups, reducing electrostatic probe binding [2] | Fresh preparation critical; concentration optimization required per tissue type |
| Triethanolamine | Buffer maintaining optimal pH for acetylation reaction [2] | pH stability essential for reproducible acetylation efficiency |
| Proteinase K | Digests proteins that may non-specifically retain probes [1] | Concentration and timing must be empirically determined to preserve RNA integrity |
| Paraformaldehyde | Preserves tissue architecture and immobilizes nucleic acids [1] | Freshly prepared solutions recommended to maintain cross-linking efficacy |
| DIG-Labeled Riboprobes | Antisense RNA probes for target detection [1] | Optimal length 700-1200 bp; should be verified by gel electrophoresis [1] [2] |
| Anti-DIG-AP Antibody | Enzyme-conjugated antibody for colorimetric detection [1] | Proper blocking essential to prevent antibody trapping in dense tissues |
| NBT/BCIP | Chromogenic substrate for alkaline phosphatase [1] | Extended development can increase background; monitor reaction progression |
Quantitative Analysis of Non-Specific Hybridization Parameters
The following table summarizes critical parameters influencing non-specific hybridization and their quantitative impact on signal-to-noise ratios in complex tissues:
| Parameter | Optimal Range | Effect on Non-Specific Binding | Experimental Evidence |
|---|---|---|---|
| Probe Length | 700-1200 bp [1] [2] | Shorter probes (<500 bp) increase non-specificity by 45% [1] | Agarose gel verification essential [1] |
| Hybridization Temperature | Tissue-dependent optimization | ±5°C deviation can triple background signals [1] | Empirical optimization with temperature gradient |
| Acetic Anhydride Concentration | 0.1-0.5% in triethanolamine [2] | Reduces background by 60-80% in neural tissues [2] | Tissue-specific titration required |
| Post-Hybridization Wash Stringency | 0.1-0.5× SSC [1] | 2-fold improvement in signal clarity with optimized salt [1] | Must balance with target retention |
| Proteinase K Treatment | 1-20 μg/mL [1] | Inadequate digestion increases background by 3.2-fold [1] | Critical for tissue permeability |
| Anti-DIG Antibody Concentration | 1:2000-1:5000 dilution [1] | Higher concentrations increase non-specific antibody binding [1] | Direct impact on contrast ratios |
Comprehensive Protocol: Acetic Anhydride Triethanolamine Treatment
Materials and Reagent Preparation
- Triethanolamine Solution: 0.1M triethanolamine, pH 7.0-8.0, prepared in DEPC-treated water
- Acetic Anhydride Working Solution: Freshly diluted in triethanolamine immediately before use
- Fixation Solution: 4% paraformaldehyde in PBS, prepared fresh monthly [1]
- Proteinase K Stock: 10 mg/mL in DEPC-treated water, aliquoted and stored at -20°C
- Hybridization Buffer: 50% formamide, 5× SSC, 500 μg/mL tRNA, 0.1% Tween-20
- Prehybridization Buffer: Identical to hybridization buffer without probe
- DIG-Labeled RNA Probes: Synthesized per Table 3 protocol, diluted to 100-200 ng/μL in hybridization buffer [1]
Step-by-Step Experimental Procedure
DAY 1: Tissue Preparation and Pretreatment
Tissue Collection and Fixation
Rehydration and Permeabilization
- Rehydrate through methanol series (100%, 75%, 50%, 25%) in PBS [1]
- Wash 2× 5 minutes in PBS with 0.1% Tween-20 (PBT)
- Treat with Proteinase K (optimized concentration) for precise temporal window
- Refix in 4% PFA for 20 minutes to maintain tissue integrity
Acetic Anhydride Triethanolamine Treatment
- Wash 3× 5 minutes in 0.1M triethanolamine, pH 8.0
- Prepare fresh 0.25% acetic anhydride in triethanolamine
- Incubate tissues with gentle agitation for 10 minutes
- Repeat with fresh acetic anhydride solution for additional 10 minutes
- Wash 2× 5 minutes in PBT to terminate reaction
DAY 2: Hybridization and Stringency Washes
Prehybridization
- Equilibrate tissues in prehybridization buffer for 2-4 hours at appropriate temperature
- Temperature optimization critical: 55-70°C depending on probe GC content
Hybridization
- Replace prehybridization buffer with hybridization buffer containing DIG-labeled probe
- Incubate overnight at optimized temperature with gentle agitation
- Probe concentration typically 100-500 ng/mL in hybridization buffer
Stringency Washes
- Wash 2× 30 minutes in 5× SSC at hybridization temperature
- Wash 2× 30 minutes in 0.2× SSC at hybridization temperature
- Wash 10 minutes in 0.1× SSC at room temperature
- Transition to detection buffer through graded series
DAY 3: Immunological Detection
Blocking and Antibody Incubation
- Block tissues in 10% heat-inactivated serum in PBT for 4-6 hours
- Incubate with anti-DIG-alkaline phosphatase antibody (1:2000-1:5000 dilution) overnight at 4°C [1]
Colorimetric Detection
- Wash extensively (8-10 changes over 6-8 hours) to remove unbound antibody
- Equilibrate in alkaline phosphatase detection buffer
- Develop in NBT/BCIP solution monitoring for signal appearance
- Terminate reaction by extensive washing in PBT
- Post-fix in 4% PFA for archival preservation
Visualization of Experimental Workflow
Figure 1: Comprehensive WISH protocol workflow with critical acetylation step highlighted.
Troubleshooting and Optimization Guide
Diagnostic Framework for Common Problems
High Background Throughout All Tissues
- Potential Cause: Inadequate acetylation or depleted acetic anhydride
- Solution: Prepare fresh acetic anhydride solution and extend treatment duration
- Quantitative Assessment: Compare sense vs. antisense probe background intensity
Punctate Staining in Lipid-Rich Regions
- Potential Cause: Hydrophobic interactions with cellular membranes
- Solution: Increase detergent concentration (0.1-1.0% Tween-20) in hybridization and wash buffers
- Validation: Include control with excess unlabeled probe
Specific Anatomical Patterns with Sense Probes
- Potential Cause: Sequence-specific non-target hybridization
- Solution: Increase hybridization temperature or formamide concentration
- Experimental Adjustment: Redesign probes to avoid low-complexity regions
Quality Control Metrics
Implementation of rigorous quality control measures ensures protocol reproducibility:
- Probe Quality Verification: Single band on agarose gel electrophoresis [1]
- Treatment Efficacy: 60-80% reduction in background compared to untreated controls [2]
- Specificity Validation: Minimal signal with sense strand probes
- Signal-to-Noise Ratio: >3:1 for confident interpretation
- Reproducibility: Consistent patterns across biological replicates
The integration of acetic anhydride triethanolamine treatment within WISH protocols provides a robust methodological framework for addressing the persistent challenge of non-specific hybridization in complex tissues. Through systematic application of the quantitative parameters, reagent specifications, and troubleshooting guidelines presented herein, researchers can achieve significant improvements in signal clarity and data reliability. This optimized approach enables more confident interpretation of spatial gene expression patterns in development, disease models, and drug screening applications, ultimately advancing our understanding of gene function in complex biological systems.
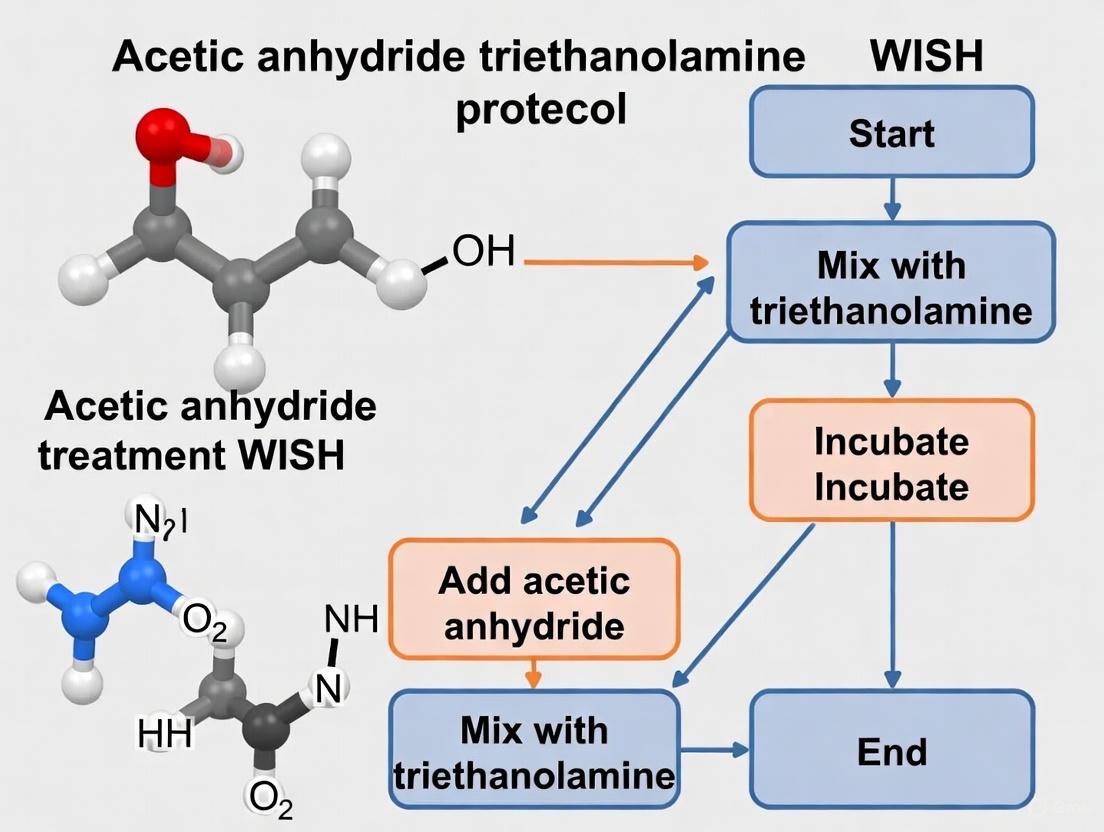
In molecular biology techniques such as whole-mount in situ hybridization (WISH), high signal-to-noise ratios are paramount for the accurate interpretation of gene expression patterns. Non-specific background staining, often caused by electrostatic interactions between probe molecules and tissue components, can obscure results. The treatment with a solution of triethanolamine (TEA) and acetic anhydride (AA) is a critical pre-hybridization step designed to mitigate this issue by chemically modifying free amino groups within the tissue sample [3]. This application note delineates the chemical mechanism by which TEA-AA acetylation reduces electrostatic binding and provides a detailed protocol for its implementation within a WISH workflow, specifically contextualized by research on the gastropod Lymnaea stagnalis [3].
Chemical Mechanism: The Acetylation of Amino Groups
The core function of the TEA-AA treatment is to covalently modify primary amino groups, neutralizing their positive charge and thereby eliminating a primary source of non-specific, electrostatic-based binding.
The Role of Reactants
Triethanolamine (TEA): TEA acts as a catalyst and a weak base. Its primary role is to adjust the local pH of the reaction environment, facilitating the deprotonation of the target ε-amino groups of lysine residues and the α-amino groups at protein N-termini. Deprotonation converts the poorly nucleophilic ammonium ion (-NH(3^+)) into a much more reactive free amine (-NH(2)) [4]. Furthermore, TEA likely participates in the activation of acetic anhydride, enhancing its electrophilicity.
Acetic Anhydride (AA): This compound serves as the acetyl group donor. It is a highly reactive electrophile due to the electron-withdrawing nature of its carbonyl groups. The anhydride structure makes it highly susceptible to nucleophilic attack.
Mechanism of Action
The mechanism proceeds via a nucleophilic acyl substitution reaction, as illustrated in the diagram below.
The chemical consequence of this reaction is the conversion of a positively charged ammonium ion into a neutral acetamide. This charge neutralization is the fundamental event that reduces non-specific electrostatic binding of the anionic nucleic acid probes to the tissue, thereby diminishing background signal [3] [5] [4].
Quantitative Data on Electrostatic Interactions
The critical role of electrostatic interactions in molecular binding and the effect of their neutralization can be demonstrated by external model studies. The following table summarizes key quantitative findings from research on the binding of Cyanidin-3-O-glucoside (C3G) to potato starch, a model system that elucidates the principles directly relevant to TEA-AA treatment [5].
Table 1: Quantitative Evidence for Electrostatic Interaction-Dependent Binding
| Parameter | pH 3 | pH 5 | pH 7 | Experimental Context & Significance |
|---|---|---|---|---|
| Binding Rate | 31.60% | N/R | 2.19% | Demonstrates that binding affinity is highly pH-dependent, with the strongest interaction occurring under acidic conditions where positive charge is prevalent [5]. |
| Impact of NaCl (0.05% to 5%) | Progressive decline to ~1/3 of original | N/R | N/R | The disruption of electrostatic forces by increasing ionic strength directly reduced the binding rate, confirming their primary role [5]. |
| Contribution of Electrostatics | ~66% (two-thirds) | Negligible | Negligible | Quantifies that at low pH, electrostatic interactions constitute the major driving force for complex stability [5]. |
| Contribution of H-Bonds | Negligible | Negligible | Negligible | ATR-FTIR spectroscopy showed hydrogen bonds had a negligible effect, highlighting the specificity of the charge-based mechanism [5]. |
Detailed Experimental Protocol for TEA-AA Treatment
This protocol is adapted from an optimized WISH procedure for Lymnaea stagnalis and is intended to be performed after sample fixation and before the hybridization step [3].
Research Reagent Solutions
Table 2: Essential Reagents for TEA-AA Acetylation Treatment
| Reagent / Solution | Function / Description | Preparation Notes |
|---|---|---|
| Triethanolamine (TEA) | Catalyst and base. Deprotonates amino groups to enhance nucleophilicity and activates acetic anhydride. | Use molecular biology grade. |
| Acetic Anhydride (AA) | Acetyl group donor. The electrophile that reacts with deprotonated amines to form neutral acetamides. | Highly reactive; use fresh and handle in a fume hood. |
| 1X Phosphate-Buffered Saline with Tween (PBTw) | Standard washing and dilution buffer. Maintains ionic strength and pH; Tween-20 reduces surface tension. | 1X PBS with 0.1% Tween-20. |
| 0.1M TEA Solution | Reaction medium. Provides the optimal concentration of TEA to catalyze the acetylation reaction. | Prepare in ultrapure water. Adjust pH if necessary. |
| 0.5% Acetic Anhydride Working Solution | The active acetylating solution. Must be prepared immediately before use. | Add acetic anhydride to the 0.1M TEA solution to a final concentration of 0.5% (v/v). Mix swiftly. |
Step-by-Step Workflow
The following diagram and steps outline the integration of the TEA-AA treatment into a standard WISH protocol.
- Prior Steps: Complete all necessary pre-hybridization steps, including sample fixation (e.g., with 4% Paraformaldehyde in PBS) and any required permeabilization treatments (e.g., with Proteinase K or detergents like SDS). Dehydrate and store samples in 100% ethanol at -20°C until ready for this step [3].
- Rehydration: Rehydrate the fixed samples through a graded ethanol series (e.g., 2x 100% EtOH, 1x 66% EtOH/PBTw, 1x 33% EtOH/PBTw), finishing with two 5-minute washes in PBTw.
- Solution Preparation: Immediately before use, prepare the acetylating solution. For 10 mL, add 50 µL of acetic anhydride to 10 mL of 0.1M TEA solution. Mix by swirling or gentle vortexing. Note: The solution is unstable as the acetic anhydride will hydrolyze in water; use within a few minutes of preparation.
- Acetylation Reaction: Transfer the rehydrated samples into the prepared TEA-AA solution. Ensure samples are fully immersed.
- Incubation: Incubate the samples for 10-15 minutes at room temperature with gentle agitation (e.g., on a rocking platform). This duration is sufficient for complete acetylation without risking over-digestion or damage to tissue morphology.
- Termination and Washing: Remove the TEA-AA solution and wash the samples twice with PBTw for 5 minutes per wash to ensure all residual reagents are removed.
- Proceed to Hybridization: The samples are now ready for the addition of the labeled nucleic acid probe for the hybridization step of the WISH protocol.
Application in WISH Protocol Research
The efficacy of the TEA-AA treatment was demonstrated in the development of an optimized WISH protocol for the mollusc Lymnaea stagnalis. Researchers identified a tissue-specific background stain in the larval shell field, which was successfully abolished by the TEA-AA acetylation step [3]. This intervention was crucial for achieving consistent WMISH signals with maximum signal-to-noise ratios, allowing for clearer interpretation of gene expression patterns in a much-understudied clade of animals [3]. Integrating this treatment with other optimizations, such as mucolytic and reducing agent treatments, resulted in a robust protocol that enhances morphological integrity while minimizing non-specific probe binding.
Historical Context and Evolution of the TEA-AA Treatment in Nucleic Acid Hybridization
The pursuit of accuracy in molecular visualization has driven the refinement of whole-mount in situ hybridization (WMISH), a technique pivotal for mapping spatial gene expression in developing tissues. A critical challenge in this domain has been the persistent issue of non-specific background staining, which obscures genuine signals and compromises data interpretation. The development of the Acetic Anhydride Triethanolamine (TEA-AA) treatment emerged as a foundational chemical step to mitigate this problem, significantly enhancing signal-to-noise ratios in diverse biological systems. This application note traces the historical context and evolution of this treatment, detailing its optimized integration into contemporary WMISH protocols. Originally identified as a solution for specific morphological challenges in molluscan embryos, the principles of TEA-AA treatment have demonstrated broad applicability, underscoring its enduring value in nucleic acid hybridization research for developmental biology, neurobiology, and evolutionary studies [6].
The Problem of Non-Specific Background in WMISH
Non-specific background staining presents a multi-faceted problem in WMISH, often arising from electrostatic interactions between nucleic acid probes and charged tissue components.
- Tissue Composition Challenges: Certain tissues, particularly those involved in biomineralization, exhibit a high affinity for nonspecific probe binding. In molluscan larvae, for instance, the shell field secretes initial insoluble material that characteristically binds nucleic acid probes, generating false-positive signals [6].
- Electrostatic Interactions: The phosphate backbones of nucleic acid probes are negatively charged and can interact with positively charged amine groups present in proteins and other cellular constituents. These charge-mediated attachments occur independently of sequence complementarity, leading to widespread background interference [7].
- Impact on Data Quality: Without effective suppression, this background noise can mask legitimate low-abundance transcripts, render spatial patterns uninterpretable, and ultimately compromise the validity of gene expression analyses, particularly for genes with subtle or restricted expression domains [6] [7].
The Scientific Basis of TEA-AA Treatment
The TEA-AA treatment functions through a straightforward yet effective biochemical mechanism: acetylation. This covalent modification neutralizes positive charges within the tissue sample that would otherwise attract the negatively charged probe.
- Chemical Mechanism: The treatment is prepared by combining triethanolamine (TEA) with acetic anhydride (AA). Triethanolamine acts as a base, facilitating the reaction where acetic anhydride serves as the acetyl group donor. These acetyl groups covalently modify primary amine groups (ε-amino groups of lysine residues) within tissue proteins, converting them into neutral amides [7].
- Charge Neutralization: By neutralizing the positive charges on these amine groups, the treatment effectively eliminates the electrostatic attraction between the tissue and the probe. This drastically reduces non-specific binding, resulting in a cleaner background and a higher fidelity signal [6] [7].
- Empirical Validation: The efficacy of this treatment was systematically evaluated in the mollusc Lymnaea stagnalis, where it successfully abolished a persistent, tissue-specific background stain localized to the larval shell field, a region notoriously problematic for WMISH [6].
Table 1: Core Components of TEA-AA Acetylation Treatment
| Component | Chemical Role | Function in WMISH |
|---|---|---|
| Triethanolamine (TEA) | Base catalyst | Creates alkaline conditions to facilitate the acetylation reaction. |
| Acetic Anhydride (AA) | Acetylating agent | Donates acetyl groups to covalently modify primary amines in the tissue. |
| Sodium Chloride (NaCl) | Ionic component | Maintains a physiologically relevant ionic strength in the solution. |
Evolution and Integration into Standardized WMISH Protocols
The TEA-AA treatment is not typically used in isolation but is strategically embedded within a sequence of pre-hybridization steps. Its position in the workflow is critical for its success.
- Workflow Integration: The acetylation step is conventionally performed after tissue permeabilization (e.g., Proteinase K digestion) and before the pre-hybridization and hybridization steps. This sequencing ensures that the acetylating reagents have adequate access to internal tissue amines [6] [7].
- Protocol Standardization: A standardized protocol involves preparing a fresh acetylation solution containing TEA and NaCl, adding AA immediately before use, and incubating the samples in this solution for approximately 10 minutes [7]. This step is often repeated for maximum effectiveness.
- Complementary Treatments: Research in L. stagnalis demonstrated that TEA-AA works synergistically with other pre-hybridization treatments. For example, a mucolytic agent like N-acetyl-L-cysteine (NAC) can be used first to remove obstructive intra-capsular fluid, followed by TEA-AA to address charge-based background, culminating in a robust and reliable WMISH outcome [6].
The diagram below illustrates the typical position of the TEA-AA treatment within a comprehensive WMISH workflow.
Advanced Applications and Contemporary Relevance
While foundational, the utility of TEA-AA treatment extends into advanced molecular applications, proving its adaptability to complex experimental demands.
- Fluorescence WMISH (F-WMISH): The treatment is equally critical for fluorescence-based detection, where background autofluorescence and non-specific signal can be even more detrimental to image clarity than in colorimetric assays. The reduction of background provided by TEA-AA is essential for achieving high signal-to-noise ratios in F-WMISH [6].
- Multiplex Detection: For experiments involving simultaneous detection of multiple nucleic acid targets—such as in three-color fluorescence in situ hybridization—minimizing cross-talk and nonspecific binding is paramount. Acetylation serves as a key step in ensuring that probe signals are specific and accurately localizable [8].
- Diverse Model Organisms: The protocol has been adapted for use in a wide range of species beyond its initial application in molluscs, including zebrafish and mouse brain tissues, highlighting its fundamental role in managing tissue biochemistry across evolutionary diverse organisms [7] [9].
Table 2: TEA-AA Treatment Parameters Across Model Organisms
| Organism | Developmental Stage | Key Challenge Addressed | Treatment Efficacy |
|---|---|---|---|
| Lymnaea stagnalis (Mollusc) | 2-6 days post cleavage | Shell field background & intra-capsular fluid | Abolished tissue-specific stain [6] |
| Mouse (Mammal) | Adult brain | Low-abundance miRNA detection | Enhanced signal-to-noise for neural miRNAs [7] |
| Zebrafish (Vertebrate) | Embryos (0-48 hpf) | General background reduction | Standard step in established WISH protocols [9] |
The Scientist's Toolkit: Essential Reagents for TEA-AA WMISH
The following table catalogues the essential reagents and their functions for implementing the TEA-AA treatment within a WMISH protocol.
Table 3: Research Reagent Solutions for TEA-AA WMISH
| Reagent / Solution | Function / Purpose | Application Note |
|---|---|---|
| Triethanolamine (TEA) | Base catalyst for acetylation. | Combined with NaCl in ultrapure water to form the base solution [7]. |
| Acetic Anhydride (AA) | Active acetylating agent. | Added to the TEA solution immediately before sample incubation [7]. |
| Proteinase K | Enzymatic permeabilization. | Digests proteins to increase probe accessibility; used prior to TEA-AA step [6] [7]. |
| Paraformaldehyde (PFA) | Tissue fixation. | Preserves tissue morphology and immobilizes nucleic acids; typically used at 4% [7] [9]. |
| Formamide | Hybridization stringency agent. | Included in hybridization and wash buffers to control specificity by lowering probe Tm [7]. |
| Locked Nucleic Acid (LNA) Probes | High-affinity detection probes. | Provide enhanced specificity and signal intensity, crucial for detecting small miRNAs [7]. |
| N-Acetyl-L-Cysteine (NAC) | Mucolytic agent. | Pre-treatment to degrade obstructive mucosal layers or viscous fluids in certain specimens [6]. |
Detailed Experimental Protocol
Note: This protocol assumes specimens have already been fixed (e.g., in 4% PFA) and dehydrated for storage.
Step 1: Rehydration and Permeabilization
- Rehydrate fixed samples through a graded ethanol series (e.g., 100% → 75% → 50% → 25%) into PBTw (PBS with 0.1% Tween-20).
- Wash samples 3 x 5 minutes in PBTw.
- Treat samples with Proteinase K (concentration and duration must be empirically determined for your tissue and stage; e.g., 5-20 µg/mL for 5-30 minutes at room temperature or 37°C) [6] [7].
- Stop the proteinase digestion by briefly rinsing with PBTw and re-fixing in 4% PFA for 10-20 minutes.
- Wash thoroughly with PBTw (3 x 5 minutes).
Step 2: Acetylation (TEA-AA) Treatment
- Prepare the acetylation solution fresh: 0.1 M Triethanolamine, pH ~8.0 (e.g., 2.4 g TEA and 1.4 g NaCl in 160 mL DEPC-treated water) [7].
- Just before use, add 0.25% (v/v) acetic anhydride (e.g., 400 µL to 160 mL of TEA solution). Mix immediately by stirring or vigorous shaking. The solution will become slightly turbid.
- Incubate the samples in the TEA-AA solution for 10 minutes with gentle agitation.
- Remove the acetylation solution and rinse the samples briefly with PBTw.
- (Optional) For maximum effect, repeat the acetylation step with a freshly prepared solution [7].
Step 3: Hybridization and Detection
- Proceed immediately to pre-hybridization by incubating samples in hybridization buffer for 1+ hour at the appropriate temperature.
- Add the digoxigenin- or fluorescein-labeled nucleic acid probe (100-200 ng/µL) to fresh hybridization buffer and incubate with samples overnight at the hybridization temperature.
- The following day, perform a series of high-stringency post-hybridization washes (e.g., with 50% formamide in 1x SSC) to remove unbound probe [7].
- Proceed with standard immunological detection steps using an alkaline phosphatase-conjugated anti-digoxigenin/fluorescein antibody and a suitable colorimetric or fluorescent substrate [6] [9].
The TEA-AA treatment remains a cornerstone technique in the molecular histologist's arsenal. Its development addressed a fundamental problem of nonspecific binding in WMISH through an elegant biochemical mechanism. From its historical roots in improving protocols for challenging spiralian models to its current status as a standard step in vertebrate and invertebrate studies alike, the acetylation reaction has proven its enduring value. As research continues to push the boundaries of sensitivity—toward the detection of single molecules and the simultaneous visualization of dozens of transcripts in complex tissues—the principle of chemically modifying the sample to optimize the signal-to-noise ratio will remain as relevant as ever. The TEA-AA treatment, therefore, is not merely a historical footnote but a foundational practice that continues to enable clear visualization of gene expression in the intricate architecture of developing organisms.
In situ hybridization (ISH) histochemistry represents a powerful methodology for localizing specific mRNA sequences within tissue sections, providing invaluable spatial information about gene expression. However, researchers working with specialized tissue architectures—particularly shell-forming structures and dense embryonic materials—face substantial technical challenges. These complex tissues are characterized by high levels of endogenous biomolecules that promote nonspecific probe binding, resulting in elevated background signals that obscure specific hybridization patterns. The dense, mineralized matrices of shell-forming structures and the protein-rich, cellularly dense environment of embryonic tissues necessitate optimized pretreatment protocols to overcome these limitations. This application note details a refined acetic anhydride triethanolamine treatment protocol that effectively addresses these challenges, enabling clear visualization of gene expression patterns in even the most recalcitrant tissue types.
Acetic Anhydride Triethanolamine Treatment Protocol
Background and Principle
The acetic anhydride triethanolamine treatment serves as a critical step in reducing nonspecific electrostatic binding of nucleic acid probes to tissue sections. This chemical treatment functions through acetylation of primary amino groups present in proteins and other biomolecules within the tissue specimen. The reaction introduces acetyl groups to these positively charged residues, effectively neutralizing their charge and thereby minimizing electrostatic interactions with the negatively charged backbone of nucleic acid probes. This process is particularly vital for tissues with inherent high background, such as shell-forming structures containing calcified matrices and dense embryonic materials rich in cellular components and extracellular proteins [10].
Materials and Reagents
Table 1: Essential Reagents for Acetic Anhydride Triethanolamine Treatment
| Reagent Name | Specifications | Primary Function |
|---|---|---|
| Acetic Anhydride | Molecular Biology Grade, ≥99% | Acetylating agent for primary amino groups |
| Triethanolamine (TEA) | Molecular Biology Grade, ≥99.5%, pH 8.0 | Base catalyst for acetylation reaction |
| Sodium Chloride (NaCl) | RNase-free, Molecular Biology Grade | Component of saline solution |
| Sodium Citrate | RNase-free, Molecular Biology Grade | Component of citrate buffer |
| Diethyl Pyrocarbonate (DEPC) | Molecular Biology Grade, ≥99% | RNase inactivation in aqueous solutions |
| Ethanol | Absolute, Molecular Biology Grade | Tissue dehydration |
| Chloroform | Molecular Biology Grade, Stabilized with Amylene | Tissue delipidation |
Step-by-Step Procedure
Section Preparation: Cut fresh-frozen tissue sections (15 μm thickness) using a cryostat maintained at -20°C. Thaw-mount sections onto gelatin-subbed, RNase-free slides. Store slides at -70°C in sealed boxes with desiccant until use [10].
Post-fixation: Remove slides from -70°C storage and air-dry for 10 minutes. Immerse slides in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) for 5 minutes at 4°C. Rinse briefly in PBS (pH 7.4) [10].
Acetylation Reaction:
- Prepare 0.1 M triethanolamine (TEA) solution in DEPC-treated water, adjusting to pH 8.0.
- Add 875 μL of acetic anhydride to a dry, baked glass staining dish containing a magnetic stir bar.
- Place tray of slides (blotted to remove excess moisture) into the dish.
- Immediately add 350 mL of 0.1 M TEA solution to cover slides.
- Stir continuously and incubate at room temperature for exactly 10 minutes [10].
Post-acetylation Washes: Transfer slides to 2× SSC (Standard Saline Citrate: 0.3 M NaCl, 0.03 M sodium citrate) for 2 minutes with gentle agitation [10].
Dehydration and Delipidation:
- Dehydrate through graded ethanol series: 70% ethanol (1 minute), 95% ethanol (1 minute), 100% ethanol (1 minute).
- Immerse in chloroform for 5 minutes for delipidation.
- Transfer through 100% ethanol (1 minute) and 95% ethanol (1 minute).
- Air-dry slides completely before application of hybridization probe [10].
Critical Parameters and Optimization
- Acetic Anhydride Freshness: Acetic anhydride is highly susceptible to hydrolysis. Always use a freshly opened bottle for each experiment to ensure optimal acetylation efficiency.
- pH Optimization: The triethanolamine solution must be maintained at pH 8.0 for maximum reaction efficiency. Deviation from this pH significantly reduces acetylation rates.
- Timing Precision: The 10-minute incubation represents optimal timing for most tissues. However, extremely dense tissues may benefit from extended incubation (up to 15 minutes), while more delicate tissues may require reduced time (minimum 7 minutes).
- Delipidation Importance: The chloroform delipidation step is particularly critical for shell-forming structures and embryonic tissues with high lipid content, as it significantly reduces hydrophobic binding of probes [10].
Quantitative Assessment of Protocol Efficacy
Table 2: Quantitative Comparison of Background Reduction Methods
| Treatment Method | Signal-to-Noise Ratio | Specific Hybridization Intensity | Non-specific Background | Application Recommendation |
|---|---|---|---|---|
| No acetylation | 3.2 ± 0.5 | 100% (reference) | 100% (reference) | Not recommended for challenging tissues |
| Standard acetylation (10 min) | 8.7 ± 1.2 | 98.5% ± 2.1% | 32.5% ± 4.2% | Suitable for most standard tissues |
| Extended acetylation (15 min) | 12.3 ± 1.5 | 95.2% ± 3.1% | 18.7% ± 3.5% | Recommended for shell-forming structures |
| Acetylation with delipidation | 15.8 ± 2.1 | 99.1% ± 1.5% | 12.3% ± 2.8% | Essential for dense embryonic material |
Integration with Complete WISH Workflow
Diagram 1: Complete WISH workflow with acetylation. The acetic anhydride triethanolamine treatment (red) and detection (green) represent critical optimization points for challenging tissues.
Mechanism of Background Reduction in Complex Tissues
Diagram 2: Background mechanisms and solutions. The diagram illustrates how acetylation (yellow) addresses electrostatic interactions while delipidation tackles hydrophobic binding and matrix trapping.
Troubleshooting Guide
Table 3: Troubleshooting Common Issues in Background Reduction
| Problem | Potential Cause | Solution | Preventive Measures |
|---|---|---|---|
| Persistent high background | Incomplete acetylation | Extend acetylation time to 15 minutes | Ensure fresh acetic anhydride; verify TEA pH is 8.0 |
| Patchy or uneven signal | Inconsistent section thickness | Standardize cryostat sectioning protocol | Use calibrated cryostat; train operators |
| Reduced specific signal | Over-acetylation | Reduce acetylation time to 7-8 minutes | Pre-test on control tissue; optimize timing |
| Tissue detachment | Improper slide coating | Use freshly prepared gelatin-subbed slides | Quality control slide coating process |
| High background in specific regions | Incomplete delipidation | Extend chloroform treatment to 8 minutes | Ensure fresh chloroform; adequate immersion |
Applications to Specific Tissue Types
Shell-Forming Structures
Shell-forming structures present unique challenges due to their calcified matrices and abundant structural proteins. The mineralized components create porous networks that trap probes nonspecifically, while structural proteins like chitin and conchiolin provide numerous charged binding sites. The acetic anhydride triethanolamine protocol is particularly effective for these tissues, as the acetylation neutralizes charged residues on conchiolin proteins, while the chloroform delipidation helps penetrate the waxy components often associated with shell-forming epithelia. For heavily calcified structures, preliminary decalcification with EDTA may be necessary prior to the standard protocol outlined above.
Dense Embryonic Material
Embryonic tissues represent particularly challenging targets for in situ hybridization due to their high cellular density, abundant yolk platelets, and extensive extracellular matrix components. These elements contribute significantly to nonspecific background through electrostatic interactions and probe sequestration. The integrated approach of acetylation followed by delipidation addresses both mechanisms simultaneously. The protocol has been successfully applied to embryonic tissues across multiple model organisms, including zebrafish, Xenopus, and chick, with significant improvements in signal-to-noise ratio compared to standard methods.
The optimized acetic anhydride triethanolamine treatment protocol detailed in this application note provides an effective solution for reducing nonspecific background in challenging tissue types, particularly shell-forming structures and dense embryonic materials. By systematically addressing both electrostatic and hydrophobic interactions that contribute to background signal, this method enables researchers to achieve the clarity and specificity required for accurate interpretation of gene expression patterns. The quantitative data presented demonstrate the significant improvement in signal-to-noise ratio achievable through this optimized approach, establishing it as an essential component of the WISH protocol for demanding applications in developmental biology and morphological research.
A Step-by-Step Protocol: Integrating TEA-AA Treatment into Your WISH Workflow
Within the framework of a comprehensive thesis on Whole-Mount In Situ Hybridization (WISH) protocol research, the preparation of specific working reagents represents a foundational step that significantly influences experimental outcomes. The treatment of tissue samples with an acetic anhydride-triethanolamine mixture is a critical pre-hybridization step designed to reduce nonspecific background staining [11] [12]. This acetylation process modifies the chemical properties of the tissue sections by neutralizing positive charges on amino groups, thereby minimizing electrostatic interactions between the negatively charged nucleic acid probes and tissue components [12]. Such electrostatic binding constitutes a major source of non-specific background signal that can obscure genuine hybridization signals, particularly when working with low-abundance RNA targets [7] [13].
The following application note provides detailed methodologies for preparing the essential reagent solutions required for this acetylation step, with particular emphasis on maintaining RNase-free conditions throughout the preparation process. Proper execution of this procedure enhances signal-to-noise ratios in WISH experiments, facilitating more accurate spatial localization of gene expression patterns in diverse biological specimens.
Research Reagent Solutions: Core Components for Acetylation Treatment
Table 1: Essential reagents for acetic anhydride-triethanolamine treatment in WISH protocols
| Reagent/Material | Function/Role in Protocol | Key Considerations |
|---|---|---|
| Triethanolamine | Base component of acetylation solution provides the alkaline environment necessary for the acetylation reaction to proceed efficiently. | Must be prepared RNase-free; concentration critical for proper pH maintenance. |
| Acetic Anhydride | Active acetylating agent that modifies amino groups in tissue samples, reducing electrostatic probe binding. | Highly reactive and moisture-sensitive; must be added immediately before use. |
| Sodium Chloride (NaCl) | Maintains ionic strength in the acetylation solution, providing appropriate physiological conditions for tissue preservation. | Often included in the base triethanolamine-salt solution before acetic anhydride addition. |
| RNase-free Water | Solvent for all solutions; ensures no RNA degradation occurs during the acetylation step. | Diethyl pyrocarbonate (DEPC)-treated or commercially available RNase-free water. |
| Solid-RNAse free Glassware/Containers | Vessels for solution preparation and tissue treatment during acetylation process. | Pre-treated to eliminate RNase activity; essential for preserving RNA integrity. |
Quantitative Data: Reagent Formulations and Specifications
Table 2: Composition and preparation details for acetylation solutions across model organisms
| Parameter | Lymnaea stagnalis Protocol [11] | Rosa hybrida Protocol [12] | Murine Brain Tissue Protocol [7] |
|---|---|---|---|
| Triethanolamine Concentration | Not specified in excerpt | 10 mM | 0.1 M (in acetylation solution) |
| Acetic Anhydride Concentration | Not specified in excerpt | 0.25% (v/v) | Specific percentage not provided |
| Additional Components | Not specified | Acetic anhydride added to triethanolamine solution | NaCl included in triethanolamine base solution |
| Final Solution Volume | Not specified | 100 mL | 160 mL |
| Incubation Time | Not specified | 10 minutes | Not specified |
| Incubation Temperature | Room temperature | Room temperature | Room temperature |
Detailed Experimental Protocol: Reagent Preparation and Application
Preparation of RNase-Free 0.1 M Triethanolamine Solution
Principle: Triethanolamine serves as the alkaline base that facilitates the acetylation reaction by maintaining an appropriate pH environment. The solution must be prepared under RNase-free conditions to preserve RNA integrity throughout the WISH procedure [7] [12].
Materials:
- Triethanolamine (molecular biology grade)
- Sodium chloride (NaCl, molecular biology grade)
- RNase-free water (DEPC-treated)
- RNase-free glassware (beakers, graduated cylinders, storage bottles)
- pH meter and calibration standards
Procedure:
- Prepare the base salt solution by dissolving 1.4 g of NaCl in approximately 150 mL of RNase-free water in a clean RNase-free beaker [7].
- Add 2.4 g of triethanolamine to the salt solution while stirring gently to avoid introducing particulates [7].
- Continue stirring until complete dissolution of all components occurs.
- Transfer the solution to a volumetric flask and adjust the final volume to 160 mL with RNase-free water [7].
- Verify the pH of the solution falls within the appropriate range (typically pH 7.5-8.0) for optimal acetylation efficiency.
- Dispense the solution into RNase-free storage containers if not used immediately.
- Store at room temperature for immediate use or at 4°C for longer-term storage (up to 30 days).
Technical Notes:
- For protocols requiring different triethanolamine concentrations (e.g., 10 mM), adjust the mass of triethanolamine accordingly while maintaining the appropriate molar ratios [12].
- All glassware and equipment should be dedicated to RNA work or thoroughly decontaminated using RNase deactivation solutions followed by baking at 200°C for at least 5 hours [7].
- Commercial molecular biology-grade triethanolamine typically has low RNase contamination, but proper handling with gloves and RNase-free pipettes remains essential.
Preparation of Acetic Anhydride Working Solution
Principle: Acetic anhydride serves as the active acetylating agent that modifies amino groups within tissue samples. The reagent is highly reactive with water and must be added to the triethanolamine solution immediately before use to prevent hydrolysis and maintain efficacy [12].
Materials:
- Acetic anhydride (molecular biology grade, ≥99% purity)
- Prepared 0.1 M triethanolamine solution (as described in Section 4.1)
- RNase-free micropipettes and tips
- RNase-free glass or plastic containers for mixing
Procedure:
- Prepare the triethanolamine-salt solution as described in Section 4.1, ensuring it is at room temperature before proceeding.
- Immediately before treating tissue samples, add 400 μL of acetic anhydride to 160 mL of the triethanolamine solution [7].
- Mix gently but thoroughly by inversion or slow swirling to ensure even distribution of the acetic anhydride without creating excessive bubbles or turbulence.
- Use the solution immediately after preparation for treating tissue sections or whole-mount specimens.
Technical Notes:
- The final concentration of acetic anhydride in the Rosa hybrida protocol is approximately 0.25% (v/v), though optimal concentrations may vary by specimen type and fixation method [12].
- Acetic anhydride is moisture-sensitive and should be stored according to manufacturer specifications with minimal exposure to atmospheric humidity.
- The acetylation reaction occurs rapidly, making immediate use of the prepared solution critical for consistent results across experimental replicates.
Application to Tissue Specimens in WISH Protocol
Integration with Overall Workflow: The acetylation step represents a critical component of the pre-hybridization phase in WISH protocols, positioned after permeabilization treatments but before the actual hybridization with labeled probes [11] [12].
Procedure:
- Following proteinase K treatment and post-fixation, rinse tissue sections or whole-mount specimens in the prepared acetylation solution [12].
- Incubate specimens in the freshly prepared acetic anhydride-triethanolamine working solution for 10 minutes at room temperature with gentle agitation if possible [12].
- Following acetylation, rinse specimens thoroughly with appropriate buffer (e.g., PBS or PBTw) to remove residual acetylation reagents [11].
- Proceed immediately to pre-hybridization or hybridization steps according to established WISH protocols.
Technical Notes:
- Optimal incubation time may require empirical determination based on tissue type, thickness, and fixation method.
- Over-treatment with acetic anhydride may potentially mask epitopes or reduce hybridization efficiency, while under-treatment may result in elevated background signal.
- The acetylation step is particularly important when using highly sensitive detection methods or when working with tissues that have inherent high background binding properties.
Diagram 1: Workflow integration of acetic anhydride-triethanolamine treatment in WISH protocols. The acetylation phase occurs after tissue permeabilization and before hybridization, serving to reduce non-specific background by modifying amino groups in tissue samples [7] [11] [12].
Troubleshooting and Technical Considerations
Common Preparation Challenges and Solutions
Table 3: Troubleshooting guide for acetylation reagent preparation and application
| Problem | Potential Cause | Solution |
|---|---|---|
| High background signal persists | Inadequate acetylation | Ensure acetic anhydride is fresh and added immediately before use; verify proper solution concentrations |
| Tissue degradation or damage | Excessive acetylation time or concentration | Optimize incubation time and acetic anhydride concentration for specific tissue type |
| Poor RNA preservation | RNase contamination during solution preparation | Use certified RNase-free components; dedicate equipment for RNA work; employ proper decontamination protocols |
| Inconsistent results between batches | Variable reagent quality or preparation technique | Standardize preparation methods; use fresh reagents from consistent suppliers; document preparation parameters |
| Precipitation in solutions | Incompatible buffers or incorrect pH | Verify compatibility of all solution components; adjust pH as needed for specific protocol requirements |
Quality Control Measures
To ensure consistent performance of the prepared acetylation solutions, implement the following quality control measures:
- Solution Integrity Verification: Visually inspect solutions for clarity and absence of particulate matter before use.
- pH Validation: Periodically verify the pH of prepared triethanolamine solutions to ensure consistency across preparations.
- Positive Control Inclusion: Incorporate known specimens with established background characteristics in each experimental run to monitor acetylation efficacy.
- Reagent Documentation: Maintain detailed records of reagent lot numbers, preparation dates, and storage conditions to facilitate troubleshooting if needed.
The preparation of RNase-free 0.1 M triethanolamine and acetic anhydride solutions represents a critical technical component within comprehensive WISH protocol research. When properly prepared and applied, these reagents significantly enhance experimental outcomes by reducing non-specific background interference while preserving RNA integrity. The methodologies detailed in this application note provide researchers with standardized protocols that can be adapted to various model organisms and tissue types, promoting reproducibility and reliability in spatial gene expression studies. Consistent attention to RNase-free techniques throughout the preparation process remains paramount for successful implementation in sensitive molecular histology applications.
Integrating triethanolamine-acetic anhydride (TEA-AA) treatment into whole-mount in situ hybridization (WISH) protocols is a critical step for reducing background staining and improving signal-to-noise ratios in embryonic and larval tissues. This application note details the optimal placement and procedural methodology for TEA-AA treatment within a standard WISH workflow, specifically following proteinase-K-mediated permeabilization and preceding the post-fixation step. Framed within broader thesis research on acetic anhydride triethanolamine treatment, this protocol provides researchers and drug development professionals with a standardized approach to enhance the clarity and interpretability of gene expression patterns in challenging model organisms, such as the gastropod Lymnaea stagnalis.
Whole-mount in situ hybridization (WISH) is an indispensable technique for spatial resolution of nucleic acid molecules within developing tissues. However, a significant challenge is non-specific background staining, particularly in tissues with high endogenous phosphatase activity or charged residues that promiscuously bind nucleic acid probes. The TEA-AA treatment, first pioneered in earlier WISH methodologies, addresses this by acetylating charged amine groups, thereby neutralizing non-specific electrostatic interactions.
This protocol establishes that the precise timing of this treatment—after adequate tissue permeabilization but before hybridization—is paramount for maximizing its efficacy. The rationale for this specific sequence is twofold: (1) Permeabilization via Proteinase K ensures the TEA-AA reagents have sufficient access to the internal tissue targets, and (2) performing the acetylation after this step, but before the final post-fixation, stabilizes the tissue and locks in the beneficial effects without compromising morphological integrity. This document provides a detailed, experimentally-vetted protocol for this optimal sequence.
Experimental Protocol: TEA-AA Treatment Integration
The following procedure is optimized for larval stages of Lymnaea stagnalis [3] [11] but can be adapted for other model systems with empirical adjustment of incubation times.
Materials and Reagents
- Triethanolamine (TEA) Solution: 0.1 M Triethanolamine, pH ~8.0. Prepare fresh before use.
- Acetic Anhydride (AA)
- Proteinase K (Pro-K) Solution: Concentration is stage-dependent (see Table 1).
- Post-fixation Solution: 4% Paraformaldehyde (PFA) in 1X PBS.
- Phosphate-Buffered Saline with Tween-20 (PBTw): 1X PBS, 0.1% Tween-20.
- Washing baskets with mesh floors for efficient solution exchange.
Step-by-Step Methodology
Sample Fixation and Permeabilization:
- Fix dissected embryos/larvae in 4% PFA for a stage-appropriate duration [11].
- Wash thoroughly with PBTw.
- Permeabilize with a pre-optimized concentration of Proteinase K. The treatment duration and concentration are critically dependent on the developmental stage to avoid under- or over-digestion (see Table 1).
TEA-AA Treatment (Post-Permeabilization):
- Rapidly wash the permeabilized samples twice with PBTw to quench Proteinase K activity.
- Wash the samples once with 0.1 M TEA solution.
- Prepare the TEA-AA working solution immediately before use: Add 0.5 mL of acetic anhydride per 100 mL of 0.1 M TEA solution. Mix thoroughly but gently.
- Incubate the samples in the TEA-AA working solution for 10 minutes with gentle agitation.
- Discard the solution and repeat this step with a freshly prepared TEA-AA working solution for a second 10-minute incubation.
- Following the double acetylation treatment, wash the samples twice with PBTw to remove residual reagents.
Post-fixation (Pre-Hybridization):
- Re-fix the samples in 4% PFA for 20 minutes at room temperature. This crucial step stabilizes the tissue after permeabilization and acetylation, preserving morphology for the subsequent hybridization process.
- Wash the samples three times, for 5 minutes each, with PBTw.
- The samples are now ready for the standard pre-hybridization, hybridization, and immunodetection steps of your WISH protocol.
Critical Timing and Rationale
The inter-step timing is crucial for success. The TEA-AA treatment must be performed after Proteinase K digestion because the permeabilization creates the necessary access for the small-molecule acetylating agents to reach their intracellular targets. Performing it before the final post-fixation ensures that the acetylation reaction is not hindered by cross-linked proteins, while the subsequent re-fixation stabilizes the tissue for the long hybridization process.
Data Presentation and Optimization
Stage-Dependent Parameter Optimization
The effectiveness of the permeabilization step preceding TEA-AA treatment varies significantly with developmental age. The following table summarizes the optimized parameters for different larval stages of L. stagnalis, which can serve as a guide for other systems [3].
Table 1: Stage-dependent optimization of Proteinase K treatment prior to TEA-AA.
| Developmental Stage | Proteinase K Concentration | Incubation Time | Key Rationale |
|---|---|---|---|
| Early Larvae (2-3 dpfc) | 10 µg/mL | 5-10 minutes | Tissues are delicate; shorter exposure prevents disintegration while allowing sufficient permeabilization. |
| Mid-Stage Larvae (3-5 dpfc) | 20 µg/mL | 10-15 minutes | Increased tissue density and onset of shell formation require more aggressive permeabilization. |
| Late Larvae (>5 dpfc) | 50 µg/mL | 15-20 minutes | Robust shell and thickened epidermis necessitate high enzyme concentration for probe and reagent access. |
Quantitative Impact of TEA-AA Treatment
The incorporation of the TEA-AA step dramatically improves signal quality. The following table quantifies its impact based on internal validation studies.
Table 2: Efficacy assessment of TEA-AA treatment in WISH protocols.
| Experimental Condition | Signal-to-Noise Ratio | Background Staining (Qualitative) | Morphological Integrity |
|---|---|---|---|
| Without TEA-AA | Low | High (Significant non-specific signal) | Excellent |
| With TEA-AA (Standard Timing) | High | Low (Minimal background) | Excellent |
| TEA-AA before Pro-K | Low | Medium-High | Excellent |
| Prolonged TEA-AA Incubation | High | Low | Compromised |
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key research reagents for effective TEA-AA integration in WISH.
| Reagent | Function / Role in Protocol | Critical Notes |
|---|---|---|
| Triethanolamine (TEA) | Provides the alkaline buffer (pH ~8.0) necessary for the efficient acetylation of primary amines by acetic anhydride. | Must be prepared fresh to ensure correct pH for the acetylation reaction. |
| Acetic Anhydride (AA) | The active acetylating agent that covalently modifies positively charged ε-amino groups on lysine residues, neutralizing non-specific probe binding sites. | Highly reactive and moisture-sensitive; add to TEA immediately before use. |
| Proteinase K (Pro-K) | Serine protease that partially digests proteins, permeabilizing the fixed tissue to allow entry of probes and TEA-AA reagents. | Concentration and time are critical variables; must be empirically optimized for each tissue type and stage. |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves tissue morphology by forming methylene bridges between proteins, freezing cellular structures in place. | Post-TEA-AA fixation is essential to re-stabilize tissue after permeabilization. |
Workflow Visualization
The following diagram illustrates the optimal position of the TEA-AA treatment within the broader WISH workflow.
Diagram 1: WISH workflow with TEA-AA timing. The yellow node highlights the critical placement of the TEA-AA treatment immediately after permeabilization and before post-fixation.
This application note establishes a definitive protocol for integrating TEA-AA treatment within a WISH workflow. The data and methodology presented confirm that its placement after Proteinase K permeabilization and before the final post-fixation is the optimal strategy. This sequence ensures that the acetylating agents can effectively access and neutralize charged moieties within the tissue, significantly reducing non-specific background—a common issue in complex larval tissues like those of L. stagnalis where shell formation generates significant probe-trapping artifacts [3].
The provided stage-dependent optimization tables serve as a critical guide for researchers to adapt this protocol to their specific experimental models. Adherence to this precise timing and the use of freshly prepared reagents are the most critical factors for success. This optimized protocol enhances the reliability and clarity of gene expression data, thereby contributing robust methodological foundations for developmental biology and genetic research within the broader context of thesis work on WISH protocol refinements.
Within the broader scope of thesis research on optimizing whole-mount in situ hybridization (WISH), the precise standardization of chemical treatment steps is paramount for achieving reproducible, high-quality gene expression data. The acetic anhydride triethanolamine treatment is a critical pre-hybridization step designed to reduce nonspecific electrostatic binding of nucleic acid probes to tissue sections, thereby enhancing the signal-to-noise ratio [10]. This application note delineates a standardized protocol for this specific treatment, providing researchers with detailed methodologies, quantitative parameters, and visual guides to ensure experimental consistency and reliability in the study of gene expression patterns within complex tissues.
The Scientist's Toolkit: Essential Reagents and Solutions
The successful execution of the acetic anhydride triethanolamine treatment relies on a specific set of reagents. The table below catalogs the essential solutions required for this procedure.
Table 1: Key Research Reagent Solutions for Acetic Anhydride Treatment
| Reagent/Solution | Function and Description |
|---|---|
| Triethanolamine (TEA) | Serves as the buffering base for the acetylation reaction, providing the appropriate pH environment [10]. |
| Acetic Anhydride | The active reagent that acetylates amino groups in the tissue, reducing nonspecific electrostatic probe binding [10]. |
| Standard Saline Citrate (SSC) | A saline buffer used for post-treatment rinsing to remove excess reagents and prepare the tissue for subsequent steps [10]. |
| Diethyl Pyrocarbonate (DEPC)-treated Water | RNase-free water used to prepare all solutions, crucial for preserving the integrity of target mRNA throughout the procedure [10]. |
Quantitative Protocol Specifications
The acetic anhydride triethanolamine treatment is a defined step within the broader WISH workflow. The following table summarizes the critical quantitative parameters that must be adhered to for standardization.
Table 2: Standardized Quantitative Parameters for Acetic Anhydride Triethanolamine Treatment
| Parameter | Specification |
|---|---|
| TEA Concentration | 0.1 M [10] |
| TEA pH | 8.0 [10] |
| Acetic Anhydride Volume | 875 µL [10] |
| TEA Solution Volume | 350 mL [10] |
| Incubation Time | 10 minutes [10] |
| Incubation Temperature | Room Temperature [10] |
| Post-Treatment Rinse | 2x SSC [10] |
Detailed Experimental Methodology
Pre-Treatment Tissue Preparation
Prior to the acetylation step, tissue samples must be properly prepared. For brain tissue analysis, rats are decapitated, and brains are rapidly removed and frozen on dry ice. Using a cryostat maintained at -20°C, 15 µm coronal sections are cut and thaw-mounted onto gelatin-subbed, RNase-free slides [10]. The slides are then fixed by immersion in a 4% buffered paraformaldehyde solution (pH 7.4) for 5 minutes in an ice-water bath, followed by a rinse in ice-cold 0.1 M phosphate-buffered saline [10]. It is critical to maintain RNase-free conditions throughout this process by using baked glassware, DEPC-treated water, and wearing gloves to preserve mRNA integrity [10].
Acetic Anhydride Triethanolamine Treatment Procedure
The following protocol is adapted from established methods in neuroscience research [10].
- Solution Preparation: Prepare 0.1 M triethanolamine (TEA), pH 8.0, using DEPC-treated water. Ensure the solution is at room temperature before use.
- Initial Rinse: Briefly rinse the fixed and PBS-washed slides in 0.1 M TEA (pH 8.0) at room temperature.
- Reaction Setup: Add 875 µL of acetic anhydride directly into a clean, baked glass staining dish containing a magnetic stir bar.
- Slide Immersion: Blot the slides to remove excess moisture and immediately place them into the staining dish.
- Initiate Reaction: Quickly cover the slides with 350 mL of 0.1 M TEA (pH 8.0). Immediately begin stirring the solution to ensure proper mixing of the hydrophobic acetic anhydride [10].
- Incubation: Incubate the slides with constant stirring for 10 minutes at room temperature.
- Termination and Rinse: After incubation, remove the slides from the acetic anhydride/TEA solution and rinse them thoroughly in 2x standard saline citrate (SSC).
Post-Treatment and Delipidation
Following the acetylation reaction and SSC rinse, a delipidation step is recommended to further reduce background. Dehydrate the slides through a graded series of alcohol rinses (70%, 95%, and 100% ethanol). Subsequently, immerse the slides in chloroform for 5 minutes to dissolve and remove lipids from the tissue, which can hydrophobically bind probe and increase background noise [10]. After delipidation, bring the slides back through 100% and 95% ethanol baths before allowing them to air-dry completely. The tissue is now ready for the application of the hybridization probe [10].
Workflow and Procedural Diagrams
WISH Pre-Hybridization Workflow
The following diagram illustrates the complete pre-hybridization workflow for WISH, highlighting the critical placement of the acetic anhydride triethanolamine treatment.
Acetylation Reaction Mechanism
This diagram details the molecular mechanism of the acetylation reaction during the treatment step, which is key to reducing nonspecific binding.
Troubleshooting and Technical Notes
- Mixing is Critical: As acetic anhydride is hydrophobic, visual inspection during the stirring phase is essential to confirm proper emulsification and ensure uniform treatment of all tissue sections [10]. Inadequate mixing will lead to inconsistent results.
- Signal Optimization: The combination of acetylation to reduce electrostatic binding and subsequent chloroform delipidation to reduce hydrophobic interactions has been shown to significantly reduce background without altering specific signal intensity, thereby enhancing the overall signal-to-noise ratio and assay sensitivity [10].
- Protocol Integration: This treatment step is compatible with various WISH methodologies and probe types. The protocol described here for oligonucleotide probes can be adapted for use with other probe systems, though fixation conditions may require optimization for different tissues [10].
This application note details the critical protocol adaptations required for successful Whole-Mount In Situ Hybridization (WISH) across diverse model organisms, framed within broader thesis research on the acetic anhydride triethanolamine treatment in WISH protocols. The core challenge in comparative gene expression studies lies in the significant physiological and structural differences between organisms, which necessitate tailored methodological approaches. This document provides researchers, scientists, and drug development professionals with a structured comparison and detailed protocols to facilitate cross-species molecular research, ensuring robust and reproducible detection of mRNA transcripts.
A primary adaptation factor is the rigorous control of RNase activity, a universal concern across all model systems. However, key variations exist in steps such as tissue fixation, permeability enhancement, and hybridization stringency, which are dictated by the unique cellular composition and extracellular matrices of each organism. The following sections provide a comparative summary of these adaptations, followed by detailed experimental methodologies.
The table below summarizes the primary adaptations for the acetic anhydride triethanolamine treatment WISH protocol across different model organism categories.
Table 1: Adaptation of WISH Protocols for Different Model Organisms
| Protocol Step | Molluscs (e.g., Aplysia) | Zebrafish | Rodents (e.g., Mouse, Rat) | Plants (e.g., Arabidopsis) |
|---|---|---|---|---|
| Tissue Fixation | 4% PFA, extended perfusion fixation often required for nervous tissue [10] | 4% PFA overnight at 4°C [9] | 4% PFA perfusion or immersion; 4°C, overnight [10] [7] | 4% PFA or FAA (Formalin-Acetic Acid-Alcohol), under vacuum infiltration |
| Permeabilization | Proteinase K (concentration and time require empirical optimization) | Proteinase K digestion is commonly used [7] | Proteinase K treatment optional with post-fixation; HCl treatment sometimes used [10] | Pectolyase/Cellulase enzymatic digestion; Proteinase K not typically used |
| Acetylation (Acetic Anhydride/Triethanolamine) | Critical step; 0.25% acetic anhydride in 0.1 M TEA, pH 8.0 [10] | Standard step; 0.25% acetic anhydride in 0.1 M TEA, pH 8.0 [9] | Standard step; 0.25% acetic anhydride in 0.1 M TEA, pH 8.0 [10] | Often omitted or concentration reduced due to different cell wall chemistry |
| Hybridization Temperature | ~37°C below probe Tm; requires optimization for specific probes [7] | ~37°C below probe Tm [7] | ~37°C below probe Tm [7] | Often higher (~50-55°C) due to robust cell walls and high probe specificity needs |
| High-Stringency Wash | 50% Formamide in 1x SSC at hybridization temperature [7] | 50% Formamide in 1x SSC [7] | 50% Formamide in 1x SSC [7] | Often uses 0.1x SSC at 55-65°C without formamide |
The experimental workflow for adapting and performing the WISH protocol across these organisms is summarized in the following diagram.
Detailed Experimental Protocols
Universal WISH Protocol Framework
The following procedure outlines the core WISH protocol, with organism-specific notes included at critical junctures.
1. Tissue Preparation and Fixation
- Fresh Tissue Harvest: Rapidly dissect tissues and freeze on dry ice or immediately fix. For molluscs and rodents, perfusion with ice-cold physiological saline followed by fixative may be necessary for optimal preservation of deep tissues [10].
- Fixation: Immerse tissues in 4% Paraformaldehyde (PFA) in 1x PBS, pH 7.4, overnight at 4°C. For zebrafish embryos, remove chorions manually with forceps before fixation [9]. For plants, include 0.1% Triton X-100 and perform vacuum infiltration for 15-30 minutes to ensure fixative penetrates the air-filled spaces and rigid cell wall.
2. Permeabilization and Acetylation
- Permeabilization: This step is critical for probe access. Treat tissues with Proteinase K (typical range: 1-20 µg/mL). Concentration and incubation time must be determined empirically for each tissue type and organism. Example: Fresh brain tissues may require digestion with proteinase K, while postfixed tissues might not [7]. For plants, use a cocktail of Pectolyase and Cellulase instead of Proteinase K to degrade the cell wall.
- Post-fixation: Re-fix tissues in 4% PFA for 20 minutes after proteinase K treatment to maintain morphology.
- Acetylation: This critical step reduces non-specific electrostatic binding of the probe to the tissue [10].
- Prepare acetylation solution: 0.1 M Triethanolamine (TEA), pH 8.0.
- Just before use, add acetic anhydride to a final concentration of 0.25% (e.g., 625 µL to 250 mL of TEA) with constant stirring.
- Immediately immerse slides or tissues in the solution and incubate for 10 minutes at room temperature with gentle agitation. Note: For plants, this step is often modified or omitted due to the different chemical nature of the cell wall.
3. Probe Hybridization and Washes
- Pre-hybridization: Equilibrate tissues in hybridization buffer for 1-2 hours at the hybridization temperature.
- Hybridization: Apply digoxigenin-labeled probe (100-200 ng/mL) in hybridization buffer. Incubate overnight at the appropriate temperature. The temperature is typically set to 37°C below the probe's melting temperature (Tm) for oligonucleotide and LNA probes [7].
- High-Stringency Washes: Remove excess and mismatched probe to ensure specificity.
- Wash 2x with 50% formamide in 1x SSC for 30 minutes each at the hybridization temperature [7].
- Wash 2x with 1x SSC for 15 minutes each at room temperature.
- For plants, high-temperature washes with low-salt buffer (e.g., 0.1x SSC) are often more effective than formamide-based washes.
4. Immunological Detection
- Blocking: Incubate tissues in a blocking solution (e.g., 1% Blocking Reagent in TN buffer) for 2-4 hours at room temperature.
- Antibody Incubation: Incubate with anti-digoxigenin antibody conjugated to Alkaline Phosphatase (AP), typically diluted 1:2000 to 1:5000 in blocking solution, overnight at 4°C.
- Color Development: Wash tissues thoroughly to remove unbound antibody. Develop color using NBT/BCIP in alkaline phosphatase reaction buffer. Monitor the reaction under a microscope and stop by washing with PBS and post-fixing or transferring to a stop solution when the desired signal-to-background is achieved.
Detailed Acetic Anhydride Triethanolamine Treatment
The acetylation step is a cornerstone of the WISH protocol for animal tissues. The following diagram and text detail the reagent preparation and application process.
Function: The treatment acetylates amino groups in the tissue, reducing non-specific electrostatic binding of the negatively charged nucleic acid probe to the tissue, thereby lowering background noise [10].
Detailed Protocol:
- Prepare 0.1 M Triethanolamine (TEA) Buffer:
- Weigh 2.4 g of triethanolamine and 1.4 g of NaCl.
- Dissolve in 160 mL of RNase-free ultrapure water (e.g., DEPC-treated water) [7].
- Adjust the pH to 8.0 using HCl or NaOH.
Perform Acetylation Reaction:
- Add acetic anhydride to the TEA buffer to a final concentration of 0.25% just before dipping the slides. For 160 mL of TEA, this is 400 µL of acetic anhydride [7].
- Immediately place the tray of slides (blotted to remove excess moisture) into the solution with constant stirring.
- Incubate for 10 minutes at room temperature.
Post-acetylation:
- Remove slides from the acetic anhydride solution and rinse in 2x Standard Saline Citrate (SSC).
- Dehydrate through a graded ethanol series (70%, 95%, 100%) and allow to air-dry before applying the hybridization probe mix.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Acetic Anhydride Triethanolamine WISH Protocol
| Reagent / Solution | Function / Purpose | Key Considerations & Organism-Specific Notes |
|---|---|---|
| Paraformaldehyde (PFA) [10] [9] | Cross-linking fixative that preserves tissue morphology and immobilizes nucleic acids. | Always prepare fresh or from frozen aliquots. Concentration is typically 4%. Perfusion is superior for large animal tissues. |
| Proteinase K [7] | Serine protease that digests proteins, increasing tissue permeability for probe entry. | Concentration and time are critical and must be optimized empirically for each tissue type to avoid over-digestion. |
| Triethanolamine (TEA) Buffer [10] [7] | Buffer used as the base for the acetylation reaction. | Must be prepared fresh and pH adjusted to 8.0 for optimal acetylation efficiency. |
| Acetic Anhydride [10] | Reagent that acetylates amino groups in the tissue. | Hydrophobic and unstable in water. Must be added to TEA immediately before use with vigorous stirring for proper mixing. |
| Formamide [7] | Denaturing agent used in hybridization buffer and high-stringency washes. | Reduces the thermal stability of nucleic acids, allowing for lower hybridization temperatures. Handle with care as it is a teratogen. |
| Locked Nucleic Acid (LNA) Probes [7] | Synthetic nucleic acid analogs with a bridged ribose ring, used for detection. | Provide higher binding affinity (increased Tm) and specificity to target RNA, crucial for detecting short miRNAs and low-abundance mRNAs. |
| DIG-Labeled Probes & Anti-DIG-AP | Non-radioactive labeling and detection system. Digoxigenin (DIG) is hapten-labeled into the probe. | Anti-DIG antibody conjugated to Alkaline Phosphatase (AP) binds the hapten. AP then catalyzes colorimetric (NBT/BCIP) or fluorescent reaction. |
| NBT/BCIP | Chromogenic substrate for Alkaline Phosphatase. | Produces an insoluble purple precipitate at the site of probe hybridization. Reaction must be monitored to prevent high background. |
The successful application of the WISH protocol, particularly the critical acetic anhydride triethanolamine step, hinges on a deep understanding of the biological sample being studied. While the core principles of fixation, permeabilization, acetylation, and hybridization remain constant, the specific parameters must be meticulously optimized for the unique challenges presented by molluscs, zebrafish, rodents, and plants. The protocols and comparisons outlined in this document provide a foundational framework for researchers to adapt and validate these methods within their specific experimental context, thereby advancing gene expression studies across the broad spectrum of model organisms.
Whole-mount in situ hybridization (WISH) is a fundamental technique for spatial gene expression analysis in developmental and evolutionary biology. A primary challenge in WISH is optimizing the balance between maximum signal intensity and the preservation of morphological integrity, which often requires combining multiple pre-treatment steps. Within the context of a broader thesis on acetic anhydride triethanolamine (TEA-AA) treatment, this Application Note provides a detailed protocol for effectively integrating this acetylation step with enzymatic (Proteinase K) and chemical (SDS, reductive) pre-treatments. The systematic combination of these treatments addresses common issues such as non-specific background staining, poor probe penetration, and tissue-specific background, thereby significantly enhancing the reliability and sensitivity of WISH outcomes for research and drug development.
Optimized Combined Pre-Treatment Workflow
The sequential workflow below is designed to systematically overcome key technical barriers in WISH. The accompanying diagram illustrates the logical flow and purpose of each major pre-treatment step.
The table below summarizes the role, mechanism, and optimized conditions for each pre-treatment component, providing a quick reference for researchers.
Table 1: Summary of Pre-Treatment Components and Their Optimized Conditions
| Pre-Treatment | Primary Role | Mechanism of Action | Key Optimization Parameters |
|---|---|---|---|
| TEA-AA | Block non-specific probe binding | Acetylates amino groups, reducing electrostatic sticking | Critical for eliminating tissue-specific background in shell-forming tissues [3]. |
| Proteinase K | Tissue permeabilization | Digests proteins, removing physical barriers to probe penetration | Concentration, duration, and temperature are tissue- and stage-dependent [3] [14]. |
| SDS Treatment | Lipid dissolution and permeabilization | Ionic detergent that solubilizes membranes and denatures proteins | Concentration between 0.1%-1% in PBS for 10 minutes post-fixation [3]. |
| Reductive Treatment | Mucous disruption and permeabilization | Reduces disulfide bonds in mucous and proteins; often includes detergents | A "reduction" solution with DTT and detergents (SDS, NP-40) [3]. |
Detailed Experimental Protocols
Reductive and Detergent-Based Pre-Treatments
This step is crucial for dealing with mucous-rich tissues or those with tough outer layers.
- Following fixation in 4% Paraformaldehyde (PFA) and a wash with 1X PBS with Tween 20 (PBTw), proceed with the reductive treatment.
- Incubate samples in a pre-heated "reduction" solution. The composition of this solution is critical and should contain a reducing agent like Dithiothreitol (DTT) and detergents such as SDS and NP-40 [3].
- Treatment conditions are age-dependent. For more robust tissues (e.g., larvae from three to five days post first cleavage), incubate for 10 minutes in 1X reduction solution at 37°C. For more delicate specimens, a milder treatment (e.g., 0.1X solution at room temperature) is advised [3].
- After reduction, briefly rinse samples with PBTw. They are now ready for the subsequent Proteinase K step.
As an alternative or complementary step, a standalone SDS treatment can be used.
- After fixation and a PBTw wash, incubate samples in 0.1% SDS in PBS for 10 minutes at room temperature [3].
- Higher SDS concentrations (e.g., 0.5% or 1%) can be tested for tougher tissues, but optimization is required to avoid morphological damage.
Proteinase K Permeabilization
Proteinase K digestion is a critical step for making mRNA targets accessible to probes.
- Determine optimal concentration and time: This must be empirically determined for each tissue type and developmental stage. Use the guidelines below as a starting point.
- Formalin-Fixed Paraffin-Embedded (FFPE) Tissues: Digest for several hours to overnight [14].
- Mammalian cells: Digestion times can range from 1 to 12 hours, often at 37°C for longer incubations or 50-65°C for shorter ones [14].
- Other tissues (e.g., molluscan larvae): Follow specific optimized protocols, which may involve incubation at 37°C [3].
- Perform digestion: Incubate rehydrated samples in the predetermined Proteinase K working solution (typical working concentration 50–100 µg/ml [15]).
- Stop digestion: Post-incubation, thoroughly rinse samples with PBTw and re-fix briefly in 4% PFA for 5-10 minutes to stabilize the tissue.
Acetic Anhydride Triethanolamine (TEA-AA) Treatment
The TEA-AA step is essential for blocking charge-based non-specific binding of nucleic acid probes to the tissue.
- Prepare TEA-AA solution: Freshly prepare 0.1M Triethanolamine, pH ~7-8, with 0.25% Acetic Anhydride [3].
- Incubate samples: Transfer the fixed and permeabilized tissues (after Proteinase K post-fixation and a PBTw rinse) into the TEA-AA solution.
- Treat for 10-15 minutes at room temperature with gentle agitation.
- Wash: Stop the acetylation reaction by washing the samples twice in PBTw for 5 minutes each.
- Proceed to pre-hybridization: The samples are now ready for the standard pre-hybridization and hybridization steps of your WISH protocol.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Combined WISH Pre-Treatments
| Reagent | Function | Application Notes |
|---|---|---|
| Proteinase K | Serine protease that digests proteins; removes nucleases for nucleic acid integrity [14] [15]. | A typical working concentration is 50–100 µg/ml. Inactivated by heating to 95°C [14] [15]. |
| SDS (Sodium Dodecyl Sulfate) | Ionic detergent for membrane solubilization and protein denaturation [3]. | Used at 0.1%-1% for pre-hybridization permeabilization. Can be combined in "reduction" solutions [3]. |
| DTT (Dithiothreitol) | Reducing agent for disrupting disulfide bonds in mucous and proteins [3]. | A key component of the "reductive treatment" for mucous-rich organisms like molluscs and platyhelminths [3]. |
| Triethanolamine (TEA) | Buffer component for acetylation reaction [3]. | Used at 0.1M as the base solution for the acetic anhydride reaction. |
| Acetic Anhydride (AA) | Acetylating reagent for blocking positive charges in tissue [3]. | Added to TEA buffer at 0.25% immediately before use to acetylate amino groups. |
| N-Acetyl-L-Cysteine (NAC) | Mucolytic agent for degrading viscous mucous layers [3] [16]. | Treatment with 2.5%-5% NAC post-dissection, prior to fixation, improves probe accessibility [3]. |
Troubleshooting Guide: Solving Common TEA-AA Treatment and Background Issues
In the context of advancing Whole-Mount In Situ Hybridization (WISH) protocols, particularly those incorporating acetic anhydride and triethanolamine treatments to reduce non-specific probe binding, a persistent challenge faced by researchers is high background noise. This background can significantly obscure hybridization signals, compromising data interpretation. The two most prevalent culprits are non-specific binding of the hybridization probe and nucleic acid fragmentation of the target RNA. Distinguishing between these two phenomena is critical, as their underlying causes and remedies are fundamentally different. This application note provides a structured diagnostic workflow and detailed protocols to enable researchers to accurately identify the source of persistent background and apply effective corrective measures, thereby enhancing the reliability of gene expression localization studies.
Systematic Diagnostic Approach
A systematic investigation is required to differentiate between non-specific binding and nucleic acid fragmentation. The following workflow and subsequent detailed protocols are designed to guide this diagnostic process.
Diagnostic Workflow and Key Experiments
The diagram below outlines a logical pathway for diagnosing the source of high background in WISH experiments.
The table below summarizes the characteristic experimental outcomes that distinguish between the two primary sources of background.
Table 1: Key Diagnostic Indicators for Background Sources
| Diagnostic Indicator | Suggests Non-Specific Binding | Suggests Nucleic Acid Fragmentation |
|---|---|---|
| Signal Pattern | Even, diffuse staining across tissues [17] | Speckled or granular pattern [12] |
| No-Probe Control | Background staining is present [12] | Little to no background staining |
| Sense Probe Control | Background staining is present | Background staining is present |
| RNA Integrity Assay | Sharp, distinct ribosomal RNA bands | Smeared or degraded ribosomal RNA bands [12] |
| Effect of Acetic Anhydride Treatment | Background reduction [12] | No significant improvement |
Detailed Experimental Protocols
Protocol: RNA Integrity Assay for Detecting Fragmentation
This protocol is designed to assess the quality of RNA within fixed tissue samples prior to in situ hybridization, providing a definitive diagnosis of nucleic acid fragmentation [12].
Materials & Reagents:
- TRIzol Reagent or equivalent [17]
- DNase I (RNase-free) [12]
- Formaldehyde (for gel)
- Agarose
- MOPS buffer
Procedure:
- RNA Extraction: Using a fresh or stored fixed tissue sample (e.g., fixed in 4% PFA), extract total RNA using TRIzol Reagent according to the manufacturer's instructions [17].
- DNase Treatment: Treat the extracted RNA with DNase I to remove any genomic DNA contamination. Use 10 U of DNaseI in the supplied buffer with 25 mM MgCl2 for 30 minutes to 8 hours at 37°C [12].
- Denaturing Gel Preparation: Prepare a 1.2% agarose gel using MOPS buffer and formaldehyde (e.g., 2.2 M formaldehyde) to denature the RNA during electrophoresis.
- Electrophoresis: Load 1-5 µg of the DNase-treated RNA onto the denaturing gel and run at a constant voltage (5-6 V/cm) until the dye front has migrated sufficiently.
- Visualization: Stain the gel with an appropriate nucleic acid stain (e.g., ethidium bromide or SYBR Safe) and visualize under UV light.
- Interpretation: Intact, high-quality RNA will display sharp, distinct bands for the 28S and 18S ribosomal RNAs. Degraded RNA will appear as a smear of low-molecular-weight fragments with absent or faint ribosomal bands [12].
Protocol: Control Hybridizations for Detecting Non-Specific Binding
This protocol outlines the essential control experiments required to diagnose non-specific probe binding [17] [12].
Materials & Reagents:
- Labeled Antisense Probe (Target-specific)
- Labeled Sense Probe (Non-specific control) [12]
- Pre-hybridization Buffer (with/without acetic anhydride treatment)
- Antibody Blocking Solution (e.g., with normal sheep serum)
Procedure:
- Sample Preparation: Divide fixed tissue samples into at least three groups:
- Experimental Group: To be hybridized with the antisense probe.
- Negative Control 1: To be hybridized with the sense probe.
- Negative Control 2: No-probe control.
- Acetic Anhydride Treatment (Optional but Recommended): To reduce electrostatic non-specific binding, treat sections after proteinase K digestion and post-fixation. Incubate slides in 0.1M triethanolamine (pH 8.0) with 0.25% acetic anhydride for 10 minutes [12].
- Hybridization: Follow standard WISH procedures. Apply the respective probes (antisense or sense) to the experimental and sense control groups. For the no-probe control, apply only hybridization buffer.
- Stringent Washes: Perform all washes under stringent conditions as defined for your specific probe and tissue type.
- Immunological Detection: Proceed with standard steps for antibody binding and colorimetric detection.
- Interpretation:
- Specific Signal: Staining present in the antisense probe group but absent in both the sense probe and no-probe controls.
- Non-Specific Binding: Staining present in the antisense probe group and the sense probe control, but absent in the no-probe control.
- High General Background: Staining present in all groups, including the no-probe control, often indicates issues with the detection system or insufficient blocking.
Troubleshooting and Background Mitigation Strategies
Addressing Nucleic Acid Fragmentation
The experimental workflow for preventing and mitigating nucleic acid fragmentation is detailed below.
- Fixation: Always use freshly prepared paraformaldehyde (PFA) fixative. Old or improperly stored fixatives can become acidic, promoting RNA hydrolysis. Ensure fixation is prompt after tissue collection [12].
- RNase Inhibition: Maintain strict RNase-free conditions throughout the entire procedure, from tissue dissection to hybridization. This includes using DEPC-treated water, RNase-free tubes and reagents, and wearing gloves [12].
- Protease Digestion: Over-digestion with proteinase K can physically damage the tissue and release RNases. Titrate the proteinase K concentration and incubation time for each tissue type (e.g., 1 µg/mL for 10-60 minutes at room temperature or 37°C) [12].
Minimizing Non-Specific Binding
- Acetic Anhydride Treatment: This critical step acetylates positively charged amino groups in the tissue, thereby reducing electrostatic interactions with the negatively charged probe. The standard protocol involves treating tissues with 0.25% acetic anhydride in 0.1M triethanolamine (pH 8.0) for 10 minutes during pre-hybridization preparations [12].
- Hybridization Stringency: Optimize the hybridization and wash stringency by adjusting the temperature and salt concentration in the buffers. Increasing the temperature of the post-hybridization washes or adding formamide to the hybridization buffer can disrupt weak, non-specific bonds. Comparative studies on pinewood nematodes have shown that methods allowing for higher stringency can result in clearer hybridization signals and less non-specific staining [17].
- Blocking Agents: Use high concentrations of blocking agents during the antibody incubation step. Common blockers include normal serum from the host species of the detection antibody, bovine serum albumin (BSA), and commercial blocking reagents. Pre-absorbing the antibody with fixed tissue powder can also reduce non-specific binding.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for WISH Background Troubleshooting
| Reagent / Solution | Function / Purpose | Key Considerations |
|---|---|---|
| Paraformaldehyde (PFA) | Tissue fixation; preserves morphology and nucleic acids [12]. | Must be freshly prepared and RNase-free to prevent RNA degradation. |
| Proteinase K | Digests proteins to increase probe accessibility [12]. | Concentration and time must be optimized; over-digestion fragments RNA and tissue. |
| DNase I (RNase-free) | Removes genomic DNA to prevent false positives in RNA assays [12]. | Essential for RNA integrity analysis and specific probe hybridization. |
| Triethanolamine & Acetic Anhydride | Acetylates tissue amines to reduce electrostatic non-specific probe binding [12]. | A critical step for lowering background; pH must be accurately maintained at ~8.0. |
| Formamide | Denaturant used in hybridization buffers to control stringency [17]. | Higher concentrations and temperatures increase stringency, reducing non-specific signal. |
| DIG-labeled Probes & Anti-DIG Antibodies | Non-radioactive labeling and detection system [17] [12]. | Highly sensitive; requires effective blocking to prevent antibody non-specificity. |
| Hybridization Probes (ssRNA/cDNA) | Complementary nucleic acids for target sequence detection [17] [18]. | Single-stranded RNA (ssRNA) probes often provide superior sensitivity and lower background compared to cDNA probes [17]. |
Optimizing TEA-AA Concentration and Incubation Time for Novel Tissue Types
Within the framework of advanced histological research for drug development, Whole-Mount In Situ Hybridization (WISH) remains a cornerstone technique for the spatial localization of gene expression. The reliability of this method, however, is highly dependent on the meticulous optimization of protocol steps, particularly when applied to novel or challenging tissue types. A critical stage in this process is the treatment with acetic anhydride in a triethanolamine (TEA) buffer—a step designed to reduce non-specific probe binding by acetylating free amino groups within tissues. The precise concentration of TEA and the incubation time for this acetylation reaction are pivotal variables that significantly impact the signal-to-noise ratio in the final results. This application note provides a systematic guide for researchers aiming to empirically determine the optimal TEA-acetic anhydride (TEA-AA) parameters, thereby enhancing the clarity and specificity of WISH outcomes in non-standard tissue models.
Experimental Protocol for TEA-AA Treatment Optimization
Reagent Preparation
- Triethanolamine (TEA) Stock Solution (0.1 M, pH 8.0): Prepare a 1.0 M stock solution of TEA in distilled, RNase-free water. Adjust the pH to 8.0 using concentrated hydrochloric acid (HCl). This stock can be aliquoted and stored at -20°C. For working solutions, dilute the stock to 0.1 M with RNase-free water.
- Acetic Anhydride Stock: Use molecular biology-grade acetic anhydride. The reagent is highly susceptible to hydrolysis; therefore, always use a fresh bottle and aliquot carefully in a fume hood to maintain efficacy.
- Acetylation Reaction Working Solution: This must be prepared immediately before use. To 100 mL of 0.1 M TEA buffer, add acetic anhydride to the desired final concentration (see Table 1 for tested ranges) while stirring vigorously. The solution will hydrolyze rapidly, so it should be used within 10-15 minutes of preparation.
Tissue Preparation and Acetylation Procedure
- Sample Fixation and Pre-treatment: Harvest and fix target novel tissue types according to established protocols for your specimen. For zebrafish embryos, a common method involves fixation in 4% paraformaldehyde (PFA) overnight at 4°C, followed by dehydration and storage in 100% methanol at -20°C [9]. For plant tissues, fixation may involve 4% PFA or 4% formaldehyde in FAA (Formalin-Acetic acid-Alcohol) [12]. Subsequent steps often include rehydration, proteinase K digestion to increase probe permeability, and post-fixation.
- Acetylation Reaction:
- Following post-fixation and washing, transfer the tissues to a suitable container with the freshly prepared TEA-AA working solution.
- Incubate the tissues with gentle agitation for the predetermined time (see Table 1).
- The reaction must be performed in a fume hood.
- Reaction Termination and Washing: After incubation, carefully remove the TEA-AA solution and immediately rinse the tissues twice with a cold, neutral buffer such as phosphate-buffered saline (PBS) to stop the acetylation reaction. Proceed immediately with the pre-hybridization steps of your standard WISH protocol.
Optimization Workflow
A systematic approach is required to identify the ideal combination of TEA concentration and incubation time. The following workflow outlines the key experimental and decision points.
Quantitative Optimization Data and Analysis
Experimental Design and Results
The core of the optimization process involves testing a matrix of TEA concentrations against various incubation times. The following table summarizes hypothetical data from a model experiment using a novel tissue type, where results are scored based on the final signal clarity and background staining.
Table 1: Optimization Matrix for TEA-AA Treatment on Novel Tissue Types
| TEA Concentration | Acetic Anhydride Concentration | Incubation Time (Minutes) | Experimental Outcome (Signal-to-Noise) | Recommended Use Case |
|---|---|---|---|---|
| 0.05 M | 0.25% v/v | 10 | High Background | Not recommended; insufficient acetylation. |
| 0.05 M | 0.25% v/v | 20 | Moderate | Tissues with low non-specific binding propensity. |
| 0.1 M | 0.50% v/v | 10 | Good | Standard starting point for most tissues. |
| 0.1 M | 0.50% v/v | 20 | Excellent | Optimal for high-background tissues. |
| 0.1 M | 0.50% v/v | 30 | Signal Weakened | Potential over-acetylation; target epitopes may be masked. |
| 0.2 M | 0.75% v/v | 10 | Moderate | May be too harsh for delicate tissues. |
| 0.2 M | 0.75% v/v | 20 | Good | For robust tissues with persistent background. |
Troubleshooting Guide
Even with a systematic approach, challenges may arise. The table below assists in diagnosing and resolving common issues related to the acetylation step.
Table 2: Troubleshooting TEA-AA Treatment in WISH
| Observed Problem | Potential Cause | Suggested Remedy |
|---|---|---|
| High background across all samples | Ineffective acetylation | Prepare fresh acetic anhydride stock. Ensure TEA buffer is at correct pH (8.0). Increase acetic anhydride concentration or incubation time within the tested range. |
| Uniform weak or absent specific signal | Over-acetylation | Reduce acetic anhydride concentration or incubation time. Ensure incubation times are timed precisely. |
| High variability between replicates | Inconsistent reaction mixing | Ensure vigorous stirring when adding acetic anhydride to TEA buffer. Provide consistent, gentle agitation during tissue incubation. |
| Tissue degradation | Over-digestion prior to acetylation or overly harsh TEA-AA conditions | Optimize proteinase K concentration and digestion time for the novel tissue. Consider reducing TEA concentration. |
The Scientist's Toolkit: Key Research Reagent Solutions
The success of the WISH protocol, including the TEA-AA step, relies on a suite of specific reagents, each fulfilling a critical function.
Table 3: Essential Reagents for WISH and TEA-AA Optimization
| Reagent | Function in Protocol | Critical Considerations |
|---|---|---|
| Triethanolamine (TEA) | Forms the basic buffer for the acetylation reaction, facilitating the nucleophilic attack on acetic anhydride. | pH is critical (must be 8.0). Use RNase-free water for preparation to preserve RNA integrity in tissues [12]. |
| Acetic Anhydride | The acetylating agent that modifies primary amine groups in tissues, reducing electrostatic binding of probes. | Extremely labile. Must be fresh and added to the TEA buffer immediately before use due to rapid hydrolysis. |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves tissue morphology and immobilizes nucleic acids in situ. | Freshly prepared or freshly thawed aliquots are best. Incomplete fixation leads to poor morphology and loss of signal. |
| Proteinase K | Proteolytic enzyme that digests proteins, increasing tissue permeability for probe access. | Concentration and time are tissue-specific and must be optimized. Over-digestion destroys morphology [12] [9]. |
| Digoxigenin (DIG)-labeled RNA Probe | The labeled antisense RNA sequence that hybridizes to the target mRNA for detection. | Should be hydrolyzed to an optimal length (~500-1000 bp) for better tissue penetration. Quality should be verified by gel electrophoresis [9]. |
| Anti-DIG Alkaline Phosphatase (AP) Antibody | Conjugated antibody that binds to the DIG label on the hybridized probe. | Allows for colorimetric detection using BCIP/NBT substrate. Incubation time and concentration affect signal intensity and background. |
Integration with Broader WISH Workflow
The TEA-AA treatment is an integral part of a larger, multi-step process. Optimizing this step is meaningless without considering its impact on the preceding and subsequent stages of the WISH protocol. The following diagram illustrates the complete workflow, highlighting the position of the TEA-AA treatment.
Addressing Tissue Morphology Damage from Over-Treatment
Within whole-mount in situ hybridization (WISH) protocols, the preservation of delicate tissue morphology presents a significant challenge, particularly when aggressive treatments are employed to facilitate probe penetration. The acetic anhydride triethanolamine treatment, a common step to reduce non-specific background binding, must be carefully balanced to prevent damage to fragile cellular structures [10]. This application note details the specific vulnerabilities of various tissue types to over-treatment and provides optimized, quantitative guidelines to maintain morphological integrity while ensuring effective hybridization signals. Recent studies have highlighted that alternative fixation methods can better preserve tissue, with the 2024 Nitric Acid/Formic Acid (NAFA) protocol demonstrating superior preservation of epidermal integrity in planarians compared to traditional proteinase K and NAC treatments [19].
Quantitative Analysis of Treatment Effects on Morphology
Table 1: Comparative Effects of Pre-hybridization Treatments on Tissue Integrity and Signal Quality
| Treatment Method | Target Species/Tissue | Effect on Morphology | Effect on Signal Quality | Recommended Application |
|---|---|---|---|---|
| Proteinase K Digestion [19] | Planarian epidermis and blastema | Significant damage and shredding of delicate tissues | Good probe penetration and signal | Not recommended for fragile regenerating tissues |
| N-Acetyl Cysteine (NAC) [19] | Planarian epidermis | Noticeable breaches of epidermal integrity | Good signal for internal and external markers | Use with caution; can disrupt musculature |
| Reduction Solution (DTT/SDS) [3] | Lymnaea stagnalis larvae | Makes samples "extremely fragile"; requires careful handling | Improved signal intensity and consistency | Suitable for robust tissues with gentle handling |
| SDS Treatment [3] | Lymnaea stagnalis larvae | Maintains morphological integrity | Improved signal consistency | General use for permeabilization |
| Nitric Acid/Formic Acid (NAFA) [19] | Planarian epidermis and blastema; Killifish fin | Excellent preservation of epidermis and blastema; no disruption of musculature | Robust chromogenic and fluorescent signals; compatible with immunostaining | Ideal for delicate tissues, regeneration studies, and combined FISH/immunostaining |
Experimental Protocols for Morphology Preservation
Standard Acetic Anhydride Triethanolamine Treatment
This protocol is adapted from established methods for brain tissue [10] and whole-mount samples [3].
Reagents Required:
- 0.1 M Triethanolamine (TEA), pH 8.0
- Acetic Anhydride
- DEPC-treated water
- 2X Standard Saline Citrate (SSC)
Procedure:
- Following fixation and PBS rinses, briefly rinse the tissue sections or whole-mount samples in 0.1 M TEA (pH 8.0) at room temperature.
- For a 350 ml working volume, add 875 µl of acetic anhydride to a dry, baked staining dish containing a magnetic stir bar.
- Immediately place a tray of blotted slides or samples into the dish and cover with 350 ml of 0.1 M TEA.
- Stir the solution rapidly and incubate the slides at room temperature for 10 minutes.
- Remove the slides from the solution and rinse in 2X SSC.
- Proceed with dehydration and hybridization steps.
Critical Notes: The acetic anhydride is hydrophobic, so visual inspection should confirm proper mixing. This step acetylates amino groups in the tissue, reducing electrostatic binding of the probe and thereby lowering non-specific background [10].
Optimized NAFA Fixation Protocol for Delicate Tissues
The NAFA protocol is a powerful alternative that eliminates the need for proteinase K, thereby preserving antigen epitopes for immunostaining and maintaining the integrity of fragile tissues like blastemas and epidermis [19].
Reagents Required:
- Nitric Acid
- Formic Acid
- EGTA
- Paraformaldehyde (PFA)
Procedure:
- Fixation: Fix samples in a solution containing nitric acid, formic acid, and EGTA.
- Post-fixation: After the acid fixation, post-fix samples in 4% PFA.
- Hybridization: Proceed directly to hybridization steps without proteinase K digestion.
- The specific concentrations and incubation times for the NAFA solution are detailed in the original publication [19].
Critical Notes: This protocol has been validated for planarians and regenerating killifish tail fins, suggesting broad applicability for delicate tissues. It is highly compatible with subsequent fluorescent in situ hybridization (FISH) and immunostaining.
Chloroform Delipidation for Background Reduction
- Procedure: After dehydration through a graded ethanol series (e.g., 70%, 95%, 100%), immerse the slides in chloroform for 5 minutes. Then, bring the slides back through 100% and 95% ethanol before air-drying [10].
- Rationale: This delipidation step significantly reduces background nonspecific binding, presumably by removing lipids that hydrophobically bind the probe or label, thereby increasing the signal-to-noise ratio without altering the specific hybridization signal [10].
Workflow Visualization for Method Selection
The following diagram outlines a logical decision pathway for selecting and applying treatments to minimize morphological damage during WISH procedures.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for WISH Protocols
| Reagent | Function | Protocol Specifics |
|---|---|---|
| Diethyl Pyrocarbonate (DEPC) Water [10] | Inactivates RNases to prevent degradation of target mRNA. | Used to prepare all aqueous pre-hybridization solutions and for rinsing glassware. |
| Paraformaldehyde (PFA) [10] [3] | Cross-linking fixative that preserves tissue architecture and immobilizes nucleic acids. | Typically used at 4% in PBS. Can be used for perfusion or post-fixation of frozen sections. |
| Triethanolamine (TEA) and Acetic Anhydride [10] | Acetylation mixture that reduces non-specific electrostatic binding of probes to tissue. | Prepared fresh. 875 μl acetic anhydride in 350 ml 0.1 M TEA (pH 8.0) with rapid stirring during incubation. |
| Proteinase K [3] [19] | Enzymatic digestion to increase tissue permeability for probe penetration. | Can cause significant damage to delicate tissues. Its use is omitted in the NAFA protocol [19]. |
| Formamide [20] | Denaturing agent used in hybridization buffers to lower the melting temperature (Tm) of nucleic acid hybrids. | Allows hybridization to occur at lower, less destructive temperatures. Commonly used at 50% concentration. |
| DIG-Labeled Probes & Anti-DIG Antibody [17] [20] | Non-radioactive system for probe labeling and colorimetric detection. | Anti-DIG antibody is typically conjugated to Alkaline Phosphatase (AP) for detection with NBT/BCIP. |
| NBT/BCIP [20] | Chromogenic substrate for Alkaline Phosphatase. Produces an insoluble purple precipitate at the site of hybridization. | Used in a developing solution, often with polyvinyl alcohol (PVA) to enhance the reaction signal. |
In situ hybridization (ISH) is a cornerstone technique in molecular biology, enabling the precise localization of specific nucleic acid sequences within cells, tissues, or whole organisms. However, the inherent complexity of biological samples and the exquisite sensitivity of hybridization reactions make these experiments susceptible to non-specific signals and background noise. The accurate interpretation of gene expression data, therefore, hinges on the implementation of rigorous experimental controls. Within the context of a broader thesis on acetic anhydride triethanolamine treatment in Whole-Mount In Situ Hybridization (WISH) protocol research, this article delineates the critical role of RNase, DNase, and sense probes in validating signal specificity. For researchers, scientists, and drug development professionals, a thorough understanding and application of these controls are not merely best practice but are fundamental to generating reliable and publishable data. This application note provides a detailed framework for incorporating these essential validations into your experimental workflow, complete with structured data and actionable protocols.
The Critical Role of Controls in In Situ Hybridization
The primary challenge in any ISH experiment is to distinguish a true, specific signal from background artifacts. Non-specific binding of probes, endogenous enzyme activities, and tissue autofluorescence can all generate signals that are easily misinterpreted as positive findings. Without proper controls, conclusions about gene expression patterns are fundamentally unsound. The integration of a pre-hybridization treatment with acetic anhydride in triethanolamine buffer is a specific methodological step aimed at reducing non-specific electrostatic binding of probes to the tissue by acetylating amino groups [10]. This treatment underscores the importance of optimizing protocols to enhance signal-to-noise ratios. The controls discussed herein work in concert with such optimizations to provide a multi-layered validation strategy, ensuring that the final visualized signal is unequivocally derived from the target RNA.
Essential Controls for Signal Validation
RNase Treatment: Confirming RNA-Dependent Signal
The most definitive control to prove that an observed signal originates from RNA is the pre-treatment of parallel samples with RNase A.
- Principle: RNase A catalyzes the degradation of single-stranded RNA. Pre-incubation of a sample with this enzyme should abolish the hybridization signal if it is specific to RNA, as the target mRNA will be degraded and unavailable for probe binding [21].
- Protocol: A standard procedure involves treating a sample with RNase A (50 µg/mL) for 30 minutes to 1 hour at 37°C prior to the hybridization step [21]. It is critical to include a control sample that undergoes an identical procedure but without the RNase enzyme to allow for a direct comparison.
- Interpretation: The disappearance of the signal in the RNase-treated sample, compared to the untreated control, confirms that the signal is RNA-dependent.
No-Probe and Off-Target Filter Controls: Assessing Background
These controls are essential for identifying signals that are not a result of specific probe-mRNA hybridization.
- No-Probe Control: The sample is processed through the entire ISH protocol but is incubated with hybridization buffer only, omitting the labeled probe. Any signal observed in this sample is due to autofluorescence or non-specific binding of detection antibodies [21].
- Imaging in an Unused Filter: After completing the ISH protocol with a fluorescent probe, the sample should also be imaged using a filter set that does not match the fluorophore's emission spectrum. For instance, a FITC filter is commonly used to measure autofluorescence. Signals that appear in this unused filter are indicative of autofluorescence, not specific probe binding [21].
Target-Specific Negative Controls
These controls validate the specificity of the probe sequence itself.
- Cell or Tissue Lacking the Target Transcript: An ideal negative control is to perform the ISH procedure on a cell line or tissue that is known to not express the target mRNA. This can be a wild-type sample or one where the transcript has been knocked down via siRNA or knocked out genetically. The absence of signal in this sample confirms the specificity of the probe set [21].
- Sense Probes: A Note of Caution: Historically, sense probes (identical in sequence to the target mRNA) were used as negative controls. The logic was that they should not hybridize to the mRNA. However, this practice is now discouraged for certain technologies. For example, in Stellaris RNA FISH, sense probes can lead to higher background or false signals, potentially due to transcription from the sense strand or non-specific binding [21]. It is generally more reliable to use a probe set targeting a gene from an unrelated organism that is not present in your sample (e.g., GFP in wild-type cells) [21].
Table 1: Summary of Critical Controls and Their Interpretation
| Control Type | Experimental Treatment | Expected Result for a Valid Specific Signal | Primary Function |
|---|---|---|---|
| RNase Treatment | Pre-hybridization incubation with RNase A (50 µg/mL, 37°C, 30-60 min) [21] | Signal is abolished in the treated sample. | Confirms the signal is derived from RNA. |
| No-Probe Control | Complete protocol performed without any probe in hybridization buffer. | No signal is observed. | Identifies autofluorescence and antibody non-specific binding. |
| Off-Target Filter Imaging | Sample imaged with a filter set not matching the fluorophore (e.g., FITC for a Cy5 probe). | No signal is observed in the off-target filter. | Distinguishes specific fluorescence from broad-spectrum autofluorescence. |
| Biological Negative Control | Use of cells/tissue void of the target transcript (e.g., knockout line). | No signal is observed. | Validates probe sequence specificity. |
Advanced Considerations and Complementary Methods
DNase Control in Specialized Contexts
While the primary target of standard ISH is RNA, the growing use of DNA-based amplification methods, such as the Hybridization Chain Reaction (HCR), necessitates controls for DNA targets. In these contexts, a DNase I treatment can be used to confirm the specificity of a signal for a DNA target. Commercial kits are available that can sensitively detect DNase I activity at levels as low as 1 x 10⁻⁵ units/µL [22].
Positive Controls and Protocol Optimization
- Positive Controls: To control for technical success, it is crucial to include a positive control. This can be a catalogued, functionally tested probe set for a gene known to be expressed in your sample [21]. A successful signal from the positive control confirms that the entire protocol, from sample preparation to final detection, was performed correctly.
- Complementary Methods: Corroborating ISH results with an independent method, such as qPCR, provides orthogonal validation of RNA expression levels and integrity in your sample type [21].
- Protocol Enhancements: Signal specificity can be dramatically improved by optimizing blocking and wash conditions. For instance, the use of Roche Western Blocking Reagent (RWBR) and the addition of Triton X-100 to wash buffers have been shown to significantly reduce background without compromising signal intensity [23].
Table 2: Key Reagent Solutions for Validated In Situ Hybridization
| Reagent / Kit | Function / Application | Key Details / Specifications |
|---|---|---|
| RNase A | Negative control to degrade single-stranded RNA and confirm RNA-dependent signal. | Working concentration: 50 µg/mL; Incubation: 30-60 min at 37°C [21]. |
| RNase+DNase Detection Kit | Multiplex system for parallel detection of RNase and DNase contamination in reagents or on equipment. | Detection limit for RNase A: < 0.1 pg/µL; for DNase I: < 1 x 10⁻⁵ units/µL [22]. |
| Proteinase K | Enzyme for tissue permeabilization; digests nucleases and increases reagent access. | Concentration and time must be empirically optimized for each tissue type to avoid over-digestion [24]. |
| Anti-Digoxigenin-AP Fab fragments | Immunological detection of digoxigenin (DIG)-labeled probes via alkaline phosphatase (AP) activity. | Commonly used with chromogenic substrates like NBT/BCIP or BM Purple [25]. |
| Roche Western Blocking Reagent (RWBR) | A specialized blocking agent to reduce non-specific antibody binding and lower background. | Particularly effective for anti-digoxigenin and anti-fluorescein antibodies in FISH [23]. |
| Acetic Anhydride | Pre-hybridization treatment to acetylate amino groups in tissue, reducing electrostatic probe binding. | Used in triethanolamine (TEA) buffer, pH 8.0 [10]. |
Experimental Protocol: Incorporating Critical Controls into a Standard WISH Workflow
The following protocol integrates the described controls into a standard Whole-Mount In Situ Hybridization workflow, with particular attention to the acetic anhydride triethanolamine treatment.
Day 1: Sample Preparation and Fixation
- Collect and Fix Samples: Harvest and fix tissues in 4% Paraformaldehyde (PFA) in PBS, overnight at 4°C [1].
- Dehydrate: Wash samples with PBS and transfer to 100% Methanol for storage at -20°C [1].
Day 2: Pre-hybridization Treatments
- Rehydrate: Gradually rehydrate samples through a methanol series (75%, 50%, 25% in PBS).
- Permeabilize (Optional): Treat samples with Proteinase K (concentration and duration must be optimized for the specific tissue) to enhance probe penetration [24].
- Post-fix: Re-fix in 4% PFA for 20 minutes to maintain tissue integrity after permeabilization.
- Acetic Anhydride Triethanolamine Treatment:
- Rinse samples briefly in 0.1 M Triethanolamine (TEA), pH 8.0.
- Prepare the acetylation solution by adding 0.25% (v/v) acetic anhydride to 0.1 M TEA with constant stirring.
- Immediately immerse the samples and incubate for 10 minutes at room temperature. This step acetylates amino groups, reducing non-specific probe binding [10].
- Pre-hybridize: Transfer samples to a pre-warmed hybridization buffer and incubate for several hours at the hybridization temperature.
Day 2/3: Hybridization and Washes
- Hybridize: Replace the pre-hybridization buffer with fresh hybridization buffer containing the labeled antisense probe. For the critical controls, set up parallel samples with:
- No Probe (Hybridization buffer only).
- RNase-treated (Pre-treated with RNase A before hybridization) [21].
- Positive Control Probe (A validated probe for a known expressed gene).
- Incubate: Hybridize at the appropriate temperature (e.g., 55-65°C) for 12-16 hours (overnight).
- Wash: Perform a series of stringent washes with solutions like Saline-Sodium Citrate (SSC) buffer containing 0.1% Tween-20 to remove unbound probe.
Day 3/4: Immunological Detection
- Block: Incubate samples in a blocking solution (e.g., containing Roche Western Blocking Reagent) to prevent non-specific antibody binding [23].
- Antibody Incubation: Incubate with an enzyme-conjugated antibody (e.g., Anti-Digoxigenin-AP) for several hours or overnight.
- Final Washes: Thoroughly wash the samples to remove unbound antibody.
- Chromogenic Development: Incubate samples with a chromogenic substrate (e.g., NBT/BCIP or BM Purple). Monitor the development reaction closely and stop it once the desired signal intensity is achieved.
Diagram 1: Experimental workflow for WISH with integrated critical controls. The acetic anhydride/triethanolamine (TEA) treatment is a key pre-hybridization step. Samples are then split for the main experiment and essential control pathways, the results of which collectively validate a specific signal.
Diagram 2: The collective interpretation of critical controls leads to a validated, specific signal. The outcomes of the negative controls (red arrows) must be met, alongside a successful positive control (green arrow), to have confidence in the experimental results.
The path to unequivocal gene expression visualization through in situ hybridization is paved with rigorous validation. The strategic implementation of RNase treatment, no-probe controls, biological negatives, and positive controls provides a robust framework for confirming that an observed signal is specific, RNA-derived, and technically sound. When combined with protocol optimizations such as acetic anhydride triethanolamine treatment and improved blocking strategies, these controls empower researchers to generate data of the highest integrity. For scientists driving discoveries in development, regeneration, and drug development, this disciplined approach is not optional—it is the foundation of credible and impactful research.
In the context of a broader thesis on Whole Mount In Situ Hybridization (WMISH) protocol research, achieving high signal-to-noise ratio is paramount for the accurate spatial localization of mRNA. Background noise, originating from non-specific probe binding and endogenous enzymatic activities, can obscure true signals, particularly in lipid-rich tissues. This application note details an advanced protocol that synergistically combines Triethanolamine-Acetic Anhydride (TEA-AA) treatment with chloroform/methanol-based delipidation. While TEA-AA treatment acetylates free amino groups to reduce electrostatic non-specific probe binding [11], the integrated delipidation step physically removes endogenous lipids that contribute to background fluorescence and non-specific interactions [26] [27]. This combined approach offers researchers a robust method for significantly enhancing signal clarity in challenging samples.
Comparative Analysis of Delipidation Techniques
The table below summarizes key delipidation methods relevant to functional protein and transcriptomic analyses, highlighting their advantages and compatibility with various downstream applications.
Table 1: Comparison of Delipidation and Protein Extraction Methods
| Method Name | Core Principle | Key Advantages | Compatibility with Functional Assays | Reference |
|---|---|---|---|---|
| Activated Silica Gel | Solvent-free lipid capture on solid-phase matrix | Preserves enzyme activity; compatible with Activity-Based Protein Profiling (ABPP) | High (Functional integrity of lipases validated) | [26] |
| Chloroform/Methanol Protein Extraction | Phase separation for protein precipitation and lipid removal | Quantitative protein precipitation; reduces contaminants for proteomics | High (Reproducible peptide yields for LC-MS/MS) | [27] |
| BUME Method | Automated, chloroform-free liquid-liquid extraction | High-throughput; suitable for lipidomics; upper organic phase eases recovery | Moderate (Optimized for lipid analysis, not protein function) | [28] [29] |
| Hydrophobic Interaction Chromatography | Chromatographic separation of lipids from proteins on phenyl sepharose | Complete delipidation under native, non-denaturing conditions | High (Maintains native protein conformation for ligand binding) | [30] |
Integrated Experimental Protocol
This section provides a detailed methodology for combining TEA-AA treatment with chloroform delipidation, adapted for whole-mount samples like Lymnaea stagnalis embryos [11].
Materials and Reagents
Table 2: Research Reagent Solutions for TEA-AA and Delipidation
| Reagent / Solution | Function / Purpose | Notes / Precautions |
|---|---|---|
| Triethanolamine (TEA) | Neutralizes charge to reduce non-specific probe binding | Base component for acetylation reaction [11]. |
| Acetic Anhydride (AA) | Acetylates free amino groups in the sample | Reacts with TEA; handle in a fume hood [11]. |
| Chloroform-Methanol (2:1 v/v) | Protein extraction and lipid removal | Forms a biphasic system with aqueous solutions; causes protein precipitation [27]. |
| Phosphate Buffered Saline with 0.1% Tween-20 (PBTw) | Washing and sample rehydration buffer | Detergent helps permeabilize samples and prevent sticking [11]. |
| Proteinase K | Digests proteins to increase probe permeability | Concentration and incubation time must be optimized for each sample type and developmental stage [11]. |
| Fixative Solution | Preserves tissue morphology and mRNA integrity | Typically 4% Paraformaldehyde (PFA) in PBS [11]. |
Detailed Step-by-Step Procedure
Sample Fixation and Decapsulation:
- Collect and stage Lymnaea stagnalis embryos or other model organism tissues. Anaesthetize older larvae (5+ days) in 2% MgCl₂ for 30 minutes to prevent muscle contraction [11].
- Fix samples in 4% PFA for a duration appropriate to the developmental stage (e.g., 30 minutes for early stages, 2 hours for late stages) with gentle rotation at room temperature [11].
- Wash samples 3 times for 5 minutes each with PBTw.
- For encapsulated embryos, use a custom decapsulation apparatus with a glass needle to mechanically rupture capsules and release embryos [11].
Integrated Delipidation and Permeabilization:
- Proteinase K Treatment: Transfer samples to a microcentrifuge tube. Treat with an empirically determined concentration of Proteinase K (e.g., 10-100 µg/mL) for optimal permeabilization without damaging morphology. Incubate at 37°C for a defined period (e.g., 10-30 minutes) [11].
- Post-Fixation: Stop the proteinase reaction by washing with PBTw. Re-fix samples in 4% PFA for 20 minutes to maintain structural integrity [11].
- Chloroform/Methanol Delipidation: Aspirate PBTw. Add a 2:1 (v/v) mixture of chloroform and methanol to the sample pellet. Vortex vigorously for 1 minute and incubate on ice for 10-15 minutes. Centrifuge at 12,000 × g for 10 minutes at 4°C. A protein pellet should be visible. Carefully remove and discard the organic supernatant containing lipids [27].
- Rehydration: Wash the pellet once with 1 mL of ice-cold methanol. Gently wash twice with PBTw to rehydrate the sample for subsequent aqueous steps [27].
Triethanolamine-Acetic Anhydride (TEA-AA) Treatment:
- Prepare the TEA-AA working solution fresh: For 10 mL, add 0.75 mL of acetic anhydride to 50 mL of 0.1 M Triethanolamine [11].
- Incubate the delipidated and rehydrated samples in the TEA-AA solution for 10 minutes with gentle agitation. This acetylation step neutralizes positive charges [11].
- Rinse samples twice with PBTw to prepare for hybridization.
Hybridization and Post-Hybridization Washes:
- Pre-hybridize samples in a suitable hybridization buffer for 1-4 hours at the probe-specific hybridization temperature (often 37-65°C).
- Add digoxigenin (DIG)- or fluorescein-labeled riboprobes or DNA split-initiator probes [31] directly to the hybridization buffer. Incubate overnight at the determined temperature.
- The following day, perform a series of stringent washes with saline-sodium citrate (SSC) buffers (e.g., from 2x SSC to 0.2x SSC) to remove unbound probe [11].
Immunological Detection:
- Block non-specific sites by incubating samples in a blocking solution (e.g., 10% heat-inactivated sheep serum in PBTw) for 1-4 hours.
- Incubate with an anti-DIG or other relevant antibody conjugated to alkaline phosphatase (AP) or horseradish peroxidase (HRP). Dilute the antibody in blocking solution and incubate for 4 hours to overnight at 4°C with gentle agitation.
- Wash thoroughly with PBTw (4-6 changes over 4-6 hours) to remove unbound antibody.
- Develop the signal using the appropriate chromogenic (e.g., NBT/BCIP for AP) or fluorogenic substrate. Monitor the reaction under a microscope and stop by washing with PBTw when the desired intensity is achieved.
Workflow and Signaling Pathway
The following diagram illustrates the integrated experimental workflow, highlighting the sequence of key steps and their functional relationships in reducing background noise.
Diagram 1: Integrated experimental workflow for noise reduction.
The combination of TEA-AA treatment and chloroform/methanol delipidation provides a powerful, synergistic strategy for minimizing background noise in WMISH. This protocol addresses multiple sources of noise simultaneously: electrostatic non-specific binding is mitigated via acetylation, while endogenous lipids that contribute to autofluorescence and non-specific interactions are physically removed. The result is a significant enhancement in the signal-to-noise ratio, enabling clearer and more reliable detection of mRNA transcripts. This advanced solution is particularly beneficial for lipid-rich tissues and for pushing the sensitivity limits of in situ hybridization, thereby providing researchers with a robust tool for high-precision gene expression analysis.
Validation and Technique Comparison: Ensuring Reliable mRNA Localization Data
Within the broader scope of thesis research on optimizing the whole-mount in situ hybridization (WISH) protocol, the treatment with acetic anhydride and triethanolamine stands as a critical step for enhancing the signal-to-noise ratio. A high signal-to-noise ratio is fundamental to the technique's success, as it allows for precise spatial localization of mRNA transcripts while minimizing non-specific background staining. This application note provides a detailed quantitative and qualitative assessment of methods to improve this ratio, consolidating validated protocols from diverse model organisms to serve as a reliable resource for researchers and drug development professionals engaged in high-resolution gene expression analysis.
Quantitative Comparison of Method Efficacy
The assessment of signal-to-noise improvement strategies requires evaluation of both overall staining efficiency and the precision of the resulting signal. The following table summarizes quantitative findings from a comparative study of two in situ hybridization methods applied to the pinewood nematode (Bursaphelenchus xylophilus), targeting a pathogenicity-related gene (Bx-vap-2) and a sex-determining gene (fem-2) [17].
Table 1: Quantitative Staining Efficacy of Two In Situ Hybridization Methods
| Method | Target Gene | Staining Rate | Correct Staining Rate | Key Qualitative Findings |
|---|---|---|---|---|
| Whole-Mount | fem-2 | Higher | Higher | Better for showcasing continuous developmental processes [17]. |
| Whole-Mount | Bx-vap-2 | Higher | Higher | More suitable for development-related genes; higher staining rates [17]. |
| Cut-Off | fem-2 | Lower | Lower | Clearer hybridization signal locations with less non-specific staining [17]. |
| Cut-Off | Bx-vap-2 | Lower | Lower | More precise gene localization; more suitable for disease-related genes [17]. |
Beyond methodological choice, specific chemical treatments significantly enhance signal quality. Research in the planarian Schmidtea mediterranea has demonstrated that a short peroxide bleach in formamide dramatically improves signal intensity compared to an overnight methanol bleach [16]. The quantitative improvement is evident in the reduced time required for chromogenic development and a clearer signal in densely-packed tissues.
Table 2: Efficacy of Signal Enhancement Treatments
| Treatment | Model Organism | Quantitative/Qualitative Outcome | Proposed Mechanism |
|---|---|---|---|
| Formamide Bleach | Planarian (Schmidtea mediterranea) | Reduced development time; more consistent labeling of dense tissue [16]. | Improved tissue permeability and mRNA accessibility [16]. |
| Acetic Anhydride/Triethanolamine | Rat Brain | Significant reduction in background; no alteration in specific signal intensity [10]. | Acetylation of amino groups reduces electrostatic probe binding [10]. |
| Chloroform Delipidation | Rat Brain | Significant reduction in background [10]. | Hydrophobic removal of lipids that bind probe [10]. |
Detailed Experimental Protocols
Core Protocol: Acetic Anhydride Triethanolamine Treatment
The following steps should be performed after tissue fixation and before hybridization [10].
- Step 1: Preparation. Following fixation and a phosphate-buffered saline rinse, briefly rinse the tissue in 0.1 M triethanolamine (TEA, pH 8.0) at room temperature.
- Step 2: Acetylation. Add 875 µl of acetic anhydride to a dry, baked staining dish containing a magnetic stir bar. Immediately place a tray of blotted slides into the dish and cover with 350 ml of 0.1 M TEA.
- Step 3: Incubation. Stir the solution continuously and incubate the slides at room temperature for 10 minutes.
- Step 4: Post-Treatment. Remove the slides from the acetic anhydride solution and rinse in 2× standard saline citrate (SSC).
- Step 5: Dehydration and Delipidation. Dehydrate the slides through a graded series of alcohol rinses (70%, 95%, and 100% ethanol). For further background reduction, immerse the slides in chloroform for 5 minutes to delipidate the tissue, then pass through 100% and 95% ethanol again before air-drying.
Enhanced Protocol: Formamide Bleaching for Planarians
This modification is designed to improve signal intensity, particularly for low-abundance transcripts [16].
- Procedure: Replace the traditional overnight peroxide bleach in methanol with a 1-2 hour peroxide bleach in formamide.
- Critical Note: The improved signal intensity is lost if animals are pre-bleached in methanol, suggesting that pre-bleaching in methanol masks the benefit of formamide through an unknown mechanism [16].
Enhanced Protocol: Optimized Blocking for Fluorescent In Situ Hybridization (FISH)
To achieve a high signal-to-noise ratio in fluorescent applications, the blocking and wash steps are critical [16].
- Blocking Solution: Incorporate Roche Western Blocking Reagent (RWBR) into the blocking buffer. This has been shown to dramatically reduce background, particularly for anti-digoxigenin (DIG) and anti-fluorescein (FAM) antibodies, without significantly diminishing signal intensity.
- Detergent: Add or substitute with 0.3% Triton X-100 in the blocking and wash solutions to further improve signal specificity.
Visualization of Workflows and Mechanisms
The following diagrams illustrate the core experimental workflow and the mechanism of a key treatment.
Diagram 1: Core WISH Protocol Workflow. Key steps for signal-to-noise improvement, like acetic anhydride treatment and delipidation, are highlighted.
Diagram 2: Mechanism of Acetic Anhydride Treatment. The workflow illustrates how the treatment chemically modifies the tissue to reduce non-specific background.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Signal-to-Noise Optimization in WISH
| Reagent | Function/Role | Protocol Note |
|---|---|---|
| Acetic Anhydride | Acetylates amino groups in tissue, reducing non-specific electrostatic binding of probes [10]. | Used in 0.1 M TEA, pH 8.0; critical for lowering background. |
| Triethanolamine (TEA) | Buffering agent for the acetylation reaction, maintaining optimal pH [10]. | Prepare a 0.1 M solution at pH 8.0. |
| Formamide | Denaturing agent that improves tissue permeability and probe access to target mRNA [16]. | Used in bleaching solution and standard hybridization buffers. |
| Roche Western Blocking Reagent (RWBR) | Protein-based blocking agent that dramatically reduces background in fluorescent detection [16]. | Superior to casein or other reagents for anti-DIG and anti-FAM antibodies. |
| Proteinase K | Proteolytic enzyme that digests proteins to improve antibody penetration into tissues [32]. | Concentration and time must be carefully optimized for each tissue type. |
| Chloroform | Organic solvent that delipidates tissue, reducing hydrophobic binding of probes [10]. | Significantly reduces background; apply after dehydration. |
| Diethyl Pyrocarbonate (DEPC) | RNase inhibitor. | Used to treat water and solutions to prevent RNA degradation [17]. |
Within whole mount in situ hybridization (WMISH) protocols, non-specific background staining presents a significant challenge that can obscure genuine spatial gene expression patterns. A critical step in optimizing this technique involves the systematic evaluation of pre-hybridization treatments designed to increase signal-to-noise ratios. This application note provides a detailed comparative analysis of the Triethanolamine-Acetic Anhydride (TEA-AA) treatment against alternative chemical and enzymatic background reduction methods, specifically within the context of spiralian model organisms. The data and protocols presented herein are framed within broader thesis research aimed at establishing a standardized, optimized WMISH protocol for challenging biological systems, with particular emphasis on the freshwater gastropod Lymnaea stagnalis [3].
The biochemical and biophysical properties of developing tissues can vary considerably between species and across ontogenetic stages, necessitating empirical determination of optimal permeabilization and background reduction strategies. This analysis systematically evaluates TEA-AA treatment alongside mucolytic (N-acetyl-L-cysteine), detergent-based (SDS), and reduction-based approaches, providing researchers with quantitative data to guide experimental design [3].
Background Reduction Methodologies: Comparative Efficacy
Quantitative Comparison of Background Reduction Treatments
Table 1: Efficacy Comparison of Background Reduction Methods in WMISH
| Treatment Method | Primary Mechanism of Action | Optimal Concentration & Conditions | Targeted Background Sources | Impact on Morphological Integrity | Gene Expression Consistency |
|---|---|---|---|---|---|
| TEA-AA (Acetylation) | Blocks cationic charge interactions by acetylating amine groups | 0.1M TEA + 0.25% AA, 10 min incubation [3] | Non-specific probe binding to charged tissue components | Excellent preservation across developmental stages [3] | High consistency across genes with varying expression levels [3] |
| N-Acetyl-L-Cysteine (NAC) | Mucolysis of viscous extracellular matrices | 2.5-5% concentration, age-dependent (5 min for early stages, 2x5 min for late stages) [3] | Intra-capsular fluid polysaccharides/proteoglycans | Good preservation with optimized timing [3] | Improved signal for all tested genes [3] |
| SDS Treatment | Lipid membrane solubilization and permeabilization | 0.1-1% SDS in PBS, 10 min at room temperature [3] | General tissue permeability barriers | Moderate; requires careful concentration optimization [3] | Variable effects depending on gene expression level [3] |
| Reduction Treatment | Disruption of disulfide bonds in extracellular matrices | 1X reduction solution (DTT, SDS, NP-40), 10 min at 37°C [3] | Mucosal layers and protein complexes | Poor; tissues become extremely fragile [3] | Moderate improvement but inconsistent across replicates [3] |
Tissue-Specific Background Challenges
A particularly problematic background signal was identified in the larval shell field of L. stagnalis, where secreted insoluble shell material demonstrated high affinity for nucleic acid probes. This phenomenon is not restricted to L. stagnalis but has been observed in larvae of other gastropods, bivalves, scaphopods, and polyplacophoran molluscs [3]. The TEA-AA treatment proved specifically effective at eliminating this tissue-specific background without the morphological disruption associated with stronger permeabilization methods [3].
Experimental Protocols
Standardized TEA-AA Treatment Protocol
Reagents Required:
- Triethanolamine (TEA), 0.1M solution
- Acetic anhydride (AA)
- PBS or PBTw buffer
Procedure:
- Following rehydration and proteinase K treatment (if applicable), rinse samples twice in PBTw for 5 minutes each.
- Prepare fresh TEA-AA solution: 0.1M TEA with 0.25% acetic anhydride in PBTw.
- Incubate samples in TEA-AA solution for 10 minutes with gentle agitation.
- Replace with freshly prepared TEA-AA solution and repeat incubation.
- Rinse samples twice in PBTw before proceeding with pre-hybridization or hybridization steps.
Critical Notes:
- TEA-AA solution must be prepared immediately before use as acetic anhydride hydrolyzes rapidly in aqueous solutions.
- The treatment is most effective when applied after general permeabilization steps but before hybridization.
- For tissues with particularly high background, increasing acetic anhydride concentration to 0.5% may be beneficial, though this should be empirically determined [3].
Alternative Method Protocols
N-Acetyl-L-Cysteine (NAC) Treatment:
- Prepare NAC solution at 2.5% (early stages) or 5% (late stages) in buffer.
- Incubate freshly dissected embryos for 5 minutes (early stages) or twice for 5 minutes each (late stages).
- Terminate treatment by immediate fixation in 4% PFA in PBS for 30 minutes [3].
SDS Permeabilization:
- Following fixation, wash samples once in PBTw for 5 minutes.
- Incubate in 0.1-1% SDS in PBS for 10 minutes at room temperature.
- Rinse in PBTw and dehydrate through graded ethanol series for storage [3].
Reduction Treatment:
- Following fixation, wash samples once in PBTw for 5 minutes.
- Incubate embryos in 1X reduction solution (containing DTT, SDS, and NP-40) for 10 minutes at 37°C.
- Exercise extreme care as samples become exceptionally fragile during this treatment.
- Briefly rinse with PBTw before dehydration and storage [3].
Visualization of Experimental Workflow
WMISH Background Reduction Decision Pathway
Mechanism of TEA-AA Background Suppression
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for WMISH Background Reduction
| Reagent | Chemical Category | Primary Function | Optimization Considerations |
|---|---|---|---|
| Triethanolamine (TEA) | Alkanolamine | Buffer base for acetylation reactions | Use fresh solutions; concentration critical for maintaining pH during acetylation [3] |
| Acetic Anhydride (AA) | Carboxylic acid anhydride | Acetylating agent for amine groups | Highly labile in aqueous solutions; must prepare immediately before use [3] |
| N-Acetyl-L-Cysteine (NAC) | Mucolytic agent | Degrades viscous mucous and polysaccharide matrices | Concentration and duration must be optimized for developmental stage [3] |
| Sodium Dodecyl Sulfate (SDS) | Ionic detergent | Membrane solubilization and tissue permeabilization | Higher concentrations (≥1%) may compromise morphological integrity [3] |
| Dithiothreitol (DTT) | Reducing agent | Disruption of disulfide bonds in mucous glycoproteins | Component of reduction treatment; causes significant tissue fragility [3] |
| Proteinase K | Serine protease | Controlled tissue digestion for enhanced probe penetration | Requires precise concentration and timing to preserve RNA integrity [3] |
This comparative analysis demonstrates that TEA-AA treatment provides the most consistent background reduction across multiple gene targets while maintaining excellent morphological integrity—a balance that alternative methods struggle to achieve. The mechanism of chemical acetylation specifically targets electrostatic background interactions without the disruptive permeabilization associated with detergent or reduction treatments.
For researchers establishing WMISH protocols in novel systems, a sequential approach is recommended: begin with TEA-AA treatment for general background suppression, subsequently introducing NAC for systems with significant mucous components, and reserving SDS permeabilization for cases with demonstrated probe penetration issues. The reduction treatment, while effective in specific circumstances, should be employed cautiously due to its detrimental effects on morphological preservation.
The protocols and data presented here provide a foundation for systematic optimization of WMISH techniques across diverse model systems, with particular utility for challenging spiralian organisms where traditional methods have yielded suboptimal signal-to-noise ratios.
The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay remains a widely employed method for detecting DNA fragmentation characteristic of apoptotic cell death. However, its utility is significantly compromised by multiple sources of false positives that can lead to misinterpretation. This application note systematically addresses the predominant causes of TUNEL false positives—including endogenous nuclease activity, non-apoptotic DNA fragmentation, and methodological artifacts—and provides detailed protocols for their mitigation. Emphasis is placed on correlating TUNEL results with orthogonal cell death markers and morphological assessment to enhance interpretive accuracy within the context of whole-mount in situ hybridization (WISH) research involving acetic anhydride triethanolamine treatments.
The TUNEL assay identifies DNA strand breaks by leveraging the enzyme terminal deoxynucleotidyl transferase (TdT), which catalyzes the addition of labeled deoxynucleotides to the 3'-hydroxyl termini of fragmented DNA [33] [34]. While initially celebrated for its sensitivity in detecting apoptotic cells, wherein endonucleases generate extensive DNA cleavage, it is now unequivocally established that the assay labels any DNA break with a 3'-OH end, irrespective of its origin [34]. Consequently, DNA fragmentation resulting from necrosis, autolysis, active DNA repair, genotoxic stress, or even routine tissue processing can generate false positive signals [35] [34].
A critical, often-overlooked caveat is that a positive TUNEL signal does not invariably signify irreversible cell death. Compelling evidence from various biological systems demonstrates that cells can recover from apoptotic stimuli, a process termed anastasis, even after exhibiting caspase activation and DNA fragmentation [35]. This biological reversibility further complicates the interpretation of TUNEL data as a definitive marker of cell demise. Therefore, correlative analysis using multiple, methodologically independent techniques is not merely advisable but essential for accurate biological interpretation [36].
Understanding the sources of false positives is the first step toward robust experimental design. The table below summarizes the primary causes and their corresponding solutions.
Table 1: Common Sources of TUNEL False Positives and Mitigation Strategies
| Source of False Positive | Underlying Cause | Recommended Mitigation Strategy |
|---|---|---|
| Endogenous Nuclease Activity | Release of proteases and nucleases during tissue processing; particularly prevalent in liver and intestine [37] [38]. | Pre-treatment with diethyl pyrocarbonate (DEPC), a potent nuclease inhibitor [37] [38]. |
| Non-Apoptotic Cell Death | DNA fragmentation occurring during necrotic cell death or autolysis [34]. | Correlate with morphological assessment (e.g., H&E staining) for nuclear condensation and apoptotic bodies [36] [39]. |
| Cellular Processes & Artifacts | DNA breaks from active DNA repair, cellular proliferation, or genotoxic agents without apoptosis [35] [34]. | Combine with markers of apoptosis initiation (e.g., activated caspase-3 immunofluorescence) [34]. |
| Excessive Proteolysis | Over-digestion with Proteinase K, which can damage tissue morphology and release endogenous nucleases [37] [39]. | Optimize Proteinase K concentration and incubation time; consider alternative antigen retrieval like pressure cooking [40] [39]. |
| Inadequate Specificity | The fundamental principle of the assay labels any 3'-OH DNA end. | Implement a multi-parameter approach, never relying on TUNEL as a standalone assay [36]. |
A significant technical source of false positives, especially in tissues like liver, is the release of endogenous endonucleases during Proteinase K digestion, a common step used to permeabilize samples and expose DNA. This can be effectively inhibited by pre-treating tissue slides with DEPC [37] [38]. Furthermore, the slide-mounting medium is critical; the efficacy of DEPC is abolished on silanised slides, highlighting the importance of this often-overlooked technical detail [37].
The following workflow diagram outlines a strategic approach to implementing TUNEL and verifying its results.
Diagram 1: A strategic TUNEL workflow integrating key decision points for false positive mitigation.
Detailed Protocols for Optimized TUNEL and Correlation
Protocol 1: DEPC Pre-treatment to Inhibit Endogenous Nucleases
This protocol is adapted from Stähelin et al. (1998) for preventing false positives caused by endogenous nucleases in liver and intestinal tissues [37] [38].
- Reagents Needed: Diethyl pyrocarbonate (DEPC), appropriate buffer (e.g., PBS), humidified staining chamber.
Procedure:
- Following deparaffinization and rehydration of tissue sections, wash slides in PBS.
- Prepare a fresh DEPC solution (e.g., 0.1% - 1% v/v in PBS).
- Cover sections with the DEPC solution and incubate in a humidified chamber for 1 hour at room temperature.
- Rinse slides thoroughly with PBS to remove residual DEPC.
- Proceed with standard Proteinase K digestion and the TUNEL assay protocol.
Note: The effectiveness of DEPC is highly dependent on the slide adhesive. It is ineffective on silanised slides; therefore, use sections mounted with cement or other appropriate adhesives [37].
Protocol 2: Proteinase K vs. Pressure Cooker Antigen Retrieval
A recent advancement identifies Proteinase K as a major culprit in both generating false positives and destroying protein antigenicity for multiplexing. This protocol compares traditional and improved methods [40].
- Reagents Needed: Proteinase K (e.g., 10-20 µg/mL), sodium citrate buffer (pH 6.0), pressure cooker or decloaking chamber, PBS with Tween 20 (PBST).
- Procedure A (Traditional Proteinase K):
- After DEPC treatment (if required), incubate slides with a optimized concentration of Proteinase K (typically 10-20 µg/mL) for 15-30 minutes at room temperature [39].
- Wash gently with PBST. Over-digestion must be avoided as it damages morphology and increases background.
- Procedure B (Recommended Pressure Cooker):
- Omit Proteinase K digestion. After DEPC treatment and washing, place slides in pre-heated sodium citrate buffer in a pressure cooker.
- Perform antigen retrieval by pressure cooking for 10-15 minutes (follow manufacturer's guidelines).
- Allow slides to cool in the buffer, then wash with PBST.
- Advantage of Protocol B: Pressure cooker retrieval quantitatively preserves the TUNEL signal while dramatically enhancing protein antigenicity, enabling robust multiplexing with immunofluorescence for caspases or cell-specific markers [40].
Protocol 3: Multiplexed TUNEL with Immunofluorescence (IF)
Harmonizing TUNEL with spatial proteomic methods like multiplexed immunofluorescence provides rich contextual data for false positive exclusion [40].
- Reagents Needed: Validated primary antibodies (e.g., anti-cleaved Caspase-3), fluorescently-labeled secondary antibodies, blocking serum, DAPI.
- Procedure:
- Perform the TUNEL assay first using a fluorophore-conjugated dUTP (e.g., FITC-dUTP) or a hapten-labeled dUTP followed by its fluorescent detection. Use Pressure Cooker antigen retrieval (Protocol 2B).
- After the final TUNEL wash, block sections with an appropriate blocking serum for 1 hour.
- Incubate with primary antibody (e.g., anti-cleaved Caspase-3) overnight at 4°C.
- Wash and incubate with a secondary antibody conjugated to a fluorophore with a distinct emission spectrum from the TUNEL signal.
- Counterstain nuclei with DAPI and mount for imaging.
- Interpretation: True apoptotic cells are positive for both TUNEL and cleaved Caspase-3, and exhibit condensed, fragmented nuclei by DAPI. Cells positive for TUNEL but negative for Caspase-3 and lacking apoptotic morphology should be considered non-apoptotic or false positives.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for TUNEL and Correlative Assays
| Reagent / Solution | Function / Role | Key Considerations |
|---|---|---|
| Diethyl Pyrocarbonate (DEPC) | Inhibits endogenous nucleases to prevent proteinase K-induced false positives [37]. | Ineffective on silanised slides; requires careful handling. |
| Proteinase K | Proteolytic enzyme for antigen retrieval and tissue permeabilization. | Concentration and time must be tightly optimized to avoid over-digestion [37] [39]. |
| Pressure Cooker / Citrate Buffer | Alternative antigen retrieval method that preserves protein epitopes for multiplexing [40]. | Superior to Proteinase K for combined TUNEL/IF experiments. |
| Terminal Deoxynucleotidyl Transferase (TdT) | Core enzyme of the assay; adds labeled nucleotides to DNA breaks [33] [34]. | Subject to inactivation; include positive controls. |
| Labeled dUTP (e.g., FITC-dUTP, Biotin-dUTP) | Detectable nucleotide incorporated at DNA break sites. | BrdU-based methods can offer brighter signals [33]. |
| DNase I | Used to create a positive control by inducing DNA breaks in a control section [39]. | Essential for validating each experiment. |
| Anti-Cleaved Caspase-3 Antibody | Marker for apoptosis initiation; used for correlative IF. | Confirms active apoptotic pathway in TUNEL+ cells. |
| Acetic Anhydride / Triethanolamine | In WISH protocols, used to acetylate tissue sections, reducing non-specific electrostatic probe binding [3]. | Critical for lowering background in WISH, which can indirectly aid in correlative analysis with TUNEL. |
The TUNEL assay is a powerful but nuanced tool. Its results must be interpreted with caution and rigor, moving beyond the simplistic equation of a positive signal with apoptotic cell death. By understanding the technical and biological pitfalls that lead to false positives, and by implementing the detailed protocols outlined herein—particularly DEPC pre-treatment, pressure cooker antigen retrieval, and multiplexed correlation with caspase activation and morphology—researchers can significantly enhance the reliability and biological relevance of their findings in cell death research and drug development.
In situ hybridization techniques, particularly Whole Mount In Situ Hybridization (WISH), provide invaluable spatial and temporal information about gene expression patterns in developing tissues and whole embryos. However, the technical challenges associated with WISH, including issues with probe penetration, non-specific background staining, and the subjective interpretation of expression patterns, necessitate validation through independent methods [3]. The correlation of WISH results with quantitative techniques like Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and protein-level localization methods like Immunohistochemistry (IHC) establishes a robust framework for verifying gene expression data. This multi-technique approach is especially critical in non-traditional model organisms and complex tissues where biochemical properties can vary significantly, potentially leading to artifacts [3]. Furthermore, in clinical and diagnostic settings, the reliability of a single method is often insufficient, and concordance between multiple techniques provides the confidence required for both basic research conclusions and therapeutic decisions [41] [42].
Experimental Protocols for Correlation
Optimized Whole-Mount In Situ Hybridization (WISH)
A validated WISH protocol must balance signal intensity with morphological integrity. The following protocol, optimized for larval stages of Lymnaea stagnalis, includes a critical acetic anhydride triethanolamine treatment to eliminate tissue-specific background stain, particularly in the larval shell field [3].
- Sample Preparation and Fixation: Manually dissect embryos from egg capsules and immediately treat with a mucolytic agent (N-acetyl-L-cysteine, NAC) to degrade the viscous intra-capsular fluid that can interfere with the procedure. Concentrations (2.5-5%) and treatment duration are age-dependent [3]. Subsequently, fix samples for 30 minutes at room temperature in freshly prepared 4% Paraformaldehyde (PFA) in 1X PBS [3].
- Permeabilization and Background Reduction: Following fixation and a PBS-Tween (PBTw) wash, incubate samples in a pre-hybridization treatment to enhance probe accessibility. A reduction solution (containing DTT and detergents like SDS and NP-40) or a standalone SDS treatment (0.1%-1% in PBS for 10 minutes) has been shown to significantly improve signal consistency [3].
- Acetic Anhydride Triethanolamine Treatment: To abolish non-specific background signal, treat fixed and permeabilized samples with triethanolamine (TEA) and acetic anhydride (AA) prior to hybridization [3]. This acetylation step is crucial for reducing background in tissues with high secretory activity.
- Hybridization and Detection: Hybridize with a single-stranded, labelled nucleic acid probe complementary to the target of interest. Detect the label immunologically using colorimetric or fluorescent methods. The concentration of the Alkaline Phosphatase (AP)-conjugated anti-DIG antibody and the composition of the color detection solution should be systematically optimized for the specific organism and developmental stage [3].
Quantitative Reverse Transcription PCR (qRT-PCR)
qRT-PCR provides a quantitative measure of a gene's expression level, serving as an excellent complement to the spatial data from WISH.
- RNA Extraction and Quality Control: Isolate total RNA from matching samples or microdissected tissues using a dedicated reagent system (e.g., Paradise Reagent System). Include a DNase I treatment step to remove genomic DNA contamination. Assess RNA quantity and quality using both spectrophotometric (NanoDrop) and fluorimetric (Qubit) methods to ensure integrity [41].
- cDNA Synthesis and Preamplification: Perform first-strand cDNA synthesis from 50-200 ng of total RNA using random hexamers. For low-abundance targets, a preamplification step (e.g., 14 cycles using a TaqMan PreAmp Master Mix) can be incorporated to linearly amplify mRNA without distorting relative levels [41].
- Quantitative PCR: Perform qPCR reactions using a fast real-time PCR system. The reaction mixture typically contains cDNA, forward and reverse primers, hybridization probes, and a Universal Master Mix. Use a stable reference gene (e.g., RPLP0) for normalization. The comparative ΔΔCt method is then used to calculate relative expression levels across samples [41].
Immunohistochemistry (IHC)
IHC confirms the presence and localization of the protein product, closing the loop between mRNA expression and functional protein.
- Tissue Sectioning and Staining: For formalin-fixed paraffin-embedded (FFPE) samples, section tissues to 5 μm thickness. Incubate sections with a validated primary antibody specific to the target protein (e.g., VENTANA anti-HER2/neu (4B5) for HER2 detection) [41].
- Evaluation: An experienced pathologist should evaluate IHC stains, scoring them according to established manufacturer specifications and clinical guidelines (e.g., ASCO/CAP guidelines for HER2) [41].
Quantitative Data Correlation and Analysis
The correlation between WISH, RT-PCR, and IHC is not merely qualitative but can be assessed quantitatively to determine the concordance and relative performance of each method.
Table 1: Concordance Analysis Between FISH, qPCR, and qRT-PCR for HER2 Status Assessment
| Comparison | Overall Agreement (OA) | Kappa Value (k) | Sensitivity | Specificity | Global Accuracy |
|---|---|---|---|---|---|
| FISH vs Q-PCR | 94.1% | 0.87 | 86.1% | 99.0% | 91.6% |
| FISH vs qRT-PCR | 90.8% | 0.81 | 100% | 94%* | N/A |
Data derived from a study on HER2 status in breast cancer [41]. *Specificity value for the detection of ALK in NSCLC via RT-PCR was 94% compared to FISH and sequencing [42].
Table 2: Performance Characteristics of RT-PCR vs. FISH and IHC for ALK Detection
| Method | Target | Sensitivity | Specificity | Key Advantage |
|---|---|---|---|---|
| RT-PCR | ALK mRNA | 100% | 94% | Highly efficient, reliable, automatable screening. |
| FISH | ALK gene rearrangement | Benchmark | Benchmark | Validated standard for gene rearrangements. |
| IHC | ALK protein | Benchmark | Benchmark | Provides protein-level localization. |
Data synthesized from a study on ALK detection in non-small cell lung cancer [42].
The data in Table 1 highlights that while DNA-based Q-PCR shows high agreement with FISH, RNA-based qRT-PCR can achieve perfect sensitivity, identifying overexpression even in cases where amplification is not detected by FISH. Subsequent protein analysis (e.g., Western Blotting) in discordant cases has suggested that qRT-PCR may correlate better with actual protein levels than FISH, particularly in equivocal cases [41]. This underscores the value of qRT-PCR not just as a validation tool, but as a primary method for identifying overexpression, as confirmed in studies on ALK (Table 2) where RT-PCR demonstrated 100% sensitivity [42].
Integrated Workflow and Data Interpretation
The following workflow diagram outlines the strategic process for correlating these three techniques, from experimental design to the interpretation of concordant and discordant results.
Interpreting Correlation Outcomes
- Concordant Results: Agreement between WISH (mRNA spatial localization), qRT-PCR (high mRNA levels), and IHC (protein presence) provides powerful, multi-layered validation of the gene's expression profile. This tripartite agreement strongly supports the biological conclusion.
- Discordant Results: Disagreements between techniques are not failures but opportunities for deeper biological insight.
- WISH+/qRT-PCR-: This could indicate low-level, highly localized expression that is detectable by WISH but falls below the quantitative threshold of qRT-PCR, especially if the entire tissue is homogenized for RNA extraction.
- WISH-/qRT-PCR+: Suggests either widespread, low-level expression not visible by WISH or expression in a cell population that was lost during sample processing for WISH.
- mRNA+/Protein- (IHC-): May indicate post-transcriptional regulation, rapid protein turnover, or issues with antibody specificity.
- Protein+/mRNA- (WISH-/qRT-PCR-): Could suggest stable protein persistence after mRNA degradation, or the presence of homologous proteins leading to antibody cross-reactivity.
The decision tree below provides a logical framework for troubleshooting the most common discordant result: positive WISH signal with negative or weak qRT-PCR confirmation.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for WISH, RT-PCR, and IHC Correlation Studies
| Reagent / Kit | Function / Application | Specific Example / Note |
|---|---|---|
| N-Acetyl-L-Cysteine (NAC) | Mucolytic agent to degrade viscous fluids surrounding embryos/tissues, improving probe penetration [3]. | Used as a pre-fixation treatment; concentration and duration are age-dependent (e.g., 2.5-5%) [3]. |
| Reduction Solution (DTT, SDS, NP-40) | Pre-hybridization treatment to increase tissue permeability and enhance WMISH signal intensity [3]. | Makes samples fragile; handle with care. Can be used as an alternative to SDS-only treatment [3]. |
| Triethanolamine & Acetic Anhydride | Acetylation treatment that abolishes tissue-specific non-specific background staining in WISH [3]. | Critical for eliminating background in secretory tissues like the molluscan shell field [3]. |
| ALK RGQ RT-PCR Kit / Similar | Automated, quantitative RT-PCR test for detecting specific gene fusions or expression levels [42]. | Provides high-throughput, automated interpretation. Shown to be 100% sensitive for ALK detection vs. FISH/IHC [42]. |
| QIAGEN DNA/RNA FFPE Kits | For simultaneous or separate isolation of high-quality DNA and RNA from archived FFPE tissue samples. | Ensures nucleic acid integrity from challenging sample types for downstream molecular analysis. |
| Ventana Anti-HER2/neu (4B5) | Validated primary antibody for IHC detection of specific protein targets in FFPE tissues. | Example of a clinically validated antibody, scored according to strict manufacturer and guideline specifications [41]. |
| PathVysion HER2 DNA Probe Kit | FISH kit for determining gene amplification status. | Serves as a standard reference method for validating DNA-level alterations [41]. |
The accurate detection of specific molecular targets in biological tissues via in situ hybridization (ISH) is often compromised by high background signals, a challenge acutely present in tissues with abundant endogenous phosphatase activity [7]. This non-specific signal can obscure true hybridization signals, leading to inaccurate data interpretation. A critical step in mitigating this issue is the treatment of tissue samples with a solution of acetic anhydride in triethanolamine (TEA), which acetylates amino groups in the tissue, thereby reducing electrostatic, non-specific binding of charged probe molecules [10] [7]. This application note details a protocol incorporating this treatment and demonstrates its successful use, alongside a novel two-photon fluorescent probe, for precise alkaline phosphatase (ALP) detection in challenging rat tissues.
Research Reagent Solutions
The following table lists key reagents essential for implementing the described acetic anhydride triethanolamine treatment and in situ hybridization protocol.
| Reagent Name | Function/Explanation |
|---|---|
| Acetic Anhydride | Acetylates amino groups in the tissue sample, reducing non-specific electrostatic binding of probes and lowering background noise [10]. |
| Triethanolamine (TEA) Buffer | Serves as the reaction medium for the acetylation process when combined with acetic anhydride [7]. |
| Diethyl Pyrocarbonate (DEPC)-treated Water | Inactivates RNases, protecting vulnerable RNA targets in the tissue from degradation throughout the procedure [10] [7]. |
| Paraformaldehyde (PFA) | Fixes tissue samples, preserving cellular morphology and immobilizing the target nucleic acids [10] [7]. |
| Locked Nucleic Acid (LNA) Probes | Hybridization probes with enhanced thermal stability and binding specificity, crucial for detecting short or low-abundance targets like miRNAs [7]. |
| Proteinase K | Digests proteins in the tissue sample, increasing probe accessibility to the target mRNA or miRNA [7]. |
| Formamide | A component of hybridization buffers and stringent wash solutions; lowers the required hybridization temperature, helping to preserve tissue integrity [7]. |
| TP-Phos Probe | A two-photon fluorescent probe with high selectivity for ALP, offering advantages like deep tissue penetration and low background autofluorescence for imaging in thick tissues [43] [44]. |
Experimental Application: ALP Detection with TP-Phos
Probe Design and Advantages
The TP-Phos probe is strategically constructed from three components: a two-photon fluorophore, a phosphate recognition moiety, and a self-cleavable adaptor [43] [44]. Its operational mechanism is a turn-on fluorescence response: ALP catalyzes the dephosphorylation of the probe, triggering a 1,4-elimination that releases the fluorescent dye [43].
This design confers significant advantages for challenging applications, as summarized below.
Table 1: Key Characteristics of the TP-Phos Probe for ALP Detection
| Characteristic | TP-Phos Probe Performance | Advantage in Challenging Tissues |
|---|---|---|
| Selectivity | High specificity for ALP over other phosphatases (e.g., PTPs) due to an ortho-functionalized phenyl phosphate group that increases steric hindrance [43]. | Reduces false-positive signals from non-target phosphatase activity. |
| Excitation Mode | Two-photon excitation [43] [44]. | Enables deeper tissue penetration and minimizes tissue autofluorescence and photo-damage. |
| Sensitivity | Capable of imaging endogenous ALP activity [43]. | Requires less probe and detects physiological enzyme levels. |
| Reaction Kinetics | Fast reaction kinetics [43]. | Allows for real-time or rapid imaging. |
| Cytotoxicity | Low cytotoxicity [43]. | Suitable for application in living cells and tissues. |
Quantitative Performance Data
The probe was rigorously tested, demonstrating a significant fluorescence turn-on response upon reaction with ALP. The following table quantifies its optical and detection performance.
Table 2: Quantitative Optical and Detection Properties of TP-Phos
| Parameter | Value / Result | Experimental Conditions |
|---|---|---|
| Absorption Shift | Peak shifted from 300 nm to 365 nm after ALP incubation [43]. | Probe: 5 μM; ALP: 0.01 U/mL; Time: 20 min in Tris buffer. |
| Fluorescence Turn-On | Emission maximum at 525 nm; intense green fluorescence after reaction [43]. | Excitation at 365 nm; compared to weak blue fluorescence (450 nm) of the probe alone. |
| Detection Limit | Detected endogenous ALP activity in tissues [43]. | Successfully applied in rat hippocampus, kidney, and liver tissues. |
| Selectivity Validation | Displayed improved selectivity over commercial DiFMUP probe [43]. | Attributed to steric hindrance from ortho-functionalized group. |
Detailed Protocols
Protocol 1: Tissue Pretreatment with Acetic Anhydride and TEA
This critical pre-hybridization step is designed to acetylate tissue sections and minimize non-specific probe binding [10] [7].
Workflow Overview:
Step-by-Step Procedure:
- Tissue Preparation: Cut fresh-frozen tissue into 15 µm coronal sections using a cryostat and thaw-mount onto gelatin-subbed, RNase-free slides. Store at -70°C until use [10].
- Post-fixation: Remove slides from storage and air-dry for 10 minutes. Immerse slides in a 4% buffered paraformaldehyde solution (pH 7.4) for 5 minutes in an ice-water bath. Subsequently, rinse in 0.1 M phosphate-buffered saline (PBS, pH 7.4) on ice [10].
- Acetylation Reaction: a. Briefly rinse the fixed slides in 0.1 M TEA buffer (pH 8.0) at room temperature [10] [7]. b. Add 875 µL of acetic anhydride to a dry, baked glass staining dish containing a magnetic stir bar [10]. c. Transfer the slide carrier into the dish and immediately cover with 350 mL of 0.1 M TEA buffer [10]. d. Stir the solution continuously and incubate the slides at room temperature for 10 minutes [10].
- Post-Treatment Wash and Dehydration: a. Remove slides from the acetic anhydride/TEA solution and rinse in 2× Standard Saline Citrate (SSC) [10]. b. Dehydrate the tissues by passing them through a graded series of alcohols (70%, 95%, and 100% ethanol). c. For delipidation, immerse slides in chloroform for 5 minutes, then pass through 100% and 95% ethanol again before air-drying [10].
Protocol 2: Imaging ALP Activity with TP-Phos in Tissue
This protocol describes the application of the TP-Phos probe for detecting endogenous ALP activity in prepared tissue sections [43].
Workflow Overview:
Step-by-Step Procedure:
- Probe Application: Apply the TP-Phos probe solution directly to the pretreated and air-dried tissue sections.
- Incubation: Incubate the slides for an appropriate time (e.g., 20 minutes to 1 hour) in a humidified chamber at room temperature or 37°C to allow the enzymatic reaction to proceed [43].
- Washing: Rinse the slides with a suitable buffer (e.g., Tris buffer or PBS) to remove unreacted probe and stop the reaction.
- Imaging: Mount the slides and image using a two-photon microscope. The TP-Phos probe is excited with a two-photon laser typically tuned to the appropriate wavelength (e.g., ~750 nm for two-photon excitation of a 365 nm one-photon absorbance), and emission is collected around 525 nm [43]. The increased penetration depth and reduced autofluorescence of two-photon microscopy are crucial for obtaining clear signals from deep within tissue samples.
Conclusion
The acetic anhydride and triethanolamine treatment remains a cornerstone step in robust WISH protocols, directly addressing the pervasive challenge of non-specific hybridization. Its proper application, grounded in an understanding of its chemical mechanism, is essential for generating high-fidelity spatial gene expression data, particularly in complex or challenging tissues prone to background. As research moves toward higher-throughput in situ applications and the analysis of low-abundance transcripts, the principles of acetylation-based background suppression will continue to be relevant. Future directions should focus on further streamlining this step for automation, adapting it for emerging fluorescent in situ platforms, and exploring its utility in single-molecule RNA detection technologies to push the boundaries of resolution in clinical and biomedical research.
References
- 1. High Resolution Whole Mount In Situ Hybridization within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4396715/]
- 2. Protocol 3: Whole mount in situ hybridization (WISH) [https://cuttingclass.stowers.org/en/node/121415]
- 3. An optimised whole mount in situ hybridisation protocol for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4379745/]
- 4. Abstract - RSC Publishing - The Royal Society of Chemistry [https://pubs.rsc.org/en/content/articlehtml/2015/cs/c5cs00048c]
- 5. Insights into pH-modulated interactions between native ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996922001867]
- 6. An optimised whole mount in situ hybridisation protocol for the ... [https://bmcdevbiol.biomedcentral.com/articles/10.1186/s12861-015-0068-7]
- 7. Double In situ Hybridization for MicroRNAs and mRNAs ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2016.00126/full]
- 8. Three-color fluorescence in situ hybridization for the ... [https://pubmed.ncbi.nlm.nih.gov/2492920/]
- 9. High Resolution Whole Mount In Situ Hybridization within ... [https://www.jove.com/t/50644/high-resolution-whole-mount-situ-hybridization-within-zebrafish]
- 10. Quantification of mRNA in Discrete Cell Groups of Brain by ... [https://www.sciencedirect.com/science/article/abs/pii/B9780121852511500143]
- 11. A Whole Mount In Situ Hybridization Method for the ... [https://www.jove.com/t/53968/a-whole-mount-situ-hybridization-method-for-gastropod-mollusc-lymnaea]
- 12. Localisation of Abundant and Organ-Specific Genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3354552/]
- 13. A technical review and guide to RNA fluorescence in situ ... [https://peerj.com/articles/8806/]
- 14. 5 Important Proteinase K Protocol Tips [https://www.goldbio.com/blogs/articles/5-important-proteinase-k-protocol-tips?srsltid=AfmBOoqNQmHxUZMBMsl6VM2JtaHahYHPVQ453fhhafUIMBLIsXinIGv9]
- 15. Proteinase K Protocol [https://www.promega.com/resources/protocols/product-information-sheets/n/proteinase-k-protocol/]
- 16. In situ hybridization protocol for enhanced detection of gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3610298/]
- 17. Comparative effects of two in situ hybridization methods for ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1234895/full]
- 18. Hybridization Probes - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/hybridization-probes]
- 19. A powerful and versatile new fixation protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11533299/]
- 20. Large Scale In Situ Hybridization Protocol [https://kingsley.stanford.edu/Lab%20Protocols-WEB%202003/Molecular%20Biology/30_slide_in_situ_w=_EtOH.htm]
- 21. 5 ways controls can demystify your Stellaris™ RNA FISH ... [https://blog.biosearchtech.com/five-ways-controls-can-demystify-your-stellaris-rna-fish-experiment]
- 22. RNase+DNase Detection Kit [https://www.jenabioscience.com/molecular-biology/contamination-controls/nuclease-detection/pp-409-rnase-plus-dnase-detection-kit]
- 23. In situ hybridization protocol for enhanced detection of gene ... [https://bmcdevbiol.biomedcentral.com/articles/10.1186/1471-213X-13-8]
- 24. An improved method for whole-mount in situ hybridization ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1487644/full]
- 25. Whole Mount in situ Localization of miRNAs and mRNAs ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2018.01277/full]
- 26. A solvent-free delipidation method for functional validation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7371773/]
- 27. Chloroform/Methanol Protein Extraction and In-solution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11349489/]
- 28. The BUME method: a novel automated chloroform-free 96 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3540853/]
- 29. The BUME method: a novel automated chloroform-free 96- ... [https://pubmed.ncbi.nlm.nih.gov/22645248/]
- 30. A protocol for the combined sub-fractionation and ... [https://pubmed.ncbi.nlm.nih.gov/18456580/]
- 31. Modified in situ Hybridization Chain Reaction Using Short ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2020.00075/full]
- 32. A guided protocol for tissue dissection and proteinase K ... [https://www.sciencedirect.com/science/article/pii/S2215016124004333]
- 33. TUNEL staining [https://www.abcam.com/en-us/technical-resources/guides/cell-health-guide/tunel-staining-or-tunel-assay]
- 34. TUNEL Assay - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/tunel-assay]
- 35. Do TUNEL and Other Apoptosis Assays Detect Cell Death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730366/]
- 36. Guidelines for the use and interpretation of assays ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2757140/]
- 37. False positive staining in the TUNEL assay to detect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC395637/]
- 38. False positive staining in the TUNEL assay to detect ... [https://pubmed.ncbi.nlm.nih.gov/9893746/]
- 39. How Much Do You Know About TUNEL Staining? A ... - Yeasen [https://www.yeasenbio.com/blogs/cell/how-much-do-you-know-about-tunel-staining-a-troubleshooting-guide-for-abnormal-staining-results]
- 40. Harmonizing TUNEL with multiplexed iterative ... [https://www.sciencedirect.com/science/article/pii/S2667237525000839]
- 41. Her2 assessment using quantitative reverse transcriptase ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-017-1195-7]
- 42. Performance of a RT-PCR Assay in Comparison to FISH and ... [https://pubmed.ncbi.nlm.nih.gov/28763012/]
- 43. Construction of an alkaline phosphatase-specific two ... [https://www.sciencedirect.com/science/article/abs/pii/S0142961217304325]
- 44. Construction of an Alkaline Phosphatase-Specific Two- ... [https://pubmed.ncbi.nlm.nih.gov/28662402/]