Proteinase K vs. Acetone Permeabilization: A Comprehensive Guide for Method Selection in Biomedical Research
This article provides a systematic comparison of proteinase K (enzymatic) and acetone (solvent) permeabilization methods, two fundamental techniques for enabling antibody access to intracellular targets.
Proteinase K vs. Acetone Permeabilization: A Comprehensive Guide for Method Selection in Biomedical Research
Abstract
This article provides a systematic comparison of proteinase K (enzymatic) and acetone (solvent) permeabilization methods, two fundamental techniques for enabling antibody access to intracellular targets. Tailored for researchers and drug development professionals, it covers the core principles, optimal applications, and limitations of each method. Drawing on recent studies, the guide offers detailed protocols, troubleshooting strategies for common issues like poor signal and morphology loss, and validation data on their effects on downstream analyses like transcriptomics and multi-omics. The goal is to empower scientists to select and optimize the most appropriate permeabilization strategy for their specific experimental models, from standard immunoassays to advanced single-cell techniques.
Core Principles: How Proteinase K and Acetone Work at the Cellular Level
In the realm of cell biology and diagnostic research, the ability to detect intracellular targets—whether proteins, RNA, or DNA—is fundamental to understanding cellular function and disease mechanisms. This process almost universally requires cell permeabilization, a technique that creates openings in the cell membrane to allow detection reagents to access internal structures. The selection of permeabilization method represents a critical juncture in experimental design, balancing the competing demands of target accessibility, structural preservation, and epitope integrity. Within this landscape, two distinct approaches have emerged as valuable tools: the enzymatic action of proteinase K and the chemical properties of organic solvents like acetone.
This guide provides an objective comparison of these methods, presenting experimental data and detailed protocols to inform researchers in their selection of optimal permeabilization strategies for specific applications.
Mechanisms of Action: A Tale of Two Methods
Permeabilization methods function through fundamentally different mechanisms, which directly influence their applications and outcomes in experimental settings.
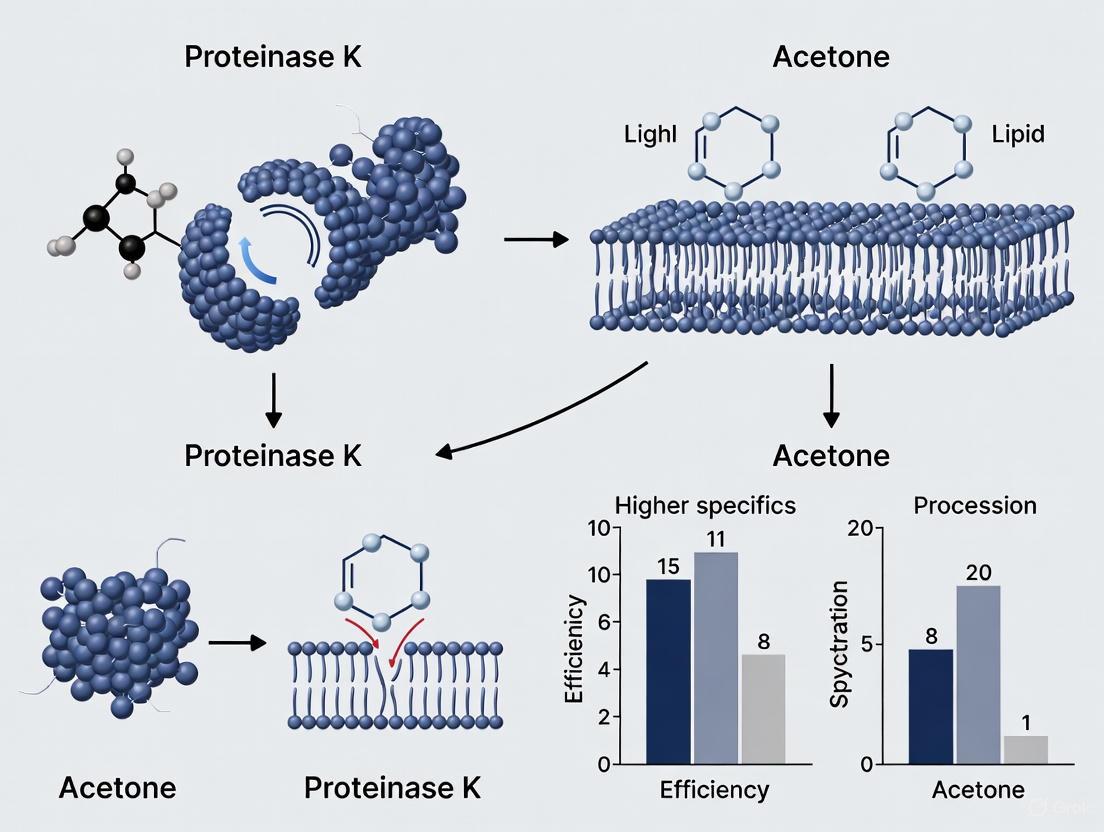
The diagram above illustrates the core mechanisms of each method. Proteinase K, a broad-spectrum serine protease, enzymatically digests proteins integral to membrane structure, creating defined access points while preserving certain cellular components [1]. In contrast, acetone and other organic solvents like methanol physically dissolve lipid bilayers through chemical disruption, simultaneously permeabilizing and fixing cells by precipitating cellular components [2].
Performance Comparison: Quantitative Experimental Data
Direct comparative studies on permeabilization methods provide valuable insights into their performance characteristics. Research investigating intracellular 18S rRNA detection in HeLa cells offers objective metrics for evaluation.
Table 1: Performance Metrics of Permeabilization Methods for 18S rRNA Detection
| Permeabilization Method | Optimal Concentration | Incubation Conditions | Cell Frequency (%) | Fluorescence Intensity | Morphology Preservation |
|---|---|---|---|---|---|
| Tween-20 | 0.2% | 30 min at 25°C | 97.9% | Highest | Good |
| Proteinase K | 0.01-0.1 µg/mL | 5-15 min at 37°C | Variable | Moderate | Moderate |
| Saponin | 0.1-0.5% | 10-30 min at 25°C | Moderate | Low to Moderate | Excellent |
| Triton X-100 | 0.1-0.2% | 5-10 min at 25°C | Moderate | Moderate | Fair |
| Streptolysin O | 0.2-1 µg/mL | 5-10 min on ice | Low | Low | Good |
Source: Adapted from Mousavi et al. (2014) [3]
The data reveals that Tween-20 demonstrated superior performance for RNA detection, achieving the highest cell frequency (97.9%) and fluorescence intensity in this particular application [3]. Proteinase K showed more variable results, highly dependent on concentration and incubation time. The performance characteristics of acetone, while not included in this specific study, are documented in other applications as providing rapid permeabilization but potentially harsher treatment that can compromise some cellular targets [2].
Application-Specific Considerations and Protocols
The optimal permeabilization method varies significantly depending on the target molecule and research application. Below are detailed protocols for implementing these techniques in specific experimental contexts.
Proteinase K for RNA In Situ Hybridization
Proteinase K is particularly valuable for RNA detection where protein barriers must be removed to allow probe access [4].
Detailed Protocol:
- Fixation: Begin with cells fixed in 4% paraformaldehyde for 10-20 minutes at room temperature or 4°C [5].
- Permeabilization: Treat with Proteinase K at 50 μg/mL for 1 hour at room temperature [4].
- Post-fixation: Re-fix with 4% paraformaldehyde for 30 minutes to maintain structural integrity after permeabilization.
- Hybridization: Proceed with standard RNA in situ hybridization protocols.
Considerations: Excessive Proteinase K concentration or incubation time can damage cellular morphology and reduce signal. Optimization is essential for different cell types [4].
Acetone for Immunofluorescence
Acetone provides rapid fixation and permeabilization, particularly suitable for certain protein targets.
Detailed Protocol:
- Preparation: Chill 100% acetone to -20°C.
- Fixation/Permeabilization: Incubate cells in cold acetone for 5-10 minutes at room temperature [5].
- Rehydration: Wash with PBS to remove acetone and rehydrate samples.
- Staining: Proceed with antibody staining protocols.
Considerations: Acetone can denature some protein epitopes and destroy membrane structures, making it unsuitable for membrane-associated proteins [5] [2].
Combined Approaches for Challenging Targets
For difficult targets such as those in dense tissues or subcellular compartments, researchers have developed hybrid approaches:
Alternative Permeabilization for Protein-RNA Co-detection: When performing simultaneous protein immunofluorescence and RNA FISH (IF/FISH), standard Proteinase K treatment often damages protein epitopes. An effective alternative combines:
- Organic Solvent Treatment: Xylenes and ethanol before rehydration
- Detergent Application: RIPA buffer after rehydration [4]
This combined approach preserves protein antigenicity while allowing sufficient RNA probe penetration, balancing the competing needs of target accessibility and epitope preservation [4].
Advanced Research Applications and Data Quality Impacts
Recent studies in spatial transcriptomics have revealed significant implications of permeabilization choice on data quality, particularly for Proteinase K applications.
Table 2: Proteinase K Concentration Effects on Spatial Transcriptomics Data
| Proteinase K Concentration | Total Reads | Negative Probe Counts | Signal-to-Noise Ratio | Genes Detected Above Background |
|---|---|---|---|---|
| Lower (0.1 μg/mL) | Baseline | Baseline | Baseline | Baseline |
| Higher (1 μg/mL) | 2-4x Increase | 2-12x Increase | 10-70% Lower | 50-80% Lower |
Source: Adapted from Delorey et al. (2024) [6]
The data demonstrates a critical trade-off: while higher Proteinase K concentrations increase total reads, they substantially elevate background noise and reduce usable data output [6]. These effects vary across tissue types, emphasizing the need for tissue-specific protocol optimization.
The Researcher's Toolkit: Essential Permeabilization Reagents
Successful permeabilization requires more than just primary agents. Below are essential laboratory reagents for implementing these techniques effectively.
Table 3: Essential Reagents for Permeabilization Protocols
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Fixatives | 4% Paraformaldehyde, Methanol, Acetone | Stabilize cellular structures and preserve morphology prior to permeabilization [5] [2] |
| Detergents | Tween-20, Triton X-100, Saponin, NP-40 | Create pores in membrane structures through lipid displacement or cholesterol binding [3] |
| Enzymes | Proteinase K, Streptolysin O | Digest specific membrane components to create access points [3] |
| Buffers & Solutions | PBS, SSC, Permeabilization Buffer Sets | Maintain pH and osmolarity while supporting reagent activity [7] |
| Blocking Agents | BSA, Normal Serum, Glycine | Reduce non-specific binding after permeabilization [5] |
The choice between proteinase K and acetone permeabilization is not a matter of superior versus inferior, but rather appropriate application based on experimental requirements. Proteinase K excels in RNA detection applications where protein barriers significantly hinder access, though it requires careful optimization to balance access with morphological preservation [3] [4]. Acetone provides rapid simultaneous fixation and permeabilization for certain protein targets but may compromise membrane structures and denature sensitive epitopes [5] [2].
The most effective permeabilization strategy aligns method selection with specific research goals, considering the nature of the target molecule, cellular localization, and required preservation of cellular architecture. As spatial biology and multi-omics approaches advance, the precision of permeabilization techniques will continue to play a pivotal role in generating high-quality, reproducible data across diverse research applications.
In life sciences research, the ability to access the interior of cells is fundamental. For techniques ranging from nucleic acid purification to immunostaining, scientists must first overcome the barrier of the cellular membrane. Two principal methods to achieve this are enzymatic digestion, using agents like proteinase K, and solvent-based permeabilization, using agents like acetone. Proteinase K, a broad-spectrum serine protease, penetrates tissues by systematically digesting proteins, thereby breaking down structural components and releasing intracellular materials [8] [9]. In contrast, organic solvents like acetone act as dehydrating agents that precipitate cellular proteins and dissolve lipids, physically creating pores in the membrane [10] [11]. This guide provides an objective comparison of these methods, focusing on their mechanisms, applications, and performance data to inform methodological choices in research and drug development.
Mechanism of Action: A Biochemical versus Physical Approach
Enzymatic Digestion with Proteinase K
Proteinase K is an endopeptidase belonging to the subtilisin group of serine proteases. Its catalytic mechanism relies on a catalytic triad consisting of Serine 224, Histidine 69, and Aspartic acid 39 [9]. This enzyme preferentially cleaves peptide bonds adjacent to the carboxyl group of hydrophobic and aromatic amino acids, leading to the extensive digestion of proteins [8] [9].
- Subcellular Targeting: By digesting histone and non-histone proteins, it disrupts the nuclear membrane and protein cross-links, facilitating the release of nucleic acids and other intracellular components [12].
- Stability and Enhancement: A key advantage is its stability under harsh conditions, including elevated temperatures (37–65°C) and the presence of denaturants like SDS (sodium dodecyl sulfate) and urea. These denaturants can enhance its activity by up to 313% by unfolding protein substrates and making cleavage sites more accessible [8] [12].
The following diagram illustrates the enzymatic mechanism of proteinase K and its role in tissue penetration for nucleic acid isolation.
Solvent-Based Permeabilization with Acetone
Acetone, a precipitating fixative and permeabilization agent, operates through a physical mechanism. It acts as a strong dehydrant, removing water from cells and leading to the precipitation of cellular proteins [10] [11]. Simultaneously, it dissolves membrane lipids, thereby creating pores in the cellular membrane [11].
- Application and Considerations: Standard protocols involve incubating cells with chilled acetone (-20°C) for 5–10 minutes [10]. As a fixative, it preserves cellular architecture but can remove small soluble molecules and lipids. A significant drawback is that it may denature overexpressed fluorescent proteins (e.g., GFP) and is highly volatile and flammable [11].
The table below provides a direct comparison of these core mechanisms and properties.
Table 1: Fundamental Comparison of Proteinase K and Acetone
| Feature | Proteinase K | Acetone |
|---|---|---|
| Mechanism of Action | Enzymatic hydrolysis of peptide bonds [8] [9] | Physical precipitation of proteins and dissolution of lipids [10] [11] |
| Primary Application | Nucleic acid isolation; antigen retrieval [13] [12] | Cell fixation and permeabilization for ICC/IF [10] [11] |
| Typical Working Concentration | 100-200 µg/mL [12] | 95-100% [10] |
| Key Advantage | Digests contaminating nucleases; stable with denaturants [8] [12] | Rapid action; no separate permeabilization step needed [10] [11] |
| Key Limitation | Requires heat inactivation and subsequent removal [12] | Can denature proteins and antigens; highly volatile [11] |
Experimental Performance and Benchmarking Data
Proteinase K in Nucleic Acid Isolation from FFPE Tissue
Optimizing the proteinase K digestion protocol is critical for recovering high-quality nucleic acids from Formalin-Fixed, Paraffin-Embedded (FFPE) tissue, where proteins and nucleic acids are extensively cross-linked.
A 2020 study systematically evaluated different proteinase K digest protocols using 54 clinical FFPE tumor biospecimens [13]. The researchers compared the manufacturer's standard protocol (20 µl proteinase K for 24 hours) against two optimized ones: one with doubled enzyme quantity and another with an extended 72-hour digestion [13].
Table 2: Proteinase K Protocol Performance in DNA Yield from FFPE Tissue [13]
| Digest Protocol | Median DNA Yield | Key Finding |
|---|---|---|
| Protocol 1 (Standard)20 µl proteinase K for 24 hr | Baseline | Reference yield for comparison |
| Protocol 2 (Doubled Enzyme)20 µl for 5 hr, then a further 20 µl for 19 hr | 96% increase vs. Protocol 1 | Doubling the quantity of proteinase K nearly doubled the DNA yield |
| Protocol 3 (Extended Time)20 µl proteinase K for 72 hr | Not specifically reported | Increases in yield were generally accompanied by increases in integrity |
The study concluded that optimization, primarily by increasing the volume of proteinase K, reduced the sample failure rate for whole genome sequencing from 33% to just 7% [13]. Furthermore, increases in DNA yield were generally accompanied by improvements in DNA integrity, as measured by the ability to amplify 400 bp PCR amplicons [13].
Performance in Fixed Single-Cell RNA Sequencing
The effectiveness of proteinase K also extends to modern single-cell genomics. A 2021 study developed FD-seq, a method for sequencing RNA from paraformaldehyde (PFA)-fixed single cells, which relies on proteinase K for cross-link reversal inside droplets [14].
The researchers found that adding proteinase K at an optimal concentration of 40 U/mL in the lysis buffer during a 1-hour incubation at 56°C efficiently reversed PFA cross-links without significantly compromising RNA integrity [14]. When they compared FD-seq on PFA-fixed cells to standard Drop-seq on live cells, they found:
- The number of genes detected was comparable (median of 640 vs. 675 for human cells).
- The relative gene expression levels were strongly correlated.
- The cross-droplet contamination rate was minimal and similar (~1% for fixed cells) [14].
This demonstrates that proteinase K treatment can be effectively integrated into complex, high-throughput workflows without sacrificing data quality.
Detailed Experimental Protocols
Optimized Proteinase K Digestion for FFPE Tissue Sections
The following protocol is adapted from a 2020 study that successfully optimized DNA yield from FFPE tissues [13].
Materials:
- QIAamp DNA FFPE Tissue Kit (or equivalent)
- Proteinase K (20 mg/ml)
- Histoclear (xylene substitute)
- 100% Ethanol
- Heating block
Method:
- Deparaffinization: Place 10x 4µm tissue sections in a 1.5 ml tube. Vortex in 1 ml Histoclear for 10 seconds and centrifuge for 2 minutes to pellet tissue. Remove supernatant and repeat. Wash with 1 ml 100% ethanol, vortex, centrifuge, and remove supernatant. Air-dry the pellet for 10 minutes [13].
- Proteinase K Digestion: Select one of the following protocols for the digest step [13]:
- Standard Protocol: Digest with 20 µl proteinase K for 24 hours at 56°C.
- Optimized (Doubled Enzyme) Protocol: Digest with 20 µl proteinase K for 5 hours at 56°C, then add a further 20 µl of enzyme and continue digestion for another 19 hours (24 hours total).
- Extended Time Protocol: Digest with 20 µl proteinase K for 72 hours at 56°C.
- Post-Digestion and Purification: Follow the manufacturer's instructions for the remainder of the purification kit, which typically includes a step to inactivate the proteinase K by heating (e.g., 95-100°C for 10-15 minutes) and subsequent binding/washing of DNA on a silica column [13] [12].
Acetone Permeabilization for Immunocytochemistry (ICC)
This standard protocol is used for permeabilizing cells prior to antibody staining for intracellular targets [10] [11].
Materials:
- Ice-cold Acetone (95-100%)
- Phosphate-Buffered Saline (PBS)
- Fixed cell sample (e.g., on a coverslip)
Method:
- Fixation: Fix cells with your chosen fixative (e.g., 4% PFA for 10-20 minutes at room temperature). If PFA is used, permeabilization is required [10].
- Wash: Wash cells three times with PBS to remove residual fixative [10].
- Permeabilization: Incubate cells with ice-cold acetone (95-100%) for 5–10 minutes at -20°C [10].
- Wash: Wash cells three times with PBS to remove the acetone [10].
- Proceed to Staining: The cells are now ready for blocking and antibody incubation steps [10].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their functions in experiments utilizing proteinase K and acetone permeabilization.
Table 3: Essential Reagents for Tissue Penetration and Permeabilization Experiments
| Reagent | Function | Example Use Case |
|---|---|---|
| Proteinase K | Broad-spectrum serine protease that digests proteins and inactivates nucleases [8] [12]. | DNA/RNA isolation from tissues and fixed cells [13] [12]. |
| SDS (Sodium Dodecyl Sulfate) | Denaturing detergent that disrupts membranes and enhances Proteinase K activity [8]. | Lysis buffer component for efficient nucleic acid release [12]. |
| Acetone | Organic solvent that precipitates proteins and dissolves lipids for permeabilization [10] [11]. | Cell permeabilization for immunocytochemistry [10]. |
| Triton X-100 | Non-ionic detergent that permeabilizes all lipid bilayers, including the nuclear membrane [10] [11]. | A common alternative to acetone for permeabilizing cells for antibody staining [14]. |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves cellular morphology by creating protein networks [10] [11]. | Tissue and cell fixation prior to permeabilization with acetone or digestion with Proteinase K [13] [14]. |
| EDTA | Chelating agent that binds calcium ions, destabilizing Proteinase K but inhibiting metal-dependent nucleases [8]. | Added to digestion buffers to protect nucleic acids from degradation [8]. |
Integrated Workflow for Sample Processing
The choice between proteinase K and acetone is dictated by the final analytical goal. The diagram below maps out the decision-making workflow for sample processing based on the desired application.
Acetone is a versatile solvent widely employed in biomedical research for its dual capacity to dehydrate biological samples and precipitate key biomolecules, namely lipids and proteins. Its effectiveness stems from its physicochemical properties as a polar aprotic solvent, which allows it to readily mix with water and other organic solvents while disrupting hydrophobic interactions essential for biomolecule solubility [15]. In the context of a broader thesis comparing sample preparation techniques, understanding acetone's mechanism of action provides a critical foundation for evaluating its performance against enzymatic methods like proteinase K permeabilization.
The core action of acetone centers on its ability to disrupt the solvation layer surrounding proteins and lipids. In an aqueous environment, biomolecules are stabilized by a hydration shell. When introduced to a sample, acetone competes for hydrogen bonds with water molecules, effectively stripping this protective layer. This disruption reduces the dielectric constant of the solvent environment, leading to decreased biomolecule solubility and subsequent aggregation and precipitation [15]. For lipids, particularly neutral lipids, acetone serves as an effective extraction solvent due to its relative polarity, though it is less effective for polar lipids which often require solvent mixtures with greater polarity or ionic strength [16]. This mechanistic understanding provides the basis for its application in various experimental protocols and its comparative performance against alternative methods.
Acetone in Protein Precipitation
Practical Applications and Protocols
In practical laboratory settings, acetone precipitation is a cornerstone technique for concentrating proteins and removing contaminants. A standard protocol involves adding at least four volumes of cold acetone (-20°C) to a liquid protein sample, incubating the mixture at -20°C for several hours (or overnight for maximum recovery), and then centrifuging at high speed (e.g., 10,000-15,000 × g) for 10-15 minutes to pellet the precipitated proteins [15]. The supernatant is carefully decanted, and the pellet is allowed to air-dry to evaporate residual acetone before being resuspended in an appropriate buffer. The use of pre-chilled acetone is critical as it enhances precipitation efficiency and helps maintain protein stability.
Comparative studies have quantified acetone's performance against other common precipitation methods. In research utilizing Chinese hamster ovary (CHO) cell homogenates, acetone precipitation demonstrated superior protein recovery rates compared to other common techniques. When enhanced with an ultrasonic bath or the addition of NaOH, recovery reached approximately 104%, significantly outperforming methanol-chloroform (94%) and trichloroacetic acid (TCA)-acetone (78%) methods [17]. This high recovery rate, coupled with minimal disruption to protein band patterns on SDS-PAGE, makes acetone particularly valuable for proteomic workflows where maintaining the original complexity of cellular composition is paramount [17].
Table 1: Comparison of Protein Precipitation Methods for CHO Cell Homogenates
| Precipitation Method | Protein Recovery (%) | Effect on Protein Pattern | Key Practical Notes |
|---|---|---|---|
| Acetone (with ultrasonic bath) | 104.18 ± 2.67 | Similar to cellular homogenates | High recovery, preserves complexity |
| Acetone (with NaOH) | 103.12 ± 5.74 | Similar to cellular homogenates | High recovery, easy protocol |
| Methanol-Chloroform (with homogenization) | 94.22 ± 4.86 | No negative effect on pattern | Intermediate recovery |
| TCA-Acetone | 77.91 ± 8.79 | Altered pattern, difficult solubilization | Low recovery, negatively affects analysis |
Comparison with Proteinase K Digestion
Within the specific thesis context of comparing permeabilization methods, acetone and proteinase K represent fundamentally different approaches. Proteinase K is a broad-spectrum serine protease used to digest proteins and permeabilize tissues for molecular biology applications, particularly in in situ hybridization (ISH) and fluorescent ISH (FISH) protocols [4]. Its enzymatic action cleaves peptide bonds, degrading cellular proteins and thereby increasing accessibility for nucleic acid probes.
However, this proteolytic activity presents a significant drawback for experiments requiring simultaneous protein detection. In dual protein-RNA labeling (IF/FISH) procedures, proteinase K treatment damages protein epitopes, resulting in weak or nonexistent protein signals during subsequent immunofluorescence staining [4]. This limitation has driven the development of alternative permeabilization strategies that preserve protein integrity. In such applications, acetone serves as a non-enzymatic permeabilizing agent that can fix and permeabilize tissues simultaneously, often through cold incubation, making it suitable for protocols where antigen preservation is critical [18].
Table 2: Acetone vs. Proteinase K for Sample Preparation
| Parameter | Acetone | Proteinase K |
|---|---|---|
| Primary Mechanism | Solvent action, dehydrates and precipitates | Enzymatic digestion, cleaves peptide bonds |
| Impact on Proteins | Precipitates/denatures proteins | Degrades proteins |
| Impact on Lipids | Dissolves and extracts neutral lipids | Minimal direct effect |
| Key Applications | Protein precipitation, lipid extraction, fixation/permeabilization for IHC/IF | Tissue permeabilization for ISH/FISH, protein depletion |
| Compatibility with Protein Detection | Compatible with many antibodies post-fixation | Often incompatible (destroys epitopes) |
| Typical Conditions | Cold incubation (-20°C), minutes to hours | 37°C incubation, minutes to hours (e.g., 50 μg/ml for 1h [4]) |
Acetone in Lipid Extraction
Role in Lipidomics and Biodiesel Research
In lipid research, acetone occupies a specific niche as an effective solvent for extracting neutral lipids, including triacylglycerols (TAGs) and sterol esters [16]. Its moderate polarity makes it particularly suitable for disrupting hydrophobic associations and solubilizing non-polar lipid species. However, for comprehensive lipidomic analysis that includes polar lipids such as phospholipids, acetone is often less effective as a standalone solvent. The classical Folch [chloroform:methanol (2:1, v/v)] and Bligh & Dyer methods, which use chloroform-methanol mixtures, typically provide more complete extraction of diverse lipid classes, including glycerophospholipids [16].
The efficiency of acetone for lipid extraction is significantly enhanced by cell disruption pretreatments, especially when working with robust biological materials like microalgae, fungi, or plant tissues that possess complex cell walls. Physical methods such as grinding, bead milling, ultrasonication, or osmotic shock create pathways for solvent penetration, dramatically improving lipid yield [16]. For instance, one comparative study on thraustochytrids demonstrated that grinding with liquid nitrogen combined with chloroform/methanol (2:1) extraction yielded the highest lipid recovery, underscoring the importance of combining mechanical disruption with optimized solvent selection [16].
Advanced and Greener Solvent Applications
The evolving landscape of green chemistry has prompted investigation into acetone as a potentially more environmentally friendly alternative to traditional solvents like acetonitrile in certain analytical workflows. While acetonitrile remains prevalent in protein and lipid analyses, its recognized toxicity and environmental impact have driven the search for substitutes. Research in solid-phase extraction of low molecular weight proteins from biological fluids has shown that ethanol (60%, v/v) can provide comparable extraction recovery to acetonitrile (75%, v/v) [19], establishing a precedent for solvent replacement. Although acetone itself was not the primary focus of this particular study, it exists within the same paradigm of developing safer, more sustainable solvent systems for biomolecule analysis.
Experimental Protocols and Research Toolkit
Detailed Methodologies for Key Applications
Protein Precipitation from Serum/Plasma:
- Sample Preparation: Begin with 100 μl of clear serum or plasma.
- Precipitation: Add 900 μl of HPLC-grade acetone (pre-cooled to -20°C). Vortex vigorously for 30-60 seconds to ensure complete mixing.
- Incubation: Allow the mixture to stand at -20°C for a minimum of 1 hour. Extended incubation (overnight) can improve precipitation efficiency.
- Pellet Formation: Centrifuge at 12,000 × g for 10-15 minutes at 4°C. A visible pellet should form at the bottom of the tube.
- Supernatant Removal: Carefully decant or pipette off the supernatant without disturbing the pellet.
- Drying: Air-dry the pellet for 5-10 minutes to evaporate residual acetone. Do not over-dry, as this can make resolubilization difficult.
- Resolubilization: Resuspend the protein pellet in an appropriate buffer (e.g., SDS-PAGE sample buffer, MS-compatible buffer) by vortexing and gentle heating if necessary [20].
Tissue Permeabilization for Immunohistochemistry (IHC):
- Fixation: Tissue sections are typically fixed first using formalin, paraformaldehyde, or in some cases, cold acetone itself.
- Permeabilization: Incubate fixed tissues with 0.1-0.5% detergent (e.g., Tween 20, Triton X-100) in PBS for 10-30 minutes. Alternatively, for certain applications, cold acetone (-20°C) can be used as a combined fixative and permeabilizing agent by incubating tissue sections for 5-10 minutes at -20°C.
- Washing: Rinse thoroughly with PBS to remove residual solvent before proceeding with antibody staining [18].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions and Their Functions
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| HPLC-Grade Acetone | High-purity solvent for precipitation and extraction | Low water content ensures efficient precipitation; often pre-cooled to -20°C |
| Proteinase K | Serine protease for enzymatic permeabilization and protein digestion | Typical working concentration: 20-100 μg/ml; incubation at 37°C [4] |
| Chloroform-Methanol (2:1) | Classic lipid extraction solvent mixture | Higher efficiency for polar lipids compared to acetone alone [16] |
| Ammonium Sulfate | Salt for "salting out" protein precipitation | Alternative to organic solvents; follows Hofmeister series [15] |
| Tris-EDTA Buffer | Common buffer for molecular biology | Used in antigen retrieval and sample storage |
| Phosphate-Buffered Saline (PBS) | Isotonic buffer for washing and dilution | Maintains pH and osmolarity for biological samples |
Signaling Pathways and Experimental Workflows
The following diagrams illustrate key experimental workflows and the mechanism of acetone action, providing visual references for the methodologies discussed.
Protein Precipitation Workflow - A standard protocol for concentrating proteins using acetone.
Acetone Precipitation Mechanism - Molecular-level action of acetone on proteins.
Acetone serves as a powerful tool in the researcher's arsenal, offering effective dehydration and precipitation capabilities for both proteins and lipids. Its key advantage lies in its simplicity and rapid action, providing a straightforward method for concentrating proteins and extracting neutral lipids without the need for complex instrumentation. When compared directly to proteinase K, acetone presents a complementary approach—while proteinase K excels in tissue permeabilization for nucleic acid detection through enzymatic digestion, acetone preserves protein epitopes, making it indispensable for immunohistochemistry and dual detection methodologies [4] [18].
Quantitative assessments confirm acetone's strong performance in protein precipitation, with recovery rates exceeding 100% in optimized protocols for CHO cell homogenates [17]. For lipid extraction, its efficiency is well-established for neutral lipids but may require combination with more polar solvents for comprehensive lipidomic profiles. The choice between acetone and alternative methods ultimately depends on the specific research objectives: acetone for speed, simplicity, and protein preservation, proteinase K for effective nucleic acid accessibility in complex tissues, and chloroform-based methods for comprehensive lipid recovery. As green chemistry principles continue to influence laboratory practices, acetone's role as a potentially more sustainable alternative to more hazardous solvents warrants further investigation and method development.
The efficient release of intracellular products is a critical step in bioprocessing and pharmaceutical development. Within this context, the selection of a cell disruption or permeabilization method can significantly impact the yield, stability, and activity of the target compound. This guide provides an objective comparison between two principal disruption strategies: enzymatic methods, with a focus on proteinase K, and solvent-based methods, utilizing acetone as a representative agent. The comparison is framed within a broader research thesis exploring the optimal conditions for isolating sensitive biological products, providing researchers and drug development professionals with experimental data and protocols to inform their methodology selection.
Comparative Mechanism and Performance
The core distinction between enzymatic and solvent-based disruption lies in their fundamental mechanisms. Enzymatic methods operate on a principle of biological specificity, where lytic enzymes selectively degrade key structural components of the cell wall or membrane [21]. In contrast, solvent-based methods rely on chemical action, where solvents dissolve or destabilize the lipid bilayer and permeabilize the cell through physicochemical forces [18] [21].
The table below summarizes the key characteristics, supported by experimental data, for a direct comparison.
Table 1: Comparative analysis of enzymatic and solvent-based disruption methods.
| Aspect | Enzymatic Disruption (e.g., Proteinase K) | Solvent-Based Disruption (e.g., Acetone) |
|---|---|---|
| Core Mechanism | Selective, catalytic hydrolysis of peptide bonds in cell wall/membrane proteins [21]. | Non-selective dissolution of membrane lipids and dehydration, leading to permeabilization [18]. |
| Primary Use Case | Gentle release of intracellular components; antigen retrieval in IHC [22] [18]. | Cell permeabilization for IHC; precipitation and purification of proteins in conjunction with other methods [23] [18]. |
| Typical Efficiency | High; can achieve 90-95% product release under optimized conditions [21]. | Variable; used as a permeabilization agent or in purification. Acetone precipitation is a key step in purifying a fibrinolytic enzyme with a 13.38-fold purification [23]. |
| Operational Conditions | Mild; typically 37°C for 5-30 minutes in a neutral buffer [22] [18]. | Mild for permeabilization (incubation for 10 minutes at room temperature); requires cold temperatures (-20°C) for precipitation [23] [18]. |
| Key Advantage | High biological specificity; preserves cell morphology; low shear stress [21]. | Rapid action; effective for a wide range of cells; also functions as a fixative [18]. |
| Key Limitation | High cost of enzymes; requires optimization for different cell types; potential for product degradation [21]. | Can denature sensitive proteins; requires careful removal post-treatment; less specific [18] [24]. |
| Impact on Product | Generally maintains protein activity due to gentle, specific action [21]. | Risk of protein inactivation or denaturation due to solvent interaction [24]. |
Detailed Experimental Protocols
Proteinase K Permeabilization Protocol
This protocol is adapted from methods used for permeabilizing zebrafish embryos for in situ hybridization and for antigen retrieval in Immunohistochemistry (IHC) [22] [18].
Materials:
- Fixed cell sample or tissue section.
- Phosphate-Buffered Saline with Tween 20 (PBTween): 1x PBS, 0.1% Tween 20.
- Proteinase K Stock Solution.
- Permeabilization Buffer: 10 µg/mL Proteinase K in PBTween.
- 4% Paraformaldehyde (PFA) in PBS for post-fixation (optional).
Procedure:
- Rehydration: If samples are stored in methanol, rehydrate through a graded series of methanol and PBTween washes.
- Permeabilization: Incubate the samples in the permeabilization buffer (10 µg/mL Proteinase K) for 5 minutes at room temperature [22]. Note: The incubation time is critical and may require optimization (from 5 to 30 minutes) depending on sample thickness and fixation level [18].
- Re-fixation (Optional): To halt Proteinase K activity and preserve structure, briefly post-fix the samples in 4% PFA for 20 minutes at room temperature [22].
- Washing: Rinse the samples thoroughly with PBTween to remove the enzyme and any cellular debris.
Acetone Permeabilization and Precipitation Protocol
This protocol outlines the use of acetone for cell permeabilization and its application in protein precipitation during purification, based on IHC and enzyme purification studies [23] [18].
Materials:
- Cell suspension or purified protein solution.
- Cold Acetone (pre-chilled to -20°C).
- Centrifuge and appropriate tubes.
- Suitable buffer for resuspending the precipitated protein (e.g., Tris-HCl).
Procedure: A. Acetone Permeabilization for IHC [18]:
- Incubation: Apply cold acetone to the air-dried sample and incubate for 10 minutes at room temperature.
- Evaporation: Allow the acetone to fully evaporate. The sample is now permeabilized and can proceed to antibody staining.
B. Acetone Precipitation for Protein Purification [23]:
- Mixing: Add a predetermined volume of ice-cold acetone to the protein solution. The optimal ratio for a fibrinolytic enzyme was reported at 1:1.5 (v/v) [23].
- Precipitation: Incubate the mixture at -20°C for 2 hours to allow protein precipitation.
- Pellet Collection: Centrifuge the mixture at 12,000 × g for 10 minutes to pellet the precipitated protein.
- Drying: Carefully decant the acetone and allow the pellet to air-dry to remove residual solvent.
- Resuspension: Resuspend the dried protein pellet in an appropriate buffer for downstream applications.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential reagents and their functions in the context of cell disruption and permeabilization protocols.
Table 2: Key research reagents and their functions in disruption and permeabilization protocols.
| Reagent/Solution | Function in Protocol |
|---|---|
| Proteinase K | A broad-spectrum serine protease that digests proteins and permeabilizes cell membranes by hydrolyzing peptide bonds [22] [18]. |
| Acetone | A solvent used for cell permeabilization by dissolving lipids and as a precipitating agent for protein purification and concentration [23] [18]. |
| Paraformaldehyde (PFA) | A cross-linking fixative used to preserve cellular structure by forming covalent bonds between proteins, immobilizing antigens [22] [18]. |
| Phosphate-Buffered Saline (PBS) | An isotonic buffer used to maintain a stable pH and osmotic balance, preventing osmotic shock to cells during washing and incubation steps [22]. |
| Tween 20 | A mild, non-ionic detergent used in buffers (e.g., PBTween) to reduce non-specific binding and aid in washing steps [22]. |
| Cetyltrimethylammonium Bromide (CTAB) | A cationic surfactant used in reverse micelle extraction systems for the purification of enzymes, such as fibrinolytic enzymes [23]. |
| Phenylmethylsulfonyl Fluoride (PMSF) | A serine protease inhibitor added to cell lysates and homogenates to prevent proteolytic degradation of the target protein after cell disruption [25]. |
Experimental Workflow and Pathway Visualization
The following diagram illustrates the logical workflow for selecting and applying either an enzymatic or solvent-based permeabilization method, highlighting the key decision points and procedural steps involved in a typical biomolecular research pipeline.
Permeabilization Method Selection Workflow
The mechanistic pathways through which proteinase K and acetone achieve permeabilization operate on fundamentally different principles, as summarized below.
Mechanism of Action Comparison
In cell biology and drug development, permeabilization is a critical sample preparation step that enables researchers to detect intracellular targets, from nucleic acids to proteins. The process involves creating holes in cellular membranes to allow entry of detection probes, such as antibodies or nucleic acid sequences, without completely destroying cellular architecture. The fundamental challenge lies in the inherent trade-off between achieving sufficient permeabilization strength for probe access and maintaining optimal structural preservation for accurate biological interpretation. Among the numerous available methods, proteinase K (an enzymatic approach) and acetone (an organic solvent) represent two philosophically and mechanistically distinct strategies. This guide objectively compares these methods based on experimental data, providing researchers with a framework for selecting appropriate protocols for their specific applications.
Mechanisms of Action: A Fundamental Divide
Understanding the core mechanisms by which proteinase K and acetone achieve permeabilization is essential for predicting their performance in experimental settings. The diagram below illustrates their distinct modes of action and the subsequent trade-offs.
The fundamental difference in mechanism leads directly to divergent experimental outcomes. Proteinase K enzymatically digests peptide bonds in proteins that constitute the membrane structure, creating precise, protein-sized channels for probe entry while largely preserving the lipid bilayer [3] [22]. In contrast, acetone acts as a strong dehydrating agent that rapidly dissolves lipids and precipitates cellular proteins, leading to a more generalized and physically disruptive breakdown of all membrane structures [26].
Performance Comparison: Quantitative and Qualitative Data
The mechanistic differences manifest in distinct performance profiles, which can be quantified and qualified across several key parameters. The following table summarizes the comparative experimental data for proteinase K and acetone permeabilization methods.
| Performance Parameter | Proteinase K | Acetone |
|---|---|---|
| Permeabilization Strength | Moderate, target-specific | Strong, non-selective |
| Structural Preservation | High for lipid architecture | Moderate; can damage membranes and microtubules [26] |
| Typical Working Concentration | 0.01 - 0.1 µg/ml [3] or 10 µg/ml [22] | 100% (pure solvent) [26] |
| Incubation Time/Temperature | 5-15 min at 37°C [3] [22] | 10-20 min at 4°C (ice-cold) [26] |
| Impact on Epitopes | Can digest target protein epitopes | Can denature/alter protein structure, damaging epitopes [26] |
| Ideal Application | Intracellular nucleic acid detection (e.g., RNA-FISH) [3] [22] | Staining of robust intracellular antigens (e.g., cytoskeletal components); not recommended for overexpressed fluorescent proteins (e.g., GFP) [26] |
| Compatibility with Multi-omics | Negative impact on whole transcriptome detection [27] | Data not available in search results |
| Key Advantage | Can be finely tuned via concentration and time for specific access | Simultaneously fixes and permeabilizes; no separate permeabilization step needed [26] |
The data reveals a clear trade-off. Proteinase K offers a more tunable and targeted approach, which is reflected in its successful application for detecting intracellular 18S rRNA in HeLa cells, where it provided measurable, though not optimal, fluorescence signals [3]. Its enzymatic nature allows researchers to fine-tune the level of permeabilization by adjusting concentration and incubation time. Conversely, acetone's strength lies in its simplicity and power, functioning as a combined fixative and permeabilization agent. However, this power comes at the cost of selectivity, as it can damage cell membranes, microtubules, and organelles, and is not suitable for preserving labile epitopes or overexpressed fluorescent proteins [26].
Detailed Experimental Protocols
To ensure experimental reproducibility, the following sections outline standardized protocols for each method as described in the literature.
Proteinase K Permeabilization Protocol
This protocol is adapted from intracellular RNA detection studies in HeLa cells and zebrafish embryos [3] [22].
- Fixation: Fix cells in 2-4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 15-20 minutes at room temperature.
- Washing: Wash the fixed cells twice with 1X PBS to remove residual fixative.
- Permeabilization: Prepare a proteinase K solution in a defined buffer. For HeLa cells, a concentration of 0.01-0.1 µg/ml in Tris-HCl (20 mM) with CaCl₂ (2 mM) was used [3]. For zebrafish embryos, a concentration of 10 µg/ml in PBTween (PBS with 0.1% Tween 20) is typical [22].
- Incubation: Incubate the cells with the proteinase K solution. Typical incubation times are 5-15 minutes at 37°C. Note: The time and concentration are critical and must be optimized empirically to prevent over-digestion.
- Termination: Wash the cells thoroughly with 1X PBS to remove all proteinase K from the medium.
- Post-fixation (Optional): Some protocols, particularly for delicate tissues like zebrafish embryos, include a second fixation step in 4% PFA for 20 minutes after permeabilization to re-stabilize the cells [22].
- Proceed to Hybridization/Staining: The cells are now ready for downstream applications like in situ hybridization or immunofluorescence.
Acetone Permeabilization Protocol
This protocol is based on standard immunocytochemistry guidelines [26].
- Chilling: Chill pure (100%) acetone to -20°C or 4°C. Using ice-cold acetone is standard practice.
- Application: For adherent cells cultured on a glass slide or coverslip, carefully remove the culture medium and immediately add enough ice-cold acetone to completely cover the cells. Alternatively, cells can be immersed in a container of acetone.
- Incubation: Incubate for 5-10 minutes at 4°C (on ice) or for 10-20 minutes at -20°C. The incubation is typically performed in a sealed container to prevent acetone evaporation.
- Removal and Drying: Carefully remove the acetone and allow the cells to air-dry completely. This drying step is integral to the fixation and permeabilization process.
- Rehydration and Washing: Gently rehydrate and wash the cells several times with PBS or a similar buffer to remove residual acetone.
- Proceed to Staining: The cells are now fixed and permeabilized and can be used for immunostaining. Note: No separate permeabilization step is required.
The Scientist's Toolkit: Essential Reagent Solutions
The following table catalogs key reagents used in the featured permeabilization methods, providing researchers with a concise overview of their primary functions.
| Reagent / Solution | Function / Purpose |
|---|---|
| Paraformaldehyde (PFA) | A cross-linking fixative that preserves cellular morphology by creating covalent bonds between proteins. It is the standard initial fixative when using proteinase K [3] [26]. |
| Proteinase K | A broad-spectrum serine protease that permeabilizes cells by selectively digesting proteins in the cellular membrane and interior. It requires optimization of concentration and time [3] [22]. |
| Acetone | An organic solvent that acts as a precipitating fixative and permeabilizing agent simultaneously. It works by dehydrating cells and precipitating biomolecules [26]. |
| Phosphate-Buffered Saline (PBS) | An isotonic buffer used for washing cells and preparing reagent solutions to maintain a stable pH and osmotic balance [3] [26]. |
| Tris-HCl Buffer | A common buffer used in enzymatic protocols, such as with proteinase K, to maintain optimal pH for enzyme activity [3]. |
| Tween-20 | A non-ionic detergent used in alternative permeabilization protocols and in wash buffers (PBTween) to reduce non-specific binding [3] [22]. |
The choice between proteinase K and acetone permeabilization is not a matter of identifying a universally superior method, but rather of aligning the technique with the specific experimental goals and constraints.
Choose Proteinase K when your target is an intracellular nucleic acid (e.g., for RNA FISH) [3], when you need to preserve lipid architecture, or when your protocol requires fine-tuning the level of membrane access. Researchers should be cautious of its potential to digest protein epitopes of interest and its documented negative impact on transcriptome integrity in single-cell multi-omics workflows [27].
Choose Acetone for its simplicity and speed when a combined fixation/permeabilization step is desirable, or when staining for robust intracellular antigens that are resistant to solvent-induced denaturation. It is a powerful but harsh method that should be avoided when studying membrane integrity, labile protein epitopes, or overexpressed fluorescent proteins like GFP [26].
Ultimately, the inherent trade-off between permeabilization strength and structural preservation demands a carefully considered experimental strategy. For novel targets or systems, empirical testing and optimization of both methods are highly recommended to achieve the delicate balance required for high-quality, interpretable data.
Practical Protocols: When and How to Apply Each Method Effectively
Proteinase K (EC 3.4.21.14) is a broad-spectrum serine protease derived from the fungus Engyodontium album [28]. It is a cornerstone reagent in molecular biology, primarily valued for its ability to digest unwanted proteins and inactivate nucleases during nucleic acid purification, thereby protecting DNA and RNA from degradation [29] [30]. Its remarkable stability in the presence of denaturants like SDS and urea, and at elevated temperatures, makes it exceptionally versatile for a range of applications from genomics to proteomics [31] [28].
This guide objectively compares the performance of proteinase K-based methods against acetone permeabilization, a technique often employed in immunohistochemistry (IHC) and in situ hybridization (FISH) for its epitope-preserving qualities [4] [32]. The broader thesis explores how these methods balance conflicting needs: efficient digestion or permeabilization versus the preservation of macromolecular integrity for downstream analysis.
Standardized Proteinase K Protocol
A standardized protocol for proteinase K is essential for achieving consistent, reproducible results across different laboratories and applications. The following section details the preparation and use of proteinase K, summarizing key parameters for easy reference.
Stock Solution Preparation and Storage
To prepare a stock solution, dissolve proteinase K powder in a compatible buffer such as Tris-HCl, TE buffer, or PBS to a final concentration of 10-100 mg/mL [31] [30]. The solution should be mixed well by vortexing or pipetting and can be aliquoted for long-term storage at -20 °C or below to maintain stability and activity [31] [30]. Under these conditions, a stock solution is stable for up to one year, while lyophilized powder can be stored desiccated at -20 °C for up to two years [30].
Key Parameters for Application
The table below summarizes the critical operational parameters for proteinase K to ensure optimal activity in various experimental procedures.
Table 1: Standardized Operational Parameters for Proteinase K
| Parameter | Optimal Range | Details & Considerations |
|---|---|---|
| Working Concentration | 50-100 µg/mL [4] | Specific protocols may require higher concentrations; e.g., DNA extraction from whole blood may use ~100 µg/mL [33]. |
| Incubation Temperature | 50-65 °C [29] [30] | Higher temperatures promote protein unfolding, enhancing activity. Active from ~20-37 °C, but less efficient [31] [30]. |
| Incubation Time | 30 minutes to several hours/overnight [31] | Duration depends on sample type and quantity. For FFPE tissues, extended digestion (24-72 hours) improves yield [13]. |
| Optimal pH | 7.5 - 9.0 [31] [30] | The enzyme is active over a broad pH range (4.0-12.0), but neutral to slightly basic pH yields highest activity [31]. |
| Activators & Inhibitors | Activators: SDS, urea [30].Inhibitors: PMSF, AEBSF, high SDS concentrations [31] [30]. | EDTA does not directly inhibit activity but chelates calcium, reducing enzyme stability [30]. |
Inactivation and Compatibility
After digestion, proteinase K can be inactivated by heating to 95 °C for 10 minutes, though this may not result in complete inactivation [29] [30]. Protease inhibitors like PMSF (phenylmethylsulfonyl fluoride) or AEBSF provide more permanent inactivation [31] [30]. Proteinase K is compatible with various buffers and salts, but its activity can be reduced by high concentrations of specific detergents like Triton X-100 or Tween 20 [31].
Proteinase K vs. Acetone Permeabilization: A Direct Comparison
The choice between proteinase K digestion and acetone permeabilization is dictated by the experimental goal. The table below provides a direct, data-driven comparison of the two methods.
Table 2: Performance Comparison of Proteinase K and Acetone Permeabilization
| Characteristic | Proteinase K Method | Acetone Permeabilization |
|---|---|---|
| Primary Mechanism | Enzymatic digestion of proteins and peptides [29]. | Solvent-based lipid dissolution and protein precipitation [32]. |
| Key Applications | Nucleic acid extraction; prion research; nuclease inactivation; protein digestion in proteomics [28] [30]. | Immunohistochemistry (IHC); immunofluorescence (IF); preservation of protein epitopes [4] [32]. |
| Impact on Proteins | Digests and removes proteins, including nucleases and antigens [4] [30]. | Fixes and permeabilizes without digesting, preserving antigenicity for antibody binding [32]. |
| Impact on Nucleic Acids | Protects and liberates intact DNA/RNA by degrading nucleases [29] [30]. | No specific protective effect; may not adequately expose nucleic acids for probe binding in FISH [4]. |
| Experimental Data (ISH/FISH) | Strong, specific signal for both germline (gurken) and follicle cell (broad) transcripts after 15-45 minute color reaction [4]. | Weak, variable signal; broad transcript detection was extremely weak even after 5.5-hour color reaction [4]. |
| Experimental Data (DNA Yield) | Doubling proteinase K volume in FFPE DNA extraction increased yield by 96% [13]. | Data not available for acetone in this context. |
| Tissue Morphology | Can be compromised if over-digested, requiring careful optimization of concentration and time [31] [4]. | Generally well-preserved due to its fixing properties [32]. |
Detailed Experimental Protocols
Proteinase K for DNA Extraction from Whole Blood
The SDS-proteinase K (SDS-PK) method is a common, non-hazardous alternative to phenol-chloroform for extracting genomic DNA [33]. The following optimized protocol highlights steps crucial for obtaining high-quality DNA.
Figure 1: Workflow for DNA extraction from whole blood using the SDS-proteinase K method.
Technical Points from Optimized Research:
- RBC Lysis: Performing the RBC lysis and wash step three times, instead of two, was critical for achieving a high A260/A280 ratio and reducing protein contamination [33].
- WBC Lysis: The use of 100 µg/mL proteinase K and incubation at 50°C for two hours was identified as optimal for cell lysis and protein digestion. If the solution is not clear after this period, incubation should be extended [33].
- DNA Washing: The 70% ethanol wash is crucial for removing salts like ammonium acetate, which can significantly impact the A260/A230 ratio and indicate contamination [33].
Acetone Permeabilization for Immunofluorescence
Acetone is used as a fixative and permeabilization agent, particularly for IHC and IF protocols where preserving protein antigenicity is paramount.
Standard Protocol:
- Fixation and Permeabilization: Immerse tissue samples or cells in pre-chilled -20°C acetone for 5-10 minutes [32].
- Washing: Remove the acetone and allow the sample to air dry completely.
- Rehydration: Wash the sample with phosphate-buffered saline (PBS) or a similar buffer to rehydrate before proceeding with antibody staining.
Technical Considerations:
- Acetone fixation and permeabilization are simultaneous, making the protocol quick and simple.
- It is a harsher treatment that can damage delicate cellular structures but is effective for many cytoskeletal, viral, and enzyme antigens [32].
- As demonstrated in FISH experiments, acetone permeabilization alone is often insufficient for nucleic acid probe penetration, leading to weak signals compared to proteinase K [4].
Hybrid Protocol for IF/FISH
For experiments requiring simultaneous detection of proteins and RNA (IF/FISH), a hybrid approach that avoids proteinase K is necessary to preserve protein epitopes.
Figure 2: A hybrid IF/FISH workflow that performs immunofluorescence before FISH, using alternative permeabilization to preserve protein epitopes.
Key Methodological Insight:
- Traditional methods perform ISH before IF, but reversing the order—conducting the entire protein IF staining before FISH—markedly improves protein detection [4]. This is followed by a post-fixation step to cross-link the antibodies before proceeding with FISH.
- Proteinase K is omitted from the FISH portion of the protocol as it is detrimental to the already-bound antibodies. Instead, permeabilization is achieved using a combination of xylenes and detergents (RIPA), which allows adequate probe penetration while maintaining a strong protein signal [4].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents used in the proteinase K and permeabilization protocols discussed, along with their critical functions.
Table 3: Essential Reagents for Permeabilization and Digestion Protocols
| Reagent | Function/Description | Example Application |
|---|---|---|
| Proteinase K | Broad-spectrum serine protease that digests proteins and inactivates nucleases. | DNA/RNA extraction; general protein digestion [31] [30]. |
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that denatures proteins and acts as an activator for proteinase K. | Cell lysis buffer in DNA extraction protocols [33] [30]. |
| Acetone | Organic solvent that fixes and permeabilizes cells by dissolving lipids and precipitating proteins. | Permeabilization for IHC/IF; preserving protein antigens [32]. |
| Xylenes | Organic solvent efficient at dissolving and removing paraffin wax and permeabilizing tissue. | Deparaffinization of FFPE samples; alternative permeabilization for IF/FISH [4] [13]. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelating agent that binds metal ions, inhibiting metal-dependent nucleases. | Component of lysis and TE buffers for nucleic acid stability [31] [33]. |
| PMSF (Phenylmethylsulfonyl fluoride) | Serine protease inhibitor that permanently inactivates proteinase K. | Halting proteinase K digestion after completion [31] [30]. |
| Tris-HCl Buffer | Common buffering agent used to maintain a stable pH (typically ~8.0) for proteinase K activity. | Solvent for proteinase K stock solution; component of lysis buffers [31]. |
This guide has detailed the standardized use of proteinase K and directly compared its performance to acetone permeabilization. The experimental data and protocols underscore a clear dichotomy: proteinase K is unparalleled for efficient digestion and nucleic acid purification, while acetone and other solvents are superior for epitope preservation in protein detection.
The selection between these methods is not a matter of superiority but of strategic alignment with experimental objectives. For workflows requiring the simultaneous detection of proteins and nucleic acids, hybrid protocols that leverage the strengths of both chemical permeabilization and enzymatic digestion offer a powerful solution. Ultimately, a deep understanding of these mechanisms enables researchers to optimize protocols for the highest data quality and reproducibility.
In the study of cellular and subcellular structures, the choice of fixation and permeabilization method is a critical determinant of experimental success. These processes preserve cellular morphology and allow detection reagents access to intracellular targets, but they often involve a trade-off between optimal structural preservation and adequate antibody accessibility. This guide focuses on a central comparison in this field: the use of proteinase K, an enzymatic permeabilization agent, versus acetone, an organic solvent. While proteinase K digests proteins to unmask antigens and facilitate entry, acetone operates by precipitating cellular proteins and dissolving lipids to permeabilize the membrane. The selection between these methods can significantly impact the outcome of experiments in immunohistochemistry (IHC), flow cytometry, and other detection protocols. This article provides a objective, data-driven comparison of these techniques, equipping researchers with the information needed to select and optimize the right protocol for their specific application.
Methodological Comparison: Proteinase K vs. Acetone
The following table summarizes the core characteristics, applications, and performance data of proteinase K and acetone permeabilization methods based on published experimental findings.
Table 1: Direct Comparison of Proteinase K and Acetone Permeabilization Methods
| Feature | Proteinase K (Enzymatic) | Acetone (Organic Solvent) |
|---|---|---|
| Mechanism of Action | Proteolytic digestion of proteins; reverses cross-links and unmasks antigens [18] [14]. | Precipitates proteins and dissolves lipids, simultaneously fixing and permeabilizing [18] [34]. |
| Primary Application Context | Antigen retrieval in cross-linked samples (e.g., PFA-fixed); intracellular RNA detection in fixed single cells [18] [14]. | Permeabilization following (or combined with) organic solvent fixation; often used for large protein antigens like immunoglobulins [18] [34]. |
| Typical Concentration | 0.01 - 0.1 µg/mL for flow cytometry [3]; 40 U/mL for cross-link reversal in FD-seq [14]; 20 µg/mL for IHC antigen retrieval [35]. | 100% (often ice-cold) for IHC [34]; used in 1:1 mixture with methanol for fixation [36] [35]. |
| Typical Incubation Duration | 5 - 15 minutes at 37°C for flow cytometry [3]; 10-20 minutes at 37°C for IHC [35]; 1-hour incubation for single-cell protocol [14]. | 5 - 10 minutes for fixed cells [35]; 10 minutes for bacterial fixation [36]. |
| Key Advantages | - Effectively reverses PFA cross-linking, enabling RNA-seq in fixed cells [14].- Can be finely tuned by varying concentration and time. | - Rapid and simple protocol [35].- Acts as both a fixative and permeabilizer, streamlining workflow [18].- Excellent for certain large protein antigens [34]. |
| Documented Limitations & Risks | - Excessive digestion can damage tissue morphology and cell surface structures [18].- Requires careful optimization of concentration and time to avoid degradation [3] [14]. | - Can extract cellular lipids and cause significant cell shrinkage, compromising ultrastructural preservation [36] [34].- Less effective for preserving fine surface filaments like flagella and pili [36]. |
Experimental Protocols from Cited Studies
Proteinase K Permeabilization for Flow Cytometry (18S rRNA Detection)
This protocol is adapted from a study that optimized permeabilization methods for the flow cytometric detection of intracellular 18S rRNA in HeLa cells [3].
- Fixation: Fix HeLa cells (2x10^6 cells/mL) in 2% cold, freshly prepared paraformaldehyde (PFA) in PBS. Incubate at room temperature for 15 minutes with slow shaking.
- Washing: Wash the cells with 1X PBS to remove excess fixative, then centrifuge at 500 g for 5 minutes.
- Permeabilization: Resuspend the cell pellet in 200 µL of Proteinase K solution. The tested concentrations were 0.01, 0.05, and 0.1 µg/mL in a buffer containing 20 mM Tris-HCl and 2 mM CaCl2. Incubate for 5, 10, or 15 minutes at 37°C.
- Washing: Wash the cells with 1X PBS to remove the proteinase K.
- Hybridization: The cells are now ready for subsequent staining or in situ hybridization protocols. In the cited study, cells were subjected to in situ hybridization with FITC-labeled probes to detect 18S ribosomal RNAs [3].
Acetone Fixation and Permeabilization for Microscopy
This protocol outlines a standard method for using acetone as a combined fixative and permeabilizing agent, commonly used in preparing samples for immunofluorescence [35].
- Sample Preparation: Attach adherent cells to microscope slides. For tissue samples, cut into 2 mm blocks.
- Fixation/Permeabilization: Immerse the samples in -20°C acetone. Incubate for 5-10 minutes for isolated cells. For larger tissue samples, extend the fixation time to an hour or more.
- Rinsing: Rinse the samples a few times with PBS to prepare them for staining.
It is noted that acetone fixation will also permeabilize the cells, so no separate permeabilization step is required [18].
Workflow and Decision Pathways
The following diagrams illustrate the standard experimental workflows for the two methods and a logical framework for selecting the appropriate protocol.
Proteinase K Experimental Workflow
Acetone Experimental Workflow
Permeabilization Method Selection Guide
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their functions in fixation and permeabilization protocols, as discussed in the cited literature.
Table 2: Key Reagents for Fixation and Permeabilization Protocols
| Reagent | Function | Example Use Case |
|---|---|---|
| Paraformaldehyde (PFA) | Cross-linking fixative; preserves morphology by creating covalent bonds between proteins [36] [35]. | Standard primary fixative for electron microscopy and IHC; often requires subsequent permeabilization [36] [34]. |
| Proteinase K | Serine protease; digests proteins to unmask antigens or reverse cross-links for nucleic acid access [3] [14]. | Antigen retrieval in IHC [18]; reversing PFA cross-links in single-cell RNA-seq of fixed cells (FD-seq) [14]. |
| Acetone | Organic solvent; precipitates proteins and dissolves lipids, acting as both fixative and permeabilizer [18] [34]. | Fixing and permeabilizing cells for IHC detection of large protein antigens like immunoglobulins [34]. |
| Glutaraldehyde | Strong cross-linking fixative; provides superior structural preservation but can mask antigens more heavily [36] [34]. | Primary fixative for electron microscopy when ultrastructural detail is paramount [36]. |
| Tween-20 | Mild, non-ionic detergent; permeabilizes lipid membranes without dissolving them [3] [18]. | Effective permeabilization for intracellular 18S rRNA detection in flow cytometry, yielding high fluorescence intensity [3]. |
| Triton X-100 | Harsh, non-ionic detergent; efficiently solubilizes membranes but can disrupt protein-protein interactions [18]. | Permeabilization of PFA-fixed cells prior to droplet-based single-cell RNA sequencing (FD-seq) [14]. |
The accurate determination of protein subcellular localization is a fundamental aspect of cell biology research, enabling scientists to understand protein function, interaction networks, and implications in disease mechanisms. The selection of appropriate permeabilization methods is critical for successful immunolocalization, as it controls antibody access to intracellular epitopes. This guide objectively compares proteinase K and acetone permeabilization methods within the broader context of optimizing immunostaining protocols for different protein targets, providing researchers with experimental data to inform their methodological choices.
The Permeabilization Principle in Protein Localization
Permeabilization is an essential technical step required for antibodies to access the inside of cells to detect target antigens, including intracellular proteins and cytoplasmic epitopes of transmembrane proteins [18]. This process involves the use of solvents or detergents to create temporary openings in cellular membranes without complete structural disintegration, thereby enabling macromolecular probes to reach their intracellular targets.
The fundamental challenge in permeabilization lies in balancing sufficient membrane disruption with preservation of cellular architecture and antigen integrity. The optimal approach varies significantly depending on whether the target is an intracellular protein residing within organelles or the cytosol, or a transmembrane protein embedded within lipid bilayers with specific topological orientations [37]. For transmembrane proteins, additional considerations include preserving the antigenic sites on cytoplasmic domains while maintaining membrane integrity for proper topological context.
Methodological Comparison: Proteinase K vs. Acetone Permeabilization
Proteinase K Permeabilization
Mechanism and Protocol: Proteinase K permeabilization employs enzymatic digestion to create access points in cellular structures. The standard protocol involves digesting samples in 10μg/ml proteinase K in phosphate-buffered saline with Tween 20 (PBTween) for approximately 5 minutes, followed by fixation in 4% paraformaldehyde for 20 minutes at room temperature and washing before hybridization [22]. This method is particularly valuable when epitopes are obscured by protein cross-linking or when working with dense tissues.
Experimental Applications: In comparative studies on zebrafish embryos, proteinase K treatment was evaluated against acetone permeabilization for in situ hybridization applications [22]. The enzymatic action helps expose hidden epitopes but requires precise timing control to prevent excessive tissue damage or antigen degradation.
Acetone Permeabilization
Mechanism and Protocol: Acetone permeabilization functions through solvent action, extracting lipids from cellular membranes to create pores. The standard approach involves treating samples with 80% acetone/20% water at room temperature for 20 minutes, followed by washing in PBTween before hybridization [22]. Acetone also serves a dual purpose as it fixes samples simultaneously through dehydration and protein precipitation.
Experimental Applications: Acetone is generally recommended for cytoskeletal, viral, and some enzyme antigens [18]. Its effectiveness was directly compared to proteinase K in zebrafish embryo studies, with researchers evaluating permeabilization efficiency through subsequent staining quality and signal-to-noise ratios [22].
Comparative Performance Data
Table 1: Direct Comparison of Proteinase K and Acetone Permeabilization Methods
| Parameter | Proteinase K | Acetone |
|---|---|---|
| Mechanism | Enzymatic proteolysis | Solvent-based lipid extraction |
| Concentration | 10μg/ml [22] | 80% solution [22] |
| Incubation Time | 5 minutes [22] | 20 minutes [22] |
| Typical Temperature | Room temperature [22] | Room temperature [22] |
| Tissue Morphology Preservation | Moderate (risk of over-digestion) | High [18] |
| Antigen Preservation | Variable (risk of epitope damage) | High for many targets [18] |
| Best Applications | Cross-linked epitopes, dense tissues | Cytoskeletal proteins, viral antigens [18] |
| Simultaneous Fixation | No (separate fixation required) | Yes [18] |
| Protocol Complexity | Moderate (requires precise timing) | Simple |
Diagram 1: Permeabilization Method Selection Framework for Different Protein Targets
Experimental Workflows and Technical Considerations
Comprehensive Immunostaining Workflow
The overall immunostaining process consists of multiple interdependent steps where permeabilization represents one critical juncture. The complete pathway includes sample preparation, fixation, antigen retrieval, permeabilization, blocking, antibody incubation, washing, and detection [38]. Each step presents potential roadblocks including epitope masking, background signal, protein relocation, and structural damage that can compromise experimental outcomes.
Diagram 2: Comprehensive Immunostaining Workflow with Permeabilization Decision Point
Method-Specific Optimization Strategies
Proteinase K Optimization: For proteinase K-based methods, concentration and timing precision is critical. Researchers should conduct preliminary titration experiments ranging from 5-20μg/ml with time courses from 2-10 minutes to identify optimal conditions for specific sample types [22] [18]. Including negative controls without enzymatic treatment helps assess specific versus non-specific signal. The enzymatic reaction should be terminated promptly by fixation or specific inhibitors to prevent excessive digestion.
Acetone Permeabilization Optimization: Acetone concentration (typically 80-100%) and exposure time should be calibrated based on sample thickness and membrane density [22] [18]. While acetone generally preserves antigenicity well for many targets, researchers should verify that the solvent action doesn't extract or denature the target antigen. Subsequent rehydration steps may be necessary for some applications.
Research Reagent Solutions
Table 2: Essential Research Reagents for Permeabilization Studies
| Reagent | Function | Application Notes |
|---|---|---|
| Proteinase K | Enzymatic permeabilization | Working concentration: 10μg/ml; incubation: 5 minutes [22] |
| Acetone | Solvent-based permeabilization | 80% concentration; 20-minute incubation; provides simultaneous fixation [22] |
| Paraformaldehyde | Sample fixation | Typically 4% solution; stabilizes cellular structures before permeabilization [22] |
| Tween 20 | Surfactant | Reduces background in washing buffers (PBTween) [22] |
| Proteinase K Inhibitors | Reaction termination | Critical for controlling digestion extent after incubation |
| Phosphate-Buffered Saline (PBS) | Buffer system | Maintains physiological pH and osmolarity during processing |
| Normal Serum | Blocking agent | Reduces non-specific antibody binding; used before primary antibody incubation |
The comparative analysis of proteinase K versus acetone permeabilization reveals a clear distinction in their mechanisms, applications, and optimal use cases. Proteinase K offers targeted access to obscured epitopes through enzymatic action but requires precise control to prevent structural damage. Acetone provides gentler, more generalized permeabilization through solvent action while simultaneously fixing cellular structures.
For transmembrane proteins, where preserving membrane context and domain orientation is crucial, acetone permeabilization generally offers superior results by maintaining lipid bilayer integrity while allowing antibody access to cytoplasmic domains [18] [37]. For intracellular targets, particularly in densely cross-linked samples or when epitopes are deeply buried, proteinase K may provide necessary access despite its more aggressive mechanism.
The methodological decision should be guided by target protein characteristics, sample type, and specific research questions. Researchers are encouraged to perform preliminary side-by-side comparisons using both methods when investigating novel targets to establish the optimal protocol for their specific application. As spatial proteomics advances, with techniques now capable of mapping over 7,600 proteins across 19 subcellular structures [39], the importance of optimized permeabilization only grows more critical for generating high-quality localization data.
Selecting and optimizing for different biological model systems is a fundamental step in the design of robust and reproducible scientific experiments. The choice between using cell lines, tissue sections, or whole mounts significantly influences downstream protocols, particularly for techniques that require the internalization of probes, such as in situ hybridization or immunofluorescence. Among the most critical protocol parameters to optimize are the methods for permeabilization, which render biological membranes accessible to reagents. Within the context of comparing proteinase K and acetone permeabilization methods, this guide objectively evaluates their performance across different model systems, supported by experimental data.
Permeabilization techniques can be broadly categorized as chemical/detergent-based or enzymatic. The choice of method directly impacts the preservation of cellular morphology, the accessibility of intracellular targets, and the final experimental readout.
Table 1: Core Characteristics of Proteinase K and Acetone Permeabilization
| Feature | Proteinase K (Enzymatic) | Acetone (Chemical) |
|---|---|---|
| Mechanism | Proteolytic digestion of proteins, reversing cross-links and unmasking epitopes. [18] | Solvent action; dehydration and precipitation of cellular components. [18] [40] |
| Primary Application | Antigen retrieval (PIER) for fixed samples; access to nucleic acids. [3] [18] | Simultaneous fixation and permeabilization. [18] |
| Typical Conditions | 0.01-0.1 µg/ml, 5-15 min at 37°C. [3] [18] | 100%, 10 minutes at -20°C. [18] |
| Impact on Morphology | Risk of tissue damage with over-digestion; requires careful optimization. [18] | Can distort morphology and scatter profiles; may reduce surface antigen signal. [41] |
| Compatibility | Effective for dense tissues and whole mounts; used in FD-seq for fixed single-cell RNA-seq. [14] | Best for cell lines and cryosections; suitable for cytoskeletal and some viral antigens. [18] |
Performance Across Model Systems
Experimental data demonstrates that the optimal permeabilization method is highly dependent on the model system and the target of interest.
Cell Lines
For single-cell suspensions like HeLa cells, mild detergent-based permeabilization often outperforms both enzymatic and solvent methods for intracellular RNA detection. A systematic comparison of six permeabilization methods for flow cytometric in situ hybridization of 18S rRNA found that treatment with 0.2% Tween-20 for 30 minutes yielded the highest fluorescence intensity, significantly outperforming Proteinase K, saponin, Triton X-100, NP40, and streptolysin O. [3] This suggests that for many cytoplasmic targets in cultured cells, controlled detergent use offers a superior balance of permeability and morphology preservation.
In contrast, acetone (and methanol) fixation and permeabilization can be detrimental to certain surface markers. Studies on T regulatory cells showed that alcohol-based methods dramatically decreased the signal for pan-leukocyte marker CD45 and altered light scatter properties, whereas specialized commercial buffer sets preserved these markers. [41] Furthermore, for single-cell RNA sequencing, a protocol using Proteinase K (40 U/mL) during the cross-link reversal step was crucial for achieving high RNA yield and quality from paraformaldehyde-fixed cells, enabling the method called FD-seq. [14]
Tissue Sections
In formalin-fixed paraffin-embedded (FFPE) tissue sections, antigen masking due to protein cross-linking is a major challenge. Here, Proteinase-Induced Epitope Retrieval (PIER) is a widely adopted solution. Proteinase K digestion is particularly useful for epitopes that are difficult to retrieve, as it enzymatically digests proteins and reverses cross-links that obscure antibody-binding sites. [18]
The competing method, Heat-Induced Epitope Retrieval (HIER), is generally gentler, but the optimal technique must be determined experimentally for each antigen. [18] While acetone is less common for standard FFPE immunohistochemistry, it is a recognized permeabilization agent, though its solvent action carries a higher risk of morphological damage compared to controlled enzymatic treatment. [18]
Whole Mounts
Whole-mount samples present a unique challenge due to their density and the presence of barriers like the plant cuticle. Successful permeabilization requires a balance between antibody penetration and structural integrity. A universal plant immunolocalization protocol highlights the use of a non-ionic detergent (IGEPAL CA-630) combined with DMSO for effective permeabilization of thick specimens like ovules and roots. [42] For particularly dense plant organs, a pre-treatment with hot methanol was implemented to permeabilize the waxy cuticle effectively. [42] In these complex systems, Proteinase K is more typically reserved for specific antigen retrieval in fixed samples rather than general permeabilization, while acetone is not recommended due to its harsh effects on the intricate tissue morphology.
Experimental Protocols for Key Studies
This protocol is designed to find the optimal permeabilization method for detecting intracellular 18S rRNA via flow cytometry.
- Fixation: Harvest and wash HeLa cells. Fix cells in 2% paraformaldehyde in PBS for 15 minutes at room temperature.
- Permeabilization (Testing): Wash cells with PBS to remove fixative. Divide cells into aliquots and treat with different permeabilizing agents:
- Tween-20: 0.2% for 30 minutes at 25°C.
- Proteinase K: 0.01-0.1 µg/ml in Tris-HCl/CaCl2 for 5-15 minutes at 37°C.
- Other agents (Saponin, Triton X-100, etc.) at various concentrations and times.
- In Situ Hybridization: Wash cells to remove the permeabilization agent. Resuspend cell pellet in 50 µl of hybridization buffer containing a FITC-labeled antisense probe targeting 18S rRNA.
- Hybridization: Incubate at 40°C with gentle shaking overnight.
- Washing: Pellet cells and wash successively with 2×SSC and 0.1×SSC for 30 minutes each to remove nonspecific binding.
- Analysis: Resuspend cells in PBS and analyze on a flow cytometer (e.g., FACSCalibur), acquiring at least 10,000 events per sample.
This improved protocol ensures effective permeabilization for antibody penetration in dense whole-mount plant specimens.
- Fixation: Place plant explants in 2% formaldehyde in microtubule-stabilizing buffer (MTSB) supplemented with 0.1% Triton X-100. Apply vacuum infiltration for 20-30 minutes to ensure fixative penetration, then incubate for an additional 50-60 minutes.
- Permeabilization: Incubate the fixed samples in a permeabilization buffer containing 3% non-ionic detergent IGEPAL CA-630 and 10% DMSO in MTSB for 60 minutes. For tissues with a tough cuticle (e.g., siliques), an additional step using hot methanol may be required.
- Cell Wall Digestion (Optional): For further penetration, digest with a cocktail of 0.2% Driselase and 0.15% Macerozyme in MES buffer (pH 5.0) for 30-60 minutes.
- Blocking and Staining: Incubate samples in a blocking solution (e.g., 2% BSA) for 1-2 hours, then proceed with primary and secondary antibody staining.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Permeabilization and Related Protocols
| Reagent / Solution | Function | Example Use Case |
|---|---|---|
| Proteinase K | Proteolytic enzyme for antigen retrieval and reversing PFA cross-links. [3] [14] [18] | PIER for FFPE tissues; RNA extraction from fixed cells in FD-seq. [14] [18] |
| Acetone | Solvent that simultaneously fixes and permeabilizes by dehydrating samples. [18] | Fixation and permeabilization of cell lines or cryosections for cytoskeletal antigens. [18] |
| Tween-20 | Non-ionic detergent for creating pores in lipid membranes. [3] | Optimal permeabilization for intracellular 18S rRNA detection in HeLa cells. [3] |
| Triton X-100 | Non-ionic detergent for stronger permeabilization. [14] [18] [42] | Standard permeabilization agent in many IF protocols; component of fixation buffers. [42] [40] |
| IGEPAL CA-630 | Non-ionic detergent similar to NP-40. [42] | Key component of permeabilization buffer for whole-mount plant samples. [42] |
| Saponin | Mild detergent that perforates membranes by complexing with cholesterol. [41] [18] | Permeabilization for sensitive intracellular targets with less membrane disruption. [18] |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves cellular structure by creating protein networks. [3] [14] [42] | Standard fixation for most cell lines and tissues prior to permeabilization. [3] [14] |
| Methanol | Denaturing alcohol fixative that precipitates cellular components. [14] [40] | Alternative fixation/permeabilization method; can unmask specific epitopes. [40] |
The optimization of permeabilization methods is a critical, model-dependent process. The experimental data clearly shows that there is no universal "best" method. The choice between Proteinase K, acetone, and various detergents must be guided by the biological system (cell line, tissue, or whole mount), the nature of the target (RNA, protein, or specific epitope), and the required balance between penetration and preservation. Researchers are encouraged to empirically test multiple methods, using the protocols and data presented here as a starting point, to establish the most reliable and reproducible conditions for their specific experimental questions.
In biomedical research, the choice of fixation method is a critical determinant of experimental success, profoundly impacting both the preservation of cellular architecture and the accessibility of target antigens. Fixation techniques primarily fall into two categories: cross-linking methods using agents like paraformaldehyde (PFA) and organic solvent methods employing substances such as methanol or acetone. Within the specific context of comparing proteinase K and acetone permeabilization methods, understanding how these approaches interact with various fixation protocols becomes paramount. This guide provides a comprehensive, evidence-based comparison of common fixatives, detailing their mechanisms, applications, and compatibility with downstream processing steps to empower researchers in making informed methodological decisions.
Fundamental Fixation Mechanisms and Characteristics
Cross-Linking Fixatives (PFA/Formalin)
Paraformaldehyde (PFA) and its aqueous counterpart, formalin, function by creating covalent chemical bonds (cross-links) between proteins in the tissue and their immediate cellular environment [35]. This molecular scaffolding anchors proteins within the cellular architecture, thereby imparting significant structural rigidity to biological samples [35]. The primary advantage of this method is its superior preservation of tissue integrity and subcellular ultrastructure compared to organic solvent methods [35]. However, a significant drawback is that this extensive cross-linking can physically block antibody recognition sites, a phenomenon known as antigen masking [35]. This often necessitates subsequent antigen retrieval steps, such as heat-induced epitope retrieval or enzymatic treatment, to reverse the masking and enable successful antibody binding [35].
Organic Solvent Fixatives (Methanol, Acetone)
Organic solvent fixatives like methanol and acetone operate through a fundamentally different mechanism. They preserve samples by removing lipids, dehydrating the tissue, and denaturing and precipitating the proteins [35]. This process typically results in less physical distortion of the sample but does not provide the same level of structural reinforcement as cross-linking fixatives. A key advantage is that they generally cause less antigen masking, making them preferable for certain sensitive epitopes that are disrupted by PFA cross-linking [43]. For instance, a comparative study found that cold acetone was superior to formalin for preserving the antigenic activity of carcinoembryonic antigen (CEA) and keratin in permanently embedded tissues [43].
Table 1: Core Characteristics of Primary Fixative Types
| Fixative Type | Example Agents | Primary Mechanism | Key Advantages | Key Disadvantages |
|---|---|---|---|---|
| Cross-linking | PFA, Formaldehyde | Creates covalent bonds between proteins [35] | Superior tissue and subcellular structure preservation [35] | Can cause antigen masking, often requires retrieval steps [35] |
| Organic Solvent | Methanol, Acetone | Removes lipids, dehydrates tissue, precipitates proteins [35] | Less antigen masking, good for sensitive epitopes, often requires no retrieval [43] | Inferior structural preservation compared to cross-linking [35] |
Comparative Experimental Data and Performance
Direct Comparison of PFA vs. Acetone Fixation
Empirical evidence directly comparing these fixatives reveals context-dependent performance. The study by Kaku et al. provides a clear example, evaluating the detection of CEA and keratin using a peroxidase-antiperoxidase (PAP) immunohistochemical procedure [43]. The results demonstrated that cold acetone fixation was superior to buffered formalin for preserving the antigenic activity of both target molecules across a range of benign and malignant tissues [43]. While pronase treatment (a proteolytic enzyme) enhanced staining in formalin-fixed tissues, the acetone-based protocol still yielded superior results for many tissues, leading to the recommendation of the cold acetone method for optimal retention of antigenic activity in permanently embedded tissues [43].
Impact of Methanol on Protein Structure and Expression
Methanol's role extends beyond a simple fixative; it is also a critical inducer of recombinant protein expression in yeast systems like Pichia pastoris and Pichia methanolica, which utilize methanol-inducible promoters [44] [45]. The concentration of methanol is a critical variable. In Pichia methanolica, maximum recombinant gene expression was observed at a 0.7% (v/v) methanol concentration, while higher concentrations (1.0%) favored maximum cellular growth [44]. Furthermore, under oxygen-limited cultures of Pichia pastoris, higher methanol concentrations (up to 3%) were required to achieve high product concentrations of a single-chain antibody fragment (scFv), compensating for the lack of oxygen [45].
At a structural level, methanol induces conformational changes in proteins. Studies on β-lactoglobulin, cytochrome c, and ubiquitin show that methanol can induce the formation of partially unfolded intermediates with α-helix structure [46]. At around 35% concentration, intermediate states display high flexibility and can consist of a mixture of rapidly interconverting conformers [46].
Table 2: Quantitative Experimental Data from Fixative and Methanol Studies
| Study Focus | System/Model | Key Parameter Tested | Optimal Condition/Finding | Outcome/Performance |
|---|---|---|---|---|
| Antigen Preservation [43] | Human tissues (CEA, Keratin) | Fixative: Acetone vs. Formalin | Cold Acetone | Superior antigen retention vs. Formalin (with/without pronase) |
| Recombinant Protein Yield [44] | Pichia methanolica | Methanol Concentration | 0.7% (v/v) | Maximum recombinant protein expression (450 mg/L) |
| Recombinant Protein Yield under O₂ Limitation [45] | Pichia pastoris (scFv) | Methanol Concentration | 3.0% (v/v) | High product concentration (350 mg/L); prevents fragment accumulation |
| Protein Conformation [46] | β-lactoglobulin, Cytochrome c, Ubiquitin | Methanol Concentration | ~35% | Induces flexible, partially unfolded intermediates with α-helix structure |
Detailed Experimental Protocols
Standard PFA Fixation and Immunostaining Protocol
The following protocol is adapted from standardized immunocytochemistry procedures [47]:
- Fixation: Wash cells briefly with PBS. Fix cells with a solution of 4% PFA in PBS (often supplemented with 4% sucrose) for 15-20 minutes at room temperature (RT) [47].
- Washing: Wash the fixed cells three times with PBS for 10 minutes each [47].
- Permeabilization and Blocking: Incubate for 30 minutes with a blocking buffer (e.g., 10% normal serum, 0.1% Triton X-100 in PBS). The normal serum should ideally be from the host species of the secondary antibodies [47].
- Primary Antibody Incubation: Incubate in an incubation buffer (e.g., 5% normal serum, with or without 0.1% Triton X-100 in PBS) containing the primary antibody for 2 hours at RT (dilution as per datasheet) [47].
- Secondary Antibody Incubation: Incubate with the fluorophore-conjugated secondary antibody in incubation buffer for 1 hour at RT, protected from light [47].
- Final Wash and Mounting: Wash three times with PBS for 10 minutes each. Mount coverslips using an appropriate mounting medium [47].
Organic Solvent Fixation and Permeabilization Methods
Organic solvent methods can serve as both fixatives and permeabilization agents. Key protocols include [35]:
- Acetone/Methanol Fixation: Fix isolated cells with -20°C acetone, methanol, or a 1:1 acetone/methanol mixture for 5-10 minutes. For tissue samples, fixation time should be extended to an hour or more. Rinse several times with PBS before proceeding [35].
- Acetone Permeabilization for ISH: An alternative permeabilization method for in situ hybridization (ISH) involves treating samples with 80% acetone in water for 20 minutes at room temperature, followed by washing before hybridization [22]. This is contrasted with the more common proteinase K method.
- Proteinase K Permeabilization for ISH: The standard permeabilization method involves digesting rehydrated samples (e.g., zebrafish embryos) in 10 µg/ml proteinase K for a brief period (e.g., 5 minutes), followed by a post-fixation step in 4% PFA for 20 minutes before hybridization [22].
Research Reagent Solutions Toolkit
Table 3: Essential Reagents for Fixation and Permeabilization Protocols
| Reagent / Solution | Primary Function | Example Application & Notes |
|---|---|---|
| Paraformaldehyde (PFA) | Cross-linking fixative [35] | Typically used at 3-4% in PBS for cell and tissue fixation. Superior for structural preservation [35]. |
| Methanol & Acetone | Organic solvent fixative & permeabilizer [35] | Used cold (-20°C). Good for sensitive epitopes; can be used as a mix [35]. |
| Proteinase K | Enzymatic permeabilization & antigen retrieval [22] [35] | Digests proteins to permeabilize tissue/embryos (e.g., 10 µg/ml for 5 min) [22] or unmask antigens (e.g., 20 µg/ml for 10-20 min) [35]. |
| Triton X-100 | Detergent for permeabilization [47] | Used at low concentrations (e.g., 0.1%) in blocking or incubation buffers after cross-linking fixation [47]. |
| Normal Serum | Blocking agent [47] | Reduces non-specific antibody binding. Use serum from the host species of the secondary antibodies [47]. |
| Glycine-HCl | Antibody Elution [22] | Low pH buffer (e.g., 0.1 M, pH 2.2) used in double staining to remove primary antibodies between rounds [22]. |
Fixation and Permeabilization Workflow
The following diagram illustrates the key decision points and pathways in selecting and optimizing fixation and permeabilization methods:
The selection of an appropriate fixation and permeabilization strategy is a critical, antibody- and application-specific decision in experimental pathology and cell biology. The data consistently show that no single method is universally superior. The core trade-off lies between excellent morphological preservation (achieved by PFA) and optimal antigen accessibility (often achieved by organic solvents like acetone and methanol).
PFA fixation is the gold standard when maintaining cytoarchitecture is the highest priority. However, researchers must be prepared to invest time in optimizing antigen retrieval techniques, such as heat-induced or enzymatic methods, to overcome the inevitable antigen masking caused by protein cross-linking [35]. Organic solvent fixation offers a powerful alternative, particularly for sensitive epitopes that are denatured or masked by PFA, as evidenced by its superior performance in detecting CEA and keratin [43]. Its inherent permeabilizing action can also streamline protocols.
The choice between proteinase K and acetone for permeabilization is equally nuanced. Proteinase K digestion is a robust, widely adopted method, especially effective for challenging samples like whole-mount embryos [22]. However, acetone permeabilization offers a simpler, non-enzymatic alternative that can be equally effective in certain contexts and may be gentler on some antigen epitopes [22].
In conclusion, researchers are advised to adopt an empirical, systematic approach to optimization. Begin by testing a small panel of fixatives and permeabilization methods, including cross-linking, organic solvents, and their respective retrieval techniques, on control samples that express the target antigen. This evidence-based strategy is the most reliable path to achieving the dual goals of impeccable tissue preservation and a strong, specific signal, thereby ensuring the validity and impact of scientific findings.
Sequential staining is an advanced technique in biomedical research that enables the detection of a large number of targets on a single biological sample through multiple rounds of staining, imaging, and signal removal. This approach has gained considerable interest for its ability to provide comprehensive protein localization data while preserving precious tissue samples [48]. The method is particularly valuable for characterizing complex microenvironments, such as those found in tumors, where understanding the spatial relationships between different cell types is crucial for both research and clinical applications [49].
A critical application of sequential staining involves combining the detection of extracellular (surface) markers with intracellular targets, a technically challenging process that requires careful optimization of fixation, permeabilization, and staining protocols. The success of these multiplexed approaches depends heavily on the preservation of antigenicity across multiple staining cycles and the effectiveness of antibody removal procedures between cycles [48] [49]. This guide focuses specifically on comparing sequential staining methodologies, with particular attention to how permeabilization strategies—including proteinase K and acetone-based methods—impact experimental outcomes for researchers and drug development professionals.
Comparative Analysis of Sequential Staining Methodologies
Core Sequential Staining Technologies
Table 1: Comparison of Sequential Staining Platforms and Methods
| Methodology | Multiplexing Capacity | Key Steps | Permeabilization Approaches | Instrumentation Requirements | Typical Duration |
|---|---|---|---|---|---|
| Manual Sequential IF [48] | >30 markers | Antibody incubation, imaging, antibody stripping (chemical elution) | Detergents (Triton X-100, saponin) | Standard fluorescent scanners | Several days |
| Automated seqIF (COMET) [49] | 40+ markers | Iterative staining, imaging, elution with microfluidics | Integrated in automated protocol | COMET platform with integrated microscope | <24 hours |
| Sequential IHC/IF [48] | Limited by chromogen/fluorophore | Hybrid methods using enzymatic precipitation | Varies with protocol | Standard brightfield/fluorescence microscopes | 1-2 days |
| Flow Cytometry Sequential Staining [50] | Limited by fluorochrome panel | Surface staining → fixation → permeabilization → intracellular staining | Acetone, methanol, detergents (Triton X-100, saponin) | Flow cytometer | 4-6 hours |
Performance Metrics for Sequential Staining
Table 2: Quantitative Performance Assessment of Sequential Staining
| Performance Parameter | Manual Sequential IF [48] | Automated seqIF [49] | Flow Cytometry Sequential [50] |
|---|---|---|---|
| Maximum Demonstrated Multiplex | >30 antibodies | 40 markers | Typically 5-10 intracellular targets |
| Elution Efficiency | Chemical stripping (disulfide cleavage/chaotropic salts) | >95% epitope retention post-elution | Not applicable (no elution required) |
| Signal Reproducibility | CV <10% with optimized protocols | High repeatability and reproducibility demonstrated | Dependent on fixation/permeabilization consistency |
| Tissue Preservation | Maintains tissue architecture through >30 cycles | No tissue damage after 40 cycles | Cells remain intact for analysis |
| Antigen Retrieval | Required after chemical stripping | Built into automated protocol | Not typically performed |
Experimental Protocols for Sequential Staining
Automated Sequential Immunofluorescence (seqIF)
The COMET platform implements a fully automated sequential immunofluorescence workflow that enables hyperplex spatial proteomics [49]. The process begins with tissue autofluorescence acquisition, which can be used for background subtraction in post-processing. The iterative protocol then proceeds with brief incubations of primary antibodies and fluorescently labeled secondary antibodies, followed by imaging and gentle elution to remove antibody complexes [49].
Key protocol specifications:
- Microfluidic Technology: Enables rapid reagent exchange (less than 1 second) and temperature control (reaching 50°C in <30 seconds) through a 50µm chamber height [49]
- Parallel Processing: Simultaneous staining and imaging cycles across multiple slides reduce total protocol time by 50%
- Imaging Capabilities: Integrated widefield microscope with DAPI, TRITC, and Cy5 channels, 20× objective providing 0.45µm resolution [49]
- Antibody Compatibility: Works with off-the-shelf antibodies without requiring custom conjugation
Manual Sequential Immunofluorescence
For laboratories without access to automated platforms, manual sequential IF provides a robust alternative with high multiplexing capabilities [48]. The validated method involves cycles of four-color indirect immunofluorescence, image acquisition, and antibody removal before applying subsequent stains.
Critical steps include:
- Antibody Removal: Achieved through disulfide cleavage with detergent or chaotropic salt treatment followed by antigen refolding [48]
- Image Processing: Digital registration of images and autofluorescence subtraction
- Antibody Validation: Primary antibodies screened for sensitivity and specificity at 1 and 0.1 µg/ml concentrations in TBS with 2% BSA, 0.05% sodium azide, and 100 mM trehalose [48]
- Antigen Retrieval: Microwave irradiation in EDTA-Tris buffer (pH 8) for 8 minutes at full power followed by 20 minutes of intermittent radiation [48]
Sequential Staining for Flow Cytometry
For intracellular flow cytometry applications, a different sequential approach is employed that does not require antibody elution but rather careful staging of staining procedures [50]. This method involves detecting extracellular and intracellular markers in the same experiment through sequential staining before and after permeabilization.
Optimal protocol:
- Surface Staining First: Block and stain for surface markers prior to fixation and permeabilization
- Fixation Considerations: Formaldehyde preserves cellular morphology and reduces soluble protein loss; methanol simultaneously fixes and permeabilizes but may disrupt epitopes [50]
- Permeabilization Agents: Triton X-100 permeabilizes plasma and intracellular membranes; saponin only permeabilizes plasma membrane and is reversible [50]
- Fluorophore Selection: Avoid protein fluorophores like PE and APC when using methanol permeabilization, as they will be denatured [50]
Figure 1: Sequential Staining Workflow Decision Tree. This diagram outlines the key decision points in designing sequential staining experiments, particularly the choice of permeabilization method based on experimental goals.
Permeabilization Methods: Proteinase K vs. Acetone
Proteinase K Permeabilization
Proteinase K is a broad-spectrum serine protease that digests proteins and is commonly used in antigen retrieval and permeabilization protocols. While not explicitly detailed in the search results for sequential staining, proteinase K treatment falls within the category of enzymatic permeabilization methods that can be employed for epitope recovery in challenging fixation conditions.
Applications in sequential staining:
- Epitope Unmasking: Digests cross-linked proteins that may obscure antigen binding sites
- Combined Retrieval: Can perform simultaneous antigen retrieval and permeabilization
- Sequential Application: May be applied between staining cycles to recover antigens affected by previous fixation steps
Acetone and Methanol Permeabilization
Acetone and methanol are denaturing fixatives and permeabilization agents that work by precipitating cellular proteins and extracting lipids from membranes [51] [50]. These methods are particularly useful for certain applications in sequential staining protocols.
Key characteristics:
- Rapid Action: Acetone fixation at -20°C for 10 minutes is sufficient for many targets [51]
- Simultaneous Fixation/Permeabilization: Methanol offers the advantage of both fixing and permeabilizing cells in a single step [50]
- Epitope Considerations: These denaturing methods may destroy some epitopes while revealing others, requiring empirical optimization [50]
- Temperature Sensitivity: Cold acetone (-20°C) is typically required for optimal results [51]
Table 3: Proteinase K vs. Acetone/Methanol Permeabilization for Sequential Staining
| Parameter | Proteinase K | Acetone/Methanol |
|---|---|---|
| Mechanism of Action | Proteolytic digestion of proteins | Protein precipitation and lipid extraction |
| Optimal Temperature | 20-37°C | -20°C to 4°C |
| Tissue Preservation | Potential over-digestion concerns | Better preservation of cellular structure |
| Compatibility with Sequential Cycles | May require optimization between cycles | Generally compatible with multiple cycles |
| Best Applications | Formalin-fixed tissues, cross-linked epitopes | Cytoskeletal, viral, and some enzyme antigens [51] |
| Limitations | Potential destruction of some epitopes | May disrupt protein conformation and function |
Research Reagent Solutions for Sequential Staining
Table 4: Essential Research Reagents for Sequential Staining Experiments
| Reagent Category | Specific Examples | Function in Sequential Staining | Optimization Tips |
|---|---|---|---|
| Fixatives | Formaldehyde, Paraformaldehyde, Methanol, Acetone [51] [50] | Preserve cellular architecture and antigen position | Cross-linking fixatives better preserve structure; denaturing fixatives may enhance some epitopes [50] |
| Permeabilization Agents | Triton X-100, Saponin, Tween-20, Proteinase K, Acetone [50] | Enable antibody access to intracellular targets | Triton X-100 permeabilizes all membranes; saponin is reversible and plasma membrane-specific [50] |
| Antibody Elution Reagents | Disulfide cleavage agents, Chaotropic salts, Low pH buffers [48] [49] | Remove antibodies between staining cycles | Efficiency must be balanced with epitope preservation; >95% removal achievable [49] |
| Blocking Agents | BSA, Normal serum, Casein, Commercial blocking buffers | Reduce non-specific antibody binding | Should be included in antibody diluents when using reversible permeabilization like saponin [50] |
| Antigen Retrieval Solutions | EDTA-Tris buffer, Citrate buffer, Proteinase K [48] | Reverse cross-linking and expose hidden epitopes | Microwave irradiation enhances retrieval efficiency [48] |
Figure 2: Antibody Binding and Elution Cycle. This diagram illustrates the core process of antibody binding, detection, and elution that enables multiple rounds of staining in sequential immunofluorescence protocols.
Implementation Guidelines and Best Practices
Panel Design and Experimental Planning
Successful sequential staining requires meticulous panel design that accounts for the specific challenges of multiple staining cycles. Researchers should prioritize markers based on abundance and antibody performance, assigning brighter fluorophores to less abundant targets [50]. When combining extracellular and intracellular marker detection, it is essential to validate that permeabilization methods do not compromise surface epitopes or vice versa.
Critical considerations include:
- Antibody Validation: Thoroughly test each antibody individually before multiplexing to determine optimal concentrations and staining conditions [48]
- Species Compatibility: Plan primary antibody host species to minimize cross-reactivity in subsequent rounds
- Fluorophore Stability: Select fluorophores that can withstand elution conditions or plan for spectral compatibility across cycles
- Control Samples: Include appropriate controls for autofluorescence, antibody specificity, and elution efficiency
Troubleshooting Common Challenges
Even with optimized protocols, sequential staining experiments can encounter technical challenges that require systematic troubleshooting:
- Diminished Signal in Later Cycles: This may indicate incomplete antigen retrieval or epitope damage from repeated elution steps. Solution: Optimize elution conditions and include additional antigen retrieval steps if needed [48]
- High Background Fluorescence: Often caused by insufficient blocking or incomplete antibody removal. Solution: Increase blocking agent concentration, extend washing steps, and verify elution efficiency [38]
- Tissue Degradation: Multiple processing cycles can compromise tissue integrity. Solution: Monitor fixation times carefully and consider using tissue stabilization methods [48]
- Incomplete Antibody Removal: Residual antibodies cause carryover between cycles. Solution: Optimize elution buffer composition and incubation times [48] [49]
Sequential staining represents a powerful methodological approach for comprehensive cellular characterization, enabling researchers to extract maximal information from limited biological samples. The integration of extracellular marker detection with intracellular staining requires careful balancing of fixation, permeabilization, and elution conditions to preserve both structural integrity and antigenicity across multiple cycles.
As the field advances, automated platforms like the COMET system are addressing many technical challenges through standardized workflows and optimized reagent delivery [49]. However, manual methods continue to offer flexibility and accessibility for laboratories developing custom multiplexing panels. The choice between permeabilization methods—including proteinase K, acetone, methanol, or detergents—remains application-dependent, requiring empirical validation for each experimental context.
For researchers embarking on sequential staining experiments, success hinges on systematic validation of each component in the workflow, from antibody performance to elution efficiency. When properly optimized, these methods provide unprecedented insights into cellular organization and function, with particular value for drug development applications requiring deep characterization of complex biological systems.
Solving Common Problems and Enhancing Signal-to-Noise Ratio
In protein research, particularly within immunofluorescence and immunohistochemistry, the twin challenges of over-fixation and inadequate permeabilization represent significant bottlenecks that can compromise experimental outcomes. Over-fixation causes excessive protein cross-linking that masks epitopes, while insufficient permeabilization creates physical barriers that prevent antibody access to intracellular targets [52] [18]. Within the broader context of comparing proteinase K and acetone permeabilization methodologies, this guide objectively examines their performance characteristics, supported by experimental data. These methods offer divergent approaches to antigen recovery—proteinase K employing enzymatic retrieval of masked epitopes, while acetone utilizes solvent-based permeabilization that simultaneously fixes and permeabilizes cellular structures [18]. Understanding their comparative effectiveness, limitations, and optimal application scenarios empowers researchers to make informed methodological choices that enhance signal detection while preserving cellular morphology.
Methodological Comparison: Proteinase K vs. Acetone Permeabilization
The fundamental differences between proteinase K and acetone permeabilization methods stem from their distinct mechanisms of action. Proteinase K, a broad-spectrum serine protease, functions through enzymatic digestion of proteins that cross-link during fixation, thereby retrieving masked epitopes and enhancing antibody accessibility [53] [18]. This method is particularly valuable for difficult-to-retrieve epitopes and is typically performed at 37°C for 5-30 minutes, commonly 10-15 minutes, in neutral buffer solutions [18]. In contrast, acetone permeabilization operates through solvent action, extracting lipids from cellular membranes to create pores for antibody penetration while simultaneously fixing tissues through dehydration and protein precipitation [18]. This dual fixation-permeabilization action occurs rapidly, typically requiring only 10 minutes of treatment time.
The experimental workflow for evaluating these methods involves standardized sample processing followed by quantitative signal assessment. As demonstrated in spatial transcriptomics benchmarking studies, consistent sample preparation across comparison groups is essential for valid methodological comparisons [54]. Following permeabilization with either proteinase K or acetone, samples undergo standard immunostaining procedures with carefully optimized antibody concentrations, followed by imaging and signal quantification using standardized microscopy parameters to ensure comparable results across experimental conditions.
Comparative Performance Data
Table 1: Direct Comparison of Proteinase K and Acetone Permeabilization Methods
| Parameter | Proteinase K | Acetone |
|---|---|---|
| Mechanism of Action | Enzymatic digestion of cross-linking proteins | Solvent-based lipid extraction and protein precipitation |
| Typical Concentration | 7.5 µg/mL [53] | 100% (pure solvent) [18] |
| Incubation Time | 30 minutes [53] | 10 minutes [18] |
| Temperature | 37°C [53] [18] | Room temperature [18] |
| Simultaneous Fixation | No | Yes [18] |
| Tissue Morphology Preservation | Risk of damage with over-treatment [18] | Good to excellent [18] |
| Best Applications | Difficult-to-retrieve epitopes, heavily cross-linked samples [18] | Cytoskeletal antigens, viral antigens, some enzyme antigens [18] |
| Primary Limitations | Potential tissue damage with extended use [18] | Less effective for nuclear antigens, may not reverse over-fixation [18] |
Table 2: Experimental Outcomes from Methodological Applications
| Experimental Context | Proteinase K Performance | Acetone Performance | Reference |
|---|---|---|---|
| On-chip RT-LAMP reactions | Enabled polymerase and reverse transcriptase penetration through digested cell membranes [53] | Not applicable in this context | [53] |
| Intracellular antigen detection | Effective for epitope retrieval after formaldehyde fixation | Recommended for cytoskeletal, viral, and some enzyme antigens [18] | [18] |
| Spatial transcriptomics | Used in sample preparation for subcellular resolution platforms | Not typically used in these workflows | [54] |
| Flow cytometry intracellular staining | Part of optimized fixation/permeabilization protocols | Alternative method using ice-cold acetone [55] | [55] |
Experimental Protocols for Method Comparison
Proteinase K Permeabilization Protocol
The proteinase K permeabilization method is particularly valuable for recovering epitopes masked by aldehyde-based fixation. The following protocol has been optimized for tissue sections and cellular samples:
- Sample Preparation: Begin with fixed cells or tissue sections. For formaldehyde-fixed samples, a post-fixation wash with PBS is recommended to remove residual fixative.
- Proteinase K Solution Preparation: Prepare a working solution of 7.5 µg/mL proteinase K in an appropriate buffer (typically Tris-EDTA or PBS) [53].
- Permeabilization: Apply the proteinase K solution to completely cover the samples. Incubate at 37°C for 30 minutes in a humidified chamber to prevent evaporation [53].
- Termination: Carefully remove the proteinase K solution and rinse samples with PBS containing protease inhibitors to halt enzymatic activity.
- Validation: Proceed with standard immunostaining protocols. Include controls without primary antibody to assess non-specific signal.
This enzymatic approach was successfully implemented in pixelated spatial gene expression analysis, where proteinase K treatment (7.5 µg/mL for 30 minutes) effectively permeabilized pixelated tissue sections while preserving RNA integrity for subsequent on-chip amplification [53].
Acetone Permeabilization Protocol
Acetone permeabilization offers a simultaneous fixation-permeabilization approach particularly beneficial for certain antigen classes:
- Sample Preparation: Start with fresh or lightly fixed cells or tissue sections.
- Acetone Application: Apply pre-chilled acetone (-20°C) to completely cover samples. Incubate for 10 minutes at room temperature [18].
- Removal: Carefully remove acetone and allow samples to air-dide completely.
- Rehydration: Rehydrate samples with PBS or appropriate buffer before proceeding with immunostaining.
- Validation: Implement appropriate controls including unstained samples and isotype controls.
As noted in technical resources, acetone fixation inherently permeabilizes tissues, making it suitable for antigens sensitive to detergent-based methods [18]. This method is particularly recommended for cytoskeletal components, viral antigens, and certain enzymes where detergent-induced redistribution might occur.
Technical Workflow and Pathway Analysis
The decision pathway for addressing low signal issues related to fixation and permeabilization involves systematic troubleshooting with method selection based on specific experimental parameters. The following workflow diagram illustrates the logical relationship between problem identification, method selection, and optimization steps:
Diagram 1: Permeabilization Method Decision Pathway - This workflow guides researchers through systematic troubleshooting of low signal issues, directing appropriate method selection based on specific experimental parameters and targets.
Research Reagent Solutions
The following table details essential reagents for implementing these permeabilization methods, along with their specific functions in experimental protocols:
Table 3: Key Research Reagents for Permeabilization Protocols
| Reagent | Function | Application Notes |
|---|---|---|
| Proteinase K | Serine protease that digests proteins; recovers masked epitopes by breaking cross-links | Use at 7.5 µg/mL for 30 min at 37°C; concentration and time require optimization for specific samples [53] |
| Acetone | Organic solvent that fixes by dehydration/precipitation and permeabilizes by lipid extraction | Apply pure pre-chilled acetone for 10 min at room temperature; suitable for simultaneous fixation and permeabilization [18] |
| Formaldehyde | Cross-linking fixative that preserves structure but may mask epitopes | Typically used at 4% concentration; may require antigen retrieval methods like proteinase K for epitope recovery [55] |
| Triton X-100 | Non-ionic detergent for membrane permeabilization | Use 0.1-0.2% in PBS for 10 min only; harsher than mild detergents like Tween 20 [18] |
| Tween 20 | Mild non-ionic detergent for gentle permeabilization | Use 0.2-0.5% for 10-30 min; less disruptive to protein structures than harsher detergents [18] |
| Saponin | Mild detergent that complexes with cholesterol in membranes | Suitable for reversible permeabilization; often used at 0.2-0.5% concentration [18] |
Discussion and Comparative Analysis
Contextual Method Selection
The experimental data reveals that method selection between proteinase K and acetone permeabilization must be guided by multiple factors, including fixation history, antigen location, and morphological requirements. Proteinase K demonstrates particular effectiveness for reversing over-fixation effects, as evidenced by its implementation in spatial transcriptomics workflows where it enabled enzyme penetration through digested cell membranes without compromising tissue architecture [53]. Conversely, acetone permeabilization offers advantages for delicate cellular structures and specific antigen classes, serving as the preferred method for cytoskeletal elements where detergent-induced redistribution might occur [18].
The comparative analysis extends to practical laboratory considerations. Proteinase K protocols require precise concentration and timing optimization to balance epitope recovery against potential tissue damage [18]. Acetone treatment, while faster and simpler, may prove insufficient for nuclear antigens or heavily cross-linked samples. Technical resources note that acetone's simultaneous fixation and permeabilization can be beneficial for certain applications but may not adequately reverse over-fixation artifacts [18].
Integration in Multiplexed Workflows
In advanced applications such as high-parameter spatial biology, permeabilization methods must be compatible with subsequent detection technologies. Recent benchmarking studies of subcellular resolution platforms highlight how permeabilization optimization contributes to data quality. Research indicates that proteinase K treatment effectively supports sophisticated multiplexed protein detection using technologies like CODEX when properly optimized [54]. The integration of appropriate permeabilization methods with subsequent detection steps underscores the importance of considering entire workflow compatibility when selecting between proteinase K and acetone approaches.
The comparative analysis of proteinase K versus acetone permeabilization methods reveals a nuanced landscape where neither approach universally supersedes the other. Proteinase K demonstrates superior capability in addressing over-fixation issues through enzymatic retrieval of masked epitopes, making it invaluable for nuclear targets and heavily cross-linked samples. Acetone permeabilization offers distinct advantages for specific antigen classes including cytoskeletal components, while providing simultaneous fixation and permeabilization. The methodological selection should be guided by experimental priorities—whether epitope accessibility, morphological preservation, or workflow efficiency. As spatial biology advances toward increasingly multiplexed detection, optimal permeabilization will remain fundamental to data quality, requiring researchers to maintain both methods in their technical repertoire and apply them discriminatively based on experimental context and target characteristics.
In protein localization studies, the permeabilization step is critical for providing antibody access to intracellular targets. However, this very step is a major source of high background staining resulting from non-specific antibody binding and trapping. The choice between enzymatic methods (like proteinase K) and organic solvent methods (like acetone) involves significant trade-offs between epitope preservation, cellular morphology, and background reduction [56] [18]. This guide objectively compares proteinase K and acetone permeabilization, providing experimental data and protocols to inform method selection for specific research applications.
Permeabilization works by disrupting cellular membranes to allow antibodies access to intracellular compartments. Proteinase K, a proteolytic enzyme, digests proteins and cleaves cross-links formed during aldehyde fixation, thereby physically opening the cellular structure and unmasking epitopes [4] [18]. In contrast, acetone is an organic solvent that dehydrates samples and precipitates cellular proteins, simultaneously fixing and permeabilizing by dissolving lipids in cell membranes [18] [57].
The table below summarizes the core characteristics of these two distinct permeabilization strategies.
Table 1: Fundamental Characteristics of Proteinase K and Acetone Permeabilization
| Characteristic | Proteinase K | Acetone |
|---|---|---|
| Primary Mechanism | Enzymatic digestion of proteins and cross-links [18] | Solvent-based dehydration and lipid dissolution [18] [57] |
| Typical Application | Follows aldehyde fixation (e.g., PFA) [4] | Often used as a simultaneous fixative and permeabilizer [57] |
| Impact on Epitopes | Can unmask epitopes masked by cross-linking [18] | Can denature proteins, potentially destroying some epitopes [40] |
| Impact on Morphology | Risk of damaging tissue morphology if over-used [18] | Generally good preservation of cellular structure [57] |
Figure 1. Mechanisms of Action for Permeabilization Methods. This diagram illustrates the distinct pathways through which proteinase K and acetone treatment facilitate antibody access while influencing background signal.
Direct Performance Comparison
Evaluating permeabilization agents requires balancing signal quality with specimen integrity. The following table synthesizes comparative experimental data from model organism studies and controlled cell line experiments.
Table 2: Experimental Performance Comparison in Model Systems
| Performance Metric | Proteinase K | Acetone | Experimental Context & Notes |
|---|---|---|---|
| Signal Strength | Strong and consistent for target mRNAs [4] | Weak and variable signal for target mRNAs [4] | Experimental Context: RNA FISH in Drosophila ovaries. Proteinase K yielded strong signals within 15-45 min, while acetone required longer (5.5h) and produced weak staining [4]. |
| Background Level | Controlled via precise optimization | Generally low, but signal may also be low | N/A |
| Morphology Preservation | Reproducible but can be damaged by over-treatment [4] [18] | Good structural preservation [57] | Experimental Context: Comparison of permeabilization methods for tissue. Optimal proteinase K concentration and time are critical to avoid tissue damage [4]. |
| Immunofluorescence Compatibility | Can be too harsh for protein epitopes, leading to weak or lost signal [4] | Compatible; often used in combined IF/FISH protocols [4] | Experimental Context: Dual protein-RNA labeling (IF/FISH). Proteinase K treatment post-IF was detrimental to protein signal, whereas solvent-based methods preserved it [4]. |
| Recommended For | Unmasking difficult epitopes in cross-linked samples; RNA detection [4] [18] | Preserving protein antigenicity; simultaneous fixation & permeabilization; cytoskeletal targets [4] [18] [57] | N/A |
For flow cytometry applications, a systematic study on detecting intracellular 18S rRNA in HeLa cells found that optimal permeabilization is agent-dependent. While proteinase K was one of several methods tested, the highest fluorescence signals were achieved with detergent-based permeabilization using Tween-20 [3].
Detailed Experimental Protocols
Proteinase K Permeabilization for Tissues
This protocol, optimized for Drosophila ovaries, balances permeabilization with morphology preservation [4].
- Fixation: Fix dissected tissues in 4% paraformaldehyde (PFA) with 1% DMSO for 1 hour at room temperature.
- Dehydration/Rehydration: Pass tissues through a series of increasing ethanol concentrations (50%, 70%, 90%, 100%) for storage or pause points, then rehydrate through a decreasing series (90%, 70%, 50%).
- Permeabilization:
- Prepare a solution of 50 µg/mL Proteinase K in an appropriate buffer (e.g., Tris-HCl with CaCl₂).
- Incubate tissues for 1 hour at room temperature.
- Critical Step: The activity of proteinase K can vary between lots. This concentration and time serve as a starting point and may require optimization to prevent over-digestion and tissue damage [4] [18].
- Post-Fixation: Re-fix tissues in 4% PFA for 30 minutes to maintain structural integrity during subsequent high-temperature washes.
- Washing: Rinse tissues thoroughly with PBS containing 0.1% Tween 20 to terminate proteinase K activity.
Acetone Permeabilization for Cells
This protocol is commonly used for adherent cells and can also act as a simultaneous fixation and permeabilization method [18] [57].
- Preparation: Culture cells on glass coverslips until they reach the desired confluency.
- Fixation/Permeabilization:
- Aspirate culture medium and gently rinse cells with pre-warmed PBS.
- Incubate cells in -20°C Cold Acetone for 5-10 minutes at room temperature or -20°C.
- Note: Acetone fixation can make cells brittle. Handle with care to avoid detaching them from the coverslip.
- Drying and Rehydration: Allow the acetone to evaporate completely, leaving the cells dried. Then, rehydrate the cells by washing several times with PBS or a suitable buffer before proceeding to immunostaining.
- Alternative: For post-aldehyde fixation, cells can first be fixed with 4% PFA for 10-15 minutes, rinsed, and then treated with cold acetone as above [18].
The Scientist's Toolkit: Essential Research Reagents
Successful permeabilization requires more than just the primary agent. The table below lists key reagents and their functions in optimizing these protocols.
Table 3: Key Reagents for Permeabilization and Background Reduction
| Reagent | Function | Consideration |
|---|---|---|
| Proteinase K | Serine protease that digests proteins; unmasks cross-linked epitopes [4] [18] | Concentration and time must be tightly optimized to avoid destroying morphology and antigens [4]. |
| Acetone | Organic solvent that dehydrates and dissolves lipids; co-fixative [18] [57] | Can denature sensitive epitopes; often requires no separate permeabilization step [40]. |
| Paraformaldehyde (PFA) | Cross-linking fixative; standard pre-treatment for proteinase K [4] | Can mask epitopes, making subsequent retrieval (e.g., with proteinase K) necessary [18]. |
| Triton X-100 | Non-ionic detergent for permeabilization after aldehyde fixation [40] [14] | Creates pores in all membranes; can be harsh. Often used at 0.1-0.5% for 10-30 min [18]. |
| Tween-20 | Mild non-ionic detergent for permeabilization and washing [3] | Can be effective for intracellular nucleic acid detection; often included in wash buffers to reduce background [3]. |
| Saponin | Mild detergent that complexes with cholesterol to create pores [40] [18] | Requires presence in all subsequent antibodies and wash buffers, as pores are transient [57]. |
Figure 2. Experimental Workflow for Immunostaining. This flowchart outlines the key steps in a standard immunostaining procedure, highlighting the point at which a permeabilization method is chosen. The choice directly influences the need for prior fixation and the effectiveness of subsequent blocking and antibody binding.
The choice between proteinase K and acetone permeabilization is not one of superiority but of context. Proteinase K is a powerful tool for recovering signals from heavily cross-linked samples and is often essential for RNA in situ hybridization protocols [4]. However, its aggressive nature requires careful titration and poses a significant risk to protein antigenicity and tissue morphology. Acetone offers a milder alternative for protein detection, providing adequate permeabilization with good structural preservation, making it suitable for dual protein-RNA labeling and for use with denaturation-sensitive antibodies [4] [40].
The decision framework should be guided by the primary target and sample type:
- For challenging epitopes masked by aldehyde fixation, particularly in structured tissues for RNA detection, proteinase K is often indispensable, provided its usage is meticulously optimized.
- For routine intracellular protein detection, especially when combining with immunofluorescence or when working with sensitive epitopes, acetone or other mild detergents (e.g., saponin, Tween-20) are generally recommended as a starting point [4] [3].
Ultimately, reducing high background requires a holistic view of the entire immunostaining protocol. The permeabilization step is deeply intertwined with the choice of fixative and the nature of the target antigen. Researchers are encouraged to use this comparative data as a starting point for empirical optimization in their specific experimental systems.
In molecular biology and histology, the ability to access intracellular components without compromising cellular structure is paramount. Proteinase K, a potent broad-spectrum serine protease, and organic solvents like acetone, are two fundamental tools used for tissue permeabilization, each with distinct mechanisms and outcomes. While Proteinase K enzymatically digests proteins and degrades nucleases to facilitate nucleic acid extraction, acetone and other solvents physically dissolve lipids from cell membranes. The choice between these methods often hinges on a critical trade-off: the need for efficient penetration and extraction versus the imperative to preserve original tissue morphology and antigen integrity for accurate analysis. This guide provides an objective comparison of their performance, grounded in experimental data, to inform researchers developing robust, reproducible protocols.
Mechanism of Action: A Fundamental Comparison
Understanding the core mechanism of each agent is key to predicting its interaction with biological samples.
Proteinase K (Enzymatic Digestion): This enzyme catalyzes the cleavage of peptide bonds adjacent to the carboxyl group of aliphatic and aromatic amino acids. In molecular biology, it is routinely used to inactivate nucleases that could degrade DNA or RNA during extraction. Its activity is often performed in the presence of SDS and EDTA, which denature proteins and chelate metal ions required for nuclease activity, respectively. A crucial characteristic is its stability under harsh conditions, including elevated temperatures (37-65°C) and the presence of SDS [58]. The very potency that makes it effective also makes over-digestion a significant risk, potentially leading to the loss of antigenic epitopes and morphological damage [4].
Acetone (Solvent-Based Precipitation): As a precipitating fixative and permeabilizing agent, acetone acts as a strong dehydrant. It rapidly removes water from tissues and precipitates cellular proteins, thereby preserving cellular architecture while dissolving membrane lipids to create pores [59]. A standard protocol involves treating cells with ice-cold 100% acetone for 5-10 minutes at -20°C. Unlike cross-linking fixatives, it does not create molecular bridges, which can help preserve some antigen epitopes that are sensitive to formaldehyde. However, it can also remove some soluble molecules and lipids [59].
Table 1: Core Characteristics and Mechanisms of Action
| Feature | Proteinase K | Acetone |
|---|---|---|
| Primary Mechanism | Enzymatic proteolysis | Lipid dissolution & protein precipitation |
| Key Reagents | Proteinase K, SDS, EDTA | 100% ice-cold Acetone |
| Standard Working Concentration | 50 µg/mL - 100 µg/mL [4] | 100% |
| Typical Incubation | 15 min to several hours/overnight [60] | 5-10 minutes [59] |
| Inactivation/Removal | Heat inactivation (95°C for 10 min) [58] | Evaporation and washing |
| Key Advantage | Highly effective nuclease inactivation; digests contaminants. | Rapid action; no cross-linking; can fix and permeabilize simultaneously. |
| Key Disadvantage | High risk of over-digestion and antigen damage if unchecked. | Can remove lipids and soluble molecules; may denature some proteins. |
Performance and Experimental Data
The choice between these methods is rarely absolute but is dictated by the specific application and the cellular targets of interest. Experimental data from various models highlights the performance trade-offs.
Nucleic Acid Integrity and Yield
The primary strength of Proteinase K is its unparalleled effectiveness in nucleic acid applications. In forensic science, tissue samples preserved in solid sodium chloride and subsequently treated with Proteinase K yielded high-quality DNA suitable for STR typing even after one year of room-temperature storage, with quantities of 397.9 ± 131.5 ng of autosomal DNA/mg of tissue [61]. This underscores its power to protect and release high-molecular-weight DNA by efficiently inactivating nucleases.
Antigen and Morphology Preservation
For applications requiring the simultaneous detection of proteins and RNA, the detrimental effect of Proteinase K on protein epitopes becomes a major limitation. Research on Drosophila ovaries directly compared permeabilization methods for protein immunofluorescence (IF) and RNA FISH double labeling (IF/FISH). The study found that Proteinase K treatment, even at a reduced concentration of 20 µg/mL, resulted in little or no protein signal for tested antibodies [4]. In contrast, permeabilization with alternative methods, including acetone, resulted in a strong and specific protein signal [4].
Signal Strength and Background
The same study on Drosophila ovaries provided a clear comparison of signal strength for RNA detection. While Proteinase K permeabilization resulted in a strong ISH signal within 15-45 minutes, permeabilization with acetone required a much longer development time (2-5.5 hours) and produced an extremely weak and variable signal for the transcript being tested [4]. This indicates that while gentler on proteins, solvent-based permeabilization may not be sufficiently aggressive for optimal probe penetration in some dense tissue models.
Table 2: Experimental Performance Comparison in Different Applications
| Application | Proteinase K Performance | Acetone Performance | Supporting Data |
|---|---|---|---|
| Genomic DNA Extraction | Excellent. Yields high-quality, high-molecular-weight DNA suitable for advanced typing. | Not typically used as a primary agent for DNA extraction. | Yields of ~400 ng DNA/mg tissue; successful STR profiling [61]. |
| RNA-Protein Co-detection (IF/FISH) | Poor. Destroys protein antigenicity. | Good. Preserves protein epitopes for antibody binding. | Proteinase K: "little or no protein signal". Acetone: "strong and specific" signal [4]. |
| RNA In Situ Hybridization (ISH) | Strong and fast signal. The gold standard for many ISH protocols. | Weak, slow, and variable signal. | Proteinase K: strong signal in 15-45 min. Acetone: weak signal even after 5.5 hours [4]. |
| Cellular Morphology | Risk of damage with over-digestion; can create a "cloudy liquid" from digested tissue [58]. | Good structural preservation; acts as a fixative. | Acetone and methanol are recognized as precipitating fixatives that preserve architecture [59]. |
Detailed Experimental Protocols
To ensure reproducibility, below are generalized protocols for each method, as cited in the literature.
Protocol 1: Proteinase K Permeabilization for ISH
This protocol, optimized for Drosophila ovaries, balances permeabilization with morphology preservation [4].
- Fixation: Fix dissected tissue in 4% paraformaldehyde with 1% DMSO for 1 hour.
- Dehydration/Rehydration: Pass the tissue through a series of increasing concentrations of ethanol (a pause point for storage), then rehydrate through a series of decreasing concentrations.
- Permeabilization: Treat the rehydrated tissue with 50 µg/mL Proteinase K in an appropriate buffer (e.g., PBTween) for 1 hour.
- Post-fixation: Re-fix the tissue in 4% paraformaldehyde for 30 minutes to halt Proteinase K activity and stabilize the permeabilized tissue.
- Hybridization: Proceed with prehybridization and hybridization with labeled RNA probes.
Protocol 2: Acetone Permeabilization for IF/FISH
This protocol is designed to preserve protein antigens for immunofluorescence while allowing subsequent RNA FISH [4].
- Initial Fixation: Fix dissected tissue in 4% paraformaldehyde for 20 minutes.
- Protein Immunofluorescence (IF): Perform the entire IF procedure (blocking, primary antibody, secondary antibody) first.
- Post-IF Fixation: Re-fix the tissue to cross-link the antibodies and preserve the protein signal through subsequent steps.
- Solvent Permeabilization: Permeabilize the tissue by treatment with xylenes and ethanol, or with 100% acetone for 20 minutes, as an alternative to Proteinase K.
- FISH: Continue with the standard FISH protocol for RNA detection.
Visual Workflow for Method Selection
The following diagram illustrates the logical decision-making process for choosing between these permeabilization methods based on experimental goals.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their functions in the context of these permeabilization workflows.
Table 3: Essential Reagents for Permeabilization and Tissue Processing
| Reagent | Primary Function | Key Considerations |
|---|---|---|
| Proteinase K | Enzymatic digestion of proteins; inactivation of nucleases for nucleic acid isolation. | Concentration & time are critical to prevent over-digestion. Stable at high temps and in SDS [58]. |
| Acetone | Precipitating fixative and permeabilizing agent; dissolves lipids. | Use ice-cold. Rapid action but can denature some fluorescent proteins [59]. |
| Paraformaldehyde (PFA) | Cross-linking fixative; preserves morphology by creating protein networks. | The gold standard for morphology; can mask some epitopes. A 4% solution is standard [4] [59]. |
| Triton X-100 | Non-ionic detergent for permeabilization; dissolves lipid bilayers. | Common concentration: 0.1-0.4%. Non-selective; can lyse cells at high conc./long times [59]. |
| SDS (Sodium Dodecyl Sulfate) | Ionic detergent; denatures proteins and aids in cell lysis. | Often used in Proteinase K buffers to denature contaminants and enhance enzyme activity [58]. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelating agent; binds Mg²⁺ and Ca²⁺ ions. | Inactivates metal-dependent nucleases, protecting DNA and RNA during extraction [58]. |
The data clearly shows that neither Proteinase K nor acetone is universally superior. The choice is application-dependent. Proteinase K is the unequivocal choice for nucleic acid-focused workflows where maximizing DNA/RNA yield and integrity is the priority, provided digestion time and concentration are carefully optimized to prevent morphological havoc. In contrast, acetone and other solvents are better suited for protein-detection and multiplexing experiments, such as IF/FISH, where the preservation of antigenicity is paramount, even if it may come at the cost of a slower or weaker signal for nucleic acid probes in challenging tissues.
For researchers aiming to mitigate the risks of Proteinase K over-digestion, key strategies include:
- Empirical Titration: Always conduct a pilot experiment to determine the minimum effective concentration and incubation time for your specific tissue type.
- Monitor Digestion: Visually inspect samples for a transition to a clear lysed solution as an indicator of complete digestion [60].
- Sequential Protocols: In IF/FISH, perform immunofluorescence first, then post-fix, and use a mild detergent or solvent for the FISH permeabilization to avoid Proteinase K's damaging effects on proteins [4]. By understanding the mechanisms and trade-offs outlined in this guide, scientists can make informed decisions to preserve the delicate balance between access and integrity in their experimental samples.
The process of formalin fixation and paraffin embedding (FFPE) has been the cornerstone of pathological specimen preservation for over a century, providing exceptional morphological detail and long-term sample stability. However, this process creates methylene bridges between proteins, particularly targeting lysine residues, which can mask antigenic epitopes and restrict antibody binding during immunohistochemistry (IHC) and immunofluorescence (IF) applications [62] [63]. This epitope masking presents a significant challenge for researchers and diagnosticians, necessitating the development of effective antigen retrieval techniques to reveal hidden epitopes without compromising tissue integrity.
Antigen retrieval has emerged as a critical preparatory step for successfully localizing proteins in fixed tissues, enabling accurate visualization of cellular components and expanding our understanding of disease mechanisms. The two primary approaches—heat-induced epitope retrieval (HIER) and protease-induced epitope retrieval (PIER)—employ fundamentally different mechanisms to reverse the cross-linking effects of formalin fixation [18] [63]. While HIER uses heated buffers to break methylene bridges, PIER utilizes enzymatic digestion with proteases like proteinase K to cleave peptides masking antigenic sites [64] [63].
The choice between these methods carries significant implications for research outcomes, particularly in studies requiring precise protein localization or multiplex staining. This guide provides a comprehensive comparison of proteinase K-based enzymatic retrieval and acetone-based permeabilization methods, offering experimental data and protocols to inform methodological selection for specific research applications in drug development and basic science.
Fundamental Principles of Epitope Masking and Retrieval
Chemical Basis of Formalin-Induced Epitope Masking
Formalin fixation primarily works through non-coagulant cross-linking, creating covalent bonds between protein molecules, especially at lysine residues [63]. This cross-linking stabilizes tissue architecture but simultaneously creates a network of bridged proteins that physically obstruct antibody access to epitopes. The formation of methylene bridges represents the fundamental mechanism behind epitope masking, transforming soluble cellular proteins into insoluble networks while preserving structural relationships [62] [63].
The extent of epitope masking depends on several factors during fixation, including formalin concentration, fixation duration, and temperature [63]. Over-fixation can produce particularly challenging masking effects, while under-fixation risks cellular degradation and loss of morphological detail. This balance necessitates careful optimization of both fixation and subsequent retrieval protocols for different tissue types and target antigens.
Permeabilization Mechanisms in Antigen Retrieval
Beyond reversing chemical cross-links, effective antigen retrieval must also address the need for antibody penetration into cellular compartments. While cross-linking fixatives like formalin preserve structure, they create dense protein networks that impede antibody movement. Permeabilization techniques address this challenge through different physical and chemical mechanisms:
Detergent-based permeabilization: Surfactants like Triton X-100 or Tween-20 insert into lipid bilayers, creating pores that allow antibody passage. Concentration and incubation time must be optimized to balance access with membrane preservation [18] [41].
Solvent-based permeabilization: Organic solvents like acetone and methanol dissolve lipid components of cellular membranes, effectively permeabilizing tissues but potentially extracting some cellular components [63] [41].
Enzyme-mediated permeabilization: Proteases like proteinase K directly cleave peptide bonds, eroding physical barriers to antibody access while simultaneously unmasking epitopes [64].
The following diagram illustrates the fundamental workflow for antigen retrieval and the decision points between primary methodological approaches:
Comparative Analysis: Proteinase K vs. Acetone Methods
Proteinase K Enzymatic Antigen Retrieval
Proteinase K belongs to the family of serine proteases and demonstrates broad substrate specificity, cleaving peptide bonds at the carboxyl side of aliphatic, aromatic, or hydrophobic amino acids. In antigen retrieval applications, this enzymatic activity digests proteins that mask epitopes, effectively reversing the cross-linking effects of formalin fixation [64]. The effectiveness of proteinase K retrieval depends heavily on concentration optimization, as excessive digestion damages tissue morphology while insufficient treatment fails to adequately expose epitopes [64].
Research on skeletal tissues demonstrates that proteinase K concentration must be carefully calibrated for different applications. For in situ hybridization (ISH) on FFPE sections of rat distal femurs, optimal results were achieved at 10 μg/mL for 15 minutes, whereas standard concentrations of 100 μg/mL produced inconsistent results with impaired morphology [64]. Similarly, for immunohistochemistry (IHC) and immunofluorescence (IF) on formalin-fixed, decalcified bone tissue, mild proteinase K digestion improved detection of targets like GFP and osteocalcin while maintaining tissue integrity [64].
A significant advantage of proteinase K-based retrieval is its gentleness on tissue morphology compared to heat-based methods, making it particularly valuable for tissues with poor adhesion to slides, such as bone and cartilage [64]. The enzymatic approach also avoids the tissue detachment problems associated with microwave-based HIER methods and represents a more definable, controllable parameter for epitope retrieval [18] [64].
Acetone-Based Permeabilization Methods
Acetone permeabilization operates through a fundamentally different mechanism, classified as a coagulant or precipitating fixative that dehydrates samples and disrupts lipid membranes [63]. Unlike cross-linking fixatives, acetone and other alcohols (methanol, ethanol) cause protein denaturation and precipitation, which can simultaneously fix and permeabilize tissues, particularly in frozen sections or cytologic smears [63].
The permeabilization effect occurs through acetone's capacity to dissolve membrane lipids, creating pores that allow antibody penetration. This method is particularly effective for larger proteins, such as immunoglobulins, and is often employed in combination with acetone fixation for rapid processing of frozen tissues [63]. However, the dehydrating effect can alter protein conformation, potentially destroying some epitopes while revealing others.
Studies comparing fixation and permeabilization methods for flow cytometry demonstrate that acetone and methanol-based approaches can significantly alter light scatter properties and surface marker detection [41]. For instance, in intracellular staining of FoxP3 in T regulatory cells, alcohol-based permeabilization methods diminished CD45 and CD3 staining intensity compared to detergent-based approaches [41]. These findings highlight the method-dependent effects on antigen detection that must be considered during experimental design.
Direct Method Comparison and Experimental Data
The table below summarizes key comparative parameters between proteinase K and acetone-based antigen retrieval methods:
Table 1: Direct Comparison of Proteinase K vs. Acetone Antigen Retrieval Methods
| Parameter | Proteinase K (PIER) | Acetone Permeabilization |
|---|---|---|
| Mechanism of Action | Enzymatic cleavage of peptide bonds masking epitopes [64] | Solvent extraction of lipids and dehydration [63] |
| Optimal Concentration | 10 μg/mL for skeletal tissues [64] | 100% concentration typically used [41] |
| Incubation Time | 5-30 minutes (commonly 10-15 minutes) [18] | 10-15 minutes typically sufficient [41] |
| Temperature Conditions | Typically 37°C [18] | -20°C to room temperature [41] |
| Tissue Morphology Preservation | Good with optimized concentration [64] | Variable; can alter scatter properties [41] |
| Impact on Surface Epitopes | Minimal effect when properly optimized | Can diminish detection of some surface markers [41] |
| Best Applications | FFPE tissues, heat-sensitive antigens, skeletal tissues [64] | Frozen sections, cytologic smears, large proteins [63] |
The following diagram illustrates the decision-making workflow for selecting between these antigen retrieval methods based on experimental requirements:
Experimental Protocols and Methodological Implementation
Proteinase K Antigen Retrieval Protocol for FFPE Tissues
Materials Required:
- Proteinase K (commercially available as lyophilized powder)
- TE buffer or PBS (pH 7.4-8.0)
- Water bath or incubator (37°C)
- Coplin jars or humidity chambers
- FFPE tissue sections mounted on slides
Step-by-Step Procedure:
- Deparaffinization and Hydration: Process FFPE sections through xylene and graded alcohol series to water following standard protocols.
- Proteinase K Solution Preparation: Dilute proteinase K in TE buffer or PBS to achieve working concentration of 5-20 μg/mL. Optimal concentration must be determined empirically for each tissue type and antigen [64].
- Enzymatic Digestion: Apply proteinase K solution to cover tissue sections completely. Incubate at 37°C for 10-30 minutes in a humidity chamber to prevent evaporation.
- Enzyme Inactivation: Rinse slides thoroughly with PBS or distilled water to remove proteinase K and terminate digestion.
- Immunostaining Proceedure: Continue with standard immunohistochemistry or immunofluorescence protocols including blocking, antibody incubation, and detection steps.
Critical Optimization Parameters:
- Concentration Titration: Test a range from 1-50 μg/mL to identify ideal concentration [64].
- Time Optimization: Evaluate digestion times from 5-30 minutes to balance epitope retrieval against morphological preservation.
- Temperature Control: Maintain consistent 37°C temperature throughout incubation for reproducible results.
Acetone Permeabilization Protocol for Frozen Sections
Materials Required:
- High-quality acetone (analytical grade)
- Freezer (-20°C) or cold room
- Coplin jars or staining containers
- Frozen tissue sections mounted on slides
Step-by-Step Procedure:
- Section Preparation: Cut frozen sections (4-8 μm) using cryostat and mount on charged slides.
- Acetone Fixation/Permeabilization: Immerse slides in pre-chilled acetone (-20°C) for 10-15 minutes.
- Drying Step: Remove slides from acetone and air-dry for 5-10 minutes at room temperature.
- Rehydration: Rinse slides briefly in PBS or appropriate buffer to remove residual acetone.
- Immunostaining Procedure: Proceed with standard immunostaining protocols, noting that no additional permeabilization is typically required.
Methodological Variations:
- Acetone-Methanol Mixtures: Some protocols employ 1:1 acetone:methanol combinations for enhanced permeabilization.
- Temperature Variations: While -20°C is standard, some antigens may benefit from room temperature acetone treatment.
- Sequential Approaches: For challenging targets, brief acetone permeabilization may be followed by mild detergent treatment.
Troubleshooting Common Issues
Table 2: Troubleshooting Guide for Antigen Retrieval Methods
| Problem | Proteinase K Solutions | Acetone Solutions |
|---|---|---|
| Poor Signal | Increase enzyme concentration (up to 20 μg/mL) or extend incubation time (up to 30 min) [64] | Ensure acetone is fresh; extend treatment time to 15 min; try room temperature incubation |
| Tissue Damage | Reduce enzyme concentration (as low as 1 μg/mL); shorten incubation time [64] | Shorten treatment time; use colder temperature; consider milder detergents |
| High Background | Optimize concentration to prevent over-digestion; include proper blocking steps | Ensure complete drying after treatment; include additional washing steps |
| Inconsistent Results | Standardize incubation conditions; use fresh enzyme aliquots; control temperature precisely | Use fresh acetone; standardize drying time; control humidity |
| Loss of Surface Epitopes | N/A (method primarily affects intracellular epitopes) | Combine with gentle cross-linking fixatives like low-concentration PFA |
Research Reagent Solutions and Essential Materials
Successful implementation of antigen retrieval techniques requires specific research reagents optimized for each methodology. The following table outlines essential solutions and their applications:
Table 3: Essential Research Reagents for Antigen Retrieval Techniques
| Reagent/Solution | Composition/Characteristics | Primary Function | Method Application |
|---|---|---|---|
| Proteinase K | Serine protease with broad specificity (27-35 kDa) | Enzymatic cleavage of peptide bonds masking epitopes [64] | PIER (Protease-Induced Epitope Retrieval) |
| Acetone | Organic solvent (CH₃)₂CO, analytical grade | Lipid dissolution and membrane permeabilization [63] | Solvent-based permeabilization |
| TE Buffer | 10 mM Tris-Cl, 1 mM EDTA, pH 7.4-8.0 | Optimal buffer for proteinase K activity | PIER method |
| PBS | Phosphate-buffered saline, pH 7.2-7.4 | Isotonic buffer for biological applications | Both methods (dilution, washing) |
| Formalin | 37-40% formaldehyde in water with methanol stabilizer | Tissue fixation creating cross-links that require retrieval [63] | Pre-retrieval processing |
| Triton X-100 | Non-ionic detergent | Supplemental permeabilization for membrane barriers [18] | Often combined with both methods |
| Tween 20 | Mild non-ionic detergent | Reducing non-specific background staining [18] | Washing steps in both methods |
The selection between proteinase K enzymatic retrieval and acetone permeabilization methods should be guided by specific experimental parameters, including tissue processing method, antigen characteristics, and preservation requirements. Proteinase K-based PIER offers distinct advantages for FFPE tissues requiring gentle but effective epitope unmasking, particularly for heat-sensitive antigens or delicate morphological contexts [64]. Acetone permeabilization provides a rapid, straightforward approach for frozen sections and cytoplasmic targets, though with potential impacts on surface epitope detection [41].
For researchers and drug development professionals, methodological consistency remains paramount when comparing experimental results across samples or studies. The validated protocols and troubleshooting guidance provided herein serve as a foundation for optimizing antigen detection in diverse research contexts. As spatial biology and multiplexed imaging technologies advance, precisely calibrated antigen retrieval will continue to play a critical role in extracting meaningful biological insights from fixed tissue specimens.
In cellular and molecular biology, high-quality staining is a cornerstone technique for visualizing subcellular structures, proteins, and nucleic acids. The effectiveness of these techniques is highly dependent on the sample preparation process, particularly the permeabilization step, which enables reagents to access their intracellular targets. Permeabilization methods primarily involve the use of detergents or organic solvents, each with distinct mechanisms and outcomes. Proteinase K, an enzymatic permeabilization agent, and acetone, an organic solvent, represent two fundamentally different approaches. This guide objectively compares these methods and explores how additives like detergents and volume exclusion agents can be strategically used to optimize staining quality, reduce background, and enhance specific signal detection for research and drug development applications.
Mechanism of Action: How Permeabilization Agents Work
Detergent-Based Permeabilization
Detergents are amphipathic molecules containing a hydrophilic head and a hydrophobic tail. In aqueous solutions, they form micelles above a specific threshold known as the critical micelle concentration (CMC). Their cleaning and permeabilizing action arises from their ability to integrate into lipid bilayers. The hydrophobic tails interact with lipid chains and hydrophobic regions of proteins, while the hydrophilic heads remain exposed to the aqueous environment. This interaction disrupts the lipid bilayer, creating pores that allow the passage of antibodies, probes, and other staining reagents into the cell. The effectiveness of a detergent is influenced by its properties, including CMC, micelle size, and charge [65].
Detergents are categorized based on the nature of their hydrophilic head group (ionic, non-ionic, or zwitterionic) and their relative harshness. Harsh detergents, such as Triton X-100 and NP-40, are highly effective at disrupting membranes but can also extract proteins and damage epitopes, potentially affecting cell morphology and antigenicity. Milder detergents, such as Tween 20, saponin, and digitonin, create more subtle pores by complexing with cholesterol and are preferred for preserving membrane structures and labile antigens [18].
Organic Solvent Permeabilization
Organic solvents like acetone and methanol act through a different mechanism. They precipitate and dehydrate cellular components by removing water and dissolving lipids. Acetone, in particular, is a potent lipid solvent that rapidly extracts cholesterol and phospholipids from cellular membranes, thereby permeabilizing the cell. A key characteristic of acetone and methanol is that they simultaneously fix and permeabilize tissues and cells. While this can simplify protocols, it can also lead to excessive protein precipitation and epitope masking, potentially reducing staining intensity for certain targets [18].
Enzymatic Permeabilization with Proteinase K
Proteinase K is a broad-spectrum serine protease that permeabilizes tissues by hydrolyzing peptide bonds. It does not create physical pores like detergents but rather digests proteins that form a physical barrier to probe penetration. This is especially useful for densely packed tissues or when epitopes have been masked by over-fixation, particularly with cross-linking fixatives like paraformaldehyde. However, its activity must be carefully controlled, as over-digestion can damage protein epitopes, lead to cell loss, and compromise tissue morphology [4] [66].
Table 1: Core Mechanisms of Different Permeabilization Agents
| Agent Type | Example Agents | Primary Mechanism | Key Impact on Sample |
|---|---|---|---|
| Harsh Detergents | Triton X-100, NP-40 | Disrupts lipid bilayers and protein-lipid interactions | Can denature proteins, alter morphology; effective for strong staining |
| Mild Detergents | Tween-20, Saponin | Forms transient pores in membranes (e.g., by complexing with cholesterol) | Better preservation of membrane structure and antigen integrity |
| Organic Solvents | Acetone, Methanol | Dissolves lipids and precipitates cellular proteins | Simultaneously fixes and permeabilizes; can make some epitopes inaccessible |
| Enzymes | Proteinase K | Digests peptide bonds to break down physical barriers | Can retrieve masked epitopes; risk of destroying antigens and morphology |
Comparative Analysis: Proteinase K vs. Acetone Permeabilization
The choice between proteinase K and acetone permeabilization is highly target- and sample-dependent. A study on intracellular RNA detection in HeLa cells provides illustrative quantitative data. Researchers tested six permeabilization methods and evaluated the success of flow cytometric in situ hybridization based on the frequency of stained cells and fluorescence intensity [66].
In this study, treatment with 0.2% Tween-20 for 30 minutes yielded the highest percentage of positively stained cells (97.9%) and the strongest fluorescence intensity. In contrast, acetone treatment was not among the top-performing methods, suggesting it may be less effective for enabling RNA probe access in this specific context. The study concluded that Tween-20 provided the highest mean fluorescence, highlighting the importance of optimizing the permeabilization agent [66].
Proteinase K is often employed as a crucial step in protocols for difficult samples. In RNA in situ hybridization (ISH) of Drosophila ovaries, a key optimization involved using 50 μg/mL proteinase K for 1 hour to permeabilize the tissue, followed by a post-fixation step. This aggressive treatment was necessary to overcome the barriers posed by the tissue's thickness and surrounding muscle layer, allowing probes to penetrate and produce a strong, specific signal [4].
However, when the goal is dual protein and RNA detection (IF/FISH), proteinase K becomes problematic. Its protein-digesting activity damages protein epitopes, leading to a weak or absent immunofluorescence signal. For such multimodal applications, an alternative permeabilization strategy is required. The optimized IF/FISH protocol for Drosophila ovaries omits proteinase K and instead uses a combination of xylenes and detergent-based buffers (RIPA) to permeabilize the tissue after protein immunofluorescence is completed. This approach preserves protein antigenicity while still allowing sufficient RNA probe penetration [4].
Table 2: Experimental Comparison of Permeabilization Methods
| Method | Protocol Details | Application Context | Performance Outcome | Key Limitations |
|---|---|---|---|---|
| Proteinase K | 50 μg/mL for 1 hr, 37°C [4] | RNA ISH on Drosophila ovaries | Strong, specific signal for RNA detection | Can damage morphology; destroys protein antigens |
| Proteinase K | 0.01-0.1 μg/mL for 5-15 min, 37°C [66] | RNA FISH on HeLa cells | Tested but was not the top-performing method | Requires extensive optimization of concentration and time |
| Tween-20 | 0.2% for 30 min, 25°C [66] | RNA FISH on HeLa cells | Optimal: 97.9% positive cells, highest fluorescence intensity | Milder action may be insufficient for dense tissues |
| Acetone | Application as a fixative/permeabilizer [18] | General IHC | Simultaneous fixation and permeabilization | Can mask epitopes, leading to weak staining |
| Xylene + RIPA | Sequential use post-IF [4] | IF/FISH on Drosophila ovaries | Preserves protein signal while allowing RNA probe access | A multi-step, longer protocol |
The following workflow diagram illustrates the decision-making process for selecting and optimizing a permeabilization method based on the experimental goal and sample type:
The Scientist's Toolkit: Key Research Reagent Solutions
Successful staining experiments rely on a toolkit of specific reagents, each serving a defined function. The following table details essential materials used in the permeabilization and staining protocols cited in this guide.
Table 3: Essential Research Reagents for Permeabilization and Staining
| Reagent | Type/Function | Example Application |
|---|---|---|
| Proteinase K | Broad-spectrum serine protease for enzymatic permeabilization | Digests proteins to enable probe penetration in dense tissues for RNA ISH [4]. |
| Tween-20 | Non-ionic, mild detergent | Permeabilization for intracellular RNA detection in flow cytometry [66]. |
| Triton X-100 | Non-ionic, harsh detergent | Disrupts membranes for strong staining; use at 0.1-0.2% for short durations [18]. |
| Saponin | Mild detergent that complexes with cholesterol | Creates transient pores for immunostaining while preserving membrane structures [18]. |
| Acetone | Organic solvent for fixation and permeabilization | Simultaneously fixes and permeabilizes cells by dissolving lipids [18]. |
| Paraformaldehyde | Cross-linking fixative | Stabilizes cellular structure by creating protein cross-links; standard pre-permeabilization fixative [4] [66]. |
| Uranyl Formate | Heavy metal salt for negative stain EM | Stains background in EM grids to visualize protein particles against detergent micelle background [67]. |
| RIPA Buffer | Lysis buffer containing a mix of detergents | Effective permeabilization buffer for complex tissues in IF/FISH protocols [4]. |
Advanced Applications: Mitigating Detergent-Related Artifacts in Structural Biology
The challenges of using detergents extend beyond light microscopy into structural biology techniques like cryo-electron microscopy (cryo-EM). A significant pitfall in negative stain EM, often used for sample pre-screening, is the background caused by empty detergent micelles. This background can be easily mistaken for detergent-embedded protein samples, leading to misinterpretations of sample quality and homogeneity [67].
A case study on common detergents showed that most produce significant background in negative stain EM, even below their nominal critical micelle concentration (CMC). This highlights that the mere presence of a clean background in EM is not a reliable indicator of a detergent-free sample. To counteract this, researchers must include a "detergent-only" control sample during the screening process. This control is essential for accurately attributing observed particles to the protein of interest rather than to detergent micelles or other artifacts [67].
For high-resolution cryo-EM studies, the intrinsic properties of detergent-extracted samples can pose challenges during grid preparation and data processing. Detergents can reduce image contrast and form spurious micellar structures, a problem particularly acute for small membrane proteins where the micelle size can rival that of the protein itself [68].
Advanced strategies to mitigate these issues involve replacing or supplementing detergents with alternative stabilizing agents:
- Amphipols: These amphiphilic polymers adsorb onto the hydrophobic transmembrane surfaces of proteins, shielding them from water without forming large micelles. This eliminates interference from micellar structures during analysis [68].
- Nanodiscs: These are nanoscale phospholipid bilayers stabilized by a membrane scaffold protein (MSP) belt. They provide a more native-like lipid environment for membrane proteins, which can significantly enhance stability and functionality [68].
- Salipro Nanoparticles: This system uses the lipid-binding protein saposin A to stabilize and reconstitute membrane proteins into a lipid-bilayer-like environment, useful for helical inner membrane proteins [67] [68].
These advanced systems often require an initial detergent extraction but offer a superior environment for structural studies, underscoring the ongoing innovation in the use of additives for improving staining and visualization in science.
Permeabilization is a foundational step in cell and tissue analysis, enabling detection of intracellular proteins, RNA transcripts, and other biomolecules by allowing reagents to penetrate cellular membranes. The choice between enzymatic and solvent-based permeabilization methods represents a critical decision point that directly impacts experimental outcomes, signal intensity, and morphological preservation. This guide provides an empirical comparison between two widely used permeabilization techniques: the enzymatic approach using proteinase K and the solvent-based method using acetone.
The optimization of these methods requires careful titration of multiple parameters, including concentration, incubation time, and temperature. As research advances toward more complex applications such as multiplexed assays and single-cell analyses, the strategic selection and precise optimization of permeabilization conditions becomes increasingly vital for generating reliable, reproducible data. This guide synthesizes current experimental evidence to establish a framework for researchers to systematically evaluate and implement these essential techniques.
Methodological Comparison: Mechanisms and Applications
Proteinase K Permeabilization
Proteinase K is a broad-spectrum serine protease that enzymatically digests proteins, thereby disrupting cellular structures and enabling reagent penetration. This method is particularly effective for breaking down tough structural components, including collagen-rich matrices and cuticles, making it indispensable for challenging sample types.
The mechanism involves proteolytic cleavage of peptide bonds, which effectively digests cellular membranes and extracellular matrices that would otherwise block reagent access. This enzymatic action must be carefully controlled through parameter optimization to balance adequate permeabilization with preservation of cellular morphology and antigen integrity.
Acetone Permeabilization
Acetone permeabilization operates through a fundamentally different mechanism. As an organic solvent, acetone rapidly dehydrates samples and dissolves lipid components of cellular membranes, creating physical pores that allow reagent passage.
This method simultaneously fixes and permeabilizes tissues through precipitation of cellular components. The process is particularly effective for preserving certain antigen epitopes that may be damaged by proteolytic digestion, though it may compromise some structural details due to its dehydrating effects.
Quantitative Comparison of Performance Parameters
Table 1: Comprehensive Comparison of Proteinase K and Acetone Permeabilization Methods
| Parameter | Proteinase K | Acetone |
|---|---|---|
| Optimal Concentration | 20-100 μg/ml (common range); 50 μg/ml successfully used in Drosophila ovaries for 1 hour [4]; 40 U/mL determined optimal for PFA cross-link reversal in FD-seq [14] | Pure (100%) application is standard [18] |
| Incubation Time | Wide range (5 min to several hours): 1 hour for Drosophila ovaries [4]; 30-40 minutes for nematode preparations (following collagenase treatment) [69] | Typically 10 minutes or less [18] |
| Temperature Conditions | 37°C (common); Room temperature for some applications; 56°C for cross-link reversal in FD-seq [14] | Room temperature or 4°C (often used during fixation process) [18] |
| Primary Mechanism | Enzymatic digestion of proteins [4] [69] | Solvent action that dissolves lipids and dehydrates samples [18] |
| Tissue Morphology | Potential for degradation with over-digestion; nematodes showed complete degradation with extended incubation [69] | Generally good preservation, though may cause shrinkage due to dehydration [18] |
| Ideal Applications | Tough samples (nematodes, Drosophila ovaries), RNA in situ hybridization, PFA-fixed samples [4] [14] [69] | Intracellular proteins, viral antigens, some enzyme antigens [18] |
| Compatibility | Excellent with antibody staining (with optimized concentration); compatible with various fixatives including PFA [4] [14] | Excellent with many antibody-based detection methods; may not be effective for all antigens [18] |
Experimental Protocols for Method Optimization
Proteinase K Titration Protocol for Challenging Samples
Based on successful applications in recalcitrant tissues, this protocol provides a framework for optimizing proteinase K treatment:
Sample Preparation: Fix tissues appropriately (commonly with 4% PFA). For particularly resistant structures, consider preliminary treatments such as collagenase (30 minutes for nematodes) or bleach dissociation (2 minutes in 1% bleach for nematodes) [69].
Enzyme Solution Preparation: Prepare proteinase K at 50 μg/ml in an appropriate buffer (commonly PBS or TE). For PFA cross-link reversal, use 40 U/mL in lysis buffer [14].
Incubation Conditions: Apply enzyme solution to samples and incubate at 37°C for 1 hour. For less resistant tissues, begin with shorter incubation times (5-30 minutes) [18].
Termination: Remove enzyme solution and rinse samples with PBS or glycine solution to stop proteolytic activity.
Post-Fixation: For some applications, particularly when performing RNA FISH after protein immunostaining, post-fixation for 30 minutes helps preserve morphology [4].
Experimental Note: Researchers working with Drosophila ovaries found that a 1-hour treatment at 50 μg/ml produced optimal results, while nematode preparations required a combination of collagenase (30 minutes) followed by proteinase K (30-40 minutes) for effective penetration without excessive degradation [4] [69].
Acetone Permeabilization Protocol
The acetone permeabilization protocol is notably simpler but requires careful attention to timing:
Sample Preparation: Fix tissues appropriately. Note that acetone can serve as both fixative and permeabilization agent.
Application: Apply cold (-20°C) or room temperature 100% acetone to samples for 10 minutes [18].
Rehydration: Gradually rehydrate samples through a series of buffered solutions if subsequent aqueous-based assays are required.
Processing: Proceed with staining protocols. Note that no special termination step is required.
Experimental Note: Acetone permeabilization is particularly recommended for cytoskeletal, viral, and some enzyme antigens, but researchers should verify compatibility with their specific antibodies or probes [18].
Decision Framework for Method Selection
The following flowchart illustrates the strategic decision-making process for selecting between proteinase K and acetone permeabilization methods based on experimental requirements:
Advanced Applications and Integrated Workflows
Protein-RNA Co-detection Workflows
Advanced applications frequently require simultaneous detection of proteins and RNA transcripts, creating complex optimization challenges. Research on Drosophila ovaries demonstrated that reversing the traditional order of detection steps—performing protein immunofluorescence before RNA FISH—markedly improves protein signal preservation while maintaining RNA detection sensitivity [4].
For such integrated workflows, proteinase K treatment presents significant challenges for protein epitope preservation. The optimized protocol instead employs alternative permeabilization methods including xylenes and detergent combinations (RIPA buffer) to enable sufficient probe penetration while maintaining protein antigen integrity [4].
Single-Cell RNA Sequencing Applications
In high-throughput single-cell RNA sequencing of PFA-fixed cells, FD-seq methodology demonstrates that proteinase K treatment (40 U/mL) effectively reverses cross-links while preserving RNA integrity. This application requires precise optimization, as excessive proteinase K concentration reduces RNA yield and increases variability [14].
The integration of proteinase K into droplet-based single-cell sequencing workflows enables analysis of fixed cells while maintaining transcript detection sensitivity comparable to live cells, with strong correlation in gene expression levels between fixed and live samples [14].
Essential Research Reagent Solutions
Table 2: Key Reagents for Permeabilization Optimization
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Enzymatic Permeabilization | Proteinase K, Trypsin, Pepsin | Digests protein barriers; Proteinase K effective for tough samples and PFA cross-link reversal [14] [18] |
| Solvent Permeabilization | Acetone, Methanol | Dissolves lipids; Acetone recommended for cytoskeletal and viral antigens [18] |
| Detergent Permeabilization | Triton X-100, Tween-20, Saponin | Creates membrane pores; Concentration critical (0.1-0.5%) [18] |
| Fixation Reagents | Paraformaldehyde (PFA) | Standard cross-linking fixative; requires antigen retrieval or permeabilization [14] |
| Enzyme Buffers | TE buffer, PBS, Lysis buffer | Proteinase K activity depends on appropriate buffering conditions [14] |
| Hybridization Reagents | HCR probes, FISH reagents | RNA detection; require effective permeabilization for tissue penetration [4] [69] |
Troubleshooting and Quality Assessment
Evaluating Permeabilization Efficiency
Assessment of permeabilization success should include multiple validation metrics:
DAPI Staining Efficiency: In nematode studies, successful proteinase K permeabilization resulted in 100% of samples showing nuclear staining, compared to no penetration without treatment [69].
Antibody Penetration: For whole-mount samples, effective permeabilization enables antibody access to internal structures. In optimized nematode preps, 52.5% of samples showed specific antibody binding to longitudinal muscles [69].
RNA Probe Accessibility: Successful in situ HCR in nematodes demonstrated specific effector transcript detection in gland cells, with 45.4% of prepared samples showing clear signal [69].
Addressing Common Challenges
Excessive Digestion: Proteinase K concentrations too high or incubation times too long degrade tissue morphology. Nematode studies showed complete tissue degradation with extended proteinase K exposure [69].
Incomplete Permeabilization: For challenging samples, combined approaches (collagenase + proteinase K) significantly improve outcomes compared to single-enzyme treatment [69].
Antigen Damage: When protein detection is compromised by proteinase K, alternative permeabilization methods (xylenes + detergents) can preserve epitopes while allowing sufficient probe penetration [4].
The empirical optimization of permeabilization methods requires systematic titration of critical parameters including concentration, time, and temperature. Through careful experimental design and validation, researchers can select the optimal permeabilization strategy for their specific application, balancing the competing demands of sample penetration, morphological preservation, and biomolecule integrity. The continued refinement of these fundamental techniques supports increasingly sophisticated analytical approaches across biological research and drug development.
Head-to-Head Comparison and Impact on Multi-Omics Analyses
In the field of cellular and molecular biology, the successful visualization of intracellular targets via techniques such as immunofluorescence (IF) and in situ hybridization (ISH) is critically dependent on effective sample permeabilization. This step is necessary for enabling reagents like antibodies and nucleic acid probes to access their intracellular targets by disrupting the lipid bilayer of the cell membrane. The choice of permeabilization method, however, is not trivial, as it directly influences key performance metrics including staining intensity, background signal, and spatial resolution [38].
Among the numerous available techniques, permeabilization with the protease proteinase K and the organic solvent acetone represent two fundamentally different approaches. A direct comparison of their performance is essential for optimizing experimental outcomes in demanding applications such as multiplexed protein and RNA detection. This guide provides a head-to-head evaluation of proteinase K and acetone permeabilization, drawing on experimental data to outline their distinct advantages, limitations, and ideal use cases within the broader context of methodological research for cell and tissue analysis.
The core function of any permeabilization method is to create pores in the cell membrane. Proteinase K and acetone achieve this through two distinct biochemical mechanisms, which are summarized in the following diagram and table.
Table 1: Fundamental Characteristics of the Two Permeabilization Methods
| Characteristic | Proteinase K | Acetone |
|---|---|---|
| Primary Mechanism | Enzymatic digestion of proteins at the cell membrane and in the interior [4] | Solvent action that dissolves membrane lipids and simultaneously fixes [18] |
| Typical Concentration | 20–100 µg/mL [4] [69] | 100% (as a cold solution) [18] |
| Incubation Conditions | 37°C for 5 minutes to 1 hour [4] [69] | Room temperature or -20°C for 5–10 minutes [18] |
| Key Effect on Sample | Can degrade protein antigens, damaging epitopes for antibody binding [4] | Can disrupt lipid-based structures and precipitate proteins, potentially masking epitopes [18] [38] |
Direct Performance Comparison
Experimental data from optimized protocols reveals a clear performance trade-off between these methods: proteinase K generally enables superior staining intensity for nucleic acid detection, whereas acetone better preserves protein antigenicity for immunofluorescence.
Staining Intensity
Staining intensity is a direct measure of the signal derived from the target molecule, reflecting the efficiency of probe access and binding.
- RNA Detection: Proteinase K is superior for RNA in situ hybridization (ISH) and FISH applications. In a study on Drosophila ovaries, proteinase K permeabilization produced a strong, specific signal for the gurken and broad transcripts within 15-45 minutes of color reaction development. In contrast, alternative methods, including acetone, resulted in weak, variable, or undetectable RNA signals even after 2-5.5 hours of development [4].
- Protein Detection: Acetone is often preferred for immunofluorescence. As a solvent, it simultaneously fixes and permeabilizes tissues by dehydrating them and precipitating proteins, which helps preserve many protein epitopes [18]. Proteinase K's enzymatic activity often damages protein antigens, leading to a weak or absent immunofluorescence signal. This is a critical limitation for dual protein-RNA detection protocols (IF/FISH) [4].
Background Signal
Background signal refers to non-specific staining that can obscure the true signal and reduce the signal-to-noise ratio.
- Proteinase K: The enzymatic digestion can increase background by exposing more non-specific binding sites or by releasing cellular contents that contribute to autofluorescence. The optimal concentration and incubation time must be empirically determined, as over-digestion can severely damage tissue morphology and increase background [69] [38].
- Acetone: While generally clean, acetone permeabilization can sometimes lead to heightened background autofluorescence in certain tissues [38]. The background generated is often more consistent and manageable compared to the variable effects of proteinase K digestion.
Resolution
Resolution here refers to the precision of subcellular localization, which can be compromised by diffusion of reaction products or poor preservation of morphology.
- Proteinase K: In colorimetric ISH, the enzymatic reaction product can diffuse, limiting the resolution for pinpointing transcript localization [4]. For FISH, however, the fluorescent signal is confined and, when combined with proteinase K's efficient probe access, allows for high-resolution, subcellular mRNA localization [4]. A significant drawback is the potential degradation of subcellular structures, compromising morphological resolution.
- Acetone: By precipitating proteins, acetone often provides excellent preservation of cellular morphology and structural details. This makes it valuable for resolving the localization of proteins within specific compartments, such as the cytoskeleton [18].
Table 2: Direct Comparison of Performance Metrics
| Performance Metric | Proteinase K | Acetone |
|---|---|---|
| RNA Staining Intensity | High (Strong, specific signal in ISH/FISH) [4] | Low (Weak, variable signal) [4] |
| Protein Staining Intensity | Low (Can destroy protein epitopes) [4] | High (Preserves many protein epitopes) [18] |
| Typical Background | Moderate to High (Dependent on digestion optimization) [69] [38] | Low to Moderate (Can cause autofluorescence) [38] |
| Morphological Resolution | Lower (Can damage tissue ultrastructure) [4] [69] | Higher (Good preservation of structure) [18] |
| Best Application | RNA-centric techniques (e.g., FISH, ISH) [4] | Protein-centric techniques (e.g., Immunofluorescence) [18] |
Experimental Protocols for Performance Testing
To directly compare these methods, researchers can adapt the following standardized protocols. The workflow for such a comparative experiment is illustrated below.
Proteinase K Permeabilization Protocol (for Drosophila Ovaries)
This protocol, adapted from a study optimizing for Drosophila ovaries, balances permeabilization with morphology preservation [4].
- Step 1: Fixation. Fix dissected tissues in 4% (wt/vol) paraformaldehyde with 1% (vol/vol) DMSO for 1 hour at room temperature.
- Step 2: Dehydration/Rehydration. Pass tissues through a graded ethanol series (30%, 50%, 70%, 100%) to dehydrate. Samples can be stored indefinitely in 100% ethanol at -20°C at this point. For processing, rehydrate through a reverse ethanol series.
- Step 3: Permeabilization. Incubate tissues in a solution of 50 µg/mL proteinase K in PBS for 1 hour at room temperature.
- Step 4: Post-fixation. To halt proteinase K activity and re-stabilize the tissue, post-fix in 4% PFA for 30 minutes.
- Step 5: Washing. Rinse tissues thoroughly with PBS containing 0.1% Tween 20 (PBTw) to remove residual fixative and enzymes.
- Key Note: The proteinase K concentration and incubation time are critical. For more delicate tissues or specific protein targets, reduce the concentration to 20 µg/mL and/or shorten the incubation time to preserve antigenicity [4].
Acetone Permeabilization Protocol
This is a standard protocol commonly used for immunofluorescence of cultured cells or cryosections [18].
- Step 1: Fixation. Fix cells or tissues with an appropriate fixative (e.g., 4% PFA for 10-20 minutes). Note: Acetone can also act as a fixative itself when used cold.
- Step 2: Washing. Wash fixed samples thoroughly with PBS or a similar buffer to remove salts and serum that could precipitate.
- Step 3: Permeabilization. Incubate samples in pre-chilled 100% acetone at -20°C for 5-10 minutes.
- Step 4: Drying and Rehydration. Allow the acetone to fully evaporate at room temperature. This drying step is part of the permeabilization process. Subsequently, rehydrate the samples in PBS or a blocking buffer before proceeding with immunostaining.
- Key Note: Acetone can make samples brittle. Handle them with care after treatment. This method is not recommended for whole-mount tissues as it does not provide sufficient penetration [4].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials required for implementing and testing these permeabilization methods.
Table 3: Essential Research Reagents for Permeabilization Studies
| Reagent/Material | Function/Description | Example Application |
|---|---|---|
| Proteinase K | A broad-spectrum serine protease that digests proteins and creates pores in tissues. | Permeabilization for RNA FISH in thick tissues like Drosophila ovaries [4]. |
| Acetone | An organic solvent that dissolves lipids and dehydrates/precipitates proteins. | Rapid permeabilization and fixation for cell monolayer immunofluorescence [18]. |
| Paraformaldehyde (PFA) | A cross-linking fixative that preserves cellular structure by creating covalent bonds between proteins. | Standard primary fixative for both protocols prior to permeabilization [4]. |
| Triton X-100 / Tween 20 | Detergents used for mild permeabilization and as additives in wash buffers to reduce non-specific binding. | Can be used as a standalone mild permeabilization agent or in combination with other methods [4] [18]. |
| Collagenase | An enzyme that specifically digests collagen, a major component of extracellular matrices and certain cuticles. | Used in tandem with proteinase K to permeabilize tough structures like nematode cuticles [69]. |
| Digoxigenin (DIG)-labeled RNA Probes | Hapten-labeled nucleic acid probes detected by anti-DIG antibodies conjugated to enzymes or fluorophores. | Standard probe for colorimetric and fluorescent RNA in situ hybridization [4]. |
| Fluorophore-conjugated Antibodies | Secondary antibodies conjugated to fluorescent dyes for target detection in IF and FISH. | Essential for visualizing bound primary antibodies in fluorescence microscopy [4] [38]. |
The choice between proteinase K and acetone permeabilization is not a matter of one being universally superior, but rather of selecting the right tool for the specific biological question and target.
- For experiments where the primary goal is the sensitive and high-resolution detection of RNA transcripts, particularly in challenging whole-mount tissues, proteinase K is the unequivocal leader. Its powerful enzymatic action ensures probe penetration and strong staining intensity, though careful optimization is required to manage background and preserve some structural integrity.
- For studies focused on protein localization via immunofluorescence, acetone is often the more reliable choice. Its solvent action effectively permeabilizes while maintaining the antigenicity of many protein targets, resulting in strong specific signal with good morphological preservation.
The emerging frontier of multiplexed experiments that require simultaneous detection of proteins and RNA (IF/FISH) presents a significant challenge. Research indicates that a sequential protocol, performing immunofluorescence first followed by FISH using a combination of xylenes and detergent permeabilization (omitting proteinase K), successfully balances the conflicting needs of epitope preservation and RNA probe penetration [4]. This integrated approach, leveraging the strengths of different methods, represents the current state-of-the-art for complex co-localization studies.
The integrity of RNA is a paramount concern in molecular biology, directly influencing the sensitivity and accuracy of advanced techniques such as RNA-FISH (Fluorescence in situ Hybridization) and single-cell RNA sequencing (scRNA-seq). The permeabilization step, which enables reagents and probes to access intracellular RNA, is a critical determinant of RNA integrity. This guide objectively compares two common permeabilization methods—proteinase K treatment and acetone/organic solvent-based methods—within a broader research thesis on optimizing RNA visualization and sequencing outcomes. The selection between these methods involves a fundamental trade-off: achieving sufficient permeability to detect target molecules while preserving RNA quality and structural morphology [4]. For single-cell sequencing, where the isolation of intact, high-quality RNA from specific cell types is notoriously challenging due to high endogenous ribonuclease activity [70], this choice is particularly crucial. The following sections provide a detailed comparison based on experimental data, outline standardized protocols, and offer guidance for method selection in different research contexts.
Comparative Analysis: Proteinase K vs. Acetone Permeabilization
Table 1: Direct Comparison of Permeabilization Methods for RNA Studies
| Feature | Proteinase K Method | Acetone/Organic Solvent Method |
|---|---|---|
| Primary Mechanism | Enzymatic digestion of proteins [4] | Solvent-based lipid dissolution and dehydration [4] [22] |
| Typical RNA Signal Strength | Strong, consistent signal [4] | Variable; can be weak to moderate [4] |
| Impact on RNA Integrity | Risk of RNA degradation with over-digestion [4] | Generally better preservation of RNA structure [4] |
| Impact on Protein Antigens | Damaging; often destroys epitopes for subsequent IF [4] | Preserved; suitable for combined IF/FISH experiments [4] |
| Tissue Morphology | Can be compromised if over-digested [4] | Better preserved [4] |
| Typical Application | Standalone RNA-FISH with high sensitivity requirements [4] | Dual protein-RNA detection (IF/FISH); scRNA-seq [4] [70] |
| Protocol Duration | Relatively long (e.g., 1-hour treatment) [4] | Relatively short (e.g., 20-minute treatment) [22] |
| Key Experimental Evidence | In Drosophila ovaries, provided strong gurken and broad transcript signals [4] | In Drosophila ovaries, enabled strong protein signal but variable FISH signal [4] |
Key Experimental Findings:
- A seminal study on Drosophila ovaries directly compared these methods for RNA-FISH. Proteinase K permeabilization (50 µg mL⁻¹ for 1 hour) resulted in a strong and specific RNA signal detectable within 15-45 minutes. In contrast, alternative methods, including acetone and xylenes, showed weak or undetectable signals for the transcript broad even after a 5.5-hour color reaction [4].
- For dual protein-RNA labeling (IF/FISH), reversing the standard order—performing immunofluorescence (IF) first, followed by FISH with organic solvent permeabilization—markedly improves protein detection while maintaining an adequate FISH signal. Proteinase K treatment in this context was found to be detrimental to protein epitopes, resulting in a weak or absent protein signal [4].
- In zebrafish embryo protocols, a brief 5-minute proteinase K digestion is standard for ISH, while acetone (80%) is noted as an alternative permeabilization method [22].
Detailed Experimental Protocols
Proteinase K-Dependent RNA-FISH Protocol
This protocol, optimized for Drosophila ovaries, balances permeabilization with morphology preservation and can be adapted for other tissues [4].
Workflow Diagram: Proteinase K RNA-FISH
Key Reagents and Solutions:
- Fixative: 4% (wt/vol) Paraformaldehyde (PFA) with 1% (vol/vol) DMSO.
- Proteinase K Solution: 50 µg mL⁻¹ proteinase K in an appropriate buffer (e.g., PBS with Tween-20).
- Hybridization Buffer: Contains 50% formamide, 1.5x SSC, 5 µg/mL heparin, 0.1% Tween-20, and 50 µg/mL yeast tRNA [4] [22].
- Detection System: Tyramide Signal Amplification (TSA) for enhanced sensitivity [4].
Organic Solvent-Based IF/FISH Protocol
This 5-day protocol for Drosophila ovaries prioritizes the preservation of protein epitopes for simultaneous detection with RNA [4].
Workflow Diagram: IF/FISH with Organic Solvents
Key Reagents and Solutions:
- Permeabilization Agents: Xylenes and detergents (e.g., RIPA buffer) in sequence [4].
- Blocking Solution for IF: 5% normal sheep serum, 2% bovine serum albumin (BSA), and 1% DMSO in PBTween [4].
- Hybridization Buffer: As in protocol 3.1 [4].
Single-Cell RNA Sequencing Tissue Processing
While not directly comparing proteinase K and acetone, scRNA-seq protocols for complex tissues like the pancreas and fruit pericarp highlight the critical importance of tailored digestion and permeabilization for RNA quality.
- Pancreas Digestion: The "DIE-RNA" protocol uses a rapid digestion with collagenase P in alternating EGTA and calcium buffers to isolate acinar and ductal cells, successfully yielding high-quality RNA (RIN > 8.4) suitable for sequencing, even during pancreatitis [70].
- Plant Tissue (Pitaya Pericarp) Optimization: scRNA-seq of senescing fruit required optimized protoplast isolation to handle high lignin and polysaccharide content, followed by careful filtration to remove debris without damaging viable cells [71].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for RNA Integrity Preservation and Detection
| Reagent/Solution | Function | Critical Consideration |
|---|---|---|
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves tissue architecture and immobilizes RNA [4]. | Concentration and fixation time must be optimized to balance morphology and probe accessibility. |
| Proteinase K | Serine protease that digests proteins to permeabilize the tissue [4]. | Activity varies by batch; concentration and time must be tightly controlled to prevent RNA degradation. |
| Formamide | Component of hybridization buffer that lowers the melting temperature of nucleic acids [4] [22]. | Allows for specific hybridization at manageable temperatures. |
| Yeast tRNA & Heparin | Blocking agents in hybridization buffer [4] [22]. | Reduce non-specific binding of probes to tissue. |
| Tyramide Signal Amplification (TSA) | Enzyme-mediated amplification system that greatly enhances FISH signal detection [4]. | Crucial for detecting low-abundance transcripts, especially with sub-optimal permeabilization. |
| Collagenase | Enzyme for tissue dissociation in single-cell protocols [70]. | Essential for breaking down fibrous tissues like pancreas to obtain viable single cells. |
| RNase Inhibitors | Chemicals or proteins that inhibit RNase activity [70]. | Vital for maintaining RNA integrity during all steps post-tissue collection, especially in RNase-rich tissues. |
Decision Framework and Concluding Recommendations
Decision Diagram: Choosing a Permeabilization Method
The choice between proteinase K and acetone/organic solvent permeabilization is not one of superiority but of application-specific suitability.
- For Maximum RNA-FISH Signal: Proteinase K is the preferred method when the sole aim is to achieve the highest possible sensitivity for RNA detection, as evidenced by its strong and consistent signal in model tissues [4].
- For Combined IF/FISH Studies: Organic solvent-based permeabilization is indispensable. It preserves protein epitopes, enabling accurate simultaneous protein localization and RNA detection, albeit with a potential compromise in RNA signal intensity that may be offset by TSA [4].
- For Single-Cell RNA Sequencing: The permeabilization debate shifts toward tissue dissociation strategies. The primary goal is to isolate intact, viable single cells with high-quality RNA. Enzymatic cocktails like collagenase are often central to this process, as demonstrated in protocols for pancreas and fruit pericarp [71] [70]. The extreme sensitivity of scRNA-seq to RNA integrity demands methods that minimize RNase exposure and mechanical stress during tissue processing.
In conclusion, the impact of permeabilization on RNA integrity is a foundational consideration that directly dictates the success of downstream RNA analyses. Researchers must align their permeabilization strategy with the primary objective of their study, whether it is ultra-sensitive RNA detection, multi-omics integration at the single-cell level, or spatiotemporal mapping of gene expression.
In immunofluorescence (IF) and immunohistochemistry (IHC), the accurate detection of intracellular targets hinges on effective permeabilization—the process of creating openings in cell membranes to allow antibodies access to their epitopes. The choice of permeabilization agent directly influences antigenicity, antibody binding efficiency, and ultimately, experimental outcomes. Among the various methods, proteinase K (an enzymatic approach) and acetone (an organic solvent) represent two fundamentally different strategies. This guide objectively compares these methods, drawing on experimental data to illustrate their performance in preserving epitope integrity and facilitating optimal antibody binding, providing researchers with evidence-based selection criteria.
Mechanisms of Action: How Proteinase K and Acetone Work
Understanding the fundamental mechanisms of proteinase K and acetone reveals why they differ significantly in their effects on protein epitopes.
Proteinase K is a broad-spectrum serine protease that permeabilizes tissue by digesting proteins and cleaving peptide bonds [18]. In immunostaining protocols, it is used in a controlled manner to break down cross-linked proteins at the cell surface, thereby exposing masked antigenic epitopes that became inaccessible during chemical fixation, particularly from aldehydes like formaldehyde and paraformaldehyde [18] [4]. Its action is primarily on proteins, and the degree of permeabilization depends on concentration and incubation time.
Acetone, as an organic solvent, functions by dehydrating the sample and precipitating cellular components [72]. It dissolves lipids from cell membranes, thus physically creating pores that allow antibodies to pass through [72]. Unlike proteinase K, it does not enzymatically cleave proteins but can alter the three-dimensional structure of proteins, potentially revealing linear epitopes hidden within the native conformation [72].
The diagram below illustrates the decision-making workflow for selecting and optimizing these permeabilization methods.
Comparative Performance Data
The selection between proteinase K and acetone involves trade-offs between signal strength, structural preservation, and epitope compatibility. The following table summarizes the key characteristics of each method based on experimental findings.
Table 1: Direct Comparison of Proteinase K and Acetone Permeabilization Methods
| Parameter | Proteinase K | Acetone |
|---|---|---|
| Mechanism of Action | Enzymatic digestion of proteins; cleaves peptide bonds to expose masked epitopes [18]. | Organic solvent that dehydrates samples, precipitates proteins, and dissolves lipids [72]. |
| Effect on Epitopes | Can reveal epitopes masked by aldehyde cross-linking [18] [4]. May destroy sensitive protein epitopes if over-used [18]. | Can disrupt protein conformation, revealing linear epitopes but potentially destroying conformational epitopes [72]. |
| Impact on Morphology | Can damage tissue morphology if used excessively or for too long [18] [4]. | Harsher on structure; can cause loss of lipids and soluble proteins, compromising cellular integrity [72]. |
| Typical Protocol | 5-30 minutes incubation at 37°C, commonly at a concentration of 50 µg/ml [18] [4]. | Often used as a combined fixative and permeabilizer by incubating chilled samples for 10 minutes at -20°C [72]. |
| Ideal Use Case | Critical for retrieving epitopes in heavily cross-linked, aldehyde-fixed tissues [18] [4]. | Effective for targets sensitive to cross-linking or for simultaneous fixation and permeabilization [72]. |
Experimental data underscores the performance differences between these methods. In a study optimizing RNA FISH in Drosophila ovaries, proteinase K treatment (50 µg/ml for 1 hour) resulted in a strong and specific signal for the transcript gurken within 15 minutes of the color reaction. In contrast, acetone-permeabilized samples showed extremely weak and variable signals for the transcript broad even after 5.5 hours, demonstrating the superior efficiency of proteinase K for challenging targets in certain tissue types [4].
Furthermore, the effect on protein epitopes is a critical consideration. While proteinase K is highly effective at unmasking epitopes, it is a destructive enzyme. Over-treatment can lead to the degradation of the target protein itself, resulting in a complete loss of signal [18]. Acetone, while less specific, poses a different risk: by precipitating and potentially denaturing proteins, it can irreversibly alter the three-dimensional structure of conformational epitopes, making them unrecognizable to their corresponding antibodies [72].
Detailed Experimental Protocols
To ensure reproducible results, adherence to detailed protocols is essential. Below are standardized methodologies for implementing proteinase K and acetone permeabilization, compiled from experimental procedures.
Proteinase K Permeabilization Protocol
This protocol is adapted from optimized in situ hybridization (ISH) procedures for Drosophila ovaries [4] and general antigen retrieval guidelines [18].
Table 2: Key Reagents for Proteinase K Permeabilization
| Reagent | Function/Description |
|---|---|
| Proteinase K | A broad-spectrum serine protease. Stock solution is typically reconstituted in water or buffer and stored at -20°C. |
| Tris-EDTA Buffer (pH 7.4) or PBS | A neutral buffer used to dilute proteinase K to the working concentration and to maintain enzyme activity. |
| Glycine (2 mg/ml in PBS) | Optional stop solution to quench proteinase K activity after the incubation period. |
| Post-fixation Solution (4% PFA) | Used to re-fix samples after permeabilization to maintain structural integrity during subsequent steps. |
- Sample Preparation: Begin with tissues or cells that have been fixed, typically with 4% paraformaldehyde (PFA). Following fixation, wash samples thoroughly with PBS.
- Permeabilization: Prepare a working solution of proteinase K (e.g., 50 µg/ml) in Tris-EDTA buffer or PBS [4]. Incubate the samples in this solution at 37°C for a defined period. The incubation time is critical and must be optimized; a typical starting range is 5 to 30 minutes, though some protocols use up to 1 hour for dense tissues [18] [4].
- Stop Reaction: Remove the proteinase K solution and wash the samples briefly with PBS. Optionally, incubate for a few minutes in a glycine solution (2 mg/ml in PBS) to inactivate any residual enzyme.
- Post-fixation: To prevent ongoing degradation and preserve morphology, re-fix the samples in 4% PFA for 10-15 minutes at room temperature [4].
- Washing: Rinse samples thoroughly with PBS before proceeding to immunostaining steps.
Acetone Permeabilization Protocol
Acetone is frequently used as a simultaneous fixative and permeabilizing agent, particularly for cell cultures [72].
- Sample Preparation: Culture cells on glass coverslips. Ensure cells are rinsed with PBS to remove culture media.
- Fixation/Permeabilization: Chill anhydrous acetone to -20°C. Incubate the coverslips in the chilled acetone for 5-10 minutes. Note: Acetone fixation will also permeabilize the cells [18].
- Drying: After incubation, allow the acetone to fully evaporate from the samples in a fume hood.
- Rehydration and Washing: Gently rehydrate and wash the cells with PBS or your immunostaining buffer before applying antibodies.
For samples that have already been cross-linked with aldehydes, acetone can be used as a secondary permeabilization step. After aldehyde fixation and a PBS wash, incubate the samples in chilled acetone for 5-10 minutes, followed by thorough PBS washing [72].
The Scientist's Toolkit: Essential Research Reagents
Successful permeabilization requires more than just the primary agent. The table below lists key reagents and their functions in the context of these protocols.
Table 3: Research Reagent Solutions for Permeabilization Studies
| Reagent | Function in Protocol |
|---|---|
| Paraformaldehyde (PFA) | A cross-linking fixative that preserves cellular structure but can mask epitopes, creating the need for enzymatic retrieval [4] [72]. |
| Triton X-100 | A non-ionic detergent used for permeabilization; it non-selectively interacts with lipids and proteins and is effective for tough membranes [18] [72]. |
| Tween 20 | A mild non-ionic detergent often used in wash buffers to reduce non-specific background binding [18]. |
| Saponin | A mild detergent that interacts with cholesterol to create pores in membranes while leaving many membrane-associated proteins in place [72]. |
| Tris-EDTA Buffer | A common buffer for diluting and maintaining the activity of proteinase K [18]. |
| Sulfo-NHS-SS-Biotin | A cell-impermeable biotinylation reagent used in surface protein labeling experiments to validate permeabilization efficiency and confirm intracellular access [73]. |
The choice between proteinase K and acetone permeabilization is not a matter of one method being universally superior, but rather of matching the method to the specific experimental context. Proteinase K is a powerful tool for recovering antigenicity in aldehyde-fixed tissues where epitopes are deeply masked, though it demands careful optimization to avoid destroying the very targets it aims to reveal. Acetone offers a rapid, combined fixation-permeabilization approach that can be ideal for certain epitopes and applications, but it risks damaging delicate cellular structures and conformational epitopes. The experimental data and protocols provided herein offer a framework for researchers to make an informed decision, laying the groundwork for rigorous and reproducible immunostaining results.
The Drosophila melanogaster ovary serves as a premier model system for investigating how signaling pathways and cellular morphologies interact to govern development in complex tissues. The precise spatial and temporal regulation of signals between germline and somatic cells ensures proper formation of the stem cell niche, differentiation of various cell types, and eventual production of mature oocytes [74] [75]. Studying these processes relies heavily on robust methodological approaches for visualizing gene expression and protein localization, with permeabilization techniques serving as a critical foundation for successful experimental outcomes.
This case study objectively compares two permeabilization methods—proteinase K enzymatic treatment and acetone-based solvent permeabilization—within the context of Drosophila ovarian research. We provide comparative data on their performance in supporting the visualization of key signaling pathways and morphological features, along with detailed experimental protocols to guide researchers in selecting the optimal approach for their specific investigations.
Experimental Approaches for Tissue Permeabilization
Proteinase K Enzymatic Permeabilization
Proteinase K is a serine protease that digests proteins and enables antibody access to epitopes by breaking protein cross-links formed during fixation [18].
Protocol: Following standard fixation and washing steps, tissues are digested with 10 µg/ml proteinase K in phosphate-buffered saline with Tween 20 (PBTween) for precisely 5 minutes at room temperature [22] [76]. The reaction is terminated by post-fixing samples in 4% paraformaldehyde for 20 minutes, followed by thorough washing with PBTween before proceeding with hybridization or immunohistochemistry [22].
Applications: This method is particularly effective for in situ hybridization protocols and for antibodies targeting antigens masked by extensive protein cross-linking [18]. It has been successfully employed in studies of larval zebrafish and Drosophila ovaries to detect mRNA expression patterns [22] [76].
Acetone Solvent-Based Permeabilization
Acetone permeabilizes tissues through solvent action, dissolving lipids and dehydrating samples to create pores in cellular membranes.
Protocol: Fixed tissues are treated with 80% acetone/20% water solution for 20 minutes at room temperature, followed by washing with PBTween before hybridization or staining [22]. Acetone fixation itself can simultaneously permeabilize tissues, streamlining the workflow [18].
Applications: This method is often recommended for cytoskeletal, viral, and some enzyme antigens, and is particularly suitable for immunohistochemistry protocols where epitope integrity is crucial [18].
Table 1: Comparison of Permeabilization Method Characteristics
| Characteristic | Proteinase K | Acetone |
|---|---|---|
| Mechanism | Enzymatic digestion | Solvent action |
| Typical Concentration | 10 µg/ml | 80% solution |
| Incubation Time | 5 minutes | 20 minutes |
| Temperature | Room temperature | Room temperature |
| Primary Applications | in situ hybridization, heavily cross-linked epitopes | Immunohistochemistry, cytoskeletal antigens |
| Tissue Morphology | Potential damage with over-digestion | Better preservation |
Impact on Signaling Pathway Visualization
The choice of permeabilization method significantly impacts the quality of signal detection for key pathways governing Drosophila oogenesis.
Critical Signaling Pathways in Drosophila Ovaries
The development and function of the Drosophila ovary is coordinated by multiple conserved signaling pathways. The following diagram illustrates the key pathways and their primary functions in niche formation and oogenesis:
Visualizing the activity of these pathways requires effective permeabilization to allow probes or antibodies access to their targets. Proteinase K treatment has proven particularly effective for detecting FGF signaling components, which are crucial for regulating apical cell proliferation and epithelial sheath formation during larval and pupal stages [77]. Similarly, studies of Hippo and JAK/STAT pathways that regulate terminal filament cell formation benefit from the epitope retrieval provided by enzymatic permeabilization [74].
Quantitative Performance Comparison
Table 2: Method Performance in Visualizing Key Signaling Pathways
| Signaling Pathway | Proteinase K Efficiency | Acetone Efficiency | Biological Role |
|---|---|---|---|
| FGF Signaling | High | Moderate | Supports fertility by regulating epithelial sheath formation [77] |
| Hippo Pathway | High | Moderate | Controls terminal filament cell proliferation [74] |
| JAK/STAT | High | Moderate | Regulates terminal filament cell formation [74] |
| Notch Signaling | High | High | Promotes cap cell formation [74] |
| BMP Pathway | Moderate | High | Maintains germline stem cells [75] |
| Wnt Signaling | Moderate | High | Regulates follicle stem cell maintenance [78] |
| EGFR Pathway | Moderate | High | Supports escort cell extensions [75] |
Morphological Analysis Outcomes
The permeabilization method directly influences the quality of morphological assessment in Drosophila ovarian tissues.
Tissue Architecture Preservation
Acetone permeabilization generally provides superior preservation of overall tissue architecture, including:
- Germarium organization with distinct regions 1, 2a, 2b, and 3
- Terminal filament structure composed of stacked cuboidal cells
- Follicle cell monolayer integrity surrounding developing cysts
- Microtubule networks and actin cytoskeleton organization [79]
In contrast, proteinase K can cause varying degrees of tissue damage if incubation times are not carefully controlled, potentially disrupting the delicate morphology of the germarium and early egg chambers [22] [18].
Subcellular Structure Visualization
For studies requiring visualization of subcellular structures, acetone permeabilization offers distinct advantages:
- Meiotic spindle morphology: Actin populations within the spindle are better preserved using acetone, crucial for studies of chromosome segregation [79].
- Cellular protrusions: Actin-based cytonemes and microtubule-rich cytocensors involved in niche-stem cell signaling maintain their structural integrity with acetone treatment [75].
- Membrane organization: Microvilli and other specialized membrane structures remain intact with solvent-based permeabilization.
Experimental Protocols for Drosophila Ovaries
Proteinase K Permeabilization Workflow
The following diagram outlines the complete experimental workflow for proteinase K-based permeabilization in Drosophila ovarian tissue:
Key Considerations:
- Precise timing is critical during proteinase K treatment to balance epitope accessibility against tissue degradation
- Post-fixation is essential to stabilize tissues after enzymatic digestion
- Concentration optimization may be needed for different ovarian stages or fixation conditions
This protocol has been successfully used to detect FGF signaling components in developing ovaries, revealing their expression within somatic cells at larval and pupal stages [77].
Acetone Permeabilization Protocol
Workflow Steps:
- Fixation: 4% paraformaldehyde for 20 minutes at room temperature
- Washing: 3 × 5 minutes with PBTween
- Permeabilization: 80% acetone/20% water for 20 minutes at room temperature
- Washing: 3 × 5 minutes with PBTween
- Staining: Proceed with immunohistochemistry or other staining protocols
Advantages:
- Simplicity: Fewer steps than enzymatic methods
- Compatibility: Can be combined with methanol fixation
- Morphology: Superior preservation of cellular structures
This method has proven effective for visualizing bioelectrical signaling components in follicular epithelium, including ion channels and gap junctions [80].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Drosophila Ovarian Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| Permabilization Agents | Proteinase K, Acetone | Enable antibody/probe access to intracellular targets | Fundamental step for IHC and ISH [22] [18] |
| Signaling Pathway Reporters | Fz3-RFP (Wnt), Stat92E (JAK/STAT) | Visualize pathway activity in live or fixed tissues | Monitoring niche signaling [78] [74] |
| Cell Type-Specific Markers | Traffic jam (somatic cells), Vasa (germ cells) | Identify specific cell populations within ovary | Lineage tracing and cell identification [81] |
| Genetically-Encoded Sensors | ArcLight (Vmem), pHluorin-Moesin (pHi) | Monitor bioelectrical signals in live tissues | Studying bioelectrical regulation [80] |
| Gal4 Drivers | c587-Gal4, tj-Gal4, nos-Gal4vp16 | Target gene expression to specific cell types | Functional studies in precise cell populations [77] [81] [74] |
| Cytoskeletal Markers | SiR-actin, Jupiter-GFP, Phalloidin | Visualize actin and microtubule networks | Studying cytoskeletal organization [79] [75] |
The choice between proteinase K and acetone permeabilization methods for Drosophila ovarian research depends primarily on the experimental goals. Proteinase K offers superior results for in situ hybridization and visualizing targets masked by extensive protein cross-linking, making it ideal for studying signaling pathways like FGF, Hippo, and JAK/STAT. Conversely, acetone permeabilization provides better preservation of tissue morphology and subcellular structures, and performs excellently for immunohistochemistry targeting cytoskeletal elements and membrane proteins.
Researchers should consider these performance characteristics when designing experiments to investigate the complex signaling and morphological outcomes in Drosophila ovaries. The protocols and comparative data provided here offer a foundation for selecting the optimal permeabilization approach to address specific research questions in developmental biology and beyond.
This guide objectively compares the performance of proteinase K and acetone permeabilization methods, detailing their compatibility and outcomes with flow cytometry, immunohistochemistry (IHC), and spatial transcriptomics technologies.
Permeabilization Mechanism and Technology Compatibility
Table 1: Core Characteristics and Technology Compatibility
| Feature | Proteinase K | Acetone |
|---|---|---|
| Primary Mechanism | Enzymatic digestion of proteins [14] [53] | Organic solvent that dissolves lipids and dehydrates cells [82] |
| Typical Application | Pre-treatment for RNA retrieval in spatial transcriptomics and sequencing of fixed cells [14] [53] | Standalone fixative and permeabilization agent, often for intracellular proteins [82] |
| Compatibility with Flow Cytometry | Requires optimization; may affect light scatter and surface epitopes [82] | Compatible; can be used for intracellular staining and DNA content analysis [82] |
| Compatibility with IHC/mIF | Used for antigen retrieval; can be combined with other methods [83] | Direct use possible; preserves some epitopes damaged by other fixatives [82] |
| Compatibility with Spatial Transcriptomics | High; widely used in protocols for FFPE and PFA-fixed tissues [14] [53] [83] | Not commonly used as a primary permeabilization agent in major spatial transcriptomics protocols [84] |
| Key Advantage | Effective reversal of cross-links for nucleic acid access; enables sequencing of fixed cells [14] | Simplicity (fixation and permeabilization in one step); good for certain phospho-antigens [82] |
| Key Limitation | Requires careful optimization of concentration and time to prevent tissue damage or RNA degradation [14] | Can destroy some epitopes; not suitable for all targets; precludes concurrent use of protein-based fluorophores in flow cytometry [82] |
Experimental Data and Performance Comparison
Performance in Spatial Transcriptomics and Single-Cell Sequencing
Table 2: Quantitative Performance in Transcriptomics Applications
| Parameter | Proteinase K Performance | Acetone Performance | Experimental Context |
|---|---|---|---|
| RNA Integrity Number (RIN) | Maintained high RIN (>8.0) after cross-link reversal [14] | Data not available in search results | Total RNA extraction from PFA-fixed cells [14] |
| Single-Cell Capture Efficiency | ~1% cross-droplet contamination (comparable to live cells) [14] | Data not available in search results | Species-mixing experiment with FD-seq [14] |
| Number of Genes Detected | Median ~640 genes (fixed) vs ~675 (live cells) [14] | Data not available in search results | Single-cell RNA sequencing with FD-seq [14] |
| Gene Expression Correlation | Strong correlation with live cell data (R values not specified) [14] | Data not available in search results | Comparison of fixed vs. live cell sequencing [14] |
| Spatial Transcriptomics Integration | Directly integrated into STcEM and Pixelated RT-LAMP workflows [53] [83] | Not featured in major spatial transcriptomics protocols [84] | MERFISH and on-chip RT-LAMP protocols [53] [83] |
Performance in Protein Detection and Imaging
Table 3: Performance in Protein-Based Applications
| Parameter | Proteinase K Performance | Acetone Performance | Experimental Context |
|---|---|---|---|
| Epitope Preservation | Can be harsh; may damage some protein epitopes [82] | Variable; can preserve epitopes damaged by PFA, but destroys others [82] | General flow cytometry and IHC experience [82] |
| Signal-to-Noise Ratio | Data not available in search results | Can provide improved signal for certain intracellular targets [82] | Intracellular staining for flow cytometry [82] |
| Compatibility with Protein Fluorophores | Compatible when used before antibody staining [82] | Denatures protein-based fluorophores (e.g., PE, APC) [82] | Flow cytometry with conjugated antibodies [82] |
| Tissue Morphology | Preserves tissue architecture for spatial analysis [53] [83] | Data not available in search results | Spatial transcriptomics-correlated EM [83] |
Detailed Experimental Protocols
Proteinase K Protocol for Spatial Transcriptomics and Fixed-Cell Sequencing
FD-seq (Fixed Droplet RNA sequencing) Workflow [14]:
- Fixation: Fix cells with 4% Paraformaldehyde (PFA).
- Permeabilization: Treat with 0.1% Triton-X-100.
- Proteinase K Treatment: Incubate fixed and permeabilized cells with Proteinase K at an optimal concentration of 40 U/mL in lysis buffer.
- Cross-link Reversal: Heat at 56°C for 1 hour to reverse PFA cross-links.
- Single-Cell Partitioning: Perform droplet-based single-cell encapsulation (e.g., using Drop-seq microfluidics).
- Library Preparation & Sequencing: Proceed with standard single-cell RNA sequencing library preparation.
Pixelated RT-LAMP Workflow [53]:
- Tissue Pixelation: Partition tissue cryosection onto a silicon chip with microwells.
- Fixation: Fix pixelated tissue with acetone for 10 minutes at room temperature.
- Proteinase K Treatment: Digest with Proteinase K (7.5 µg mL⁻¹) for 30 minutes to permeabilize cells.
- Reagent Loading: Load RT-LAMP amplification reagents into wells.
- On-Chip Amplification: Perform real-time RT-LAMP reaction on a hot plate at 65°C for 45 minutes.
Acetone Protocol for Flow Cytometry
Methanol/Acetone Fixation and Permeabilization Workflow [82]:
- Preparation: Create a single-cell suspension (1x10⁶ cells per tube).
- Wash: Wash cells in 1x PBS and pellet by centrifugation (200-300g for 2-5 minutes). Discard supernatant.
- Fixation/Permeabilization: Add 100% ice-cold acetone (100 µL per tube). Incubate for 10-15 minutes on ice.
- Note: Acetone acts as both a fixative and permeabilization agent. Polystyrene/plastic tubes are not suitable for use with acetone.
- Wash: Wash cells twice with suspension buffer (e.g., PBS with 0.5% BSA) to remove acetone.
- Antibody Staining: Proceed with intracellular antibody staining steps.
- Critical Note: Acetone denatures protein-based fluorophores (e.g., PE, APC). These antibodies must be applied after the acetone step if using this method.
Research Reagent Solutions
Table 4: Essential Materials for Permeabilization Workflows
| Reagent | Function | Example in Protocol |
|---|---|---|
| Proteinase K | Enzymatically digests proteins to reverse cross-links and access nucleic acids [14] [53] | 40 U/mL in FD-seq; 7.5 µg mL⁻¹ in Pixelated RT-LAMP [14] [53] |
| Acetone | Organic solvent that fixes and permeabilizes cells by dissolving lipids and dehydrating samples [82] | 100%, ice-cold, 10-15 minute incubation [82] |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves cellular structure; often used prior to Proteinase K [14] [82] | 4% solution for cell fixation [14] [82] |
| Triton X-100 | Non-ionic detergent for permeabilization; can be used alongside Proteinase K [14] | 0.1% for initial permeabilization in FD-seq [14] |
| Saponin | Mild detergent for permeabilization; creates pores in membranes without dissolving them [82] | 0.1-0.3% in PBS for reversible permeabilization [82] |
| Reverse Transcriptase | Enzyme for cDNA synthesis; critical for transcriptomics after permeabilization [14] [85] | Used in FD-seq and DEPICT-seq after fixation/permeabilization [14] [85] |
The choice between proteinase K and acetone permeabilization is highly dependent on the analytical technology and target molecule. Proteinase K is the strongly preferred method for spatial transcriptomics and single-cell RNA sequencing applications, as it effectively reverses cross-links from aldehyde fixation, enabling robust RNA retrieval and maintaining transcriptomic data quality comparable to live cells [14]. In contrast, acetone finds its primary application in flow cytometry and IHC for detecting certain intracellular proteins, especially those where aldehyde fixation may mask epitopes, though it is incompatible with protein-based fluorophores [82]. Researchers must align their permeabilization strategy with their downstream analytical platform to ensure optimal data quality and reliability.
This guide objectively compares the performance of proteinase K and acetone permeabilization methods in multi-omics workflows, with a specific focus on their impact on transcriptomic and proteomic data quality. The evaluation is framed within a broader thesis investigating optimal tissue preparation protocols that minimize molecular loss. Permeabilization is a critical step for enabling reagent access to intracellular targets, and the choice of method presents a significant trade-off: proteinase K effectively digests proteins for superior RNA detection but often compromises protein epitopes, while acetone and other organic solvents preserve protein integrity for immunofluorescence but offer inconsistent and often weaker RNA signal penetration [4] [18].
Quantitative data and detailed methodologies from controlled experiments are summarized below to provide a clear basis for comparison and informed protocol selection.
Quantitative Comparison of Permeabilization Methods
The following table synthesizes key experimental findings from direct comparisons of permeabilization techniques, highlighting their impact on signal quality for RNA and protein detection.
Table 1: Performance Comparison of Permeabilization Methods in Omics Studies
| Permeabilization Method | Target Omics | Reported Signal Quality (Transcriptomic) | Reported Signal Quality (Proteomic) | Key Experimental Findings |
|---|---|---|---|---|
| Proteinase K | RNA ISH / FISH [4] | Strong signal for germline (gurken) and follicle cell (broad) transcripts [4]. | Little or no protein signal; epitopes damaged [4]. | gurken signal detected in 15 min; broad in 45 min. Deemed "too aggressive" for combined IF/FISH [4]. |
| Acetone | RNA ISH / FISH [4] | Weak and variable signal; inconsistent penetration [4]. | Strong and specific signal; epitopes preserved [4]. | After 5.5 hours, broad expression was "extremely weak and variable" [4]. |
| Xylenes + RIPA | Combined IF/FISH [4] | Strong and consistent FISH signal [4]. | Strong and specific IF signal [4]. | Optimized protocol for dual RNA-protein detection; successful in Drosophila ovaries [4]. |
| RNaseH (Enzymatic) | Spatial Multi-omics (DBiT-plus) [86] | High-quality spatial transcriptome data [86]. | Intact tissue for subsequent CODEX multiplexed protein imaging [86]. | Enables sequential profiling of transcriptome and proteome from the same tissue section [86]. |
| Triton X-100 (Detergent) | Spatial Multi-omics (DBiT-plus) [86] | N/A (used after cDNA retrieval) | Used for permeabilization in CODEX protein staining workflow [86]. | Standard in multiplexed immunofluorescence protocols post-fixation [86] [18]. |
Detailed Experimental Protocols
The quantitative data in Table 1 is derived from the following rigorously optimized experimental procedures.
Protocol for Proteinase K-based Permeabilization in RNA FISH
This protocol is optimized for Drosophila ovaries but illustrates general principles for RNA-focused studies [4].
- Tissue Fixation: Fix dissected tissues in 4% paraformaldehyde (PFA) with 1% DMSO for 1 hour.
- Dehydration/Rehydration: Treat tissue with a graded ethanol series (e.g., 50%, 70%, 90%, 100%) for storage and permeabilization preparation.
- Permeabilization: Incubate tissue with 50 µg/ml of proteinase K for 1 hour at room temperature.
- Post-fixation: Re-fix tissue with 4% PFA for 30 minutes to maintain morphology after digestion.
- Pre-hybridization & Hybridization: Proceed with standard FISH procedures using digoxigenin-labeled RNA probes or TSA-based FISH.
Protocol for Solvent-based Permeabilization in IF/FISH
This dual-labeling protocol prioritizes protein antigen preservation [4].
- Initial Fixation for IF: Fix tissue with 4% PFA for 20 minutes.
- Immunofluorescence (IF): Perform complete protein IF staining with primary and secondary antibodies.
- Antibody Cross-linking: Post-fix with 4% PFA for 30 minutes to cross-link antibodies to the tissue, preserving them during subsequent FISH steps.
- Solvent Permeabilization:
- Incubate tissue in xylenes for 1 hour.
- Dehydrate in a graded ethanol series (e.g., 50%, 70%, 90%, 100%).
- Rehydrate through a reverse ethanol series.
- Detergent Permeabilization: Further permeabilize by incubating in RIPA buffer.
- FISH Procedure: Continue with standard FISH steps. Note that hybridization and wash temperatures may be adjusted for RNA probes.
Protocol for Integrated Spatial Omics (DBiT-plus)
This cutting-edge protocol uses an enzymatic approach to sequentially retrieve cDNA and image proteins from the same section [86].
- Spatial Barcoding: On-tissue section, perform in situ reverse transcription and use a microfluidic chip to deliver DNA barcodes for spatial transcriptomics (DBiT-seq).
- cDNA Retrieval (Key Step): Instead of tissue lysis, incubate the intact tissue section with Thermostable RNaseH to selectively degrade RNA in RNA-DNA hybrids, releasing the barcoded cDNA for library preparation.
- Multiplexed Protein Imaging: With the tissue section still intact, perform standard staining with a DNA-barcoded antibody panel (e.g., CODEX) and image.
- Data Integration: Use computational pipelines to register the spatial transcriptome and proteome datasets into a unified single-cell atlas.
Visualizing Experimental Workflows
The diagrams below illustrate the logical flow and critical decision points for the key protocols described.
Diagram 1: Proteinase K vs. Solvent-Based Workflows
Diagram 2: Sequential Spatial Multi-omics Workflow
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their critical functions in the permeabilization and detection workflows discussed.
Table 2: Key Reagents for Permeabilization and Multi-omics Studies
| Reagent / Solution | Function / Purpose | Application Context |
|---|---|---|
| Proteinase K | Proteolytic enzyme that digests proteins to unmask epitopes and permeabilize tissue. | RNA ISH/FISH; can be too harsh for protein antigen preservation [4] [18]. |
| Acetone / Methanol | Organic solvents that fix and permeabilize cells by dissolving lipids in cell membranes. | Common in IF protocols; preserves many protein epitopes but weak for RNA FISH [4] [18]. |
| Triton X-100 / NP-40 | Non-ionic detergents that solubilize membrane lipids. Use at 0.1-0.2% for short durations. | General permeabilization for IF; harsher than Tween 20 [18]. |
| Tween 20 / Saponin | Mild detergents for permeabilization that are less disruptive to protein structures. | Gentle permeabilization for intracellular targets; saponin is often used for cytoplasmic epitopes [18]. |
| RNaseH | Enzyme that specifically degrades the RNA strand in RNA-DNA hybrids. | Key for retrieving cDNA in spatial omics (DBiT-plus) while leaving tissue intact for protein imaging [86]. |
| Xylenes | Organic solvent that efficiently permeabilizes tissue by dissolving lipids. | Used in combination with detergents for challenging IF/FISH dual applications [4]. |
| RIPA Buffer | A lysis buffer containing detergents and salts, used for permeabilization. | Combined with xylenes in an optimized IF/FISH protocol for Drosophila ovaries [4]. |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves tissue structure by creating protein-protein bonds. | Standard fixation for both RNA and protein studies; requires antigen retrieval for masked epitopes [4] [18]. |
Conclusion
The choice between proteinase K and acetone permeabilization is not one-size-fits-all but a critical strategic decision. Proteinase K offers powerful penetration for dense tissues and is often essential for robust RNA-FISH, but it risks damaging protein epitopes and tissue morphology. Acetone provides a milder, simultaneous fixation-permeabilization that better preserves certain protein antigens and cellular structures, though it may be insufficient for deeply embedded targets. The optimal method hinges on the experimental triad: the target's subcellular location, the sample type, and the desired downstream readout, especially when integrating transcriptomic or proteomic data. Future directions point toward developing gentler, more targeted permeabilization agents and standardized protocols that minimize informational loss, thereby enhancing the fidelity of multi-omic analyses in both basic research and clinical diagnostics.
References
- 1. Autophagy-independent role of ATG9A vesicles as carriers ... [https://www.nature.com/articles/s41467-025-59605-5]
- 2. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOorYLugLVIvW1IGksMUMRbgB4xOduc4rqJVQaOUtCft7GRTsTV4r]
- 3. Assessment of Different Permeabilization Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3895578/]
- 4. Optimized RNA ISH, RNA FISH and protein-RNA double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4126239/]
- 5. ICC/IF Protocol [https://www.antibodies.com/applications/immunofluorescence/immunofluorescence-protocol]
- 6. The effect of Proteinase K treatment on GeoMx digital ... [https://www.sciencedirect.com/science/article/pii/S0031302524001910]
- 7. Staining Intracellular Antigens for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/staining-intracellular-antigens-flow-cytometry.html]
- 8. Proteinase K [https://en.wikipedia.org/wiki/Proteinase_K]
- 9. Introduction into Proteinase K [https://www.goldbio.com/blogs/articles/intro-into-pro-k?srsltid=AfmBOooP5KyBxNdb69NBsbjcQfon_HLTtXRGZwLHF5lODzR0O4stP498]
- 10. Immunocytochemistry protocol [https://www.abcam.com/en-us/technical-resources/protocols/icc-protocol]
- 11. Fixation and Permeabilization in ICC/IF [https://www.novusbio.com/support/fixation-and-permeabilization-in-icc-if?srsltid=AfmBOopiz3xwjEaoxIMDbPQ5_VmDy8CSqMJQxldETW98xOVI0d2n1u7v]
- 12. Proteinase K in Tissue Samples [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/dna-and-rna-purification/how-proteinase-k-is-used-in-tissue-samples?srsltid=AfmBOoog_lqEuvAstbED4Z1qya0mgMoqvHRumtoipRD1ymewj-AGAoaZ]
- 13. Effect of Different Proteinase K Digest Protocols and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7045302/]
- 14. High-throughput RNA sequencing of paraformaldehyde ... [https://www.nature.com/articles/s41467-021-25871-2]
- 15. Protein precipitation: A comprehensive guide [https://www.abcam.com/en-us/knowledge-center/proteins-and-protein-analysis/protein-precipitation]
- 16. Advances in Lipid Extraction Methods—A Review [https://www.mdpi.com/1422-0067/22/24/13643]
- 17. Comparison of protein precipitation methods for sample ... [https://www.sciencedirect.com/science/article/pii/S0717345820300452]
- 18. Antigen retrieval and permeabilization for IHC [https://www.abcam.com/en-us/technical-resources/guides/ihc-guide/antigen-retrieval-and-permeabilization]
- 19. Greener solvents for microelution solid phase extraction of ... [https://www.sciencedirect.com/science/article/pii/S2772582025000130]
- 20. Comparison of different serum sample extraction methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4669428/]
- 21. Cell Disruption - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/cell-disruption]
- 22. Comparative in situ hybridization protocols in zebrafish [https://pmc.ncbi.nlm.nih.gov/articles/PMC9490454/]
- 23. Purification of fibrinolytic enzyme from Bacillus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10740062/]
- 24. Enzyme Stability and Activity in Non-Aqueous Reaction ... [https://www.mdpi.com/2073-4344/6/2/32]
- 25. of Pseudomonas putida by high pressure homogenization... Disruption [https://amb-express.springeropen.com/articles/10.1186/s13568-016-0260-6]
- 26. Fixation and Permeabilization in Immunocytochemistry [https://www.bio-techne.com/applications/imaging/immunocytochemistry/fixation-and-permeabilization-in-icc-if]
- 27. Assessing the Influence of Selected Permeabilization ... [https://www.mdpi.com/2073-4468/14/1/15]
- 28. Proteinase K - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/proteinase-k]
- 29. Breaking the Bad: An Introduction to Proteinase K [https://blog.abclonal.com/blog/breaking-the-bad-an-introduction-to-proteinase-k]
- 30. 20 Answers to Important Proteinase K Questions [https://www.goldbio.com/blogs/articles/20-answers-to-important-proteinase-k-questions-plus-free-printable-fact-sheet?srsltid=AfmBOorpeNqTejU8_F0t8Z4A4po6LnCcsLd0I6M4vXwpIRF9y1gOHS90]
- 31. Guide on How to Use Proteinase K in Different Procedures [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/dna-and-rna-purification/guide-to-use-proteinase-k-procedures?srsltid=AfmBOoovoLHCHJdqyj0MxRMjmM_9pcdHwZ0Y3WFAsOVD6t8_ebczpNNP]
- 32. Antigen retrieval and permeabilization for IHC | Abcam [https://www.abcam.co.jp/technical-resources/guides/ihc-guide/antigen-retrieval-and-permeabilization]
- 33. Optimization of conditions to extract high quality DNA for PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6169501/]
- 34. Fixation Strategies and Formulations Used in IHC Staining [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/fixation-strategies-formulations.html]
- 35. Cell and Tissue Fixation 101- Top Tips For Protocol ... [https://bitesizebio.com/13459/cell-fixation-101-top-tips-for-protocol-optimization/]
- 36. Optimization of fixation methods for observation of bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3181414/]
- 37. Benchmarking subcellular localization and variant tolerance ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5865-0]
- 38. Ten Approaches That Improve Immunostaining: A Review ... [https://www.mdpi.com/1422-0067/23/3/1426]
- 39. Global organelle profiling reveals subcellular localization ... [https://www.sciencedirect.com/science/article/pii/S0092867424013448]
- 40. Successful Immunofluorescence: Fixation & Permeabilization [https://blog.cellsignal.com/successful-immunofluorescence-fixation-and-permeabilization]
- 41. Comparison of Different Fix/Permeabilization Buffers for use in ... [https://blog.td2inc.com/comparison-of-different-fix-permeabilization-buffers]
- 42. Protocol: an improved and universal procedure for whole ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-015-0094-2]
- 43. Comparison of formalin- and acetone-fixation for ... [https://pubmed.ncbi.nlm.nih.gov/6195915/]
- 44. Effects of methanol concentration on expression levels ... [https://pubmed.ncbi.nlm.nih.gov/12474251/]
- 45. Impact of methanol concentration on secreted protein ... [https://pubmed.ncbi.nlm.nih.gov/16320364/]
- 46. The methanol-induced conformational transitions of β ... [https://www.sciencedirect.com/science/article/pii/S1044030500002269]
- 47. ICC: Staining Protocol - Formaldehyde Fixation [https://www.sysy.com/protocols/protocol-icc-pfa]
- 48. Multiplex Staining by Sequential Immunostaining and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5533273/]
- 49. Fully automated sequential immunofluorescence (seqIF) ... [https://www.nature.com/articles/s41598-023-43435-w]
- 50. Staining Strategies for Intracellular Flow Cytometry [https://fluorofinder.com/staining-strategies-for-intracellular-flow-cytometry/]
- 51. The Search for an Optimal DNA, RNA, and Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3375896/]
- 52. Troubleshooting - Immunofluorescence Assays [https://ibidi.com/content/366--troubleshooting]
- 53. Pixelated spatial gene expression analysis from tissue - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5768672/]
- 54. Systematic benchmarking of high-throughput subcellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534522/]
- 55. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOop8iIr_x_NMXf90xgr18VtXwPIkiAnVv-Wd0HYwhkVmh9mPrWty]
- 56. Ten Approaches That Improve Immunostaining: A Review of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8836139/]
- 57. Guide to Fixation and Permeabilization [https://fluorofinder.com/guide-to-fixation-and-permeabilization/]
- 58. ProK: An Old 'Pro' That is Still In The Game [https://www.promegaconnections.com/prok-an-old-pro-that-is-still-in-the-game/]
- 59. Fixation and Permeabilization in ICC/IF [https://www.novusbio.com/support/fixation-and-permeabilization-in-icc-if?srsltid=AfmBOoqfYswpTvS0EKSZJhE15wLLq4aP9CXZ_4WKoO7-MwoM5jnhZs2r]
- 60. 5 Important Proteinase K Protocol Tips [https://www.goldbio.com/blogs/articles/5-important-proteinase-k-protocol-tips?srsltid=AfmBOop0C7ZYCdm5Dzi_M1HFMbin1IwCOLtfAZPz2eGoA0uvVEAf-g5F]
- 61. Long-term room temperature preservation of corpse soft tissue [https://pmc.ncbi.nlm.nih.gov/articles/PMC3170604/]
- 62. Cell fixation improves performance of in situ crosslinking ... [https://www.nature.com/articles/s41467-024-52844-y]
- 63. Choosing your fixation, embedding, and antigen retrieval ... [https://www.enzo.com/note/choosing-your-fixation-embedding-and-antigen-retrieval-method-for-successful-ihc/]
- 64. Optimized protocols for in situ hybridization ... [https://www.sciencedirect.com/science/article/abs/pii/S0065128121000696]
- 65. A Rapid FT-IR-based Method for Monitoring Detergent ... [https://www.spectroscopyonline.com/view/rapid-ft-ir-based-method-monitoring-detergent-removal-biological-samples-0]
- 66. Assessment of Different Permeabilization Methods ... [https://www.ajmb.org/article?id=140]
- 67. A case study on detergent background in negative stain ... [https://www.sciencedirect.com/science/article/abs/pii/S1047847718301321]
- 68. PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12466997/]
- 69. Whole mount multiplexed visualization of DNA, mRNA, and ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-023-01112-z]
- 70. DIE-RNA: A Reproducible Strategy for the Digestion of ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2018.00129/full]
- 71. Single-cell and spatial RNA sequencing reveal the ... [https://www.nature.com/articles/s41467-024-47329-x]
- 72. Deep Dive: Fixing and Permeabilizing for ... [https://blog.addgene.org/deep-dive-fixing-and-permeabilizing-for-immunofluorescence]
- 73. Rotavirus NSP4: Cell type-dependent transport kinetics to the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3129587/]
- 74. Niche formation and function in developing tissue: studies ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-022-01035-7]
- 75. Modulation of Cell–Cell Interactions in Drosophila Oocyte ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7072342/]
- 76. All-age whole mount in situ hybridization to reveal larval ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7413480/]
- 77. FGF signaling supports Drosophila fertility by regulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4469552/]
- 78. Quantitative microscopy of the Drosophila ovary shows ... [https://www.nature.com/articles/s41467-017-01322-9]
- 79. F-actin coordinates spindle morphology and function in ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011111]
- 80. Analysing bioelectrical phenomena in the Drosophila ovary ... [https://bmcdevbiol.biomedcentral.com/articles/10.1186/s12861-020-00220-6]
- 81. A single-cell atlas and lineage analysis of the adult Drosophila ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7648648/]
- 82. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOootEF_ufQ8ksiUPPm1MTLodTaFQ1TMkYWQjpWRLAVahwT3vntSl]
- 83. Spatial Transcriptomics-correlated Electron Microscopy ... [https://www.nature.com/articles/s41467-023-39447-9]
- 84. Spatial multi-omics analyses of the tumor immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9661775/]
- 85. DEPICT-seq: Single-Cell Transcriptomic Analysis of Rare ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11483345/]
- 86. Integration of Imaging-based and Sequencing-based Spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11661374/]