Validating Single-Cell RNA-Seq in Developmental Biology: A Comprehensive qPCR Guide
This article provides a definitive guide for researchers and drug development professionals on validating single-cell RNA sequencing (scRNA-seq) findings in developmental biology using qPCR.
Validating Single-Cell RNA-Seq in Developmental Biology: A Comprehensive qPCR Guide
Abstract
This article provides a definitive guide for researchers and drug development professionals on validating single-cell RNA sequencing (scRNA-seq) findings in developmental biology using qPCR. It explores the foundational principles of both technologies, outlines robust methodological workflows for cross-validation, and offers practical troubleshooting advice to optimize experimental success. Furthermore, it synthesizes evidence from comparative studies, demonstrating how this multi-technique approach powerfully illuminates developmental trajectories, cell fate decisions, and potency states, ultimately strengthening biological conclusions for publication and translational research.
The Synergy of scRNA-seq and qPCR in Decoding Developmental Pathways
Why Validation is Non-Negotiable in Single-Cell Developmental Studies
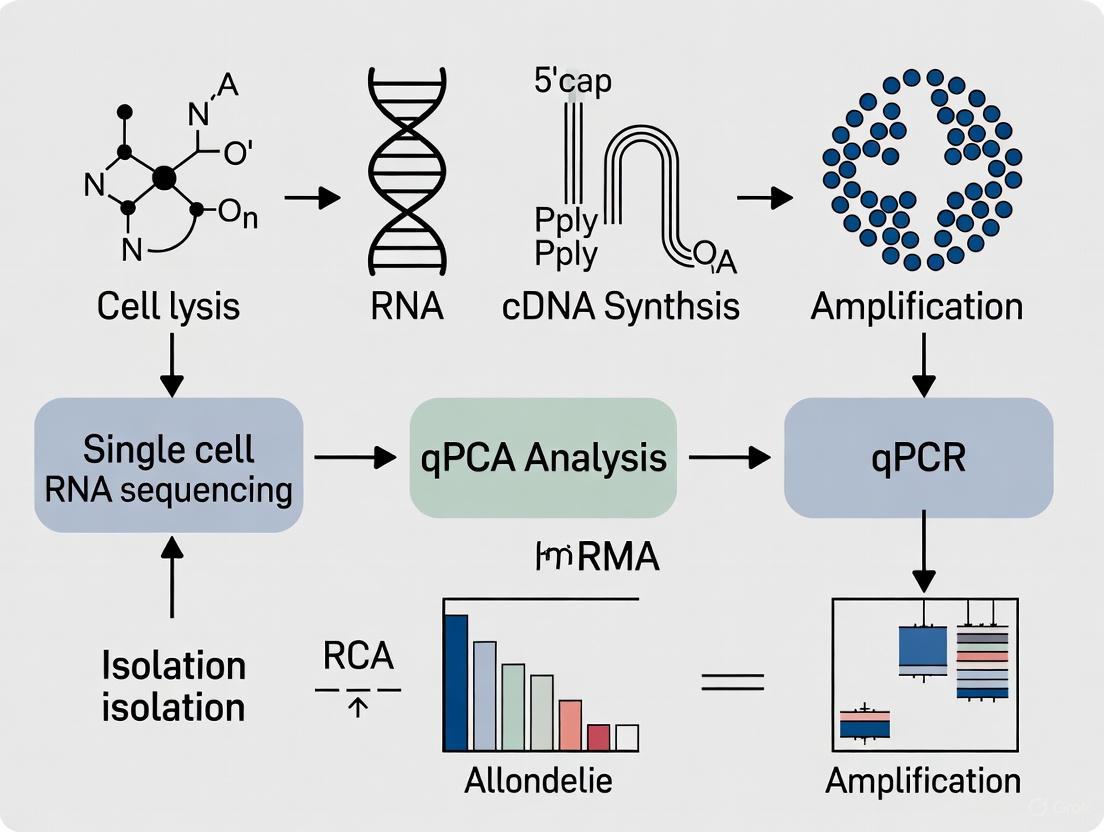
Single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology by enabling researchers to profile gene expression at unprecedented resolution, revealing cellular heterogeneity, identifying rare cell populations, and tracing lineage relationships during development [1] [2]. However, the inherent technical noise, amplification biases, and computational challenges associated with scRNA-seq necessitate rigorous validation to ensure biological conclusions are accurate and reproducible. This is particularly crucial in developmental studies where understanding precise cellular transitions can inform therapeutic strategies for developmental disorders and regenerative medicine.
Validation serves as a critical checkpoint that bridges high-dimensional screening data with biological truth. While scRNA-seq provides a comprehensive landscape of transcriptional activity across thousands of individual cells, confirmation through orthogonal methods establishes confidence in the findings, especially when investigating novel cell types, developmental trajectories, or subtle transcriptional changes that might be obscured by technical artifacts [3]. For developmental biologists studying dynamic processes from embryogenesis to tissue specialization, this validation step is non-negotiable for building accurate models of how multicellular organisms form and function.
The Validation Toolbox: Methods and Applications
Quantitative PCR (qPCR): The Gold Standard
qPCR remains the established benchmark for validating gene expression studies, including scRNA-seq data [4]. Its well-characterized workflow, sensitivity, and quantitative nature make it ideal for confirming transcriptional patterns identified in single-cell experiments.
Experimental Protocol: For validating scRNA-seq results with qPCR, researchers typically:
- Sample Preparation: Isolate RNA from a separate set of biological samples (not the same cells used for scRNA-seq) representing key developmental stages or cell populations [4]
- cDNA Synthesis: Convert RNA to cDNA using reverse transcriptase with poly(T) or random primers
- Target Amplification: Amplify genes of interest using sequence-specific primers alongside reference genes for normalization
- Quantitative Analysis: Calculate relative expression using ΔΔCt method or absolute quantification with standard curves
The correlation between scRNA-seq and qPCR measurements can be remarkably high (r > 0.84) when properly executed [5]. Notably, sample preparation volume significantly impacts accuracy, with nanoliter-volume reactions in microfluidic systems demonstrating nearly ideal regression slopes (close to 1) compared to microliter-volume preparations, highlighting the importance of reaction conditions in quantitative accuracy [5].
Spatial Validation Techniques
RNA Fluorescence In Situ Hybridization (FISH) provides spatial context to scRNA-seq findings by visualizing the precise localization of RNA molecules within tissue sections [3]. This method is particularly valuable in developmental biology for confirming the spatial distribution of cell types identified through clustering analysis.
Experimental Protocol:
- Probe Design: Design fluorescently-labeled nucleic acid probes complementary to target RNAs
- Tissue Preparation: Fix and permeabilize tissue sections while preserving RNA integrity
- Hybridization: Apply probes under conditions favoring specific binding to target sequences
- Imaging and Analysis: Visualize using fluorescence microscopy and quantify signal distribution
Immunofluorescence (IF) and Immunohistochemistry (IHC) extend validation to the protein level, confirming that transcriptional identities correspond to appropriate protein expression patterns [3]. For example, IHC validation demonstrated reduced NPTX2 protein expression in cognitively impaired individuals, aligning with scRNA-seq findings [3].
Functional Validation Approaches
Gene Manipulation Techniques including overexpression, silencing (RNA interference), and knockout (CRISPR/Cas9) provide mechanistic validation of genes identified through scRNA-seq [3]. In a study on cotton development, researchers used CRISPR/Cas9 to knockout GhLAX1 and GhLOX3 genes identified through scRNA-seq, confirming their roles in healing tissue proliferation and plant regeneration [3].
Cell Sorting and Population Validation using fluorescence-activated cell sorting (FACS) or magnetic-activated cell sorting allows physical isolation of cell populations identified computationally through scRNA-seq clustering [3]. When researchers sorted various immune cell types including macrophages, neutrophils, and NK cells using FACS, the population frequencies showed consistent alignment with scRNA-seq predictions [3].
Quantitative Comparison of scRNA-seq Validation Methods
Table 1: Validation Methods for Single-Cell RNA Sequencing in Developmental Studies
| Method | Applications | Key Advantages | Technical Considerations | Correlation with scRNA-seq |
|---|---|---|---|---|
| qPCR | Gene expression confirmation, sensitivity assessment | High sensitivity, quantitative, well-established | Requires separate biological replicates, limited to moderate number of targets | r > 0.84 with proper experimental design [5] |
| RNA FISH | Spatial localization, rare cell population confirmation | Preserves spatial context, single-molecule sensitivity | Throughput limitations, specialized imaging equipment needed | Complementary spatial information [3] |
| IF/IHC | Protein-level validation, spatial distribution | Confirms translation of transcriptional identities, widely accessible | Antibody quality dependent, semi-quantitative | Confirms protein expression patterns [3] |
| Functional Assays | Mechanistic validation, pathway interrogation | Establishes causal relationships, functional relevance | Technically demanding, time-intensive | Confirms biological significance of transcriptional findings [3] |
Case Studies in Developmental Biology
Neural Crest Development and Lineage Tracing
A compelling example of rigorous validation in developmental biology comes from studies of neural crest contributions to the enteric nervous system [6]. Researchers combined scRNA-seq with replication-incompetent avian (RIA) retrovirus lineage tracing to compare vagal and sacral neural crest contributions to gut innervation. This approach enabled transcriptional profiling of neural crest-derived cells with precise knowledge of their developmental origin, validating population-specific differentiation patterns while revealing that post-umbilical vagal neural crest more closely resembles sacral neural crest than pre-umbilical vagal neural crest [6].
Resolving Technical Artifacts
Single-cell sample preparation can introduce significant artifacts that require validation to distinguish from biological signals. Studies have shown that tissue dissociation at 37°C can induce expression of stress genes, leading to inaccurate cell type identification [7]. Performing dissociation at 4°C or utilizing single-nucleus RNA sequencing (snRNA-seq) instead minimizes these artifactual changes, but requires validation to ensure nuclear transcripts accurately represent cellular states [7].
Experimental Design and Workflow Integration
Strategic Validation Framework
Implementing an effective validation strategy requires careful planning throughout the experimental timeline:
Experimental Validation Workflow
When Validation is Essential
qPCR validation is particularly important when:
- Confirming Novel Findings: When observations challenge existing paradigms or identify previously uncharacterized cell types [4]
- Limited Replication: When scRNA-seq data is based on a small number of biological replicates, limiting statistical power [4]
- Clinical or Therapeutic Applications: When findings may inform diagnostic approaches or therapeutic development [8]
Conversely, validation may be less critical when scRNA-seq serves primarily for hypothesis generation followed by extensive functional studies, or when findings are confirmed through independent scRNA-seq experiments on larger sample sets [4].
Research Reagent Solutions
Table 2: Essential Research Tools for Single-Cell Validation Studies
| Category | Specific Examples | Applications in Validation | Key Features |
|---|---|---|---|
| Single-cell Platforms | Fluidigm C1, 10x Genomics Chromium, Dolomite Bio μEncapsulator | Platform-specific technical validation | Microfluidic handling, nanoliter reactions reduce bias [5] |
| cDNA Synthesis Kits | SMARTer Ultra Low RNA Kit, TransPlex Kit | Amplification for validation assays | High sensitivity for low input material [5] |
| Library Preparation | Nextera (Illumina), NEBNext | Sequencing library construction | Compatibility with single-cell cDNA [5] |
| Spatial Validation | RNAscope kits, Multiplexed FISH probes | Spatial confirmation of cell identities | High specificity, multiplexing capability [3] |
| Cell Isolation | FACS, magnetic bead sorting | Population purification for validation | High purity cell populations [3] |
In single-cell developmental studies, validation transcends mere technical formality—it represents a fundamental scientific imperative. The complex nature of developmental processes, combined with the technical challenges of single-cell analysis, makes independent confirmation essential for building accurate models of cellular differentiation, lineage specification, and tissue morphogenesis. As single-cell technologies continue to evolve and find applications in clinical contexts including developmental disorders and regenerative medicine, the role of validation will only grow in importance.
By implementing a rigorous, multi-faceted validation strategy that spans transcriptional, spatial, and functional confirmation, developmental biologists can ensure their findings withstand scrutiny and contribute meaningfully to our understanding of how complex organisms form and function. In the challenging but rewarding landscape of single-cell developmental biology, validation remains non-negotiable.
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed developmental biology by providing an unparalleled lens through which to observe the intricate tapestry of cellular heterogeneity. This technology enables researchers to move beyond population-level averages and capture the transcriptional states of individual cells, revealing rare cell populations, transient developmental intermediates, and dynamic lineage trajectories that were previously obscured [7] [9]. The ability to profile gene expression at single-cell resolution has made scRNA-seq an indispensable discovery engine for mapping cell states and developmental potential across diverse biological systems, from spermatogonial stem cell differentiation to T-cell specialization and cancer evolution [10] [11] [12]. By integrating scRNA-seq with complementary approaches like bulk RNA-seq and spatial transcriptomics, scientists can now construct high-resolution maps of developmental processes, identify key regulatory genes, and validate these findings through targeted experiments, thereby accelerating discovery in both basic research and drug development [13] [11] [14].
Comparative Analysis of scRNA-seq Technologies
The selection of an appropriate scRNA-seq platform is critical for experimental success, as different technologies offer distinct advantages in throughput, sensitivity, and cost. The following table summarizes the key characteristics of major scRNA-seq technologies used in developmental research.
Table 1: Comparison of scRNA-seq Technologies and Their Applications
| Technology | Throughput | Key Advantages | Transcript Coverage | Amplification Method | Ideal Developmental Biology Applications |
|---|---|---|---|---|---|
| SORT-seq | Medium (384-well plates) | Cost-effective; flexible sample size; handles large cells; suitable for small samples [15] | 3' or 5' end counting | PCR | Studies with budget constraints; projects requiring precise cell selection; cardiomyocyte research [15] |
| 10x Genomics Chromium | High (thousands of cells) | Highly standardized; well-documented; low cost per cell; integrated immune profiling [15] [9] | 3' or 5' end counting | PCR with UMIs | Large-scale atlas projects; immune cell development; tumor microenvironment studies [15] [9] |
| Smart-seq2 | Low to medium | High sensitivity; full-length transcript coverage; superior for detecting more expressed genes [7] [9] | Full-length | PCR with template switching | Alternative splicing analysis; allelic expression; detection of low-abundance transcripts [9] |
| VASA-seq | Medium (384-well plates) | Full-length total RNA; captures non-coding RNA; analyzes immature mRNA [15] | Full-length total RNA | Proprietary | Single-nucleus sequencing; non-coding RNA biology; nuclear transcriptome dynamics [15] |
| CEL-seq2/ MARS-seq | Medium to high | Low amplification noise; molecular indexing [7] [9] | 3' end counting | IVT with UMIs | Quantitative expression studies; projects requiring high quantification accuracy [9] |
The experimental workflow for scRNA-seq involves several critical steps that can significantly impact data quality. Sample preparation begins with the isolation of viable single cells from complex tissues, a process that can be achieved through fluorescence-activated cell sorting (FACS), microfluidics, or other isolation methods [7] [9]. For tissues that are difficult to dissociate or when working with frozen samples, single-nucleus RNA sequencing (snRNA-seq) provides a valuable alternative, though it primarily captures nuclear transcripts and may miss certain biological processes related to mRNA processing and metabolism [7]. Following cell isolation, library preparation involves cell lysis, reverse transcription with unique molecular identifiers (UMIs) to correct for amplification biases, cDNA amplification via PCR or in vitro transcription (IVT), and finally, deep sequencing [7] [9]. The resulting data then undergoes sophisticated computational analysis to extract biological insights.
Analytical Frameworks for Developmental Trajectories
Cell Type Identification and Marker Gene Selection
A fundamental step in scRNA-seq analysis is the identification of cell types through clustering and marker gene detection. Current benchmarking studies indicate that simple statistical methods, particularly the Wilcoxon rank-sum test and Student's t-test, often outperform more complex machine learning approaches for marker gene selection [16]. These methods effectively identify genes that exhibit large expression differences between cell types, enabling accurate annotation of biological cell types from defined clusters [16]. The Seurat and Scanpy frameworks provide widely-used implementations of these methods, facilitating the transformation of high-dimensional gene expression data into interpretable cell type classifications [11] [16].
Trajectory Inference and Cell Fate Mapping
Beyond static classification, scRNA-seq enables the reconstruction of developmental trajectories through pseudo-temporal ordering algorithms. Tools like Monocle2 model cellular transitions by arranging cells along a trajectory based on expression similarity, effectively predicting the progression from progenitor to differentiated states [11]. This approach has revealed critical insights into developmental processes, such as the dynamic expression of the Kit gene during spermatogonial stem cell differentiation and early meiosis initiation [12]. Similarly, studies of CD4+ T helper cell differentiation have demonstrated remarkable long-term program stability in circulating clones, with TCR-Track mapping revealing clear distinctions between Th1, Th17, Th22, and T regulatory subsets despite a 4-year interval between sampling [10].
Table 2: Key Computational Tools for scRNA-seq Analysis in Developmental Biology
| Tool Category | Representative Tools | Primary Function | Application in Developmental Studies |
|---|---|---|---|
| Cell Type Annotation | Seurat, Scanpy, SingleR | Cluster identification and cell type labeling [11] [17] [16] | Identifying novel cell types; characterizing heterogeneous populations [11] [17] |
| Trajectory Inference | Monocle2, PAGA, Slingshot | Reconstructing developmental pathways [11] | Mapping lineage relationships; ordering differentiation processes [11] [12] |
| Cell-Cell Communication | CellPhoneDB, NicheNet | Predicting intercellular signaling [11] | Understanding stromal-epithelial crosstalk; microenvironmental signals [11] [14] |
| Multi-omics Integration | Weighted Gene Co-expression Network Analysis (WGCNA) | Identifying co-expressed gene modules [13] [11] [12] | Linking gene networks to phenotypic traits; identifying regulator genes [13] [11] |
Diagram 1: scRNA-seq analytical workflow for developmental studies (Max Width: 760px)
Integrative Approaches with Bulk RNA-seq and Spatial Data
The combination of scRNA-seq with bulk RNA-seq data creates a powerful framework for discovery, leveraging the strengths of both approaches. While bulk RNA-seq provides higher sequencing depth and better quantification of abundant transcripts, scRNA-seq reveals cellular heterogeneity and identifies rare populations [11]. This integrative strategy has proven highly effective in multiple contexts:
In lung adenocarcinoma research, the intersection of scRNA-seq marker genes with bulk RNA-seq differentially expressed genes and WGCNA-identified hub genes enabled the construction of a robust 13-gene prognostic model that effectively stratified patient risk [11]. Similarly, in endometriosis, researchers identified mesenchymal cells as key contributors to disease pathogenesis by combining scRNA-seq and bulk RNA-seq analyses, ultimately developing an 8-gene diagnostic classifier with an AUC of 1.00 in the training cohort and 0.8125 in validation [14]. For hepatocellular carcinoma, integration of scRNA-seq with spatial transcriptomics revealed that patients with low PTM scores exhibited heightened cell proliferation and malignancy, providing insights into therapeutic responses [13].
Experimental Validation in Developmental Contexts
qPCR Validation Strategies
Validation of scRNA-seq findings is essential for establishing biological credibility, with quantitative PCR (qPCR) serving as a cornerstone validation methodology. In studies of mouse spermatogonial stem cells (SSCs), researchers employed a multi-tiered validation approach beginning with immunocytochemistry for protein-level verification of key markers including Dazl, Pou5f1 (Oct4), Gfra1, Nanog, and Kit [12]. This was followed by qPCR analysis to quantify expression differences in identified marker genes, confirming the central role of Kit in SSC differentiation and its association with retinoic acid-mediated signaling pathways [12]. Similar validation approaches in endometriosis research demonstrated consistent expression patterns for critical genes including SYNE2, TXN, NUPR1, CTSK, GSN, MGP, IER2, and CXCL12 through RT-qPCR, corroborating bioinformatics predictions from integrated scRNA-seq and bulk RNA-seq analyses [14].
Functional and Spatial Validation
Beyond transcriptional validation, functional assays are crucial for establishing biological significance. In cancer studies, cell culture models enable experimental manipulation of identified marker genes to assess their functional roles in proliferation, invasion, and drug response [11]. Spatial transcriptomics provides orthogonal validation by localizing identified cell states within tissue architecture, bridging the gap between scRNA-seq clusters and anatomical context [13]. For developmental processes, lineage tracing approaches combined with scRNA-seq offer direct validation of predicted differentiation trajectories, as demonstrated in studies of T-cell development where TCR-Track mapping confirmed the stability of Th cell functional programs over a 4-year period [10].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for scRNA-seq in Developmental Biology
| Reagent Category | Specific Examples | Function in scRNA-seq Workflow | Developmental Biology Applications |
|---|---|---|---|
| Cell Isolation Reagents | Collagenase/Dispase DNase; FACS antibodies; MACS beads [12] | Tissue dissociation; specific cell population isolation | Obtaining viable single cells from complex tissues; enriching rare progenitor populations [12] |
| Cell Culture Media | StemPro-34; N2 supplement; L-glutamine [12] | Maintaining cell viability; supporting specific cell types | Culturing spermatogonial stem cells; expanding primary tissue cells [12] |
| Library Preparation Kits | 10x Genomics Single Cell Gene Expression; Smart-seq2 reagents [15] [9] | cDNA synthesis; amplification; library construction | Generating high-quality sequencing libraries with minimal bias [15] [9] |
| qPCR Validation Reagents | SYBR Green/TAQMAN assays; reverse transcription kits; primers for marker genes [12] [14] | Validating scRNA-seq findings; quantifying gene expression | Confirming expression of key developmental regulators [12] [14] |
| Immunocytochemistry Reagents | Primary antibodies (e.g., anti-Kit, anti-Oct4); fluorescent secondary antibodies; DAPI [12] | Protein-level validation; spatial localization | Verifying protein expression of identified markers; confirming cell type identity [12] |
Signaling Pathways in Development Revealed by scRNA-seq
Diagram 2: Key signaling pathway in germ cell development (Max Width: 760px)
scRNA-seq has been instrumental in elucidating critical signaling pathways that govern developmental processes. In mouse spermatogonial stem cells, scRNA-seq analysis revealed the central role of Kit/Kitl signaling in regulating the transition from undifferentiated to differentiating spermatogonia [12]. This pathway activates four major downstream signaling cascades: (1) the PI3K/AKT pathway responsible for cell survival, adhesion, and proliferation; (2) the SRC pathway influencing cell migration; (3) the PLCG pathway essential for meiosis resumption; and (4) the MAPK cascade mediating gene transcription changes [12]. Similarly, in studies of CD4+ T helper cell differentiation, scRNA-seq combined with TCR-Track mapping has clarified the relationship between traditionally defined Th subsets and their transcriptional identities, revealing significant clonal overlap between Th1 and cytotoxic CD4+ T-cell clusters while demonstrating the clonal independence of Th1, Th2, Th17, Th22, and Treg subsets [10]. These insights resolve longstanding ambiguities in developmental immunology and provide clearer targets for therapeutic intervention.
scRNA-seq has firmly established itself as a powerful discovery engine in developmental biology, enabling researchers to map cell states and developmental potential with unprecedented resolution. The integration of scRNA-seq with complementary technologies—including bulk RNA-seq, spatial transcriptomics, and sophisticated computational algorithms—creates a robust framework for identifying novel regulatory genes, reconstructing developmental trajectories, and validating key findings through experimental approaches. As benchmarking studies continue to refine analytical methods and technology platforms evolve toward higher throughput and sensitivity, scRNA-seq is poised to deepen our understanding of developmental processes and accelerate the translation of these insights into clinical applications, particularly in regenerative medicine, infertility treatment, and cancer therapeutics [12] [14]. The continued refinement of multi-omics integration and spatial profiling technologies will further enhance our ability to map developmental pathways in their native tissue contexts, ultimately providing a comprehensive understanding of how cellular diversity emerges during development and how these processes become disrupted in disease.
In the field of developmental biology, single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to profile transcriptional heterogeneity within complex tissues, such as the migrating neural crest cell streams in chick embryos [18]. However, the identification of novel gene expression patterns from scRNA-seq data requires rigorous, precise, and quantitative validation. This is where quantitative real-time PCR (qPCR) establishes its role as the gold standard, providing an essential independent verification tool that confirms the reliability of high-throughput genomic discoveries.
This guide objectively compares the performance of qPCR with emerging PCR-based technologies, specifically digital PCR (dPCR), and provides developmental biologists with the experimental protocols and data analysis frameworks necessary to generate publication-quality validation data. Adherence to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines is emphasized throughout to ensure robust assay performance, reproducibility, and meaningful biological interpretation [19] [20].
Performance Comparison: qPCR vs. Digital PCR
While qPCR is the established technique for gene expression analysis, droplet digital PCR (ddPCR) is a newer technology that partitions a PCR reaction into thousands of nanodroplets, enabling absolute quantification without a standard curve. The choice between these technologies often depends on the specific experimental context, particularly the abundance of the target and the sample purity.
Table 1: Comparative Performance of qPCR and ddPCR for Key Assay Parameters
| Parameter | qPCR | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Quantification Method | Relative (via Cq) or absolute (requires standard curve) | Absolute, without standard curve [20] |
| Ideal Dynamic Range | Broad (typically 5-6 log orders) [19] | Excellent for low-abundance targets [20] |
| Precision with Low Abundance Targets (Cq ≥ 29) | Highly variable and susceptible to inhibitors [20] | Superior precision and reproducibility [20] |
| Effect of Sample Contaminants | Cq values and efficiency are significantly impacted; requires optimal dilution [20] | More resilient to variable levels of contaminants [20] |
| Data Acquisition | Measures fluorescence per cycle (Cq) during exponential amplification | End-point detection of positive/negative droplets [20] |
| Reaction Efficiency | Critical for accurate quantification (ideal: 90-110%) [19] | Less critical; quantification is efficiency-independent [20] |
| Multiplexing Capability | Well-established | Amenable, with careful design [20] |
A direct comparison using synthetic DNA samples demonstrated that for well-purified samples with low contamination, both technologies perform comparably. However, for the challenging samples often encountered in validation workflows—those with low target levels or variable amounts of chemical and protein contaminants (e.g., from reverse transcription reactions)—ddPCR produced more precise and reproducible data. In such cases, qPCR data showed artifactual Cq values and high variability, while ddPCR reliably quantified targets despite the contaminants [20].
Experimental Protocols for Targeted Validation
Core qPCR Workflow for scRNA-seq Validation
The following protocol is adapted from methodologies used to validate novel cell-specific signatures discovered in scRNA-seq studies of developmental models [18] [21].
- cDNA Synthesis: Convert purified total RNA from your sample (e.g., bulk tissue, sorted cells, or amplified material from single cells) into cDNA using a high-quality reverse transcription kit. Include genomic DNA removal steps.
- Assay Design: Design and validate primer pairs for your target genes (e.g., trailblazer genes from a neural crest scRNA-seq study [18]) and selected reference genes. Amplicons should typically be 70–200 bp with optimal GC content (40–60%) [19].
- Reaction Setup: Prepare reactions in triplicate using a validated qPCR master mix. A standard 20 µL reaction may contain:
- 1X qPCR Master Mix (e.g., SYBR Green I or probe-based)
- Forward and Reverse Primers (e.g., 200 nM each)
- cDNA template (e.g., 2–5 µL, depending on concentration)
- Nuclease-free water to volume.
- qPCR Run: Use the following cycling conditions on a calibrated real-time PCR instrument:
- Initial Denaturation: 95°C for 2–5 minutes.
- Amplification (40 cycles): Denature at 95°C for 10–15 seconds, then anneal/extend at 60°C for 30–60 seconds (acquire fluorescence).
- Melt Curve Analysis (for SYBR Green): 95°C for 15 sec, 60°C for 1 min, then ramp to 95°C continuously.
Data Preprocessing and Analysis Methods
The accuracy of qPCR quantification is highly dependent on data preprocessing. A study comparing analytical methods found that the "taking-the-difference" approach—which subtracts the fluorescence in one cycle from that of the subsequent cycle—outperforms traditional background subtraction by reducing estimation error [22].
Table 2: Comparison of qPCR Data Analysis Models [22]
| Model | Description | Key Finding |
|---|---|---|
| Simple Linear Regression (SLR) | Standard linear regression of log(fluorescence) vs. cycle number. | Lower accuracy and precision compared to weighted models. |
| Weighted Linear Regression (WLR) | Incorporates a weight factor (reciprocal of variance) to account for data variation. | Improved accuracy and precision over SLR; a better way to preprocess data. |
| Linear Mixed Model (LMM) | Accounts for repeated measurements (e.g., technical triplicates). | Improved precision over SLR. |
| Weighted Linear Mixed Model (WLMM) | Combines weighting for variance and random effects for replicates. | Top-performing model; offers the best precision. |
After preprocessing, the ∆∆Cq method is most commonly used for relative quantification of gene expression. For scRNA-seq validation, this involves:
- Normalization: Normalize the Cq values of your target genes to a stable reference gene (e.g.,
GAPDH,ACTB) to get ∆Cq. - Calibration: Calculate ∆∆Cq by comparing the ∆Cq of your experimental sample (e.g., trailblazer neural crest cells) to a control sample (e.g., follower cells) [18].
- Fold Change: Calculate the relative expression as Fold Change = 2^(–∆∆Cq).
Visualizing the Validation Workflow
The following diagram illustrates the logical pathway from single-cell discovery to targeted qPCR validation, a common process in developmental biology studies.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Tools for scRNA-seq Validation by qPCR
| Item | Function/Description | Example Use-Case |
|---|---|---|
| High-Efficiency RT Kit | Converts RNA to cDNA with high fidelity and yield; critical for limited input from sorted cells. | Generating cDNA from RNA of FACS-isolated neural crest subpopulations [18]. |
| qPCR Master Mix | Optimized buffer, enzymes, and dNTPs for efficient amplification. SYBR Green or probe-based. | GoTaq qPCR Systems, Luna kits [23] [19]. Used for amplifying trailblazer genes. |
| Validated Primer Assays | Sequence-specific primers for target and reference genes. Efficiency (90-110%) must be confirmed. | Primers for novel invasion signatures (e.g., ITGB5, GPC3) from bulk RNA-seq [18]. |
| Digital PCR System | For absolute quantification of low-abundance targets resistant to sample contaminants. | Quantifying difficult-to-amplify targets or targets with less than 2-fold expression differences [20]. |
| Data Analysis Software | Tools for Cq determination, efficiency calculation, and fold-change analysis. | GeneGlobe (QIAGEN), "dots in boxes" method (NEB) [24] [19]. |
| Reference Genes | Stable, constitutively expressed genes for data normalization (e.g., GAPDH, ACTB). |
Essential for accurate ∆∆Cq calculation in all qPCR experiments [24]. |
qPCR maintains its status as the gold standard for targeted gene validation due to its accessibility, well-understood workflow, and robust performance in confirming transcriptional discoveries from high-throughput techniques like scRNA-seq. Its precision is paramount for building reliable models of developmental processes, as demonstrated in the validation of neural crest cell trailblazer signatures [18].
For the vast majority of validation tasks in developmental biology, especially those with adequate target abundance and pure samples, qPCR is unrivaled in its cost-effectiveness and throughput. However, as the field increasingly focuses on rare cell populations and subtle transcriptional differences, ddPCR presents a powerful complementary technology for scenarios involving very low abundant targets or challenging sample matrices. By understanding the comparative strengths outlined in this guide, researchers can strategically apply these technologies to ensure the highest standards of data quality and reproducibility in their work.
In developmental biology, two fundamental concepts describe a cell's journey from immaturity to a specialized state: developmental potential (the capacity to differentiate into other cell types) and lineage commitment (the restriction of fate to a specific cellular pathway) [25]. Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to capture snapshots of this dynamic process, generating vast datasets from individual cells. However, deriving biologically meaningful insights from this data requires robust computational methods to predict potency and identify lineage-specific markers, followed by careful validation to confirm these predictions.
This guide objectively compares the leading methodologies for defining developmental transitions, from computational frameworks that assign potency scores to experimental approaches for identifying lineage markers. We provide direct performance comparisons and supporting experimental data to help researchers select the most appropriate tools for their investigative context.
Computational Scoring of Developmental Potential
Core Methodologies and Algorithms
Computational methods for inferring developmental potential from scRNA-seq data generally operate on the principle that a cell's transcriptional complexity correlates with its immaturity. We compare two prominent approaches in the table below.
Table 1: Comparison of Computational Methods for Developmental Potential
| Method | Underlying Principle | Input Requirements | Key Output | Interpretability |
|---|---|---|---|---|
| CytoTRACE 1 [25] | Gene counts per cell (a proxy for transcriptional diversity) | Single-cell gene expression matrix | Dataset-specific relative potency ordering | High (direct count-based metric) |
| CytoTRACE 2 [25] | Interpretable deep learning (Gene Set Binary Networks) trained on an atlas of validated potency levels | Single-cell gene expression matrix | Absolute potency score (0-1 scale) and broad potency categories | High (uses binary gene weights, provides feature importance) |
CytoTRACE 2 represents a significant advance by leveraging a deep learning framework trained on a curated atlas of human and mouse scRNA-seq datasets with experimentally validated potency levels [25]. Its key innovation is the use of a Gene Set Binary Network (GSBN), which assigns binary weights (0 or 1) to genes to identify highly discriminative gene sets for each potency category. This architecture allows the model to provide an absolute developmental potential score on a continuous scale from 1 (totipotent) to 0 (differentiated), enabling meaningful cross-dataset comparisons without requiring integration or batch correction [25].
Performance and Validation Data
The performance of these tools has been systematically benchmarked. In evaluations involving 33 datasets and 406,058 cells, CytoTRACE 2 outperformed eight state-of-the-art machine learning methods for cell potency classification, achieving a higher median multiclass F1 score and lower mean absolute error [25]. Furthermore, it demonstrated over 60% higher correlation, on average, for reconstructing known developmental hierarchies compared to eight other developmental trajectory inference methods [25].
Figure 1: A typical workflow integrating computational analysis of scRNA-seq data with qPCR validation.
Experimental Definition of Lineage Trajectories
While computational methods predict potential, lineage-specific markers are essential for isolating and characterizing distinct cell populations during differentiation, reprogramming, and in embryos [26]. The identification of these markers has been tackled through both transcriptomic and proteomic approaches.
Table 2: Comparison of Lineage Marker Identification Strategies
| Strategy | Description | Key Advantage | Limitation | Example Findings |
|---|---|---|---|---|
| Transcriptomic Profiling [27] | RNA-seq or microarrays of different stem cell lines or embryonic cells. | Comprehensive, can analyze all expressed genes. | mRNA level may not correlate with surface protein presence [26]. | Identified gene expression trajectories for primitive endoderm, trophoblast, and ectoderm [27]. |
| Cell-Surface Proteomics [26] | Direct labeling and mass spectrometry of cell-surface proteins. | Identifies proteins directly usable for live-cell isolation (e.g., FACS). | Technically challenging; may miss low-abundance proteins. | Provided a resource of 27 lineage-specific surface markers for embryo-derived stem cells (ES, EpiSC, TS, XEN) [26]. |
Large-scale cell-surface proteomics has proven particularly powerful for identifying markers that enable the prospective isolation of viable lineage progenitors. For example, one study provided a proteomic resource of signaling, adhesion, and migration proteins for four embryo-derived stem cell lines, validating 27 antibodies against lineage-specific cell-surface markers [26]. This allowed for the investigation of specific cell populations during ES-EpiSC reprogramming and the isolation of lineage progenitors directly from blastocysts [26].
The Critical Role of qPCR in Validation
Validation of scRNA-seq Findings
Quantitative PCR (qPCR) remains a cornerstone for validating gene expression findings from high-throughput technologies. The relationship between scRNA-seq and qPCR is complementary.
Figure 2: The complementary relationship between discovery and validation technologies.
- When qPCR validation is appropriate: It is crucial when a second, independent method is required to confirm an observation, such as for manuscript publication or when scRNA-seq data is based on a small number of biological replicates [4]. For the most powerful confirmation, qPCR should be performed on a new set of RNA samples, which validates both the technology and the underlying biology [4].
- When qPCR validation is less critical: When RNA-seq data is used as a primary screen to generate hypotheses for further functional tests (e.g., at the protein level), or when the same results are confirmed by generating more RNA-seq data on a new, larger set of samples [28] [4].
Studies have shown that expression values from qPCR and RNA-seq correlate well (r > 0.84), confirming that single-cell RNA-seq methods can perform quantitative transcriptome measurements consistent with this gold standard [5].
Key Validation Protocols and Considerations
For validation studies, the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines are a critical standard for ensuring the reproducibility and reliability of qPCR data [29]. Key steps in the validation workflow include:
- Assay Design: Primers and probes should be designed using specialized software (e.g., PrimerQuest, Primer3) and tested for specificity against the host genome. It is recommended to design and empirically test at least three primer and probe sets [29].
- Reference Gene Selection: A critical and often overlooked step. Reference genes for normalization must be stably expressed across all experimental conditions. Studies have shown that commonly used genes like Actb and 18S can be unstable during certain processes, such as murine heart development, and should be avoided without prior stability testing [30]. Tools like GeNorm, NormFinder, and RefFinder can be used to evaluate the stability of candidate reference genes [30].
- Accuracy and Precision: Assay validation should establish linearity, efficiency, and limits of detection. For regulated bioanalysis supporting cell and gene therapies, cross-industry recommendations provide a framework for validation parameters [29].
Furthermore, qPCR methods have been adapted for specialized applications in developmental biology, such as screening single-cell clones after genome editing. One such method exploits the sensitivity of Taq DNA polymerase to primer mismatches to accurately determine editing efficiency and genotype cell colonies [31].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents and materials essential for experiments in this field.
Table 3: Research Reagent Solutions for Developmental Studies
| Reagent/Material | Function | Example Use Case | Considerations |
|---|---|---|---|
| Cell-Surface Marker Antibodies [26] | Flow cytometric analysis and fluorescence-activated cell sorting (FACS) of live cells. | Isolation of specific lineage progenitors (e.g., EPI, PE, TE) directly from blastocysts. | Specificity must be confirmed via proteomics or other direct protein-level analysis. |
| Validated qPCR Assays (Primers/Probes) [29] | Accurate quantification of gene expression for validation. | Validating potency-associated genes (e.g., Fads1, Fads2) or lineage markers identified by scRNA-seq. | Follow MIQE guidelines; use stable reference genes for normalization [30]. |
| Stable Reference Genes [30] | Normalization of qPCR data to account for technical variation. | Accurate gene expression analysis across different developmental stages. | Stability must be empirically determined for each experimental system (e.g., Rplp0 is stable in murine heart development). |
| CRISPR/Cas9 System [3] | Functional validation via gene knockout or editing. | Testing the role of a gene (e.g., GhLAX1) in a developmental process identified by scRNA-seq. | Requires careful design of gRNAs and efficiency confirmation (e.g., via qPCR assay) [31]. |
| RNA FISH Probes [3] | Spatial validation of gene expression in tissue context. | Confirming the spatial localization of a specific cell population identified by scRNA-seq. | Provides spatial context but is lower throughput than scRNA-seq. |
Defining developmental transitions requires a synergistic combination of computational and experimental biology. Frameworks like CytoTRACE 2 provide powerful, generalizable predictions of absolute developmental potential, while direct proteomic and transcriptomic analyses deliver concrete lineage markers for cell isolation. Regardless of the discovery platform, qPCR remains an indispensable tool for validating key findings, with its utility maximized when applied to independent samples and conducted under rigorous, standardized guidelines. By understanding the strengths and applications of each technology, researchers can construct robust and reproducible experimental pipelines to decode the complexities of cell fate.
From Single-Cell Atlas to qPCR Confirmation: A Step-by-Step Protocol
In developmental biology research, single-cell RNA sequencing (scRNA-seq) has emerged as a transformative technology for dissecting cellular heterogeneity and uncovering novel cell types and states during organism development [32]. However, the high degree of technical noise, methodological variability, and biological complexity inherent to these studies necessitates rigorous experimental design to ensure valid, reproducible findings [32] [33]. A foundational thesis in this field posits that robust scRNA-seq data validation through quantitative PCR (qPCR) requires strategic integration of pilot studies and appropriate biological replication at every stage. This approach transforms exploratory findings into biologically meaningful discoveries that can withstand scientific scrutiny.
The misconception that massive data quantities from deep sequencing can compensate for poor experimental design represents a critical pitfall in modern biology [34]. In reality, biological replication—not sequencing depth—forms the cornerstone of statistical inference and rigorous validation [34]. Each biological replicate represents an independent observation of the population under study, enabling researchers to distinguish consistent biological signals from random variation or technical artifacts. For developmental studies especially, where biological variability across organisms or timepoints can be substantial, inadequate replication dooms experiments to irreproducibility regardless of the sophistication of subsequent validation methods.
The Validation Framework: From Transcriptional Profiling to Mechanistic Insights
Evaluating scRNA-seq results requires a structured approach to validation, progressing from technical reproducibility to biological meaning. Current practices support a three-tiered framework for validating scRNA-seq clusters, with each level providing increasingly stronger evidence for biological significance [32] [33].
Replicability of Expression Levels
The initial validation stage assesses whether transcriptional profiles can be reproduced across different technical replicates, sequencing batches, or analysis pipelines. This level confirms that observed expression patterns are robust to methodological variations. Researchers typically evaluate this through sample-sample correlation analyses or by examining the consistency of cluster formation across multiple experimental batches [32]. For example, in a landmark retinal cell characterization study, researchers profiled over forty thousand cells across seven batches, with each batch containing pooled tissue from multiple mice. They demonstrated that 38 of 39 identified cell clusters sampled proportionally from all batches, while one cluster failed to replicate, suggesting a technical artifact rather than a genuine biological entity [32] [33].
Generalization to Orthogonal Data
The second validation tier examines whether scRNA-seq findings generalize to data collected using different measurement techniques. This often involves comparing scRNA-seq results with bulk RNA-seq, qPCR, protein imaging, or other complementary approaches [32]. Such orthogonal validation is particularly important when scRNA-seq identifies novel cell subtypes or states during development. A comprehensive benchmark study comparing scRNA-seq to multiplexed qPCR—considered the gold standard for gene expression validation—found strong correlation (r > 0.84) between methods, confirming that scRNA-seq can provide quantitative measurements consistent with established technologies [5].
Mechanistic Validity
The highest validation tier establishes whether computationally-defined cell types or states have distinct functional properties or developmental behaviors. This might involve demonstrating that specific clusters correspond to cells with different differentiation potentials, spatial organizations, or chemical sensitivities [32]. In developmental biology, this often requires linking transcriptional profiles to functional assays or perturbation studies that test hypothesized mechanisms driving cell fate decisions.
Experimental Design Principles for scRNA-seq with qPCR Validation
Distinguishing Biological and Technical Replication
A fundamental requirement in scRNA-seq experimental design involves properly distinguishing between and implementing both biological and technical replicates, each serving distinct purposes in validation workflows [35].
Biological replicates are independent biological samples (e.g., different embryos, distinct primary tissue samples, or separately cultured organoids) that capture natural biological variation [35]. These are essential for ensuring that findings generalize beyond individual specimens and for drawing statistically valid conclusions about developmental processes. For scRNA-seq experiments with subsequent qPCR validation, a minimum of 3-5 biological replicates per condition is typically recommended, though more may be needed for heterogeneous samples or subtle biological effects [35].
Technical replicates involve multiple measurements of the same biological sample [35]. In scRNA-seq workflows, this might include processing the same cell suspension across multiple sequencing lanes or library preparation batches. Technical replicates primarily assess variability introduced by measurement processes rather than biological variation itself. While useful for optimizing protocols, technical replicates cannot substitute for biological replicates when making inferences about developmental biology [36].
The Critical Role of Pilot Studies
Pilot studies represent a cost-effective strategy for optimizing experimental parameters before committing to large-scale scRNA-seq experiments and subsequent validation work [35]. Well-designed pilot experiments directly inform multiple aspects of final experimental design:
- Estimating biological variability: Pilot data provides crucial information about within-group variance, enabling formal sample size calculations through power analysis [34].
- Optimizing sequencing depth: Pilot studies help determine the point of diminishing returns for sequencing depth, allowing researchers to balance the number of biological replicates against sequencing depth within fixed budgets [34].
- Testing cell dissociation and viability: For developmental tissues, pilot tests evaluate whether dissociation protocols yield sufficient viable single cells while minimizing stress-induced transcriptional responses [32].
- Validating qPCR assays: Pilot qPCR on a subset of samples confirms that reference genes are stable across developmental conditions and that candidate genes show appropriate expression dynamics [37].
Table 1: Key Parameters to Address in scRNA-seq Pilot Studies for Developmental Biology
| Parameter Category | Specific Metrics | Informs Final Experiment |
|---|---|---|
| Technical Quality | Cell viability after dissociation, mRNA quality, doublet rates | Sample preparation protocol optimization |
| Sequencing | Reads per cell, genes detected per cell, sequencing saturation | Required sequencing depth, cell numbers |
| Biological Variation | Within-condition variance for key marker genes | Number of biological replicates needed |
| Cluster Validation | Preliminary cluster stability, known cell type detection | Feature selection strategy, analysis pipeline |
Quantitative Comparison of scRNA-seq Method Performance
Selecting appropriate scRNA-seq methodologies is crucial for generating data that can be effectively validated through qPCR. Different platforms and protocols vary significantly in their sensitivity, accuracy, and technical performance, directly impacting downstream validation success.
A comprehensive quantitative assessment compared commercially available single-cell RNA amplification methods using bulk RNA-seq and multiplexed qPCR as benchmarks [5] [38]. The study evaluated 102 single-cell transcriptomes using tube-based methods (SMARTer Ultra Low RNA Kit, TransPlex Kit) and a microfluidic approach (Fluidigm C1 system), with all libraries constructed using Nextera and sequenced on Illumina platforms. Performance was benchmarked against qPCR measurements of 40 genes in 457 single cells, providing robust ground-truth comparisons.
Table 2: Quantitative Performance Comparison of scRNA-seq Methods Against qPCR Benchmark
| Method | Reaction Volume | Sensitivity (% of bulk RNA-seq) | Correlation with qPCR (r) | False Positive Rate | Key Advantages |
|---|---|---|---|---|---|
| Fluidigm C1 | Nanoliter | ~42-44% | 0.84-0.90 | Low | Reduced amplification bias, minimal false positives |
| SMARTer Ultra Low | Microliter | ~30-35% | 0.84-0.87 | Moderate | Established protocol, good sensitivity |
| TransPlex | Microliter | ~25-30% | 0.84-0.86 | Moderate | Compatibility with standard lab equipment |
| Ovation (NuGEN) | Microliter | ~20-25% | Not reported | Variable | High reproducibility but lower sensitivity |
The comparison revealed several critical insights for developmental biology applications. First, methods utilizing nanoliter reaction volumes (e.g., microfluidic approaches) demonstrated superior accuracy with regression slopes near 1 when compared to qPCR standards, indicating minimal systematic bias [5]. Second, these methods also showed reduced false positive signals for genes known to be absent in the cell type studied, a crucial consideration when validating novel cell types during development. Third, the distribution of expression values for housekeeping genes was notably tighter in nanoliter-volume preparations, closely matching qPCR distributions and providing more reliable normalization for validation experiments [5].
Replication Assessment in Practice: The MetaNeighbor Framework
For developmental biologists seeking to validate putative novel cell types identified through scRNA-seq, the MetaNeighbor framework provides a systematic approach for quantifying replicability across datasets [39]. This method evaluates how well cell-type-specific transcriptional profiles replicate by testing whether knowing a cell type's expression features in one dataset allows accurate identification of the same cell type in another dataset.
The MetaNeighbor workflow operates through three core steps: (1) calculating correlations between all pairs of cells across datasets based on gene set expression, (2) performing cross-dataset validation by hiding cell-type labels in test datasets while using labeled training datasets, and (3) predicting test set labels through neighbor voting based on similarity to training data [39]. Performance is quantified using the area under the receiver operator characteristic curve (AUROC), where scores >0.9 indicate excellent replication, 0.5 represents random guessing, and scores <0.3 indicate clear distinction from other types.
Application of this framework to neuronal cell types demonstrated that large sets of variably expressed genes can identify replicable cell types with high accuracy, suggesting a practical path forward for large-scale evaluation of scRNA-seq data [39]. For developmental biologists, this approach provides quantitative evidence for whether newly identified progenitor or differentiated cell states represent robust biological entities versus technical artifacts or over-interpreted clustering results.
qPCR Validation: Best Practices and Reagent Solutions
Reference Gene Selection Strategies
qPCR validation of scRNA-seq findings requires careful selection of reference genes for normalization, a step often overlooked but critical for accurate interpretation. Traditional housekeeping genes (e.g., ACTB, GAPDH) frequently show expression variability across developmental stages or cell types, potentially compromising validation accuracy [37]. Instead, systematic identification of stable reference genes directly from RNA-seq data provides superior normalization.
The Gene Selector for Validation (GSV) software implements an optimized workflow for identifying appropriate reference and validation candidate genes from transcriptomic data [37]. This tool applies five sequential filters to select optimal reference genes: (1) expression >0 TPM in all samples, (2) standard variation of log2(TPM) <1, (3) no outlier expression (>2× average log2 expression), (4) average log2 expression >5, and (5) coefficient of variation <0.2 [37]. For validation candidates, GSV selects genes with high expression (log2 average >5) and high variability (standard variation >1), ensuring selected targets are both detectable and likely to show meaningful expression differences.
Research Reagent Solutions for scRNA-seq Validation
Table 3: Essential Research Reagents and Tools for scRNA-seq Experimental Validation
| Reagent/Tool Category | Specific Examples | Function in Validation Workflow |
|---|---|---|
| scRNA-seq Platforms | Fluidigm C1, 10x Chromium, SMARTer | Single-cell capture, cDNA amplification, library prep |
| Reverse Transcription Kits | SMARTer Ultra Low, TransPlex | cDNA synthesis from limited RNA input |
| Spike-in Controls | ERCC RNA Spike-In Mix, SIRVs | Technical variation assessment, normalization |
| qPCR Master Mixes | Multiplex PCR kits, SYBR Green, TaqMan assays | Target gene quantification |
| Reference Gene Selection | GSV Software, NormFinder, GeNorm | Identification of stable normalization genes |
| Replication Assessment | MetaNeighbor R package | Cross-dataset cell type replicability analysis |
Strategic experimental design incorporating appropriate pilot studies, biological replication, and systematic validation frameworks transforms scRNA-seq from a descriptive tool into a powerful discovery engine for developmental biology. The integration of qPCR as a validation method provides an essential bridge between high-throughput screening and targeted confirmation of key findings. By implementing the principles and practices outlined here—including proper replication, method selection based on quantitative performance data, and structured validation workflows—researchers can generate scRNA-seq findings with the robustness necessary to advance our understanding of developmental mechanisms.
The expanding toolkit for replication assessment, including frameworks like MetaNeighbor and specialized software like GSV, empowers developmental biologists to critically evaluate their findings and focus subsequent mechanistic studies on the most replicable and biologically meaningful cell types and states. Through this integrated approach, scRNA-seq with qPCR validation will continue to drive fundamental discoveries in developmental biology while maintaining the rigor required for reproducible science.
Single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology by enabling researchers to probe transcriptional heterogeneity at unprecedented resolution [2]. A ubiquitous step in scRNA-seq analysis is the selection of marker genes—a small subset of genes whose expression profiles distinguish specific cell sub-populations [16]. These markers enable critical downstream applications including cell type annotation, biological interpretation of clusters, and understanding developmental trajectories [16] [40]. However, unlike general differential expression analysis, marker gene selection has distinct requirements: ideal markers exhibit large expression differences between cell types, are typically up-regulated in specific populations, and demonstrate low expression in others [16].
In developmental biology research, where subsequent validation often occurs through qPCR or spatial techniques, selecting high-confidence markers becomes paramount. The reliability of these markers directly impacts validation success and experimental efficiency. This guide provides a comprehensive comparison of computational methods for selecting marker genes, with a specific focus on identifying robust candidates for experimental validation in developmental studies.
Comprehensive Benchmarking of Marker Gene Selection Methods
Performance Evaluation of Computational Methods
Recent benchmarking efforts have systematically evaluated 59 computational methods for selecting marker genes using 14 real scRNA-seq datasets and over 170 simulated datasets [16]. Methods were assessed on multiple criteria: recovery of known marker genes, predictive performance of selected gene sets, computational efficiency, and implementation quality.
Key findings from comprehensive benchmarking: Simple statistical methods, particularly the Wilcoxon rank-sum test, Student's t-test, and logistic regression, demonstrated superior efficacy in selecting reliable marker genes [16]. These methods consistently outperformed more complex machine learning approaches in recovering expert-annotated and simulated marker genes. Surprisingly, newer methods did not comprehensively exceed the performance of these established techniques, highlighting the value of simplicity and transparency in marker gene selection.
Comparative Analysis of Major Method Categories
Table 1: Comparison of Major Marker Gene Selection Approaches
| Method Category | Representative Examples | Key Advantages | Limitations | Suitability for Validation |
|---|---|---|---|---|
| Simple Statistical Tests | Wilcoxon rank-sum, t-test [16] | Fast, interpretable, proven efficacy | May select overly abundant metabolic genes | Excellent - High confidence targets |
| Machine Learning Approaches | scGeneFit [41] | Joint optimization, hierarchy-aware | Complex parameter tuning, less interpretable | Moderate - Requires careful validation |
| Differential Expression Based | Seurat's FindAllMarkers, FindConservedMarkers [40] | Flexible thresholds, multiple testing correction | P-value inflation from using cells as replicates | Good - Useful with parameter adjustment |
| Compressive Classification | scGeneFit [41] | Minimal marker redundancy, optimized panels | Less control over individual gene selection | Good for panel design |
The benchmarking revealed that methods implementing a "one-vs-rest" strategy (comparing one cluster against all others) face specific challenges including imbalanced sample sizes and increased biological heterogeneity in the pooled "other" group [16]. These factors can impact marker quality and should be considered when selecting candidates for validation.
Experimental Protocols for Marker Gene Identification
Standardized Workflow for Marker Discovery
Table 2: Key Parameters for Marker Identification in Seurat
| Parameter | Default Value | Recommended Setting | Rationale |
|---|---|---|---|
| logfc.threshold | 0.25 | 0.25-0.5 | Balances specificity and sensitivity |
| min.pct | 0.1 | 0.25 | Filters lowly detected genes |
| min.diff.pct | - | 0.25 | Selects genes with expression percentage differences |
| only.pos | FALSE | TRUE | Selects only upregulated markers |
| test.use | Wilcoxon | Wilcoxon | Based on benchmarking results |
A typical marker identification workflow begins with quality-controlled, normalized, and clustered scRNA-seq data [40]. For studies with multiple conditions, the recommended approach uses conserved marker detection:
This function performs differential expression testing within each condition separately then combines p-values across groups, identifying markers robustly expressed regardless of experimental conditions [40]. For studies with a single condition, the FindAllMarkers() function provides an efficient alternative.
Advanced Method: scGeneFit for Marker Selection
The scGeneFit method employs a fundamentally different approach, formulating marker selection as a label-aware compressive classification problem [41]. Unlike one-vs-all methods, scGeneFit jointly identifies genes that optimally discriminate all cell labels simultaneously:
This method is particularly valuable when designing targeted validation assays, as it minimizes redundancy between selected markers while maintaining discriminatory power across multiple cell types [41].
Visualization of Marker Selection and Validation Workflows
Computational Marker Selection Process
Experimental Validation Pathway
Table 3: Key Research Reagent Solutions for Marker Gene Validation
| Reagent/Resource | Category | Primary Function | Example Applications |
|---|---|---|---|
| Smart-seq2 | Library Prep | Full-length scRNA-seq | High sensitivity for rare transcripts |
| 10x Chromium | Platform | Droplet-based scRNA-seq | High-throughput cell profiling |
| Seurat | Software | scRNA-seq analysis | Marker identification, visualization |
| Scanpy | Software | scRNA-seq analysis | Python-based analysis pipeline |
| RNA FISH Probes | Validation | Spatial confirmation | Tissue localization of markers |
| CRISPR/Cas9 | Functional Tool | Gene knockout | Validation of marker function |
| Cell Ranger | Software | Data processing | Process 10x Genomics data |
| UMI Tools | Software | Quality control | Molecular counting, deduplication |
Practical Considerations for Validation Studies
Optimizing Marker Selection Parameters
When selecting markers for qPCR validation in developmental studies, parameter tuning significantly impacts success rates. Based on empirical evidence [16] [40]:
- Fold change thresholds: Set
logfc.thresholdto 0.25-0.5 to select markers with substantial expression differences - Expression fraction: Apply
min.pct = 0.25to ensure markers are detected in sufficient cells - Specificity: Use
min.diff.pct = 0.25to select genes with differential detection rates - Multiple testing: Employ Bonferroni correction or similar methods to control false discoveries
For developmental time course experiments, conserved marker detection across timepoints identifies stable cell type markers rather than transient state associations.
Validation Techniques Beyond qPCR
While qPCR remains a gold standard for expression validation, several orthogonal approaches strengthen marker confirmation:
- RNA FISH: Provides spatial context and single-molecule resolution, ideal for confirming rare cell populations [3]
- Immunofluorescence/HIC: Protein-level validation of marker expression at tissue and cellular resolution [3]
- Flow cytometry/FACS: Enables quantification and isolation of marker-positive cells for functional assays [3]
- Spatial transcriptomics: Correlates computational predictions with spatial expression patterns [3]
Each technique addresses different aspects of marker validity, with multi-method validation providing the strongest evidence for marker utility.
Benchmarking evidence consistently supports simple statistical methods like the Wilcoxon rank-sum test as top performers for marker gene selection [16]. These methods provide an optimal balance of performance, interpretability, and computational efficiency for identifying high-confidence markers for validation.
For developmental biology applications, selection parameters should be tuned to prioritize markers with clear biological interpretability and robust expression differences. Combining computational selection with orthogonal validation techniques—particularly spatial methods like RNA FISH—strengthens the evidence for marker utility in understanding developmental processes.
Future methodology development should focus on integrating multi-omic data, addressing batch effects more effectively, and improving marker selection for rare cell populations—all critical considerations for developmental studies where cellular heterogeneity and dynamic processes present unique analytical challenges.
Single-cell reverse transcription quantitative PCR (scRT-qPCR) remains a powerful tool for investigating cellular heterogeneity, validating single-cell RNA sequencing (scRNA-Seq) findings, and profiling rare cell populations in developmental biology research. Despite the rise of high-throughput scRNA-Seq, scRT-qPCR maintains its relevance due to its precision, sensitivity, wide dynamic range, and cost effectiveness [42]. A robust scRT-qPCR workflow is crucial for generating reliable data, and its optimization begins with the critical initial steps of cell collection, lysis, and reverse transcription. This guide provides a detailed, evidence-based comparison of methodologies and reagents for establishing a reliable scRT-qPCR workflow for single-cell analysis.
The journey from a single cell to amplifiable cDNA involves a series of delicate, interconnected steps. The workflow begins with the preparation of a single-cell suspension, followed by the isolation of individual cells, their lysis, and finally, the reverse transcription of cellular RNA into complementary DNA (cDNA). Each step introduces potential variables that can impact the final results [42].
The following diagram maps this core workflow and its key decision points.
Cell Collection and Lysis
Preparation of Single-Cell Suspension
The foundation of a successful scRT-qPCR experiment is a high-quality single-cell suspension. For tissues, this involves dissociation, which must be optimized to maximize cell viability and yield while minimizing stress-induced gene expression artifacts [42]. Immediate-early gene activation can be mitigated by using low temperatures during dissociation and/or the application of transcriptional inhibitors. The use of psychrophilic proteases has been suggested as a way to maintain dissociation efficiency at low temperatures [42]. Cell viability and count should be routinely assessed using counting chambers or automated cell counters with stains like trypan blue or propidium iodide [42].
Single-Cell Collection Methods
The choice of cell collection method depends on the need for throughput, visual inspection, and spatial context. The following table compares the most established techniques.
Table 1: Comparison of Single-Cell Collection Methods
| Method | Throughput | Key Features | Best For |
|---|---|---|---|
| FACS (Fluorescence-Activated Cell Sorting) | High | Fast, suitable for fluorescence-based cell selection, requires specialized equipment. | High-throughput collection of live, pre-labeled cells from heterogeneous mixtures [42]. |
| Micromanipulation | Low | Allows for visual inspection and selection of individual live cells, labor-intensive. | Studies where direct visual identification of cell morphology is critical [42]. |
| Laser Capture Microdissection (LCM) | Low | Retains spatial information of the cell within the tissue, usually requires fixed material. | Projects where the spatial context of the collected cell is a key parameter [42]. |
Cell Lysis and Storage
For single-cell applications, RNA extraction is not recommended due to the high risk of sample loss with minimal starting material. Instead, cells should be collected directly into a lysis buffer [42].
- Lysis Buffer: A simple solution of 0.1% BSA in nuclease-free water has been shown to maintain high RNA quality, even during extended storage at room temperature (up to four hours) or through freeze-thaw cycles [42]. The buffer's role is to rupture the cell membrane, inactivate RNases, and prevent RNA from adhering to tube walls.
- Storage: Collected cells in lysis buffer should be stored in 96- or 384-well plates at -80 °C. Plates must be sealed with hardback foils designed to withstand low temperatures. While single-cell lysates are relatively stable, repeated freezing and thawing should be avoided [42].
Reverse Transcription
Reverse transcription is often considered the most critical and variable step in the entire scRT-qPCR workflow. The efficiency of this enzymatic reaction directly determines which RNA molecules are represented in the final cDNA pool and can be detected in subsequent qPCR [42].
Reverse Transcriptase Selection
The enzyme used for reverse transcription is a major factor in cDNA yield and quality. Key desired properties for single-cell work include high sensitivity, efficiency, processivity, and thermostability [42].
- Recommended Enzymes: Independent studies have identified Maxima H- minus and SuperScript IV (both from ThermoFisher) as among the most efficient reverse transcriptases for single-cell applications [42].
- Rationale: These enzymes feature high processivity (for reverse transcribing long transcripts), increased thermostability (for dealing with RNA secondary structure), high synthesis rates, robustness to inhibitors, and strand displacement activity. Their performance makes them suitable for both scRT-qPCR and scRNA-Seq [42].
Priming Strategies for cDNA Synthesis
The choice of primer for the reverse transcription reaction determines which subset of RNAs will be converted to cDNA. The optimal strategy depends on the experimental goals.
Table 2: Comparison of Reverse Transcription Priming Strategies
| Priming Method | Mechanism | Advantages | Disadvantages |
|---|---|---|---|
| Oligo(dT) | Priming from the poly-A tail of mRNA. | Generates cDNA focused on poly-adenylated mRNA; good for limited starting material. | biased towards the 3' end of transcripts; will miss non-polyadenylated RNA [43]. |
| Random Primers | Hexamers or nonamers that anneal at multiple points along all RNA transcripts. | Can anneal to all RNA (rRNA, tRNA, mRNA), including those without a poly-A tail; good for transcripts with secondary structure. | cDNA is generated from all RNAs, which can dilute the signal from mRNA of interest [43]. |
| Sequence-Specific Primers | Custom primers targeting a specific mRNA sequence. | Creates a highly specific cDNA pool; increases sensitivity for a target gene. | Limited to one gene of interest per reaction, not suitable for transcriptome-wide analysis [43]. |
| Mixed Priming (Oligo(dT) + Random) | A combination of both oligo(dT) and random primers. | Can improve reverse transcription efficiency and qPCR sensitivity by providing more comprehensive coverage. | May still combine some biases inherent to each individual method [43]. |
Technical Considerations and Data Validation
The Role of scRT-qPCR in Validating scRNA-Seq
While RNA-Seq is a robust technology, validation with an orthogonal method like scRT-qPCR can be crucial in certain scenarios [44] [28] [4].
When Validation is Appropriate:
- To Satisfy Scientific Rigor: When a key finding needs confirmation by a second, well-established method to convince reviewers or the broader scientific community [4].
- Underpowered RNA-Seq Studies: When the original scRNA-Seq experiment was performed with a small number of biological replicates, limiting statistical power. Using qPCR to analyze a larger set of samples for a few key targets can strengthen the biological validation [4].
- Focus on a Few Critical Genes: When the entire biological story hinges on the differential expression of a small number of genes, especially if the expression changes are small or the expression levels are low [44].
When Validation is Less Critical:
- RNA-Seq as a Screening Tool: When the RNA-Seq data is used primarily for hypothesis generation, with key findings being followed up with functional protein-level assays [4].
- Adequately Powered RNA-Seq: When the RNA-Seq study itself is well-designed with sufficient replicates, and the results are consistent. In such cases, a suitable validation can be performing a second, independent RNA-Seq experiment [4].
Essential Controls and Primer Design
To ensure the accuracy of scRT-qPCR data, incorporating proper controls and thoughtful primer design is non-negotiable.
- The "No-RT" Control: A minus reverse transcriptase control must be included to check for genomic DNA contamination. This control contains all reaction components except the reverse transcriptase. Any amplification signal in this control indicates the presence of contaminating DNA [43].
- qPCR Primer Design: Primers for the qPCR step should be designed to span an exon-exon junction, with one primer potentially crossing the exon-intron boundary. This design prevents the amplification of contaminating genomic DNA, as the intronic sequence disrupts primer binding. If this is not possible, treating the RNA sample with DNase is necessary [43].
The Scientist's Toolkit
This table details key reagents and materials essential for implementing a robust scRT-qPCR workflow.
Table 3: Essential Research Reagent Solutions for scRT-qPCR
| Item | Function / Application | Considerations |
|---|---|---|
| High-Efficiency Reverse Transcriptase | Enzymatic conversion of RNA to cDNA. | Select for high sensitivity, processivity, and thermal stability (e.g., Maxima H-, SuperScript IV) [42]. |
| Nuclease-Free Water | Base for lysis buffers and dilution of reagents. | Ensures no RNase or DNase activity is introduced, which is critical for RNA stability. |
| BSA (Bovine Serum Albumin) | Key component of a simple and effective cell lysis buffer (e.g., 0.1% BSA). | Stabilizes RNA, prevents adhesion to plastic, and inactivates RNases [42]. |
| qPCR Primers | Sequence-specific amplification of cDNA targets during qPCR. | Should be designed to span exon-exon junctions; purity (>80% full-length) and consistency are vital for robust performance [45] [43]. |
| RNase/DNase-Free Multi-Well Plates | Collection and storage vessel for single-cell lysates and reaction setups. | Essential for preventing sample degradation and for compatibility with high-throughput workflows. |
| Fluorescent Probes / Antibodies | For labeling and identifying specific cell populations prior to collection (e.g., via FACS). | Enables targeted collection of rare or specific cell types from a heterogeneous suspension [42]. |
A meticulously optimized scRT-qPCR workflow, from cell collection through reverse transcription, is fundamental for generating high-quality, reliable data in developmental biology and drug development research. The choice of cell collection method, a gentle yet effective lysis protocol, and the strategic selection of a high-efficiency reverse transcriptase with an appropriate priming strategy collectively form the bedrock of a successful experiment. While scRNA-Seq provides a broad, discovery-oriented view of the transcriptome, scRT-qPCR remains an indispensable tool for focused, high-precision validation and analysis, with each method reinforcing the findings of the other in a comprehensive single-cell research strategy.
Assay Design and Preamplification for Low-Input Single-Cell cDNA
Single-cell RNA sequencing (scRNA-seq) has transformed developmental biology research by enabling the transcriptomic profiling of individual cells, revealing cellular heterogeneity that drives complex biological systems [46]. A significant technological challenge in this field remains the accurate amplification of minute quantities of starting material from individual cells for reliable downstream analysis, particularly validation through quantitative PCR (qPCR). Preamplification methods serve as a critical bridge between single-cell lysates and robust cDNA libraries, ensuring that the limited RNA from a single cell (typically 2-50 pg) is sufficiently amplified for comprehensive transcriptome analysis while maintaining quantitative accuracy essential for qPCR validation [47] [5].
The development of reliable preamplification protocols is particularly vital for developmental biology studies where researchers often work with rare cell populations, such as primordial germ cells or specific progenitor cells during embryogenesis [48]. Without effective preamplification, the transcriptomic signatures of these rare cells would remain inaccessible, hindering our understanding of key developmental processes. This guide objectively compares leading preamplification technologies and their performance in generating high-quality cDNA from low-input single-cell samples, with a specific focus on applications requiring qPCR validation.
Comparative Analysis of Preamplification Technologies
Commercial preamplification systems employ different strategic approaches to amplify cDNA from minimal input material. The SuperScript IV Single Cell/Low-Input cDNA PreAmp Kit utilizes a template-switching mechanism that enables full-length transcript coverage with high sensitivity down to single-cell input levels [47]. The competing NEBNext Single Cell/Low Input cDNA Synthesis and Amplification Module employs a different methodology that yields approximately half the cDNA output from the same 2 pg Universal Human Reference RNA input according to comparative studies [47]. The SMARTer Ultra Low RNA Kit represents another alternative that has been benchmarked in comprehensive evaluations of single-cell RNA-seq methods [5].
Independent validation studies have systematically compared the quantitative accuracy of these amplification methods against multiplexed qPCR, which remains the gold standard for gene expression measurement [5]. These comparisons reveal that while all major commercial systems can generate data that correlates well with qPCR results (r > 0.84), significant differences exist in their sensitivity, precision, and amplification bias [5].
Table 1: Performance Comparison of Single-Cell/Low-Input cDNA Preamplification Kits
| Technology | Starting Input Range | Amplification Principle | Key Applications | Sensitivity | qPCR Correlation |
|---|---|---|---|---|---|
| SuperScript IV Single Cell/Low-Input cDNA PreAmp Kit | 1-1,000 cells; 2 pg-10 ng total RNA | Template-switching reverse transcription with global preamplification | RNA-seq, qPCR, full transcriptome analysis | +++ (Detects low-abundance targets from single cell) | High correlation with qPCR benchmarks [5] |
| NEBNext Single Cell/Low Input cDNA Synthesis | Not specified in results | Not specified in results | RNA-seq | ++ | Lower cDNA yield compared to SuperScript IV [47] |
| SMARTer Ultra Low RNA Kit | Single cells | Template-switching and PCR amplification | RNA-seq, transcriptome analysis | ++ | Good correlation (r > 0.84) with qPCR [5] |
| TransPlex Kit | Single cells | Whole transcriptome amplification | RNA-seq | + | Moderate correlation with qPCR [5] |
Template-Switching Mechanism
The template-switching mechanism employed by leading technologies like the SuperScript IV Single Cell/Low-Input cDNA PreAmp Kit represents a significant advancement in preamplification technology. This process involves several precise molecular steps [47]:
- Reverse Transcriptase Binding: A reverse transcriptase enzyme binds to the RNA template and RT primer duplex, initiating complementary DNA strand synthesis
- Terminal Nucleotide Addition: When the reverse transcriptase reaches the 5' end of the RNA template, its terminal deoxynucleotidyl transferase (TdT) activity adds 1-3 additional non-templated nucleotides (typically deoxycytidine) to the cDNA end
- Template Switching Oligo Binding: The template switching oligo (TSO), which contains complementary guanine or riboguanine residues at its 3' end, binds to the non-templated nucleotides
- Known Sequence Incorporation: The reverse transcriptase switches templates and continues DNA synthesis using the TSO as a template, thereby incorporating a known adapter sequence at the 3' end of the cDNA
This mechanism ensures that full-length cDNA molecules are generated with known sequences at both ends, facilitating efficient amplification and library construction while maintaining transcript integrity [47].
Figure 1: Template-Switching Mechanism for Full-Length cDNA Generation. This process enables high-efficiency cDNA synthesis from minimal input RNA by incorporating known adapter sequences essential for downstream amplification and analysis.
Experimental Design for qPCR Validation
Establishing a qPCR Validation Framework
qPCR validation of scRNA-seq data serves two critical purposes in developmental biology research: confirming observations using an orthogonal method and strengthening findings from studies with limited biological replicates [4]. When designing qPCR validation experiments for single-cell preamplification data, researchers should incorporate several key considerations:
- Independent Sample Validation: qPCR should ideally be performed on a different set of samples with proper biological replication, not merely the same RNA samples used for RNA-seq [4]
- Reference Gene Selection: Carefully select reference genes that show stable expression across the specific developmental system being studied
- Amplicon Design: Design amplicons to overlap with regions detected in RNA-seq to confirm transcript identity
- Technical Replication: Include sufficient technical replicates to account for pipetting errors and reaction variability
The simplified workflow of qPCR compared to RNA-seq reduces opportunities for introducing bias, while its maturity as a technology provides well-established protocols and expectations [4]. For single-cell studies specifically, qPCR validation becomes particularly valuable when the scRNA-seq data represents a starting point for more focused investigations of key developmental regulators or markers.
Workflow Integration: From Single Cells to Validated Data
A robust integrated workflow for single-cell cDNA preamplification and validation encompasses both laboratory procedures and computational analysis steps. The laboratory workflow begins with single-cell isolation through FACS or microfluidics, proceeds through cell lysis and reverse transcription with preamplification, and culminates in both library preparation for sequencing and qPCR assay for validation [47] [48]. The computational workflow incorporates quality control assessment, data normalization, and the application of specialized algorithms like CLEAR (coverage-based limiting-cell experiment analysis) to identify reliably quantifiable transcripts in limiting-cell RNA-seq data [48].
Figure 2: Integrated Workflow for Single-Cell cDNA Preamplification and Validation. The process incorporates both sequencing and qPCR approaches with quality control checkpoints to ensure data reliability for developmental biology applications.
Quantitative Performance Data
Sensitivity and Yield Comparisons
Independent evaluations provide critical quantitative data on the performance of different preamplification systems. In comparative testing, the SuperScript IV Single Cell/Low-Input cDNA PreAmp Kit demonstrated significantly higher cDNA yields from low-input (2 pg) Universal Human Reference RNA—almost twice the yield obtained with the NEBNext Single Cell/Low Input cDNA Synthesis and Amplification Module [47]. This enhanced yield directly translates to improved detection of low-abundance transcripts, which is particularly important in developmental biology where key regulatory genes may be expressed at low levels.
Sensitivity testing across different input levels reveals that the SuperScript IV CellsDirect cDNA Synthesis Kit maintains strong linear correlation across four mRNA targets (ACTB, BCL2, PGK1, PPIA) when using serial dilutions of HeLa S3 cells ranging from 1 to 10,000 cells [47]. This linear response across a wide dynamic range is essential for accurate quantification of transcript levels in heterogeneous cell populations typically encountered in developmental systems.
Table 2: Quantitative Performance Metrics for Low-Input cDNA Preamplification
| Performance Metric | SuperScript IV Single Cell/Low-Input | NEBNext Single Cell/Low Input | SMARTer Ultra Low |
|---|---|---|---|
| cDNA yield from 2 pg UHRR | ~2x higher than NEBNext [47] | Baseline | Not specified |
| Detection sensitivity | 1 cell or 2 pg total RNA [47] | Not specified | Single cell [5] |
| Transcriptome coverage | Uniform across transcript length [47] | Not specified | Moderate 3' bias [5] |
| qPCR correlation (r value) | >0.84 [5] | >0.84 [5] | >0.84 [5] |
| Reaction volume compatibility | Microliter and nanoliter [47] | Not specified | Microliter and nanoliter [5] |
| Applications demonstrated | RT-qPCR, RNA-seq, cloning [47] | RNA-seq [47] | RNA-seq [5] |
Reaction Volume Considerations
The volume in which preamplification reactions are performed significantly impacts data quality. Studies comparing microliter versus nanoliter reaction volumes have demonstrated that nanoliter-volume preparations (such as those enabled by microfluidic systems like the Fluidigm C1) yield fewer false positives and reduced amplification bias [5]. For example, genes including CA1 and AQP8 showed sporadic false positive signals in tube-based qPCR data but were cleanly detected in nanoliter-volume preparations [5].
The reduced bias observed in nanoliter-volume systems is attributed to increased effective concentration of reactants and reduced competition for enzymes between template and nonspecific molecules or contaminants [5]. Additionally, microfluidic systems provide more uniform reverse transcription and more efficient template switching during reverse-transcription PCR, contributing to improved quantitative accuracy.
The Scientist's Toolkit: Essential Reagents and Solutions
Successful assay design and preamplification for low-input single-cell cDNA requires carefully selected reagents and solutions. The following toolkit encompasses essential components for robust experimental outcomes:
Table 3: Research Reagent Solutions for Low-Input Single-Cell cDNA Workflows
| Reagent/Solution | Function | Example Products |
|---|---|---|
| Cell Lysis Buffer | Rapidly disrupts cell membrane while stabilizing RNA | SuperScript IV CellsDirect Lysis Buffer [47] |
| Reverse Transcriptase | Synthesizes cDNA from RNA templates | SuperScript IV Reverse Transcriptase [47] |
| Template Switching Oligo (TSO) | Enables incorporation of known sequences at cDNA 3' end | SuperScript IV TSO [47] |
| RNase Inhibitor | Protects RNA samples from degradation during processing | Included in SuperScript IV Master Mix [47] |
| Capture Oligo(dT) Primer | Initiates cDNA synthesis from polyadenylated RNA | SuperScript IV Capturing Oligo(dT) Primer [47] |
| Preamplification Master Mix | Globally amplifies cDNA while maintaining representation | SuperScript IV Template Switching RT Master Mix [47] |
| DNA Removal Reagent | Eliminates genomic DNA contamination | DNase I (included in SuperScript IV CellsDirect kit) [47] |
| Library Preparation Kit | Prepares amplified cDNA for high-throughput sequencing | Illumina Nextera [5] |
| Quality Control Assays | Assesses cDNA quality and quantity before sequencing | Bioanalyzer, Fragment Analyzer, qPCR [49] |
The integration of robust preamplification technologies with rigorous qPCR validation provides a powerful framework for advancing developmental biology research at single-cell resolution. As the field continues to evolve, improvements in reaction efficiency, particularly through microfluidic implementations that reduce volumes to nanoliter scales, promise to further enhance the accuracy and reliability of single-cell transcriptome measurements [5]. The systematic comparison of performance metrics presented in this guide enables researchers to select appropriate preamplification strategies based on their specific experimental needs, sample limitations, and validation requirements.
For developmental biologists investigating rare cell populations or dynamic processes in embryogenesis, the combination of sensitive preamplification methods with orthogonal qPCR validation represents a strategically sound approach to generating reliable, reproducible data. As single-cell technologies continue to mature, this integrated methodology will undoubtedly yield deeper insights into the transcriptional programs that orchestrate developmental processes with unprecedented cellular resolution.
A fundamental goal in developmental biology is to understand the pathways through which a small number of progenitor cells give rise to diverse, specialized tissues. Single-cell RNA sequencing (scRNA-seq) has revolutionized this field by enabling researchers to profile gene expression at unprecedented resolution [7]. However, distinguishing a cell's developmental potential or potency—its capacity to differentiate into other cell types—from its transcriptome alone remains a significant computational challenge. While numerous algorithms exist to infer developmental trajectories, most provide dataset-specific predictions that cannot be easily unified across experiments or contextualized within an absolute biological framework [25].
CytoTRACE 2 represents a substantial advancement in addressing this limitation. As an interpretable deep learning framework, it predicts both discrete potency categories and a continuous potency score (ranging from 0, differentiated, to 1, totipotent) from scRNA-seq data [50] [25]. Unlike its predecessor and other trajectory inference methods, CytoTRACE 2 provides absolute developmental potential calibrated across the full spectrum of cellular ontogeny, facilitating direct cross-dataset comparison. A critical step in establishing the reliability of such computational predictions is independent biological validation. Quantitative PCR (qPCR), long considered the gold standard for gene expression measurement, provides a powerful orthogonal method for confirming these predictions [5] [4]. This case study examines the integration of CytoTRACE 2 and qPCR to validate developmental hierarchies, objectively comparing its performance against alternative methods and detailing the experimental protocols required for robust validation.
CytoTRACE 2: Core Technology and Workflow
Architectural Innovation and Interpretability
CytoTRACE 2 employs a novel, explainable deep learning architecture called a Gene Set Binary Network (GSBN). Inspired by binarized neural networks, GSBNs assign binary weights (0 or 1) to genes, identifying highly discriminative gene sets that define each potency category [25]. This design offers a significant advantage over conventional "black box" deep learning models because the informative genes driving predictions can be easily extracted and biologically interpreted [25]. The framework was trained on an extensive atlas of human and mouse scRNA-seq datasets encompassing 33 datasets, nine platforms, 406,058 cells, and 125 standardized cell phenotypes with experimentally validated potency levels [25].
Table 1: Key Enhancements in CytoTRACE 2
| Feature | Description | Impact |
|---|---|---|
| Retrained Framework | Model retrained with performance enhancements [50] | Improved granular potency prediction and cross-platform robustness [50] |
| Expanded Ensemble | Ensemble comprises 19 models (increased from 17) [50] | Enhanced predictive power and stability [50] |
| Competing Representations | Uses both ranked expression profiles and Log2-adjusted input data [50] | Captures detailed transcriptomic signals [50] |
| Adaptive Smoothing | Employs an adaptive nearest neighbor smoothing strategy [50] | Refines potency scores based on local cell similarity [50] [25] |
Computational Workflow and Outputs
The standard workflow for using CytoTRACE 2 begins with loading a gene expression matrix, followed by executing the cytotrace2() function. The package is available in both R and Python, with the Python version accessible via PyPI for easy installation [50]. The analysis yields two primary outputs for each cell:
- Potency Category: The discrete potency level with the maximum likelihood (e.g., Totipotent, Pluripotent, Multipotent, Oligopotent, Unipotent, or Differentiated) [50] [25].
- Potency Score: A continuous value between 0 (differentiated) and 1 (totipotent), generated by integrating GSBN predictions across all categories [50] [25].
These outputs can subsequently be visualized using the package's plotData function, which generates informative plots such as UMAP embeddings colored by potency score or category [50]. The following diagram illustrates the core computational workflow and the pathway for experimental validation.
Diagram 1: The CytoTRACE 2 and qPCR validation workflow. The computational analysis of scRNA-seq data produces predictions that guide the targeted experimental validation of key genes and cell states using qPCR.
Performance Comparison with Alternative Methods
Benchmarking Against Classification and Trajectory Inference Algorithms
The performance of CytoTRACE 2 was rigorously benchmarked against a wide array of computational methods. In evaluations spanning 33 datasets, it outperformed eight state-of-the-art machine learning methods for cell potency classification, achieving a higher median multiclass F1 score and lower mean absolute error [25]. More notably, for the task of developmental hierarchy inference, CytoTRACE 2 surpassed eight other methods, including its predecessor CytoTRACE 1, in both cross-dataset (absolute) and intra-dataset (relative) performance [25]. It demonstrated over 60% higher correlation, on average, for reconstructing known developmental orderings across 57 developmental systems [25].
Table 2: CytoTRACE 2 vs. Alternative Methods for Developmental Ordering
| Method Category | Representative Methods | Key Limitation | CytoTRACE 2 Advantage |
|---|---|---|---|
| Gene Count-Based | CytoTRACE 1 [51] | Dataset-specific predictions; cannot unify results across experiments [25] | Provides absolute developmental potential calibrated across datasets [25] |
| Trajectory Inference | Monocle, PAGA, SLICER [25] | Typically infer relative, not absolute, order within a single dataset [25] | Generates a universal potency score (0-1) for cross-dataset comparison [50] [25] |
| RNA Velocity | scVelo [25] | Predicts future states but requires splicing kinetics and is not a direct potency measure [25] | Directly predicts underlying developmental potential from gene expression alone [25] |
| Machine Learning Classifiers | Multiple methods (e.g., random forests, neural networks) [25] | Often act as "black boxes" with limited biological interpretability [25] | Uses an interpretable GSBN architecture to extract biologically meaningful gene programs [25] |
Case Study: Mouse Pancreatic Epithelial Development
A practical application of CytoTRACE 2 on data from murine pancreatic epithelium (from Bastidas-Ponce et al., 2019) demonstrates its predictive accuracy [50]. The tool was run on a dataset containing 2,850 cells from various developmental stages, including multipotent pancreatic progenitors, endocrine progenitors, and mature endocrine cells (alpha, beta, delta, epsilon) [50]. As expected, CytoTRACE 2 assigned low potency scores (closer to 0) to the mature, differentiated alpha, beta, delta, and epsilon cells. Crucially, it assigned higher mid-range scores to multipotent pancreatic progenitors and correctly positioned endocrine progenitors and precursors in the lower range of the potency spectrum, closer to the differentiated cells [50]. This outcome meticulously aligned with known biology and set the stage for targeted qPCR validation of these distinct cellular states.
Experimental Validation with qPCR
The Role of qPCR in Validating scRNA-seq Findings
While scRNA-seq is a powerful discovery tool, validation using an orthogonal method is often critical for confirming key findings. qPCR is widely regarded as the gold standard for quantitative gene expression analysis due to its sensitivity, specificity, and dynamic range [5] [4]. Validating with qPCR is particularly appropriate when a second method is necessary to confirm a critical observation or when the initial scRNA-seq data is based on a small number of biological replicates [4]. This process not only controls for potential technical biases in the scRNA-seq workflow but also strengthens the biological conclusions.
Integrated Protocol: From CytoTRACE 2 to qPCR Validation
A robust validation protocol involves a series of deliberate steps, from computational prioritization to wet-lab experimentation.
Step 1: Computational Prediction with CytoTRACE 2
- Install and Load Package: Install the CytoTRACE 2 R package using
devtools::install_github("digitalcytometry/cytotrace2", subdir = "cytotrace2_r")and load it withlibrary(CytoTRACE2)[50]. - Run Analysis: Execute the main function
cytotrace2_result <- cytotrace2(expression_data)on your gene expression matrix (cells as columns, genes as rows). The input should contain raw counts or CPM/TPM normalized counts [50]. - Identify Target Cells and Genes: Use the output to:
- Identify Cell Subpopulations: Based on predicted potency categories (e.g., "Multipotent" vs. "Differentiated") [50] [25].
- Extract Discriminative Markers: Leverage the interpretable GSBN output to obtain a list of genes that are most influential for predicting each potency state [25]. These genes serve as prime candidates for qPCR validation.
Step 2: Cell Sorting and Sample Preparation
- Sort Cell Populations: Using flow cytometry or magnetic-activated cell sorting (MACS), isolate the specific cell populations identified in Step 1 (e.g., multipotent vs. differentiated cells) based on known surface markers or other identifiers [3]. This step is crucial for physically obtaining the distinct cell states for downstream analysis.
- Extract RNA and Synthesize cDNA: Extract high-quality total RNA from each sorted cell population. Convert the RNA into complementary DNA (cDNA) using a reverse transcriptase enzyme. This cDNA will serve as the template for the qPCR reactions.
Step 3: qPCR Assay and Data Analysis
- Design and Validate Primers: Design primers for the target genes identified by CytoTRACE 2, as well as for stable reference genes (e.g., GAPDH, ACTB). Ensure primer specificity and efficiency through validation assays [5].
- Perform qPCR Runs: Run the qPCR reactions in triplicate for each target gene across all cDNA samples (from different cell populations). Use a standardized qPCR protocol with a robust detection chemistry (e.g., SYBR Green).
- Analyze Quantitative Data: Calculate the relative expression levels of each target gene using a stable method like the 2^(-ΔΔCt) method. Compare the expression of potency markers (e.g., positive correlates like Fads1, Fads2) between the high-potency and low-potency cell populations [25].
Table 3: Essential Research Reagent Solutions for Validation
| Reagent / Material | Function in Workflow | Example Application |
|---|---|---|
| Cell Sorting Kit (FACS/MACS) | Isolates specific cell populations identified computationally for downstream analysis [3] | Separating multipotent hematopoietic stem cells from differentiated lymphocytes based on CD34 expression [3]. |
| High-Quality RNA Extraction Kit | Prepares pure, intact RNA from sorted cells, which is critical for accurate cDNA synthesis [5]. | Isolating RNA from sorted pancreatic progenitor and mature beta cells for transcriptomic analysis. |
| Reverse Transcription Kit | Converts RNA into stable cDNA, the template for qPCR amplification [5]. | Synthesizing cDNA from sorted cell populations to measure expression of CytoTRACE 2-derived gene signatures. |
| qPCR Master Mix & Primers | Enables sensitive and specific amplification and quantification of target gene transcripts [5] [4]. | Quantifying expression of pluripotency factors (e.g., Pou5f1, Nanog) and differentiation markers. |
| Validated Reference Genes | Serves as an internal control for normalizing qPCR data and accounting for technical variation [5]. | Using GAPDH or ACTB to ensure accurate relative quantification of target gene expression across samples. |
Supporting Validation Methodologies
While qPCR is a cornerstone of validation, other powerful techniques can provide complementary information, especially spatial context.
RNA Fluorescence In Situ Hybridization (RNA FISH) This technique uses fluorescently labeled probes to bind specific RNA sequences within intact tissues, revealing the precise spatial localization of transcripts [3] [52]. It is exceptionally valuable for validating not only the expression of marker genes identified by CytoTRACE 2 but also the location of specific cell states (e.g., a potent stem cell niche) within a tissue architecture [3] [52]. The RNAscope ISH assay is a prominent example cited as a validation tool for NGS discoveries [52].
Immunofluorescence (IF) and Immunohistochemistry (IHC) These are protein-level validation assays that operate on the principle of specific antigen-antibody binding [3]. IF uses fluorescently labeled antibodies, while IHC uses a chromogenic reaction. They are used to confirm the expression, spatial localization, and relative abundance of proteins encoded by key genes identified in the analysis, thereby connecting transcriptomic predictions to protein-level biology [3].
Functional Assays: Gene Overexpression and Knockout To move beyond correlation and establish causality, functional studies are essential. Using CRISPR/Cas9 to knock out genes that are positive correlates of potency (as identified by CytoTRACE 2's interpretable model) and observing a loss of multipotency provides the strongest possible validation of the computational prediction [25] [3]. Conversely, overexpressing these genes in differentiated cells can test their ability to induce or maintain a less differentiated state [3].
CytoTRACE 2 represents a significant leap forward in computational methods for predicting cellular developmental potential. Its interpretable deep learning framework, which provides absolute, cross-dataset comparable potency scores, has been demonstrated to outperform a wide range of existing methods in benchmarking studies [25]. The integration of its computational predictions with targeted experimental validation, particularly using qPCR and other spatial or functional techniques, creates a powerful, closed-loop workflow for developmental biologists. This synergy between cutting-edge computation and rigorous experimental validation allows researchers to not only map developmental hierarchies with greater confidence but also to uncover the underlying molecular drivers of cell potency, with profound implications for regenerative medicine and cancer research.
Solving Common Pitfalls in Single-Cell Collection and qPCR Assays
In developmental biology and drug development, single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to dissect cellular heterogeneity and lineage commitment. However, the journey from a complex tissue to a library of sequenced transcripts is fraught with potential technical artifacts that can compromise data integrity. The pre-analytical phase—encompassing cell suspension preparation and lysis—is particularly critical, as it forms the foundation upon which all subsequent data rests. Inaccuracies introduced at these stages can lead to misinterpretation of biological signals, ultimately skewing our understanding of developmental trajectories and cellular responses.
This guide focuses on the systematic comparison of cell suspension buffers and lysis conditions, framing the discussion within the essential context of scRNA-seq validation by qPCR. For researchers and drug development professionals, selecting the appropriate buffer is not merely a procedural step but a strategic decision that directly influences signal-to-noise ratio, detection sensitivity, and ultimately, the biological conclusions that can be drawn from expensive and time-consuming single-cell experiments.
Comparative Analysis of Nuclear Isolation and Lysis Buffers
Performance Comparison of Common Lysis Buffers
The choice of lysis buffer significantly impacts nuclear integrity, DNA staining quality, and the presence of inhibitory debris in downstream applications like scRNA-seq. A systematic comparison of four common nuclear isolation buffers across seven plant species with varying genome sizes and tissue types revealed clear performance differences highly relevant to single-cell workflows [53].
Table 1: Performance Comparison of Nuclear Isolation Buffers
| Buffer | Key Composition | Optimal Use Cases | Performance Notes |
|---|---|---|---|
| LB01 [53] | 15 mM Tris, 2 mM Na₂EDTA, 0.5 mM spermine.4HCl, 80 mM KCl, 20 mM NaCl, 0.1% (v/v) Triton X-100, pH 8.0 | General purpose; wide range of species and genome sizes | One of the best overall performers; high nuclei yield and quality |
| Otto's Buffer [53] | Biphasic system with citric acid and Na₂HPO₄ | Species with low DNA content | Excellent for low DNA content samples; superior to others in this category |
| Galbraith's Buffer [53] | 45 mM MgCl₂, 30 mM sodium citrate, 20 mM MOPS, 0.1% (v/v) Triton X-100, pH 7.0 | Standard species and tissues | Provided satisfactory results for most species tested |
| Tris·MgCl₂ Buffer [53] | Tris-HCl, MgCl₂ | Specific applications (e.g., Celtis australis) | Generally the worst performer, though best for C. australis |
The study found that LB01 and Otto's buffers were generally the best performers, with Otto's buffer providing superior results in species with low DNA content [53]. No single buffer worked optimally for all species, underscoring the importance of empirical testing for new cell types or tissues. The variation in performance between different days was more significant than the variation between operators, highlighting the need for strict standardization of protocols once a buffer is selected [53].
Mammalian Cell Lysis Buffers for scRNA-seq Compatibility
For mammalian cells, lysis buffer selection must balance efficient rupture of the cell membrane with the preservation of RNA integrity and compatibility with microfluidic scRNA-seq platforms.
Table 2: Mammalian Cell Lysis Buffers for Single-Cell Applications
| Buffer / Reagent | Composition | Mechanism & Specificity | Downstream Compatibility |
|---|---|---|---|
| RIPA Lysis Buffer [54] | 25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS | Harsh, denaturing; extracts cytoplasmic, membrane, and nuclear proteins | Western blot, protein purification; often too harsh for intact RNA extraction |
| IP Lysis Buffer [54] | Modified RIPA without SDS | Moderate-strength, non-denaturing; solubilizes proteins without liberating genomic DNA | Ideal for immunoprecipitation (IP), Co-IP; gentler on biomolecular complexes |
| NP-40 Lysis Buffer [54] | 50 mM Tris, pH 7.4, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 1% NP-40, 0.02% NaN₃ | Mild, non-ionic detergent; preferentially extracts cytoplasmic proteins | ELISA, western blotting, native protein analysis |
| Specialized Reagents (M-PER, T-PER) [54] | Non-denaturing detergent in 25 mM bicine, pH 7.6 | Mild, efficient lysis for soluble proteins from cells (M-PER) or tissues (T-PER) | IP, enzyme assays, reporter assays (luciferase, beta-galactosidase) |
The composition of these buffers is tailored to target specific cellular compartments. Key components include [55]:
- Detergents (e.g., SDS, NP-40, Triton X-100): Break down lipid bilayers.
- Salts (e.g., NaCl, KCl): Maintain ionic strength and stabilize protein structures.
- Chelating Agents (e.g., EDTA, EGTA): Bind metal ions to inhibit metal-dependent nucleases and proteases.
- Protease & Phosphatase Inhibitors: Prevent protein degradation and preserve post-translational modifications.
Experimental Protocols for Buffer Evaluation and scRNA-seq Validation
Protocol for Comparing Lysis Buffer Performance
This protocol adapts standardized methods for evaluating buffer efficacy in a single-cell context [53] [55].
- Sample Preparation: Harvest approximately 40-50 mg of target tissue or 1x10⁷ cultured cells. Perform all steps on ice with pre-chilled solutions to minimize enzymatic degradation.
- Buffer Application: Add 300–1000 µL of ice-cold test lysis buffer (e.g., LB01, Otto's, RIPA, NP-40). For adherent cells, add buffer directly to the culture dish, scrape with a cold plastic scraper, and transfer the suspension to a microcentrifuge tube [56] [55].
- Homogenization & Incubation: Homogenize tissues with an electric homogenizer. For cells in culture, vortex briefly. Maintain constant agitation for 30 minutes at 4°C to facilitate lysis [56] [55].
- Clarification: Centrifuge the lysate at 12,000–14,000 x g for 10–20 minutes at 4°C to pellet insoluble debris, unlysed cells, and nuclei (if not the target) [56] [55].
- Assessment Parameters:
- Nuclei/Yield Quantification: Determine the number of intact nuclei or total RNA/protein yield.
- Debris Background: Analyze the supernatant for particulate matter.
- Inhibitor Assessment: Test the lysate for the presence of PCR inhibitors in a subsequent qPCR reaction.
Protocol for scRNA-seq Data Validation using qPCR
qPCR remains the gold standard for validating gene expression measurements from scRNA-seq [5]. The following protocol ensures accurate and reproducible validation.
- Cell Processing: Obtain single-cell suspensions from the same biological source used for scRNA-seq. Use either FACS to sort individual cells into plate wells or a microfluidic system (e.g., Fluidigm C1) for capture [5].
- cDNA Synthesis: Perform reverse transcription immediately after lysis to minimize RNA degradation. Use a scRNA-seq compatible kit (e.g., SMARTer Ultra Low RNA Kit) [5].
- Multiplexed qPCR Assay: Design and run a qPCR panel targeting 40 or more genes curated from the scRNA-seq data, including highly expressed genes, lowly expressed genes, and negative controls [5].
- Data Normalization & Analysis: Normalize gene expression values (Cq) to the median expression across all measured transcripts for each cell. This generates a dimensionless number that allows for direct cross-method comparison [5].
- Correlation with scRNA-seq: Calculate the correlation coefficient (e.g., Pearson's r) between the relative expression values derived from qPCR and those from scRNA-seq. A strong correlation (r > 0.84) indicates high quantitative accuracy of the scRNA-seq method [5].
The Scientist's Toolkit: Essential Reagents for scRNA-seq
Table 3: Research Reagent Solutions for Single-Cell Workflows
| Reagent / Solution | Function | Key Considerations |
|---|---|---|
| Protease Inhibitor Cocktail [56] | Prevents protein degradation by cellular proteases released during lysis. | Essential for preserving the cellular proteome; use a broad-spectrum mix. |
| RNase Inhibitors | Protects RNA from degradation during cell lysis and nucleic acid extraction. | Critical for all RNA-seq workflows; often included in lysis and RT kits. |
| SDS (Sodium Dodecyl Sulfate) [56] | Ionic detergent that denatures proteins and linearizes nucleic acids. | Can be too harsh for native complex preservation; use concentration-dependent. |
| DTT/Beta-Mercaptoethanol [56] | Reducing agents that break disulfide bonds in proteins. | Essential for denaturing gel electrophoresis; may interfere with some assays. |
| EDTA/EGTA [53] [55] | Chelating agents that bind divalent cations (Mg²⁺, Ca²⁺). | Inhibits metal-dependent nucleases and proteases; crucial for nucleic acid stability. |
| Triton X-100 / NP-40 [53] [54] | Non-ionic, mild detergents that disrupt lipid membranes. | Ideal for extracting functional proteins and organelles with minimal denaturation. |
Workflow Visualization for Experimental Planning and Validation
scRNA-seq Validation Workflow
Diagram 1: scRNA-seq validation workflow integrating qPCR.
Lysis Buffer Selection Logic
Diagram 2: Decision tree for lysis buffer selection based on experimental goals.
Preventing technical artifacts in single-cell analyses requires a meticulous, evidence-based approach to sample preparation. The experimental data demonstrates that no single lysis buffer is universally superior; performance is highly dependent on cell type, tissue structure, and downstream application [53]. For scRNA-seq, the transition to microfluidic, nanoliter-volume reactions has provided significant advantages in reducing false positives and amplification bias, leading to more accurate quantitative measurements that show a near-perfect correlation with qPCR validation data [5].
The most robust strategy involves empirical testing of multiple buffer systems using the protocols outlined herein, prioritizing options like LB01 for general use or Otto's buffer for samples with low DNA content [53]. Furthermore, incorporating a qPCR validation step for a panel of key genes remains an indispensable practice for confirming the quantitative accuracy of scRNA-seq findings [5]. By rigorously optimizing and validating cell suspension buffers and lysis conditions, researchers in developmental biology and drug development can ensure that their high-resolution data truly reflects biological reality rather than technical variation.
In developmental biology research, the validation of single-cell RNA sequencing (scRNA-seq) data via qPCR demands meticulous optimization to ensure accuracy from minimal input. The selection of an appropriate reverse transcriptase (RTase) and the fine-tuning of the reaction volume are two pivotal factors that directly impact cDNA yield, reaction efficiency, and the fidelity of gene expression analysis. This guide provides an objective comparison of RTase alternatives and reaction volume considerations, supported by experimental data, to empower researchers in making informed decisions for their experimental workflows.
RTase Performance Comparison
The core of reverse transcription is the enzyme. While traditional RTases are widely used, newly engineered variants offer significant enhancements, particularly for challenging applications like scRNA-seq. The table below compares the performance of different RTase types based on key biochemical properties.
Table 1: Comparative Analysis of Reverse Transcriptase (RTase) Enzymes
| RTase Type / Characteristic | Wild-type M-MuLV | AI-Engineered Taq pol Variants [57] |
|---|---|---|
| Primary Activity | RNA-dependent DNA polymerase | DNA-dependent DNA polymerase & Enhanced RTase activity |
| Thermal Stability | Moderate (optimal ~37-42°C) | High (thermostable, optimal ~60-70°C) |
| Fidelity | Standard | Moderately reduced (a known trade-off) [57] |
| Tolerance to Non-Canonical Substrates | Standard | Greater tolerance (e.g., to LNA-containing substrates) [57] |
| Key Application Note | Standard for most cDNA synthesis protocols. | Effective in single-enzyme, real-time RT-PCR setups, simplifying workflows for pathogen detection and gene expression [57]. |
Optimizing the Reaction Volume
Reaction volume is a critical, yet often overlooked, parameter. Its optimization is essential for maximizing reaction efficiency, especially with low-abundance samples like single cells.
The Principles of Volume Optimization
Miniaturization of reaction volumes can lead to significant improvements. A smaller volume increases the effective concentration of all reaction components if the absolute amount is kept constant, potentially enhancing enzyme efficiency and kinetics. Furthermore, reduced volumes conserve precious samples and expensive reagents. However, the practical challenges of liquid handling, evaporation, and surface adsorption become more pronounced at very small volumes (e.g., below 10 µL). The key is to find a volume that balances these factors for a given experimental setup.
An Analytical Analogy: Contrast Media Optimization
The principle of tailoring a key parameter (like volume) based on specific sample characteristics is well-established in other analytical fields. In diagnostic imaging, for instance, the administration of contrast media has evolved from a fixed-dose protocol to one adapted to individual patient physiology.
Table 2: Contrast Media Dosing Protocols: A Conceptual Parallel to Volume Optimization [58]
| Protocol Characteristic | Fixed-Dose Protocol | Lean Body Weight (LBW)-Adapted Protocol [58] |
|---|---|---|
| Dosing Principle | One volume fits all (e.g., 120 mL for all patients). | Dose is calculated based on the patient's Lean Body Weight. |
| Outcome: CM Volume | Fixed at 120 mL | Significantly reduced (mean 103.5 ± 17.7 mL) [58] |
| Outcome: Image Quality / Enhancement | Baseline (Adequate) | Improved parenchymal enhancement and higher Signal-to-Noise Ratio in key organs [58] |
| Conceptual Takeaway | Analogous to using a standard, one-size-fits-all reaction volume. | Analogous to optimizing reaction volume based on sample quality and quantity (e.g., RNA integrity and amount) to improve output. |
Experimental Protocols for Validation
Protocol: Reference Gene Validation for RT-qPCR
A robust qPCR validation of scRNA-seq data requires stable reference genes. The following protocol uses RNA-seq data to identify them systematically [59] [60].
- RNA-Seq Data Analysis: Calculate the expression levels (in TPM - Transcripts Per Million) for all genes across your RNA-seq datasets, including those from various developmental stages or conditions [60].
- Apply Selection Criteria:
- Expression Ubiquity: The gene must be expressed in all sample types.
- Low Variance: Standard deviation of log2(TPM) should be < 1.
- No Outliers: No single log2(TPM) value should differ from the mean by more than 2.
- Adequate Expression Level: Mean log2(TPM) > 5 [60].
- Rank and Select: Rank the candidate genes by their coefficient of variation (CV); lower CV indicates higher stability.
- Experimental Validation: Select the top candidate genes and validate their stability using RT-qPCR data analyzed with algorithms like geNorm and NormFinder [59].
Workflow for identifying stable reference genes from RNA-seq data [59] [60].
Protocol: Supramolecular Enhancement of Colorimetric Assays
While not directly part of the RT-qPCR workflow, this protocol exemplifies advanced optimization for assay detection, which can be crucial for developing ancillary tests in a research pipeline [61].
- Assay Setup: Perform a standard colorimetric indicator assay that releases the pink dye resorufin (e.g., a hypochlorite sensor or an enzyme assay) [61].
- Supramolecular Complexation: After the colorimetric change is complete, add the water-soluble tetralactam macrocycle M2 to the solution.
- Color Shift: The macrocycle will encapsulate the resorufin dye, causing an instantaneous color change from pink to blue.
- Result Interpretation: The final yellow-to-blue color change provides superior visual contrast for naked-eye interpretation compared to the original yellow-to-pink change [61].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for scRNA-seq Validation Workflows
| Reagent / Tool | Function / Description | Relevance to Experiment |
|---|---|---|
| Tetralactam Macrocycle (M2) | A synthetic host molecule that binds resorufin [61]. | Acts as a supramolecular adjuvant to enhance the visual contrast of colorimetric assays by shifting the output from pink to blue [61]. |
| Iomeprol 350 (Iomeron) | Non-ionic iodinated contrast medium (350 mgI/mL) [58]. | Used in imaging studies; cited here as a model for optimized, weight-adapted dosing protocols that parallel reaction volume optimization strategies [58]. |
| Crystal Violet Solution | A 0.1% solution of cationic triphenylmethane dye in PBS or water [62]. | Used for staining and quantifying adherent cells or bacterial biofilms, a technique that may be employed in ancillary cell culture viability assays [62]. |
| Contrast Resolution Chart | A specialized test chart with a wide density range (up to 101 dB) [63]. | Used for precise measurement of a camera's ability to resolve low-contrast features, analogous to assessing the detection limit of an analytical system [63]. |
Logical Workflow for RT-qPCR Validation
The following diagram integrates the key decision points and optimization strategies discussed in this guide into a coherent workflow for validating scRNA-seq data.
Logical workflow for scRNA-seq validation via qPCR, highlighting key optimization points.
In single-cell RNA sequencing (scRNA-seq) for developmental biology, the integrity and purity of RNA are paramount. Accurate validation by qPCR hinges on the quality of the starting material, as degradation or contamination can severely skew gene expression data, leading to flawed biological interpretations. This guide compares best-practice techniques and their alternatives, providing a structured framework to safeguard your RNA throughout the experimental workflow.
Core Principles for RNA Preservation
The strategies to protect RNA integrity and purity are built on a few foundational principles. The overarching goal is to inactivate RNases immediately and maintain that state throughout the procedure, while simultaneously preventing the introduction of external contaminants or cross-contamination between samples.
The logical relationship between the primary threats to RNA, the corresponding defense strategies, and the quality control checkpoints is outlined below.
Quantitative Comparison of Key Techniques
The table below summarizes experimental data and key characteristics for different aspects of RNA handling, providing a basis for objective comparison.
Table 1: Comparison of Techniques for RNA Integrity and scRNA-seq Analysis
| Technique Aspect | Key Parameter Measured | Performance Data / Characteristics | Impact on Downstream Analysis |
|---|---|---|---|
| RNA Quality Assessment | Purity (A260/A280, A260/A230) | Significantly altered gene expression in qPCR with contaminated RNA despite good purity ratios [64]. | High risk of inaccurate qPCR validation and false conclusions [64]. |
| Sample Preparation Volume | False Positive Rate, Amplification Bias | Nanoliter-volume (C1 system) reactions showed fewer false positives and reduced bias compared to microliter volumes [65]. | Higher data accuracy in scRNA-seq; regression slope of expression near 1 vs. qPCR [65]. |
| Single-Cell Isolation | Cell Viability, Stress Response | Harsh dissociation conditions can stress cells and alter gene expression profiles [46] [8]. | Introduces biological noise; can confound true transcriptional signatures in developmental studies. |
| Cell Quality Control (QC) | Count Depth, Genes/Cell, Mitochondrial Fraction | Low counts/genes & high mitochondrial fraction indicate damaged/dying cells [66] [8]. | Essential for filtering out low-quality cells before scRNA-seq analysis to prevent skewed data [8]. |
Detailed Experimental Protocols
Protocol for Cell Quality Control in scRNA-seq
This protocol is critical for ensuring that only viable, single cells are sequenced, thereby minimizing the analysis of degraded RNA from compromised cells.
- Cell Suspension Preparation: Generate a single-cell suspension from tissue using optimized mechanical and enzymatic dissociation to maximize cell viability and minimize stress [46] [8].
- Metric Calculation: For each cell barcode, calculate three key QC metrics [66] [8]:
- Count Depth: The total number of UMIs (molecules) per cell.
- Detected Genes: The number of genes with at least one UMI count.
- Mitochondrial Fraction: The fraction of counts mapping to mitochondrial genes.
- Threshold Setting and Filtering: Jointly examine the distributions of all three metrics to set filtering thresholds [66].
- Remove low-quality cells: Filter out cells with low count depth, low numbers of detected genes, and a high fraction of mitochondrial counts, as these indicate damaged, dying, or dead cells [66] [8].
- Remove doublets: Filter out cells with an unexpectedly high count depth and high number of detected genes, which likely represent multiple cells [66] [8].
- Remove contaminants: Filter out cells expressing high levels of contaminant genes, such as hemoglobin (HBB) from red blood cells [8].
Protocol for RNA Immunoprecipitation Followed by qPCR (RIP-qPCR)
This protocol is used to validate specific RNA-protein interactions, and maintaining RNA integrity is crucial for accurate quantification.
- RNA Immunoprecipitation: Perform the RIP reaction using a validated kit (e.g., Magna RIP Kit) and an antibody specific to your RNA-binding protein of interest [67].
- RNA Isolation and DNase Treatment: Purify the immunoprecipitated RNA. Treat the RNA with RNase-free DNase to remove genomic DNA contamination [68] [67].
- Reverse Transcription (RT): Synthesize cDNA using a reverse transcriptase and random hexamers. Do not use oligo(dT) primers if the target RNA is not polyadenylated (e.g., U1 snRNA) [67].
- Quantitative PCR (qPCR):
- Reaction Setup: Add 2 μL of cDNA sample to a qPCR plate, preferably in triplicate. Prepare a master mix containing primers, SYBR Green dye, and polymerase. Add 23 μL of the master mix to each sample [67].
- Primer Design: Use primers specific to your target RNA sequence. For non-polyadenylated RNAs, ensure primers are designed against the mature RNA sequence [67].
- Amplification: Run the qPCR reaction using a standard amplification program on a real-time PCR instrument [67].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following reagents and kits are fundamental for implementing the best practices described above.
Table 2: Essential Research Reagents and Kits
| Reagent/Kit | Function/Best Practice Use |
|---|---|
| RNase Inhibitors | Added to reactions to protect RNA from degradation by ubiquitous RNases [67]. |
| RNase-free Aerosol Resistant Tips | Minimizes the risk of cross-contamination between samples during liquid handling [67]. |
| Magna RIP RNA-Binding Protein Immunoprecipitation Kit | Validated system for isolating specific RNA-protein complexes for downstream RIP-qPCR analysis [67]. |
| Single-Cell RNA-seq Library Prep Kits (e.g., 10x Genomics, Singleron) | Standardized pipelines (e.g., Cell Ranger, CeleScope) for processing raw scRNA-seq data into count matrices, incorporating QC metrics [8]. |
| Validated qPCR Assays & Hot-Start Taq Polymerase | Ensures specific and efficient amplification in qPCR validation steps, minimizing non-specific products [67]. |
| RNeasy Kit (or similar) | For efficient total RNA extraction and cleanup, including DNase digestion steps to remove genomic DNA contamination [68]. |
Workflow Integration for Developmental Biology
In developmental biology research, where samples like embryonic tissues are rare and precious, a rigorous, integrated workflow is non-negotiable. The following diagram integrates the techniques discussed into a cohesive flow for scRNA-seq sample preparation and validation, highlighting critical control points.
By adopting these best practices—from stringent wet-lab techniques to rigorous computational filtering—researchers can significantly enhance the reliability of their scRNA-seq data and the subsequent qPCR validation, leading to more robust and reproducible findings in developmental biology.
Troubleshooting Low cDNA Yield and High Background in Negative Controls
In the field of developmental biology research, the validation of single-cell RNA sequencing (scRNA-seq) data often hinges upon reliable reverse transcription quantitative PCR (RT-qPCR). This process is susceptible to two common and critical technical challenges: low cDNA yield and high background in negative controls. Low cDNA yield can compromise the detection of low-abundance transcripts, a significant concern when working with the minute RNA quantities from individual cells [69]. Concurrently, high background signals in negative controls indicate the presence of contaminating DNA or primer artifacts, which can lead to false-positive results and erroneous biological conclusions [70] [71]. These issues are particularly prevalent when adapting protocols for very small samples or single-cell inputs, where the efficiency of every reaction step is paramount and contaminants are amplified with the target. This guide objectively compares the performance of various commercial kits and methodological approaches to these challenges, providing a framework for optimizing your single-cell qPCR validation workflows.
Understanding the Root Causes
A systematic approach to troubleshooting begins with understanding the underlying causes of these common problems. The following table categorizes the primary issues, their causes, and initial diagnostic steps.
Table 1: Root Causes and Diagnostics for cDNA Synthesis Issues
| Problem | Potential Causes | Diagnostic Suggestions |
|---|---|---|
| Low cDNA Yield | Poor RNA quality or integrity [72]; Reaction inhibitors present [72]; Inefficient reverse transcriptase [73]; Too little input RNA [72]. | Visualize RNA on a denaturing gel; verify sharp 28S/18S bands and A260/A280 ratio of ~1.8-2.0 [74] [72]. Test for inhibitors by adding control RNA to the sample [72]. |
| High Background (Negative Controls) | Genomic DNA contamination [72] [73]; Primer-dimer formation [74] [72]; Contamination of reagents [72]; TS-oligo concatemerization (in TS-PCR protocols) [70]. | Perform a minus-RT control [72]. Use intron-spanning primers [71]. Design primers without complementary 3' ends [72]. |
The relationship between input RNA and successful detection is quantifiable. Research on single-cell RT-qPCR shows a direct sigmoidal relationship between the quantification cycle (Cq) value from bulk samples and the percentage of single cells in which a transcript is detected. One study established that a bulk Cq of 14.85 corresponds to detection in 50% of single cells, with the percentage dropping sharply for genes with higher (less abundant) Cq values [71]. This means transcripts with a bulk Cq above 25 are unlikely to be detected in a typical set of single cells, which can be misinterpreted as low yield if not properly understood [71].
Comparative Analysis of Methods and Kits
RNA Amplification Kit Performance
Selecting a robust RNA amplification method is critical for single-cell work. A comparative study evaluated three commercial kits using single-cell equivalent RNA inputs (25-50 pg) and Affymetrix arrays, measuring the number of genes detected and reproducibility.
Table 2: Performance Comparison of Single-Cell RNA Amplification Kits
| Kit Name | Genes Detected (Correlation) | Sensitivity (vs. Bulk RNA-seq) | Key Findings |
|---|---|---|---|
| EpiStem RNA-Amp | 2,667 genes (r=0.866) [69] | N/A (Benchmarked vs. arrays) | Showed the highest sensitivity and reproducibility in this comparison; identified 67.6% of all genes found by the other two kits combined [69]. |
| NuGEN Ovation One-Direct | 1,554 genes (r=0.723) [69] | N/A (Benchmarked vs. arrays) | Less sensitive than the RNA-Amp kit under the tested conditions [69]. |
| Miltenyi μMACS SuperAmp | 865 genes (r=0.8) [69] | N/A (Benchmarked vs. arrays) | The least sensitive kit in this comparison [69]. |
| SMARTer (C1 System) | N/A | ~42-44% of bulk [5] | Microfluidic (nanoliter) reaction volume reduced false positives and amplification bias compared to tube-based (microliter) methods [5]. |
Reverse Transcriptase and Master Mix Performance
The choice of reverse transcriptase can profoundly impact cDNA yield, especially with challenging samples. Data from Thermo Fisher Scientific demonstrates the performance of their SuperScript IV VILO Master Mix across a range of suboptimal conditions.
Table 3: Performance of SuperScript IV VILO Master Mix with Challenging Samples
| Challenge Type | Experimental Setup | Performance Result |
|---|---|---|
| Inhibitor-containing RNA | cDNA synthesis with 100 ng HeLa RNA in presence of various inhibitors [73]. | Delivered maximum cDNA yield and minimal Ct values in the presence of all tested inhibitors [73]. |
| Degraded RNA | cDNA synthesis using 50 ng of degraded (RIN<5) RNA from frozen lung tissue [73]. | Produced the highest cDNA yield and lowest Ct values compared to other RNA-to-cDNA kits [73]. |
| Linearity | Serial dilutions of total RNA (1 fg to 1 μg) reverse transcribed [73]. | Exhibited a coefficient of correlation of 0.999 and high efficiency of 94.2% across the entire range [73]. |
Detailed Experimental Protocols
Standardized Two-Step RT-qPCR Protocol
This protocol is adapted from technical documents for SYBR Green-based qPCR and is a foundational method for comparison [74].
A. Reverse Transcription (First Strand cDNA Synthesis)
- Reaction Setup: Combine the following in a nuclease-free tube:
- RNA template (1 pg to 1 µg total RNA, in a volume ≤ 8 µL) [73].
- 1 µL of Oligo(dT) primer (or 2 µL of random hexamers/gene-specific primer).
- 1 µL of 10 mM dNTP mix.
- Add nuclease-free water to a final volume of 10 µL.
- Incubation: Heat the mixture to 65°C for 5 minutes, then immediately place on ice for at least 1 minute.
- Master Mix: Prepare the following on ice:
- 4 µL of 5X First-Strand Buffer.
- 1 µL of 0.1 M DTT.
- 1 µL of RNaseOUT Recombinant RNase Inhibitor (optional but recommended).
- 1 µL of SuperScript IV Reverse Transcriptase (or other MMLV-derived RT).
- Combined Reaction: Add the 7 µL master mix to the initial 10 µL RNA/primer mix. Mix gently and pulse centrifuge.
- Incubation: Use one of the following profiles:
- For Oligo(dT) primers: 50°C for 50 minutes [73].
- For random hexamers: 25°C for 10 minutes, then 50°C for 50 minutes.
- Enzyme Inactivation: Heat to 85°C for 5 minutes. The cDNA can be stored at -20°C.
B. Quantitative PCR (qPCR)
- Reaction Setup: Prepare a master mix for multiple reactions containing:
- 10 µL of 2X SYBR Green qPCR Master Mix.
- 1 µL of 10 µM Forward Primer.
- 1 µL of 10 µM Reverse Primer.
- 6 µL of Nuclease-free water.
- 2 µL of cDNA template (use up to 10% of the first-strand reaction per 50 µL PCR) [72].
- qPCR Run: Use the following standard cycling parameters on a real-time PCR instrument:
- Initial Denaturation: 95°C for 10 minutes.
- 40 Cycles of:
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 60°C for 1 minute.
- Melting Curve: 60°C to 95°C with continuous fluorescence measurement.
Modified Template-Switching Protocol for Reduced Background
For single-cell RNA-seq methods that utilize template-switching (TS), a common source of high background is the concatemerization of the template-switching oligo (TSO) [70]. The following modification can significantly reduce this artifact.
Diagram 1: Mechanism of Background Reduction with Modified TS-oligo
Key Modification:
- Replace the standard TS oligo with a modified "iso3TS oligo" that incorporates non-natural nucleotide isomers (e.g., iso-dG and iso-dC) at its 5' end [70].
Procedure:
- Follow your standard single-cell lysis and reverse transcription protocol for template-switching assays (e.g., SMARTer-based kits).
- Substitute the standard TSO with an equimolar concentration of the iso3TS oligo.
- Proceed with the remaining cDNA synthesis and amplification steps as usual.
Expected Outcome: This modification prevents the reverse transcriptase from adding a polyC tail after incorporating the TSO. Without this tail, a second TSO cannot anneal, thereby halting the concatemerization process. This leads to a dramatic reduction in background cDNA in no-template controls and a higher proportion of usable sequence reads from biological samples [70].
The Scientist's Toolkit: Essential Reagents and Solutions
Table 4: Key Research Reagents for Optimized Single-Cell cDNA Synthesis
| Reagent / Solution | Function / Purpose | Example / Note |
|---|---|---|
| SuperScript IV RT | High-processivity reverse transcriptase for improved cDNA yield, especially from low-input or challenging samples [73]. | Shows robust performance with inhibitor-containing or degraded RNA [73]. |
| Hot-Start Taq Polymerase | Reduces non-specific amplification and primer-dimer formation by limiting polymerase activity until high temperatures are reached [74] [72]. | Critical for improving specificity in qPCR. |
| RNase Inhibitor | Protects fragile RNA templates from degradation during the reaction setup and reverse transcription steps [72]. | Essential for maintaining RNA integrity. |
| ezDNase Enzyme | Rapidly removes contaminating genomic DNA in a dedicated step prior to RT. Thermolabile, so it is inactivated during the RT step without needing EDTA [73]. | Simplifies workflow and preserves RNA integrity compared to traditional DNase I [73]. |
| Iso3TS Oligo | Modified template-switching oligo containing non-natural nucleotides to prevent concatemerization and reduce background in TS-PCR protocols [70]. | Directly addresses a major source of background in single-cell RNA-seq methods. |
| dNTPs with dUTP | dUTP replaces dTTP in the PCR mix. Allows pre-treatment with Uracil-DNA Glycosylase (UNG) to degrade carryover contamination from previous PCR products [74]. | A standard practice in diagnostic qPCR to prevent false positives. |
Troubleshooting low cDNA yield and high background requires a combination of strategic reagent selection and meticulous technique. The experimental data presented indicates that kits like EpiStem RNA-Amp can provide high sensitivity for single-cell work [69], while master mixes featuring advanced reverse transcriptases like SuperScript IV offer robustness across a wide array of challenging sample types [73]. For background issues, the root cause must be identified: genomic DNA contamination is best tackled with intron-spanning primers and dedicated DNase treatments like ezDNase [73] [71], whereas protocol-specific artifacts like TSO concatemerization require innovative solutions such as iso3TS oligos [70].
Adhering to the following best practices will significantly improve the reliability of your single-cell RT-qPCR data:
- Always include controls: No-template controls (NTC) and no-RT controls are non-negotiable for diagnosing contamination [74] [72].
- Prioritize RNA quality: The success of the entire workflow is fundamentally dependent on high-quality RNA input [74] [72].
- Validate with bulk relationships: Use the known sigmoidal relationship between bulk Cq and single-cell detection rates as a quality control metric for your single-cell data [71].
- Consider reaction volume: When possible, leveraging microfluidic systems for nanoliter-volume reactions can reduce competition and bias, leading to more accurate quantitative results [5].
Beyond qPCR: Corroborating Findings with Orthogonal Methods
When is qPCR Validation Necessary? Addressing the 'Journal Reviewer' Mindset
In the rapidly advancing field of developmental biology, single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to dissect cellular heterogeneity and identify novel cell populations. However, the high-resolution transcriptional landscapes revealed by scRNA-seq require rigorous validation to ensure biological fidelity. Quantitative PCR (qPCR) remains a cornerstone for this crucial validation step, providing the sensitivity, specificity, and quantitative rigor necessary to confirm scRNA-seq findings. Understanding when and how to implement proper qPCR validation is essential for satisfying the critical 'journal reviewer' mindset—a perspective demanding technical excellence, methodological transparency, and reproducible results. This guide examines the specific scenarios mandating qPCR validation within developmental biology research and provides a structured framework for meeting the exacting standards of peer review.
The Validation Imperative: Key Scenarios Requiring qPCR Confirmation
Table 1: When qPCR Validation is Essential in Developmental Biology Research
| Scenario | Purpose of qPCR Validation | Key Validation Parameters | Reviewer Expectations |
|---|---|---|---|
| Confirming novel cell populations | Verify unique gene expression signatures of newly identified cell types from scRNA-seq data [3]. | Specificity, efficiency, dynamic range [75] [76]. | Evidence beyond computational clustering; orthogonal confirmation of marker genes. |
| Supporting differential expression | Validate significant gene expression changes between developmental stages or experimental conditions [77]. | Precision, accuracy, proper normalization [78] [79]. | Transparency in statistical analysis and normalization strategy; MIQE guideline adherence [79]. |
| Characterizing spatial localization | Correlate transcriptional findings with physical location in developing tissues (e.g., via RNAscope) [3] [52]. | Sensitivity, limit of detection, specificity [78] [75]. | Integration of single-cell data with spatial context; methodological appropriateness. |
| Biomarker development | Transition scRNA-seq discoveries toward potential clinical or functional applications [78]. | Full analytical and clinical validation; robustness [78] [29]. | Rigorous, fit-for-purpose validation protocol; assessment of clinical performance [78]. |
| Troubleshooting discordant results | Resolve contradictions between scRNA-seq findings and expected biology or prior literature [78]. | Inclusivity, exclusivity, precision [75]. | Investigation of technical vs. biological causes; demonstration of assay reliability. |
qPCR validation becomes non-negotiable when research aims to translate scRNA-seq discoveries into biologically meaningful conclusions. The scenarios outlined in Table 1 represent critical junctures where technical validation intersects with scientific credibility. For developmental biologists, confirming novel cell populations is particularly paramount, as the field increasingly relies on computational clustering of scRNA-seq data to define cellular identities. Reviewers consistently demand orthogonal validation of marker genes that define these populations, as clustering artifacts can misleadingly suggest novel cell types [3]. Similarly, claims about key differentially expressed genes driving developmental processes require qPCR confirmation to demonstrate they are not technical artifacts of sequencing depth or normalization methods.
The 'journal reviewer mindset' prioritizes methodological rigor that ensures findings are not only statistically significant but also biologically reproducible. This is especially true for studies with potential translational impact, where analytical validation bridges the gap between research-use-only findings and clinically applicable biomarkers [78]. Furthermore, as developmental biology increasingly integrates spatial context, qPCR validation via techniques like RNAscope provides essential confirmation that computationally identified cell populations correspond to physically distinct locations within developing tissues [3] [52].
The Reviewer's Checklist: Essential qPCR Validation Parameters
Table 2: Key Analytical Performance Parameters for qPCR Validation
| Performance Parameter | Definition | Acceptance Criteria | Developmental Biology Context |
|---|---|---|---|
| Specificity | Ability to distinguish target from non-target sequences [78]. | Single peak in melt curve or appropriate probe detection [76]. | Critical for paralogous genes in gene families; confirms cell-type-specific markers. |
| Amplification Efficiency | Rate of PCR product amplification per cycle [77]. | 90-110% (R² ≥ 0.980) [75] [76]. | Affects accuracy of fold-change calculations between developmental stages. |
| Dynamic Range | Range of template concentrations with linear detection [75]. | 6-8 orders of magnitude [75]. | Essential for quantifying genes across varying expression levels in heterogeneous samples. |
| Limit of Detection (LOD) | Lowest target quantity reliably detected [75]. | ≥95% detection rate [75]. | Important for rare transcripts in limited cell populations. |
| Precision | Closeness of repeated measurements (repeatability & reproducibility) [78]. | CV < 5% for Cq values [78] [29]. | Ensures consistency across biological replicates and technical repeats. |
| Accuracy/Trueness | Closeness of measured value to true value [78]. | Recovery of 80-120% from spiked controls [29]. | Validates absolute quantification approaches. |
Journal reviewers systematically evaluate qPCR validation quality through specific analytical parameters. The criteria in Table 2 represent the minimum requirements for convincing peer reviewers of technical validity. Amplification efficiency between 90-110% with a corresponding R² value of ≥0.980 is particularly scrutinized, as efficiency deviations dramatically impact relative quantification accuracy—a common approach in developmental time-course studies [75] [76]. Similarly, demonstration of specificity is paramount when validating cell-type-specific markers identified through scRNA-seq, as off-target amplification could falsely confirm putative cellular identities.
Reviewers also assess whether the validation approach matches the biological question. For instance, when working with rare cell populations typical in developmental studies—such as stem cell niches or progenitor cells—documenting the limit of detection becomes crucial, as low-abundance transcripts approach the assay's detection capabilities [75]. Precision, encompassing both repeatability and reproducibility, provides confidence that findings are not technical artifacts, especially important when comparing subtle expression differences between developmental stages or experimental conditions [78].
Decision Framework for qPCR Validation
Experimental Protocols for Robust qPCR Validation
Sample Preparation and RNA Quality Control
Prior to reverse transcription, RNA integrity must be rigorously assessed using methods such as the Agilent Bioanalyzer [77]. For developmental biology studies involving rare cell populations, the input material is often limited, making quality assessment even more critical. For scRNA-seq validation, samples for qPCR should ideally originate from the same biological source as those used for sequencing. When using amplified cDNA from single cells, include unique molecular identifiers (UMIs) to correct for amplification biases [7]. Document RNA purity (A260/A280 ratio ≥1.8) and integrity (RIN >7 for bulk RNA) in manuscripts to satisfy reviewer requirements [79].
Reverse Transcription and Primer Validation
Use consistent reverse transcription protocols across all samples to minimize technical variation. For developmental gene expression studies, select priming strategies (random hexamers vs. oligo-dT) based on target transcript characteristics—oligo-dT is preferable for mRNA but requires intact RNA [77]. Validate primer specificity in silico using tools like NCBI Primer-BLAST against the appropriate genome assembly, then empirically test using melt curve analysis for SYBR Green assays or sequence confirmation of amplification products [29]. Test primer efficiency using a standard curve with a 6-7 point 10-fold dilution series in triplicate [75] [76].
qPCR Run Conditions and Data Analysis
Perform reactions in technical triplicates including no-template controls. Set baseline and threshold parameters consistently across all plates, with the threshold placed within the exponential amplification phase above baseline [76]. For developmental studies comparing expression across stages, use the 2^(-ΔΔCq) method only when primer efficiencies are near 100%; otherwise, apply efficiency-corrected models like the Pfaffl method [77] [79]. Select reference genes validated for stability across the specific developmental stages and tissues being studied—geometric mean of multiple reference genes is preferred [77].
Table 3: Key Research Reagent Solutions for qPCR Validation
| Reagent/Resource | Function | Considerations for Developmental Biology |
|---|---|---|
| High-Sensitivity RNA Kits | Isolate intact RNA from limited or low-input samples. | Essential for rare cell populations isolated via FACS from embryonic tissues [7]. |
| Reverse Transcription Kits | Convert RNA to cDNA with high efficiency and reproducibility. | Choose oligo-dT for polyA+ mRNA or random hexamers for degraded RNA (e.g., FFPE samples). |
| qPCR Master Mixes | Provide enzymes, dNTPs, and buffers optimized for detection chemistry. | Select SYBR Green for flexibility or probe-based for multiplexing; match to platform [29]. |
| Validated Primer Assays | Ensure specific amplification of target sequences. | Commercial assays provide interoperability; custom designs needed for novel transcripts. |
| Standard Reference Materials | Create standard curves for absolute quantification. | Use linearized plasmids or synthetic gBlocks with defined copy numbers [75]. |
| MIQE Checklist | Guide comprehensive reporting of qPCR experiments [79]. | Critical for manuscript preparation and addressing anticipated reviewer questions. |
Navigating the Publication Landscape: Data Transparency Requirements
Journal reviewers increasingly mandate comprehensive data sharing to ensure reproducibility. The MIQE guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) represent the gold standard for qPCR reporting [77] [79]. When submitting manuscripts, provide detailed descriptions of sample acquisition, RNA quality metrics, reverse transcription protocols, primer sequences, amplification efficiencies, and normalization strategies. For developmental biology studies, specifically document how tissue dissociation procedures (including duration, enzymes, and temperature) were optimized to minimize stress-induced transcriptional changes that could confound results [7].
Editors and reviewers are now requesting submission of raw qPCR data at manuscript submission [79]. The Real-time PCR Data Essential Spreadsheet (RDES) format provides a standardized template for sharing amplification curves, Cq values, and experimental metadata. This transparency allows reviewers to verify the appropriateness of analysis parameters, including baseline settings and threshold determination. For studies with substantial qPCR datasets, deposition in public repositories like Gene Expression Omnibus (GEO) is increasingly expected, paralleling requirements for sequencing data [79].
qPCR validation is not merely a technical formality but a strategic component of robust scientific discovery in developmental biology. By understanding the 'journal reviewer mindset'—with its emphasis on technical rigor, methodological transparency, and biological reproducibility—researchers can design validation experiments that withstand critical scrutiny. The decision framework, experimental protocols, and reporting standards outlined here provide a pathway for transforming scRNA-seq discoveries into validated biological insights that meet the exacting standards of high-impact publications. In an era of increasing focus on reproducibility, strategic qPCR validation serves both scientific accuracy and publication success.
In the field of developmental biology research, single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to profile transcriptional heterogeneity in complex tissues. However, the question of its quantitative accuracy remains paramount, especially when validating key findings related to cell fate decisions, lineage specification, and progenitor cell identification. Quantitative polymerase chain reaction (qPCR) has long been considered the gold standard for gene expression quantification, leading to its persistent role in validating high-throughput transcriptomic data. This guide objectively compares the performance of various scRNA-seq methods against qPCR benchmarks, providing developmental biologists with a framework for assessing technological accuracy and designing proper validation experiments.
Understanding qPCR as a Benchmark
Quantitative PCR operates through cyclic amplification of cDNA using sequence-specific primers, with fluorescence measurements quantifying accumulation of amplified products at each cycle. The cycle threshold (Cq) value provides a relative measure of initial template abundance. Its strengths include high sensitivity, a wide dynamic range, and exceptional reproducibility for measuring predefined gene sets. These characteristics have established qPCR as the reference method for transcript validation in developmental studies, such as verifying expression of key lineage-specific markers like TBX5 in first heart field progenitors [80].
scRNA-seq Technological Landscape
Single-cell RNA sequencing technologies capture the transcriptome of individual cells through cell isolation, reverse transcription, cDNA amplification, and library preparation for high-throughput sequencing. The methodologies vary significantly in their implementation, which directly impacts their quantitative performance. Key distinctions include:
- Full-transcript vs. 3'-end enriched protocols: Smart-seq2 provides full-length transcript coverage, while droplet-based methods (10X Genomics) typically capture 3' ends.
- Molecular identifiers: UMIs (Unique Molecular Identifiers) in protocols like 10X Chromium correct for amplification bias by tagging individual molecules [81].
- Reaction volumes: Microfluidic (nanoliter) implementations demonstrate reduced background and false positives compared to tube-based (microliter) reactions [65].
Head-to-Head: Quantitative Performance Comparison
Correlation of Expression Measurements
Multiple benchmarking studies have evaluated how closely scRNA-seq expression measurements align with qPCR data. In one comprehensive assessment, scRNA-seq methods showed high correlation with qPCR measurements across 40 genes, with Pearson correlation coefficients (r) exceeding 0.84 [65]. Notably, microfluidic platforms that process cells in nanoliter volumes demonstrated nearly 1:1 correlation (slope ≈1) with qPCR data, indicating superior accuracy compared to tube-based methods [65].
Table 1: Correlation Between scRNA-seq Methods and qPCR Benchmarks
| scRNA-seq Method | Reaction Volume | Correlation with qPCR (r) | Key Strengths |
|---|---|---|---|
| C1 System (SMARTer) | Nanoliter | >0.84 | Near 1:1 correlation, reduced false positives |
| SMARTer Ultra Low (Tube) | Microliter | >0.84 | Good sensitivity |
| TransPlex (Tube) | Microliter | >0.84 | Reproducibility |
| Fluidigm C1 qPCR | Nanoliter | N/A (qPCR reference) | High precision, narrow expression distribution |
Sensitivity and Detection Capabilities
Sensitivity in scRNA-seq refers to the method's ability to detect low-abundance transcripts, a critical factor when studying rare transcriptional events in developmental processes. When compared to bulk RNA-seq without amplification (the theoretical maximum), microfluidic scRNA-seq platforms can detect approximately 42-44% of genes identified by bulk sequencing [65]. This detection efficiency varies significantly across platforms, with each method exhibiting distinct sensitivity profiles that must be considered when designing developmental biology studies focusing on rare cell populations or weakly expressed lineage markers.
Differential Expression Concordance
When evaluating fold changes between samples—a common analysis in developmental time course studies—scRNA-seq methods demonstrate strong concordance with qPCR. Studies comparing MAQCA and MAQCB reference samples found approximately 85% of genes showed consistent differential expression results between RNA-seq and qPCR [82]. The alignment-based algorithms (e.g., TopHat-HTSeq) showed slightly better performance (15.1% non-concordant genes) compared to pseudoalignment methods (19.4% non-concordant genes for Salmon) [82].
Table 2: Differential Expression Concordance Between scRNA-seq and qPCR
| Analysis Workflow | Alignment Method | Concordance Rate | Non-concordant Genes |
|---|---|---|---|
| TopHat-HTSeq | Alignment-based | 84.9% | 15.1% |
| STAR-HTSeq | Alignment-based | ~84% | ~16% |
| Kallisto | Pseudoalignment | ~82% | ~18% |
| Salmon | Pseudoalignment | 80.6% | 19.4% |
Experimental Design and Protocol Considerations
Critical Experimental Parameters
Several technical factors significantly impact the quantitative accuracy of scRNA-seq relative to qPCR benchmarks:
Sequencing Depth and Spurious Inflation of Poisson Fit Shallow sequencing can mask true biological variation, making data appear more consistent with a Poisson error model. As sequencing depth increases, clear evidence of overdispersion emerges across biological systems, necessitating negative binomial models [81]. This has direct implications for studying developmental systems where true biological variation is high, such as in embryonic patterning.
Molecular Identification and Amplification Efficiency Protocols incorporating UMIs more accurately correct for amplification bias, providing counts that better reflect initial mRNA concentrations [81]. The choice of amplification chemistry (e.g., SMARTer, TransPlex) also introduces specific biases, with different methods showing variable performance across transcript abundance classes [65].
Reaction Volume and Contamination Effects Nanoliter volume reactions in microfluidic platforms demonstrate reduced background signal and fewer false positives compared to microliter volume tube-based preparations [65]. This is particularly important for detecting rare transcripts in specialized progenitor cells during development.
Benchmarking Experimental Protocol
For researchers seeking to validate scRNA-seq results with qPCR in developmental studies, the following experimental approach is recommended:
Sample Preparation
- Use the same starting cell population for both scRNA-seq and qPCR validation
- For developmental studies, carefully dissociate embryonic tissues to maximize cell viability while preserving transcriptomic integrity
- Include technical replicates for both assays and biological replicates when possible
qPCR Experimental Workflow
- Design primer pairs for 40+ target genes, including lineage-specific markers (e.g., TBX5 for first heart field [80])
- Perform reverse transcription with consistent RNA input across samples
- Run qPCR reactions using a validated SYBR Green or probe-based protocol
- Calculate relative expression using the ΔΔCq method with appropriate reference genes
scRNA-seq Experimental Workflow
- Choose a platform based on required throughput and sensitivity needs
- Incorporate UMIs to correct for amplification bias
- Include spike-in RNA controls (e.g., ERCC) to assess technical variation
- Sequence to sufficient depth (typically >50,000 reads/cell) to detect relevant transcripts
Data Analysis and Comparison
- Normalize scRNA-seq counts using appropriate methods (e.g., SCTransform for negative binomial models [81])
- Calculate relative expression values comparable to qPCR ΔΔCq metrics
- Compare expression ranks and fold changes between experimental conditions
- Identify and investigate discordant genes for technical artifacts
Statistical Frameworks for Accuracy Assessment
Error Models and Their Applications
The choice of statistical error model significantly impacts the interpretation of scRNA-seq data quality relative to qPCR benchmarks:
Poisson vs. Negative Binomial Models While Poisson models initially appear appropriate for sparse UMI-based data, rigorous testing across 59 datasets reveals clear evidence of overdispersion for sufficiently sequenced genes [81]. In developmental systems, where biological heterogeneity is inherent (e.g., differentiating progenitor cells), negative binomial models consistently outperform Poisson distributions. The degree of overdispersion varies substantially across datasets, arguing for data-driven parameter estimation rather than fixed parameters [81].
Goodness-of-Fit Testing Empirical assessments show that in deeply sequenced datasets (median >8,000 UMIs/cell), >90% of genes with average expression >1 UMI/cell deviate significantly from Poisson expectations [81]. However, downsampling to shallow sequencing depths (1,000 UMIs/cell) artificially creates the appearance of Poisson variation, with only 0.5% of genes failing goodness-of-fit tests [81]. This has critical implications for experimental design in developmental biology, where adequate sequencing depth is essential for capturing true biological variation.
When is qPCR Validation Necessary?
The decision to validate scRNA-seq results with qPCR depends on several factors:
Appropriate Scenarios for qPCR Validation
- When studying specific lineage markers in novel progenitor populations (e.g., first vs. second heart field progenitors [80])
- When scRNA-seq data is based on a small number of biological replicates
- When the scientific community requires orthogonal verification of key findings
- When extending findings to new sample cohorts not included in the original sequencing study [4]
Less Necessary Scenarios
- When scRNA-seq data is used as a screening tool to generate hypotheses for functional follow-up
- When additional scRNA-seq datasets are available for independent verification [4]
- When moving to protein-level validation approaches (e.g., immunohistochemistry)
Research Reagent Solutions for Developmental Biology Studies
Table 3: Essential Research Reagents for scRNA-seq and Validation Experiments
| Reagent Category | Specific Examples | Function in Experimental Pipeline |
|---|---|---|
| scRNA-seq Kits | SMARTer Ultra Low RNA Kit, Chromium Next GEM Single Cell 3' Kit | cDNA synthesis, library preparation from single cells |
| Cell Sorting Reagents | Fluorescent antibodies for FACS, Magnetic bead conjugation kits | Isolation of specific progenitor populations for validation |
| qPCR Reagents | SYBR Green master mixes, TaqMan assays, Reverse transcriptases | Target gene validation, expression quantification |
| Nucleic Acid Controls | ERCC RNA spike-in mixes, Synthetic RNA standards | Technical variation assessment, normalization controls |
| Gene Editing Tools | CRISPR/Cas9 systems, RNA interference reagents | Functional validation of marker genes in developmental models |
Advanced Applications in Developmental Biology
Lineage Tracing and scRNA-seq Integration
Innovative approaches combine genetic lineage tracing with scRNA-seq to validate developmental lineages. For example, a TBX5/MYL2 reporter system enabled identification of first heart field progenitors and their descendants in human iPSC differentiation models [80]. scRNA-seq confirmed the predominance of FHF differentiation (>90% left ventricular cardiomyocytes) using standard Wnt-based 2D differentiation protocols [80]. This integration provides a powerful validation framework where sequencing data corroborates lineage tracing results.
Multi-Omics Corroboration
Combining scRNA-seq with other omics technologies provides orthogonal validation while expanding biological insights. In studies of CCl4-induced liver injury, integrated analysis of ATAC-seq, RNA-seq, and scRNA-seq datasets revealed coordinated metabolic shifts during injury progression [83]. Similarly, combining scRNA-seq with spatial transcriptomics validates both the identity and localization of cell types, as demonstrated in studies of influenza-infected lung tissues identifying ADAMTS4 expression in specific stromal compartments [3].
scRNA-seq technologies demonstrate strong quantitative concordance with qPCR benchmarks when appropriately implemented, with correlation coefficients exceeding 0.84 and differential expression concordance rates of approximately 85%. The most accurate results emerge from experimental designs that consider sequencing depth, incorporate UMIs, utilize nanoliter reaction volumes when possible, and apply appropriate negative binomial error models to account for biological overdispersion.
For developmental biologists, validation strategies should be tailored to the specific research context. qPCR remains valuable for confirming key lineage markers in new sample cohorts, while multi-omics integration and functional assays provide compelling alternatives for comprehensive validation. As scRNA-seq technologies continue to evolve, their quantitative accuracy will further improve, potentially reducing but not eliminating the need for orthogonal validation in developmental research.
In the field of developmental biology, the transition from population-averaged analyses to single-cell resolution has revolutionized our understanding of embryonic development. Single-cell RNA sequencing (scRNA-seq) has been instrumental in uncovering cellular heterogeneity, yet it fundamentally lacks spatial context—information that is paramount when studying the intricate processes of embryogenesis where cell positioning dictates fate and function. This guide examines the integrated application of RNA Fluorescence In Situ Hybridization (FISH) and immunofluorescence (IF) as a powerful spatial validation toolkit for scRNA-seq findings. By correlating transcriptional profiles with precise subcellular localization of RNAs and proteins within a native tissue context, researchers can move beyond mere gene expression quantification to truly mechanistic insights in developmental systems.
Technical Comparison of Integrated Spatial Techniques
The combination of RNA FISH and IF is not a singular technique but a versatile suite of methods tailored to different biological questions, particularly in developmental contexts. The table below compares the primary integrated approaches used in contemporary research.
Table 1: Comparison of Integrated RNA FISH and Immunofluorescence Techniques
| Technique | Key Principle | Spatial Resolution | Multiplexing Capacity | Best Suited for Developmental Biology Applications |
|---|---|---|---|---|
| smRNA FISH+IF [84] [85] | Single-molecule RNA detection combined with protein immunofluorescence | Subcellular (Single RNA molecules) | Medium (Typically 1-3 RNAs with 1-2 proteins) | Validating cell type-specific markers identified by scRNA-seq; correlating protein and RNA expression heterogeneity in embryonic tissues [84]. |
| HCR RNA-FISH+IF [86] | Hybridization Chain Reaction for signal amplification with IF | Subcellular to whole-mount embryo | High (Multiple RNAs with proteins) | 3D gene expression mapping in intact embryos; analyzing organogenesis and tissue patterning [86]. |
| Sequential RNA/DNA FISH+IF [87] [88] | Sequential detection of RNA and DNA loci combined with IF | Nuclear (Precise gene locus mapping) | Low to Medium (Typically 1 gene locus with its RNA and 1 protein) | Studying nuclear organization, X-chromosome inactivation, and gene regulation dynamics in embryonic stem cells [89] [87]. |
| MERFISH+IF [90] | Multiplexed Error-Robust FISH barcoding with IF | Subcellular (With expansion) | Very High (Hundreds to thousands of RNAs) | Creating spatially resolved transcriptomic atlases of developing organs; validating complex cellular identities from scRNA-seq data [90]. |
Detailed Experimental Protocols for Key Applications
Protocol 1: Single-Molecule RNA FISH Combined with Immunofluorescence
This protocol is designed for the simultaneous detection of individual mRNA molecules and specific protein markers within single cells, providing a direct correlation between transcript abundance, localization, and protein presence [84] [85].
Key Modifications for Robust Combined Detection: The challenge in combining IF with smRNA FISH lies in the potential degradation of RNA by RNases during IF or the alteration of protein epitopes by FISH denaturation. An RNase-free modification of the standard IF protocol is critical [85]. This involves using DEPC-treated water and RNase-free buffers, coupled with the use of an anti-fade mounting medium that preserves both fluorescence signals.
Workflow Summary:
- Cell Preparation and Fixation: Grow cells on glass coverslips. Fix with 4% Paraformaldehyde (PFA) to preserve cellular architecture and immobilize biomolecules.
- Permeabilization: Treat with a mild detergent (e.g., 0.1% Triton X-100) to allow antibody and probe access while minimizing RNA loss.
- Immunofluorescence (RNase-free):
- Blocking: Incubate with a blocking solution (e.g., 0.5% UltraPure BSA in RNase-free PBS).
- Primary Antibody: Apply RNase-free primary antibody in blocking solution.
- Secondary Antibody: Apply fluorophore-conjugated secondary antibody in blocking solution [91].
- Post-fixation: Re-fix with 4% PFA after IF to anchor the antibodies and prevent their detachment during subsequent stringent FISH steps.
- smRNA FISH:
- Hybridization: Incubate with a pool of singly labeled oligonucleotide probes targeting the mRNA of interest. A set of ~20-50 probes per transcript is typical for smFISH, providing sufficient fluorescence for single-molecule detection [92].
- Stringent Washes: Remove non-specifically bound probes through a series of washes, often containing formamide to control stringency.
- Mounting and Imaging: Mount in an anti-fade medium and image using a high-resolution wide-field or confocal fluorescence microscope. For rare events, High-Speed and High-Resolution Scanning (HSHRS) fluorescence microscopy can be employed [84].
Diagram 1: smRNA FISH + IF workflow
Protocol 2: HCR RNA-FISH and IF in Whole-Mount Embryos
For developmental biologists, mapping gene expression in three dimensions within an intact embryo is invaluable. This protocol adapts HCR RNA-FISH for older chicken embryos (E3.5-E5.5), combining it with IF and tissue clearing for comprehensive 3D analysis [86].
Optimization for Whole-Mount Embryos:
- Probe Design: Use ~20 pairs of split-initiator oligonucleotides per target mRNA. This split-probe design enhances specificity by reducing non-specific amplification and background [86].
- Sample Fixation and Permeabilization: Fix embryos in 4% PFA. Permeabilization is crucial and may require optimization; extended detergent treatment or proteinase K digestion can be used for larger, older embryos.
- Simultaneous HCR and IF: After permeabilization, incubate the embryo with primary antibodies and HCR probe sets simultaneously. Follow with simultaneous incubation with secondary antibodies and HCR amplification hairpins conjugated to fluorophores.
- Tissue Clearing and 3D Imaging: Clear the labeled embryos using ethyl cinnamate (ECi), which was found to be highly effective for chicken embryos and compatible with HCR signal preservation [86]. Image the transparent embryos using light sheet fluorescence microscopy to capture gene and protein expression patterns with subcellular resolution throughout the entire embryo volume.
Table 2: Key Research Reagent Solutions for Integrated FISH-IF
| Reagent / Material | Function in Protocol | Example from Literature |
|---|---|---|
| Split-Initiator HCR Probes [86] | Binds target mRNA and initiates hybridization chain reaction for signal amplification. | Used to detect SOX10, ISL1, and SLIT2 in chicken embryos; enables multiplexing and reduces background [86]. |
| Acrydite-modified poly(dT) LNA Probes [90] | Anchors polyadenylated mRNAs to a polyacrylamide gel matrix for expansion microscopy or clearing. | Essential for MERFISH and expansion protocols to retain RNA during harsh treatments [90]. |
| RNase-free Antibodies [85] | Detects protein epitopes without degrading target RNA. | Critical for successful smRNA FISH+IF; ensures RNA integrity during immuno-staining steps [85]. |
| Ethyl Cinnamate (ECi) [86] | Clears tissue by matching refractive index, reducing light scattering for deep-tissue imaging. | Used to clear whole-mount chicken embryos after HCR RNA-FISH and IF, enabling light sheet microscopy [86]. |
| Encoding Probe Libraries [90] | Contains oligonucleotides with target-specific sequences and readout sequences for barcoding. | Allows for multiplexed detection of hundreds to thousands of RNA species in MERFISH [90]. |
Quantitative Data Supporting Technical Performance
The efficacy of integrating RNA FISH with immunofluorescence is substantiated by robust quantitative data. The tables below summarize key performance metrics from published studies.
Table 3: Quantitative Performance Metrics of Combined FISH-IF Techniques
| Technique | Reported Detection Efficiency | Spatial Resolution Achieved | Key Quantitative Validation |
|---|---|---|---|
| MERFISH + Expansion [90] | ~100% (for a high-density library of ~130 RNAs) | Subcellular (post-expansion) | Detection efficiency increased from ~20% pre-expansion to ~100% post-expansion; strong correlation with RNA-seq (r=0.6 to 0.8) [90]. |
| smRNA FISH + IF [85] | Not explicitly quantified, but enables single-molecule counting. | Single RNA molecules | Enabled direct visualization of the interaction between RNase MCPIP1 and IL-6 mRNA, and quantification of cell-to-cell heterogeneity [85]. |
| HCR RNA-FISH + IF + Clearing [86] | High signal-to-noise ratio, specific pattern reproduction. | Subcellular in whole-mount embryos (3D) | Faithfully reproduced known expression patterns (e.g., SOX10 in neural crest, ISL1 in DRGs) in 3D within cleared embryos [86]. |
Application in Single-Cell RNA-seq Validation and Developmental Biology
The integration of RNA FISH and IF provides a critical bridge between scRNA-seq discoveries and their functional, spatial context in developing tissues.
- Validating Novel Cell Clusters: A scRNA-seq analysis of a developing organ might reveal a novel cluster of cells expressing a unique combination of transcription factors. Using multiplexed HCR RNA-FISH or MERFISH for these transcripts, combined with IF for a putative marker protein, allows researchers to confirm the cluster's existence and determine its precise anatomical location and cellular morphology within the embryo [86].
- Unraveling Lineage Relationships: In studying processes like X-chromosome inactivation (XCI) in mouse embryos, sequential RNA FISH (for Xist RNA) and IF (for histone modifications) on the same samples have been indispensable. This approach has elucidated the dynamics of XCI initiation and maintenance, showing the accumulation of Xist RNA on the future inactive X chromosome and its correlation with repressive chromatin marks [89] [87] [88].
- Mapping Signaling Centers: Secreted signaling proteins create gradients that pattern embryonic tissues. While scRNA-seq can identify cells expressing genes for these signals and their receptors, it cannot reveal the gradient itself. Combining IF to detect the secreted protein with RNA FISH for the transcript can distinguish producing cells from receiving cells and map the protein distribution gradient, providing direct insight into patterning mechanisms.
Diagram 2: scRNA-seq validation workflow
The integration of RNA FISH and immunofluorescence represents a powerful and versatile approach for validating and enriching single-cell RNA sequencing data. By providing precise spatial context at the subcellular, cellular, and tissue levels, these combined techniques enable developmental biologists to move from lists of differentially expressed genes to a deeper, more mechanistic understanding of how gene expression patterns direct the complex process of embryonic development. As these methods continue to evolve, particularly in multiplexing capacity and compatibility with 3D sample analysis, their role as an essential component of the single-cell validation toolkit will only become more pronounced.
The establishment of single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology, with quantitative PCR (qPCR) serving as a cornerstone for its validation. This gold-standard approach provides high-precision, quantitative confirmation of gene expression patterns discovered in heterogeneous cellular populations [5]. However, as the field advances beyond the transcriptome to interrogate the epigenetic landscape via single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq), the validation framework must similarly expand. scATAC-seq enables the profiling of chromatin accessibility—a key marker of regulatory potential—in thousands of individual cells, revealing the epigenetic heterogeneity that underpins cell fate decisions during development [93] [94].
The inherent technical challenges of scATAC-seq, primarily its extreme data sparsity (only 1-10% of peaks detected per cell compared to 10-45% of genes in scRNA-seq), necessitate robust and multi-faceted validation strategies [93]. This guide objectively compares current scATAC-seq analysis methods and their corresponding functional validation assays, providing developmental biologists with a structured framework to confirm epigenetic discoveries within a multi-omics context.
Computational Method Comparison for scATAC-seq Analysis
The analysis of scATAC-seq data presents unique methodological challenges distinct from scRNA-seq, driving the development of specialized computational tools. These methods differ significantly in their approaches to feature definition, matrix construction, and dimensionality reduction, leading to variations in performance, scalability, and suitability for different research contexts.
Table 1: Benchmarking of scATAC-seq Computational Methods
| Method | Primary Featurization Strategy | Clustering Performance (ARI/AMI) | Key Strengths | Scalability (Cell Number) | Best Use Cases |
|---|---|---|---|---|---|
| SnapATAC | Genome binning & regression-based normalization [93] | High [93] | Only method analyzed >80,000 cells; fast processing [93] | >80,000 cells [93] | Very large datasets; genome-wide accessibility studies |
| cisTopic | Latent Dirichlet allocation (LDA) topic modeling [93] | High [93] | Identifies co-accessible regions; robust to noise [93] | Medium to Large [93] | Identifying regulatory topics; moderate coverage datasets |
| Cusanovich2018 | TF-IDF + SVD on genomic windows [93] | High [93] | Two-step clustering with in silico cell sorting [93] | Medium [93] | Standard resolution datasets; balanced performance |
| chromVAR | TF motif & k-mer frequency deviation [3] | Medium | Focus on TF binding potential; motif-centric [93] | Medium | Transcription factor activity inference |
| Gene Scoring | Accessibility near TSS with distance weighting [93] | Medium | Intuitive gene-level scores; correlates with expression [93] | Medium | Integration with scRNA-seq; gene-centric analyses |
| Cicero | Gene activity scores + co-accessibility networks [94] | Medium | Predicts enhancer-promoter connections [94] | Medium | Gene regulatory network inference |
Independent benchmarking on synthetic and real datasets has demonstrated that SnapATAC, Cusanovich2018, and cisTopic consistently outperform other methods in separating cell populations across different sequencing coverages and noise levels [93]. The choice of method directly impacts downstream biological interpretations, making selection a critical consideration for experimental design.
Figure 1: scATAC-seq Computational Workflow. The analysis pipeline involves sequential steps from raw data to biological interpretation, with method-specific approaches at each stage influencing final results.
Integrative Multi-Omics Analysis in Developmental Systems
The power of scATAC-seq is magnified when integrated with scRNA-seq data, enabling the correlation of epigenetic potential with transcriptional output. This approach has proven particularly valuable for understanding lineage commitment and cellular differentiation during development.
In studies of human fetal hematopoiesis, integrative analysis of over 8,000 immunophenotypic blood cells from fetal liver and bone marrow revealed extensive epigenetic priming in HSCs/MPPs prior to transcriptional commitment. Researchers observed opposing patterns of chromatin accessibility and differentiation that coincided with dynamic changes in the activity of distinct lineage-specific transcription factors [95]. This epigenetic priming occurred without coordinated expression of lineage-specific genes, suggesting that chromatin accessibility changes precede transcriptional commitment during blood cell differentiation [95].
Similarly, in porcine embryonic myogenesis—a valuable model for both agricultural science and human developmental disorders—researchers combined scRNA-seq and scATAC-seq to construct a differentiation trajectory of skeletal muscle ontogeny. They identified a pathogenic NTN5+LSAMP+ myoblast subpopulation enriched in growth-retarded embryos that autonomously blocked differentiation through coordinated epigenetic and metabolic dysregulation [96]. This subpopulation exhibited sustained chromatin accessibility at the cytoskeletal regulator TPM3, suppression of the muscle-specific actin depolymerization factor CFL2, and impaired glycolytic flux, with inhibition of the Hippo/TGF-β signaling pathway further exacerbating the differentiation arrest [96].
Table 2: Experimental Protocols for Multi-Omic Validation
| Technique | Experimental Protocol Summary | Key Applications in Validation | Compatibility with Developmental Models |
|---|---|---|---|
| RNA FISH | Fluorescently labeled nucleic acid probes hybridize to target RNA in situ; detection via fluorescence microscopy [3] | Spatial validation of marker gene expression; cell state localization [3] | Excellent for embryonic tissues; preserves spatial context |
| Immunofluorescence (IF) | Antibodies labeled with fluorescent pigments bind target antigens; visualization via fluorescence microscopy [3] | Protein-level validation of marker expression; spatial protein localization [3] | Suitable for whole-mount embryos; requires fixation |
| Massively Parallel Reporter Assays (MPRAs) | Library of candidate regulatory elements cloned into reporter vectors; transfected into cells; quantified via sequencing [97] | Functional validation of enhancer activity; impact of genetic variants [97] | Compatible with primary cells; medium throughput |
| CRISPR/Cas9 Knockout | Guide RNA directs Cas9 nuclease to create targeted double-strand breaks; disrupts gene function [3] | Functional validation of gene necessity; phenotypic confirmation [3] | Applicable to animal models; requires specialized expertise |
| ATAC-seq on Sorted Populations | Cell sorting based on markers → chromatin accessibility profiling on purified populations [95] | Validation of cell-type specific accessibility; technical confirmation [95] | Requires viable cells post-sorting; high resolution |
Figure 2: Multi-tiered Validation Framework. A comprehensive approach integrating computational, experimental, and functional validation strategies to confirm scATAC-seq findings.
Advanced Computational Tools for Functional Classification
Beyond cell clustering and visualization, advanced computational methods have been developed to extract deeper functional insights from scATAC-seq data. CoRE-ATAC represents a significant advancement as a deep learning framework that integrates DNA sequence with ATAC-seq cut sites and read pileups to classify the functional roles of accessible regions [97]. Trained on multiple cell types, CoRE-ATAC accurately predicts known cis-regulatory element functions—distinguishing promoters, enhancers, and insulators—with a mean average precision of 0.80 and mean F1 score of 0.70 across cell types not used in model training [97].
The ability to functionally classify accessible regions is particularly valuable for interpreting non-coding variants identified in developmental disorders. CoRE-ATAC predictions from human islet samples coincided with genetically modulated gain/loss of enhancer activity confirmed by MPRAs, demonstrating its utility for prioritizing functional variants [97]. Furthermore, models built from bulk ATAC-seq data effectively predicted cis-RE functions from aggregate single-nucleus ATAC-seq data from human blood-derived immune cells, establishing their application for studying rare cell populations without cell sorting [97].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagent Solutions for scATAC-seq Workflows
| Reagent/Kit | Primary Function | Application Context | Considerations for Developmental Studies |
|---|---|---|---|
| 10x Multiome ATAC + RNA Kit | Simultaneous profiling of chromatin accessibility and gene expression in single cells [94] | Integrative analysis of regulatory landscape and transcriptome | Optimal for rare embryonic samples; preserves paired measurements |
| Tn5 Transposase | Enzyme that cleaves accessible DNA and attaches sequencing adapters [94] | Tagmentation of accessible chromatin regions | Batch effects; activity consistency across preparations |
| Chromium Controller & Gel Beads | Microfluidic partitioning of single cells with barcoding [94] | Single-cell encapsulation and barcoding | Requires high-quality single-cell/nuclei suspensions |
| Smart-seq2 Chemistry | Full-length transcript coverage with high sensitivity [95] | scRNA-seq validation with high gene detection | Lower throughput but higher sensitivity than 10x |
| C1 Microfluidic System | Automated single-cell capture and processing in nanoliter chambers [5] | Low-volume reactions reducing amplification bias | Higher precision for quantitative applications |
The expanding toolkit for scATAC-seq analysis and validation represents a paradigm shift in developmental biology, enabling researchers to move beyond correlation to causation in understanding gene regulatory mechanisms. The most robust studies employ a multi-layered validation framework that combines computational method selection with orthogonal experimental techniques. Based on current benchmarking studies and experimental evidence, SnapATAC, cisTopic, and Cusanovich2018 provide the most consistent performance for basic cell type identification, while CoRE-ATAC offers advanced functional classification of regulatory elements [93] [97].
For developmental biologists, the integration of spatial validation techniques like RNA FISH with functional perturbations using CRISPR/Cas9 represents the gold standard for confirming developmental mechanisms inferred from scATAC-seq data. Furthermore, the demonstrated success of multi-omics integration in systems ranging from human hematopoiesis to porcine myogenesis highlights the power of combining epigenetic and transcriptional profiles to build comprehensive models of lineage commitment and tissue development [96] [95] [98]. As these technologies continue to mature, this expanded validation framework will be essential for distinguishing technical artifacts from true biological discoveries in the epigenetic regulation of development.
Interpreting Concordant and Discordant Results Between Platforms
Single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology by enabling researchers to dissect cellular heterogeneity, identify novel cell states, and unravel the complex processes of tissue formation and differentiation at unprecedented resolution [99]. However, the rapidly expanding landscape of scRNA-seq technologies, each with distinct methodological approaches and performance characteristics, presents a significant challenge for data interpretation and validation. The transition from bulk RNA sequencing to single-cell analysis introduces substantial technical variability, where the average expression level of a cell population can be strongly biased by a few cells with high expression and thus may not reflect a typical individual cell from that population [5].
Understanding the sources of concordance and discordance between different scRNA-seq platforms, and between scRNA-seq and quantitative PCR (qPCR) validation data, is essential for drawing accurate biological conclusions. This is particularly crucial in developmental biology research, where cell fate decisions often hinge on subtle changes in the expression of key transcriptional regulators observed in small, rare subpopulations of cells. Direct platform comparisons reveal that differences in sensitivity, accuracy, and technical variability can significantly impact the detection of these critical cell states [100] [5]. This guide provides a systematic framework for interpreting multi-platform scRNA-seq data, with a specific focus on experimental design considerations and analytical approaches relevant to developmental biology research.
scRNA-seq Platform Technologies and Methodologies
Major scRNA-seq Platform Technologies
Current scRNA-seq platforms employ distinct technological approaches for single-cell capture, reverse transcription, and cDNA amplification, each with characteristic strengths and limitations:
- Microfluidic-based systems (e.g., Fluidigm C1) utilize integrated fluidic circuits to isolate individual cells into nanochannels for visual examination, followed by cell lysis and cDNA conversion [100]. These systems allow for intermediate assessment of cell-capture quality and quantity but are size-restricted based on nanochannel tolerance [100].
- Droplet-based systems (e.g., 10x Genomics Chromium Controller, BioRad ddSEQ) use microfluidics to co-encapsulate single cells and barcoded beads into sub-nanoliter droplets [100]. These platforms offer high throughput, capable of processing up to 80,000 cells in a single instrument run, but typically only permit 3'- or 5'-tag profiling rather than full-length transcript analysis [100].
- Plate-based systems (e.g., WaferGen ICELL8) isolate hundreds to thousands of individual cells in nanowells on a chip, allowing for analysis of cell capture and viability while supporting both 3'-profiling and full-length transcriptome analysis [100].
Experimental Design for Platform Comparison
Robust comparison of scRNA-seq platforms requires careful experimental design to control for biological variability and enable direct technical comparisons:
- Cell line standardization: To minimize sample heterogeneity associated with dissociated tissues, comparisons should utilize well-characterized cell lines. The SUM149PT cell line treated with histone deacetylase inhibitor trichostatin A (TSA) versus untreated controls has been successfully used in multiplatform comparison studies [100].
- Spike-in controls: The use of synthetic RNA spike-ins (e.g., External RNA Controls Consortium [ERCC] and Spike-in RNA Variants [SIRVs]) is essential for assessing sensitivity and accuracy across platforms [101]. These controls span multiple concentrations, lengths, GC contents, and abundance levels, enabling quantitative benchmarking.
- Matched sample analysis: Comparing "matched" single-cell cDNA across different library preparation protocols and sequencing platforms controls for cell-to-cell variability and allows direct assessment of technical performance [101].
- Cross-platform validation: Integration of orthogonal validation methods, particularly multiplex qPCR, provides a gold standard for assessing the quantitative accuracy of scRNA-seq measurements [5].
Table 1: Key scRNA-seq Platforms and Their Characteristics
| Platform | Technology Type | Throughput (Cells) | Transcript Coverage | Cell Size Restrictions | Visual QC Potential |
|---|---|---|---|---|---|
| Fluidigm C1 | Microfluidic chip | 96 | Full-length | Yes (10-17 μm) | Yes |
| 10x Genomics Chromium | Droplet | Up to 80,000 | 3' or 5' tagging | Minimal | No |
| BioRad ddSEQ | Droplet | Hundreds to thousands | 3' tagging | Minimal | No |
| WaferGen ICELL8 | Nanowell plate | 1,000-1,800 | Full-length or 3' tagging | Minimal | Yes |
Quantitative Performance Metrics Across Platforms
Sensitivity and Accuracy Measurements
Systematic comparisons of scRNA-seq platforms reveal significant differences in performance characteristics that can substantially impact data interpretation:
- Sensitivity variations: When comparing commercially available RNA-amplification protocols at single-cell equivalent RNA inputs (25-50 pg), the EpiStem RNA-Amp kit detected 2,667 genes, significantly outperforming the NuGEN Ovation One-Direct System (1,554 genes) and Miltenyi μMACS SuperAmp (865 genes) [69].
- Molecular detection limits: Assessment of detection sensitivity using RNA spike-ins shows that different platform combinations can detect as few as 12 to 47 molecules per cell, with the BGISEQ-500 platform demonstrating slightly improved sensitivity compared to Illumina HiSeq, largely attributable to higher sequencing depth [101].
- Quantitative accuracy: Correlation analysis between scRNA-seq and qPCR data for 40 genes shows strong agreement (r > 0.84), with platforms utilizing nanoliter-volume reaction chambers (e.g., Fluidigm C1) demonstrating nearly ideal linear regression slopes close to 1, indicating superior quantitative accuracy compared to microliter-volume preparations [5].
Reproducibility and Technical Variability
The reproducibility of scRNA-seq measurements combines both technical variation and biological variability between individual cells:
- Inter-platform reproducibility: Reproducibility between replicate samples varies across platforms, ranging from 57% to 65% depending on the method used [5]. The Fluidigm C1 system demonstrates high reproducibility, with pairwise correlation of spike-in transcript abundance >0.9 (Pearson correlation coefficient) [5].
- Gene detection consistency: Comparative analysis shows that 67.6% (1,365 of 2,018 genes) of all genes identified by either SuperAmp or Ovation One-Direct were also detected by the RNA-Amp protocol, indicating substantial but incomplete overlap between platforms [69].
- Cross-platform correlation: Pseudo-bulk samples created by pooling single cells show high correlation between sequencing platforms (Illumina HiSeq and BGISEQ-500) and protocols, with accuracy correlations ranging from R = 0.66 to 0.70 regardless of scRNA-seq protocol [101].
Table 2: Performance Metrics Across scRNA-seq Platforms and Methods
| Platform/Method | Sensitivity (Genes Detected) | Reproducibility (% Overlap) | qPCR Correlation (r value) | False Positive Rate |
|---|---|---|---|---|
| EpiStem RNA-Amp | 2,667 genes | 86.6% | >0.84 | Low |
| NuGEN Ovation One-Direct | 1,554 genes | 72.3% | >0.84 | Moderate |
| Miltenyi SuperAmp | 865 genes | 80.0% | >0.84 | Moderate |
| Fluidigm C1 (nanoliter) | ~42% of bulk | >90% (spike-ins) | ~1.0 (slope) | Low |
| Tube-based (microliter) | Lower than C1 | Variable | <0.9 (slope) | Higher |
Analysis of Concordant Results: Biological Validation
Biological Interpretation of Concordant Findings
Concordant results across multiple scRNA-seq platforms and with qPCR validation provide the highest confidence in biological conclusions:
- Cell type identification: In developmental biology, concordant detection of key transcriptional regulators across platforms strongly supports their utility as lineage markers. For example, PCA analysis of single MCF7 and MCF10A cells clearly separates these cell types based on transcriptional profiles regardless of the scRNA-seq platform used, identifying 92 consistently differentially expressed genes between these lineages [69].
- Pathway activity: Consistent detection of coordinated gene expression patterns across platforms provides robust evidence for activated signaling pathways or biological processes. Studies of cancer initiating cells (CICs) have identified transcriptional signatures correlated with established stem-cell and epithelial-mesenchymal transition pathways across multiple analytical platforms [69].
- Developmental trajectories: When multiple platforms consistently identify the same rare cell populations or transitional states, confidence in these putative developmental intermediates increases substantially. Research on mouse embryonic stem cells cultured in serum and LIF has identified distinct subpopulations with different pluripotency characteristics that are detectable across sequencing platforms [101].
Technical Foundations of Concordance
Concordant results typically arise from robust biological signals combined with technically optimized experimental conditions:
- High-abundance transcripts: Genes with moderate to high expression levels (Cq < 30 in qPCR) show excellent correlation between scRNA-seq and qPCR measurements across all platforms [5].
- Optimal reaction volumes: Nanoliter-volume reaction chambers demonstrate reduced false positives and improved quantitative accuracy compared to microliter-volume preparations, leading to higher concordance with validation methods [5].
- Sequence-specific factors: Transcripts with minimal secondary structure, moderate GC content, and without closely related paralogs are more likely to generate concordant results across platforms.
Diagram 1: Technical foundations leading to concordant results and validated biological conclusions.
Investigation of Discordant Results: Technical Artifacts and Biological Insights
Discordant results between scRNA-seq platforms and qPCR validation can arise from multiple technical factors:
- Sensitivity limitations: Low-abundance transcripts detectable in bulk samples may go undetected in scRNA-seq when profiling a limited number of cells due to transcriptional bursting and the highly skewed distribution of transcripts among individual cells [71]. This is particularly relevant for key developmental regulators that may be expressed at low levels but play critical roles in cell fate decisions.
- Amplification bias: Different reverse transcription and cDNA amplification methods exhibit varying degrees of bias, with tube-based methods in microliter volumes showing wider distributions of expression values for housekeeping genes compared to nanoliter-volume platforms [5].
- Genomic DNA contamination: Primers that do not span intron-exon junctions can generate false positives by amplifying genomic DNA, particularly problematic in protocols without DNase treatment steps [71]. This can lead to overestimation of the fraction of cells expressing a gene compared to bulk samples.
- Platform-specific biases: Differences in transcript coverage, GC bias, and length bias between platforms can generate discordant results for specific genes, with some platforms preferentially detecting certain transcript classes [5].
Some discordances reflect genuine biological phenomena rather than technical artifacts:
- Cell state heterogeneity: Apparent discordances may arise when different platforms capture different cell states within a heterogeneous population. Studies of mouse embryonic stem cells have identified subpopulations with distinct pluripotency characteristics that may be differentially represented across platforms due to sampling effects [101].
- Bimodal expression patterns: Genes with "bursty" expression patterns may appear discordant when different platforms sample different proportions of expressing and non-expressing cells [71]. The relationship between bulk Cq values and the fraction of single cells expressing a gene follows a sigmoidal function, with a Cq50 of approximately 14.85 cycles corresponding to expression in 50% of cells [71].
- Alternative splicing detection: Platforms with full-length transcript coverage (e.g., Fluidigm C1, ICELL8) may detect isoforms missed by 3'-end focused methods (e.g., 10x Genomics), creating apparent discordances for specific genes [100].
Systematic Approach to Discordance Resolution
A structured workflow enables researchers to distinguish technical artifacts from biologically meaningful discordances:
- Spike-in control analysis: Assess whether discordances correlate with input molecule concentration using ERCC or SIRV spike-ins [101].
- Orthogonal validation: Implement multiplex qPCR on the same cell populations to confirm expression patterns [5].
- Cross-platform mapping: Compare results across multiple scRNA-seq platforms to identify consistent patterns [100] [101].
- Biological context evaluation: Assess whether discordant genes belong to coherent biological pathways or show correlated expression patterns.
Diagram 2: Technical and biological sources of discordant results between platforms.
qPCR Experimental Design for scRNA-seq Validation
Primer Design and Optimization
Robust qPCR validation of scRNA-seq data requires meticulous primer design and optimization:
- Primer design parameters: Design primers to generate amplicons between 70-200 bp with melting temperatures of 60-63°C (maximum 3°C difference between primers) and GC content of 40-60% [102]. The 3' end of primers should contain a G or C residue to enhance specificity [102].
- Exon-exon junction spanning: Design primers to span exon-exon junctions whenever possible to avoid amplification of genomic DNA contamination [71] [102]. This is particularly critical for single-cell workflows where DNase treatment steps are challenging to implement.
- Specificity validation: Use tools like Primer-BLAST to ensure primer specificity and avoid primer-dimer formation or secondary structures [102] [103]. The strongest total dimer should be unstable (ΔG ≥ -6.0 kcal) with no 3'-end dimers having ΔG < -2.0 kcal [104].
- Experimental optimization: Systematically optimize primer concentrations (typically 50-800 nM) and annealing temperatures using temperature gradients to achieve maximum efficiency and specificity [104] [103].
qPCR Validation Workflow
A standardized qPCR workflow ensures reliable validation of scRNA-seq findings:
- Reverse transcription consistency: Use the same reverse transcription method for both scRNA-seq and qPCR validation to minimize methodological biases [5].
- Standard curve implementation: Include standard curves covering the expected target range to assess amplification efficiency, which should fall between 90-110% (corresponding to a slope of -3.1 to -3.6) for reliable quantification [104].
- Multiplexing considerations: When performing multiplex qPCR, optimize primer concentrations to ensure balanced amplification of all targets, as highly abundant transcripts may dominate the reaction and reduce detection sensitivity for rare targets [104].
- Data normalization: Normalize expression values to the median expression across all transcripts for each cell to enable direct comparison between qPCR and scRNA-seq data [5].
Table 3: Research Reagent Solutions for scRNA-seq and Validation
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| ERCC Spike-In Mix | External RNA controls for sensitivity assessment | 92 synthetic RNA species of varying lengths and concentrations [5] [101] |
| SIRV Spike-In Set | Accuracy controls for isoform detection | 69 artificial transcripts with defined isoforms and abundances [101] |
| SMARTer Ultra Low RNA Kit | cDNA synthesis from low-input RNA | Used in multiple platforms including Fluidigm C1; enables full-length transcript coverage [100] [5] |
| Nextera XT DNA Library Prep Kit | Library preparation for sequencing | Compatible with single-cell cDNA; used across multiple platforms [100] [5] |
| Primer-BLAST | Specific primer design | NCBI tool for designing target-specific primers with exon-junction spanning capability [102] |
| Fluidigm C1 System | Automated single-cell capture and processing | Enables nanoliter-volume reactions reducing false positives and improving accuracy [100] [5] |
| 10x Genomics Chromium | High-throughput single-cell partitioning | Enables processing of up to 80,000 cells but limited to 3' or 5' tagging [100] |
Interpreting concordant and discordant results across scRNA-seq platforms requires a systematic approach that integrates technical understanding with biological knowledge. Key principles include:
- Platform selection alignment: Choose scRNA-seq platforms based on specific experimental needs—high-throughput for comprehensive atlas building versus full-length transcript coverage for isoform detection in developmental systems.
- Tiered validation approach: Prioritize validation efforts for critical findings using orthogonal methods, with particular focus on low-abundance transcripts, transcription factors, and genes showing platform-specific discrepancies.
- Contextual interpretation: Evaluate discordances within the framework of biological knowledge, considering whether technically discordant genes belong to coherent pathways or represent biologically plausible regulatory networks.
- Transparent reporting: Document all platform specifications, experimental parameters, and analysis methods to enable proper interpretation of results and facilitate cross-study comparisons.
This structured approach to platform comparison and validation enables developmental biologists to distinguish technical artifacts from genuine biological signals, ultimately leading to more robust insights into the cellular mechanisms underlying development, disease, and tissue homeostasis.
Conclusion
The integration of scRNA-seq and qPCR establishes a powerful, defensible framework for advancing developmental biology. While scRNA-seq provides an unparalleled, holistic view of cellular heterogeneity and developmental potential, targeted qPCR validation offers the precision and statistical confidence required to solidify these discoveries. This synergistic approach is crucial for accurately defining developmental trajectories, from stem cell potency to terminal differentiation, as demonstrated by tools like CytoTRACE 2. Moving forward, the field will increasingly rely on multi-omics integration and spatial techniques to contextualize transcriptional dynamics within tissue architecture. For researchers in both basic and translational science, mastering this combined methodology is key to generating robust, reproducible data that can reliably inform drug discovery and regenerative medicine strategies.
References
- 1. How Single - Cell Genomics Is Changing Evolutionary and... [https://pubmed.ncbi.nlm.nih.gov/28813177/]
- 2. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications | Genome Medicine | Full Text [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0467-4]
- 3. How To Validate Single-Cell RNA-Seq Data? - CD Genomics [https://www.spatial-omicslab.com/resource/validate-single-cell-rna-seq.html]
- 4. Do I Need to Validate My RNA - Seq Results With qPCR ? [https://www.rna-seqblog.com/do-i-need-to-validate-my-rna-seq-results-with-qpcr/]
- 5. assessment of Quantitative - single -sequencing methods... cell RNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC4022966/]
- 6. - Single profiling coupled with lineage analysis reveals vagal and... cell [https://elifesciences.org/articles/79156]
- 7. Single‐cell RNA sequencing technologies and applications: A brief overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 8. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications | Military Medical Research | Full Text [https://mmrjournal.biomedcentral.com/articles/10.1186/s40779-022-00434-8]
- 9. Navigating single-cell RNA-sequencing: protocols, tools, databases, and applications | Genomics & Informatics | Full Text [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00044-5]
- 10. Frontiers | Repertoire-based mapping and time-tracking of T helper... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1536302/full]
- 11. An integrated analysis of scRNA - seq and RNA-seq data revealed... [https://jtd.amegroups.org/article/view/99451/html]
- 12. Frontiers | Integrating microarray data and single-cell RNA-seq reveals correlation between kit and nmyc in mouse spermatogonia stem cell population [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1634347/full]
- 13. Integrating bulk RNA-seq, scRNA - seq , and spatial transcriptomics... [https://cancerci.biomedcentral.com/articles/10.1186/s12935-025-03964-y]
- 14. Integration of Single cell and Bulk RNA-Sequencing Reveals Key... [https://www.dovepress.com/integration-of-single-cell-and-bulk-rna-sequencing-reveals-key-genes-a-peer-reviewed-fulltext-article-JIR]
- 15. How to Chose the Best Single-Cell RNA Sequencing Technology [https://www.scdiscoveries.com/blog/knowledge/single-cell-sequencing-technologies/]
- 16. A comparison of marker gene selection methods for single-cell RNA... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895860/]
- 17. Identity from Mapping - Cell : A primer on computational... scRNA seq [https://pubmed.ncbi.nlm.nih.gov/40270709/]
- 18. - Single transcriptome analysis of avian neural crest migration... cell [https://elifesciences.org/articles/28415]
- 19. High-Throughput Data Analysis for qPCR | NEB | NEB [https://www.neb.com/en/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr]
- 20. Droplet Digital PCR versus qPCR for gene expression analysis with... [https://www.nature.com/articles/s41598-017-02217-x?error=cookies_not_supported&code=d6610c4f-4f0d-439a-8d25-955aafe5adb1]
- 21. Finding cell -specific expression patterns in the early Ciona embryo with... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7080732/]
- 22. Comparison of Analytic Methods for Quantitative Real-Time Polymerase Chain Reaction Data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4642823/]
- 23. qPCR Data Selector | Real-time qPCR Data | qPCR Performance Data [https://www.promega.com/resources/tools/pcr-data-selector/]
- 24. RT-qPCR data analysis [https://www.qiagen.com/us/applications/geneglobe/qpcr-data-analysis]
- 25. Improved reconstruction of single-cell developmental with... potential [https://www.nature.com/articles/s41592-025-02857-2?error=cookies_not_supported&code=4e00e8f7-c0cd-4216-94fb-2c5bef9e8b44]
- 26. Cell-Surface Proteomics Identifies Lineage-Specific Markers of Embryo-Derived Stem Cells - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3405530/]
- 27. Defining developmental and cell potency trajectories by... lineage [https://pubmed.ncbi.nlm.nih.gov/19112179/]
- 28. Validation of RNAseq Experiments by qPCR? [http://bridgeslab.sph.umich.edu/posts/validation-of-rnaseq-experiments-by-qpcr]
- 29. Recommendations for Method and Validation of Development ... qPCR [https://link.springer.com/article/10.1208/s12248-023-00880-9]
- 30. Identification of reference genes for gene expression studies among... [https://bmcdevbiol.biomedcentral.com/articles/10.1186/s12861-021-00244-6]
- 31. A qPCR for genome editing efficiency determination and... method [https://pubmed.ncbi.nlm.nih.gov/31827197/]
- 32. Single cell RNA-sequencing: replicability of cell types - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6551252/]
- 33. Single cell RNA-sequencing: replicability of cell types - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0959438818301612]
- 34. How thoughtful experimental can... | Nature Communications design [https://www.nature.com/articles/s41467-025-62616-x?error=cookies_not_supported&code=f60bfde0-62d7-499a-ada2-f4430b89aaea]
- 35. RNA-Seq Experimental Guide for Drug Discovery | Lexogen Design [https://www.lexogen.com/planning-for-success-a-strategic-design-guide-for-rna-seq-experiments-in-drug-discovery/]
- 36. 3.4 - Steps in Designing a Study | STAT 555 [https://online.stat.psu.edu/stat555/node/43/]
- 37. RNA-seq validation: software for selection of reference and variable candidate genes for RT-qPCR | BMC Genomics | Full Text [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10511-y]
- 38. assessment of Quantitative - single -sequencing methods cell RNA [https://www.nature.com/articles/nmeth.2694?error=cookies_not_supported&code=558e7248-c8fa-477a-bba4-1950386dbfd8]
- 39. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor | Nature Communications [https://www.nature.com/articles/s41467-018-03282-0]
- 40. Single-cell RNA-seq: Marker identification | Introduction to Single-cell RNA-seq - ARCHIVED [https://hbctraining.github.io/scRNA-seq/lessons/09_merged_SC_marker_identification.html]
- 41. Optimal marker gene selection for cell type discrimination in single cell analyses | Nature Communications [https://www.nature.com/articles/s41467-021-21453-4]
- 42. Tutorial: Guidelines for Single- Cell RT- qPCR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8534298/]
- 43. Basic Principles of RT-qPCR | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/basic-principles-rt-qpcr.html]
- 44. Do results obtained with RNA-sequencing require independent verification? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7823214/]
- 45. Smart PCR primers for qPCR success | The DNA Universe BLOG [https://the-dna-universe.com/2025/05/03/the-primer-puzzle-solved-how-smart-defaults-and-fast-delivery-simplify-your-qpcr-workflow/]
- 46. - Single & Cell - Low RNA-Seq | Input - Single sequencing methods cell [https://www.illumina.com/techniques/sequencing/rna-sequencing/ultra-low-input-single-cell-rna-seq.html]
- 47. Direct Reverse Transcription | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/reverse-transcription/superscript-cellsdirect.html]
- 48. CLEAR: coverage-based limiting- cell experiment analysis for RNA-seq [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-020-02247-6]
- 49. Library Preparation Quality Control and Quantification | RNA Lexicon [https://www.lexogen.com/blog/rna-lexicon-library-preparation-quality-control-and-quantification/]
- 50. GitHub - digitalcytometry/ cytotrace : 2 is an interpretable... CytoTRACE 2 [https://github.com/digitalcytometry/cytotrace2]
- 51. CytoTRACE [https://cytotrace.stanford.edu/]
- 52. NGS Validation with RNAscope ISH | Advanced Cell Diagnostics [https://acdbio.com/science/applications/research-solutions/ngsht-transcriptomic-validation-rna-seq]
- 53. Comparison of Four Nuclear Isolation Buffers for Plant DNA Flow Cytometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2803574/]
- 54. | Thermo Fisher Scientific - US Cell Lysis Buffers [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-purification-isolation/cell-lysis-fractionation/cell-lysis-total-protein-extraction.html]
- 55. Methods: A Guide to Efficient Protein Extraction | Boster Bio Cell Lysis [https://www.bosterbio.com/blog/post/cell-lysis-the-first-and-crucial-step-in-molecular-biology]
- 56. Sample preparation for western blot | Abcam [https://www.abcam.com/en-us/technical-resources/guides/western-blot-guide/sample-preparation-for-western-blot]
- 57. Frontiers | Enhancing the reverse transcriptase function in Taq... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1495267/full]
- 58. Optimization of contrast medium volume for abdominal CT in oncologic patients: prospective comparison between fixed and lean body weight-adapted dosing protocols | Insights into Imaging | Full Text [https://insightsimaging.springeropen.com/articles/10.1186/s13244-021-00980-0]
- 59. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem | Scientific Reports [https://www.nature.com/articles/srep44905]
- 60. Systematic identification and validation of the reference genes from 60 RNA-Seq libraries in the scallop Mizuhopecten yessoensis | BMC Genomics | Full Text [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5661-x]
- 61. Supramolecular optimization of the visual contrast for colorimetric indicator assays that release resorufin dye - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7429340/]
- 62. Crystal violet | Abcam staining protocol [https://www.abcam.com/en-us/knowledge-center/cell-biology/crystal-violet-staining-protocol]
- 63. Contrast Resolution chart and analysis | Imatest [https://www.imatest.com/docs/contrast-resolution/]
- 64. and degradation and its impact on... Controlled RNA contamination [https://pubmed.ncbi.nlm.nih.gov/23999274/]
- 65. Quantitative assessment of single-cell RNA-sequencing methods - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4022966/]
- 66. Current best practices in single‐cell RNA‐seq analysis: a tutorial - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 67. Immunoprecipitation RNA (RIP- qPCR ) Protocol qPCR [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/qpcr/rip-qpcr-protocol]
- 68. Comparison between qPCR and RNA-seq reveals challenges of quantifying HLA expression - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9883133/]
- 69. Evaluation and validation of a robust single -amplification... cell RNA [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-1129]
- 70. Incorporation of non-natural nucleotides into template-switching... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-11-413]
- 71. The correlation between expression profiles measured in single cells and in traditional bulk samples | Scientific Reports [https://www.nature.com/articles/srep37022]
- 72. Reverse Transcription and RACE Support - Troubleshooting [https://www.thermofisher.com/sg/en/home/technical-resources/technical-reference-library/pcr-cdna-synthesis-support-center/reverse-transcription-race-support/reverse-transcription-race-support-troubleshooting.html]
- 73. synthesis for cDNA | Thermo Fisher Scientific - DE qPCR [https://www.thermofisher.com/de/de/home/life-science/pcr/reverse-transcription/superscript-iv-vilo-master-mix.html]
- 74. Universal SYBR Green qPCR Protocol [https://www.sigmaaldrich.com/MY/en/technical-documents/protocol/genomics/qpcr/sybr-green-qpcr]
- 75. ... - VIROLOGY RESEARCH SERVICES Quantitative PCR validation [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 76. lubio.ch/blog/ensure-reliable- qpcr -results-advice-on-assay- validation [https://www.lubio.ch/blog/ensure-reliable-qpcr-results-advice-on-assay-validation]
- 77. Top 11 qPCR Papers Every Researcher Should Know [https://bitesizebio.com/62143/qpcr-papers/]
- 78. Consensus guidelines for the validation of qRT- PCR assays in clinical... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 79. Disclosing quantitative RT‐PCR raw data during manuscript submission: a call for action - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10158759/]
- 80. Combined lineage tracing and scRNA - seq reveals unexpected first... [https://elifesciences.org/articles/80075]
- 81. and evaluation of statistical error models for Comparison - scRNA seq [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02584-9]
- 82. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5431503/]
- 83. Frontiers | ScRNA - seq combined with ATAC-seq analysis to explore... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1600685/full]
- 84. Single-Cell Single-Molecule RNA - FISH Combined with... [https://pubmed.ncbi.nlm.nih.gov/38743220/]
- 85. Simultaneous detection of mRNA and protein in single cells using immunofluorescence-combined single-molecule RNA FISH - PubMed [https://pubmed.ncbi.nlm.nih.gov/26458549/]
- 86. 3D exploration of gene expression in chicken embryos through... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-024-01922-0]
- 87. Live-cell imaging combined with immunofluorescence , RNA , or DNA... [https://pubmed.ncbi.nlm.nih.gov/23979997/]
- 88. A Multifaceted FISH Approach to Study Endogenous RNAs and DNAs within Native Nuclear and Cell Structure - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3556644/]
- 89. - RNA and FISH of Mouse Preimplantation and... Immunofluorescence [https://pubmed.ncbi.nlm.nih.gov/30218367/]
- 90. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy | Scientific Reports [https://www.nature.com/articles/s41598-018-22297-7]
- 91. A hybrid RNA FISH immunofluorescence protocol on Drosophila polytene chromosomes | BMC Research Notes | Full Text [https://bmcresnotes.biomedcentral.com/articles/10.1186/s13104-023-06482-0]
- 92. Fluorescence in situ hybridization - Wikipedia [https://en.wikipedia.org/wiki/Fluorescence_in_situ_hybridization]
- 93. Assessment of computational methods for the analysis of single - cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1854-5]
- 94. 23 Single - cell sequencing — ATAC - Single best practices cell [https://www.sc-best-practices.org/chromatin_accessibility/introduction.html]
- 95. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7939551/]
- 96. - Single - cell and RNA - Seq profiling identifies... ATAC Seq [https://www.zoores.ac.cn/article/doi/10.24272/j.issn.2095-8137.2025.190]
- 97. CoRE- ATAC : A deep learning model for the functional classification of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8699717/]
- 98. Integrative single-cell RNA-seq and ATAC-seq analysis of myogenic differentiation in pig | BMC Biology | Full Text [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-023-01519-z]
- 99. Single-cell RNA sequencing for the study of development, physiology and disease - PubMed [https://pubmed.ncbi.nlm.nih.gov/29789704/]
- 100. Analysis of Comparative - Single Sequencing Cell and... RNA Platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC9258609/]
- 101. analysis of sequencing technologies for Comparative - single ... cell [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1676-5]
- 102. qPCR Primer Design: A Handy Step-by-Step Guide [https://bitesizebio.com/10041/designing-qpcr-primers/]
- 103. qPCR primer design revisited - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5702850/]
- 104. PCR Assay Optimization and Validation [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/assay-optimization-and-validation]