Validating Synthetic Signaling Patterns: From Foundational Principles to Clinical Applications in Developmental Biology
This article provides a comprehensive framework for researchers and drug development professionals to validate synthetic signaling patterns within developmental contexts.
Validating Synthetic Signaling Patterns: From Foundational Principles to Clinical Applications in Developmental Biology
Abstract
This article provides a comprehensive framework for researchers and drug development professionals to validate synthetic signaling patterns within developmental contexts. It explores the foundational principles of building synthetic signaling systems, from basic components like synthetic promoters and receptors to complex circuit design. The piece details cutting-edge methodological approaches, including computational protein design and heterologous systems, for constructing and applying these circuits. It further addresses critical troubleshooting and optimization strategies to overcome integration challenges and non-orthogonal signal crosstalk. Finally, the article establishes rigorous validation paradigms and comparative analyses against natural systems, highlighting successful case studies in cancer therapy and other biomedical applications to bridge the gap between theoretical design and reliable, predictable function in living systems.
Deconstructing the Blueprint: Core Components and Design Principles of Synthetic Signaling Systems
Synthetic signaling patterns are engineered biological systems designed to mimic, probe, or re-wire the intricate communication networks that guide developmental processes. Built from biological parts like genes, proteins, and signaling molecules, these synthetic circuits are introduced into cells or model organisms to exert control over cell fate decisions, tissue patterning, and morphogenesis. Their application in research provides a powerful, causal approach to validating hypotheses about developmental mechanisms, moving beyond mere observation to active testing and manipulation. This guide compares the leading experimental approaches for constructing and validating these patterns, providing a resource for scientists aiming to decipher the logic of development.
Comparative Analysis of Validation Methodologies
The fidelity and functional impact of a synthetic signaling pattern are typically validated through a multi-faceted approach, combining molecular biology, imaging, and computational techniques. The table below summarizes the quantitative data and key characteristics of core validation methodologies.
Table 1: Comparison of Core Validation Methodologies for Synthetic Signaling Patterns
| Methodology | Primary Measured Output(s) | Key Performance Metrics | Typical Experimental Scale | Temporal Resolution | Key Advantage |
|---|---|---|---|---|---|
| Reporter Gene Expression [1] [2] | Fluorescence/Luminescence intensity | Signal-to-Noise Ratio, Induction Fold-Change, Response Time | Population of cells or single cells | Minutes to Hours | Quantifiable, multiplexable output; enables high-throughput screening. |
| Cell Morphological/Phenotypic Analysis [3] | Cell shape, division, differentiation markers | Division Rate, Cytoskeletal Rearrangement, Marker Expression Co-localization | Single cells | Hours to Days | Directly links signaling to functional cellular outcomes in development. |
| Synthetic Community (SynCom) Benchmarking [4] | Virus-host linkage accuracy (Specificity, Sensitivity) | Specificity (e.g., 99%), Sensitivity (e.g., 62%), Abundance thresholds (e.g., 10^5 PFU/mL) [4] | Defined microbial consortia | End-point measurement | Provides empirical benchmarks for interaction reliability in complex systems. [4] |
| Synthetic Genetic Circuit Characterization [1] [2] | State of genetic switch (ON/OFF), Oscillation frequency | Leakiness, Dynamic Range, Switching Time, Robustness to Noise | Single cells | Minutes to Hours | Enables testing of network topology and logic in a living context. |
Detailed Experimental Protocols
To ensure reproducibility, below are detailed protocols for two critical experiments cited in the comparison.
Protocol 1: Validating a Synthetic Patterning Circuit with Fluorescent Reporter Genes
This protocol is used to quantify the performance of a synthetic circuit designed to create a spatial pattern, such as a French flag analog, in a population of cells.
Circuit Design and Cloning: Design the genetic circuit using a standardized visual language like SBOL Visual 2 [2]. The core components typically include:
- Inducer: A small molecule (e.g., a synthetic hormone) that can be diffused to create a gradient [1].
- Sensor: A synthetic promoter or receptor activated by the inducer [1].
- Processor: Genetic logic (e.g., AND, NOT gates) to interpret the sensor signal.
- Actuator/Reporter: Fluorescent protein genes (e.g., GFP, RFP) that produce a visible, quantifiable output [1] [2]. Assemble the circuit using molecular cloning (e.g., Golden Gate, Gibson Assembly) and integrate it into a plasmid or the host genome.
Cell Culture and Transfection: Culture the recipient cells (e.g., mammalian HEK293, synthetic cells in a chassis [3]). Introduce the constructed plasmid into the cells using an appropriate transfection method (e.g., lipofection, electroporation).
Induction and Pattern Formation: Once cells are viable, apply the inducer molecule in a spatial gradient. This can be achieved using microfluidic devices or static diffusion setups. Incubate the cells for a defined period (e.g., 12-24 hours) to allow for gene expression.
Imaging and Data Acquisition: Use confocal or fluorescence microscopy to capture high-resolution images of the cell population. Acquire images for each fluorescent channel corresponding to the different reporter proteins.
Image and Data Analysis: Quantify the fluorescence intensity of each cell using image analysis software (e.g., ImageJ, CellProfiler). Plot the fluorescence intensity against the spatial position to verify the formation of the expected pattern. Calculate key metrics like induction fold-change and signal-to-noise ratio.
Protocol 2: Benchmarking Signaling Interactions using Synthetic Communities (SynComs)
This protocol, adapted from Hi-C proximity ligation benchmarking studies, is used to empirically validate specific molecular interactions, such as virus-host linkages, which can be analogous to ligand-receptor pairs in signaling [4].
SynCom Construction: Create a defined community of interacting partners. For instance, combine four known marine bacterial strains with nine phages that have known infection relationships [4].
Cross-Linking and Hi-C Library Preparation: Culture the SynCom and treat it with formaldehyde to cross-link molecules that are in close physical proximity. Lyse the cells, digest the DNA with a restriction enzyme, and then re-ligate the cross-linked DNA fragments to form chimeric molecules. Sequence these chimeric fragments using high-throughput sequencing [4].
Bioinformatic Analysis: Map the sequenced reads to the reference genomes of all organisms in the SynCom. Identify chimeric reads that connect a viral genome to a host genome. Infer virus-host linkages based on the frequency and pattern of these chimeric reads [4].
Accuracy Assessment and Filtering: Calculate the specificity and sensitivity of the Hi-C method by comparing the inferred linkages to the known ground-truth interactions of the SynCom. Apply statistical filters, such as a Z-score threshold (e.g., Z ≥ 0.5), to improve specificity (e.g., from 26% to 99%) at the cost of some sensitivity [4].
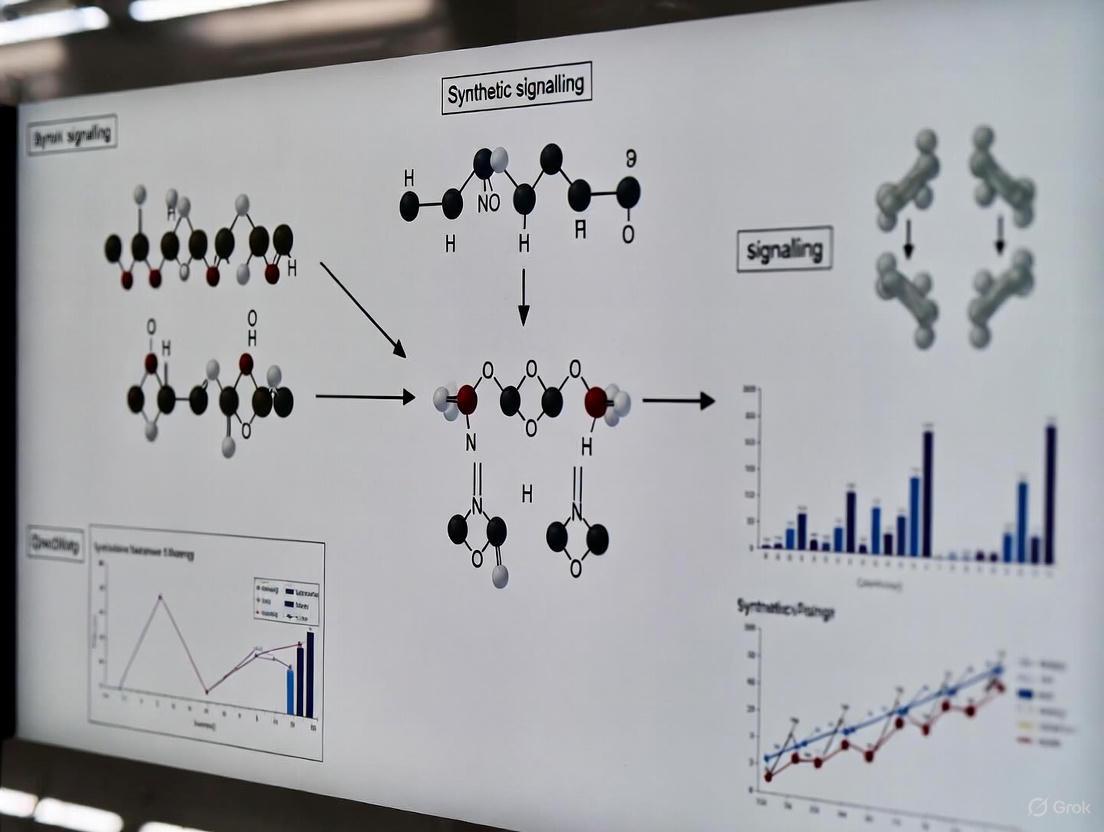
Visualizing Signaling Pathways and Workflows
Standardized diagrams are crucial for communicating the structure and function of synthetic biological systems [2]. The following diagrams, created using SBOL Visual 2 conventions, illustrate a core patterning concept and the experimental workflow for its validation [2].
Synthetic Patterning Principle
Experimental Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential materials and reagents for building and testing synthetic signaling patterns, with explanations of their specific functions in developmental contexts.
Table 2: Essential Research Reagents for Synthetic Signaling Research
| Research Reagent / Tool | Primary Function in Experimentation |
|---|---|
| Cell-Free Protein Synthesis (CFPS) Systems [3] | A chassis-free platform for rapid prototyping of genetic circuits. It allows for the in vitro expression of genetic designs from DNA templates, bypassing the need for live cells during initial testing and optimization. [3] |
| Lipid Vesicles / Polymersomes [3] | Serve as a minimal synthetic cell chassis. These compartments encapsulate synthetic gene circuits and metabolic pathways, providing a cell-like environment to study signaling in a simplified, controlled system. [3] |
| Standardized Genetic Parts (Promoters, CDS, Terminators) [2] | The modular building blocks of synthetic circuits. Using parts with standardized and predictable performance (e.g., from the SBOL Visual framework) is critical for reliable and composable design. [1] [2] |
| Fluorescent Reporter Proteins (e.g., GFP, RFP) [1] [2] | Essential visual readouts for signaling activity. By fusing reporters to promoters activated by a synthetic pathway, researchers can quantify the dynamics, spatial distribution, and intensity of the signaling pattern in real-time. [1] |
| Optogenetic Actuators [1] | Provide high spatiotemporal control over signaling initiation. By using light-sensitive proteins to activate pathways, researchers can impose precise patterns on developing systems without the diffusion limitations of chemical inducers. [1] |
| Synthetic Quorum Sensing Modules [1] | Enable engineered cell-cell communication. These modules allow populations of synthetic cells or engineered bacteria to coordinate their behaviors, mimicking the collective decision-making seen in natural developmental processes. [1] |
Synthetic biology is fundamentally reshaping our approach to developmental biology and therapeutic design by providing tools to deconstruct and reconstruct signaling processes. This field has moved beyond traditional genetic engineering by applying systematic engineering principles to create orthogonal biological systems—components that operate independently of native cellular pathways—enabling precise dissection of complex developmental mechanisms [5] [6]. The core building blocks of these synthetic systems are engineered promoters, receptors, and transcription factors that allow researchers to establish causal relationships in signaling networks that were previously only correlational.
This guide provides a comparative analysis of these foundational components, focusing on their performance characteristics, experimental validation data, and implementation protocols. By objectively evaluating these tools within the context of developmental biology research, we aim to equip scientists with the necessary information to select appropriate synthetic biology tools for validating signaling patterns in various research contexts, from basic developmental studies to therapeutic drug development.
Synthetic Promoters: Engineering Transcriptional Specificity
Definition and Design Principles
Synthetic promoters (synPs) are engineered DNA sequences designed to initiate transcription with precise temporal, spatial, and conditional control. Unlike native promoters that have evolved complex regulatory features, synPs are minimalistic sequences specifically designed to minimize background expression while maintaining strong inducible characteristics [7]. They achieve orthogonality through engineered transcription factor binding sites with minimal sequence identity to the host's endogenous promoters, thereby avoiding unintended regulation by native cellular machinery.
Natural promoters typically contain core elements (e.g., TATA box, initiator), proximal elements, and distal enhancers that work in concert to regulate transcription [8]. In contrast, synthetic promoters are systematically engineered to contain specific binding sites for synthetic transcription factors, allowing researchers to create transcriptional circuits that operate independently of native regulatory networks.
Comparative Performance Analysis
Table 1: Performance Characteristics of Synthetic Promoter Systems
| Promoter System | Basal Expression | Induced Expression | Induction Factor | Key Applications |
|---|---|---|---|---|
| synTALE-targeted synPs | Very low background | High, tunable output | Up to 400-fold | Heterologous pathway balancing |
| dCas9-targeted synPs | Minimal leakage | Wide dynamic range | Varies by guide RNA | Complex genetic circuits |
| Bacterial σ70-derived | Low uninduced | Strong activation | Context-dependent | Multi-layer circuits in E. coli |
| T7-derived systems | Repressible design | High protein yield | Orthogonal repression | Metabolic pathway engineering |
Data derived from characterization studies in S. cerevisiae and E. coli demonstrate that properly engineered synP systems can achieve induction factors of up to 400-fold with minimal background expression under uninduced conditions [7]. The expression output can be systematically tuned by modifying the number, arrangement, and affinity of transcription factor binding sites within the promoter architecture.
Experimental Protocol: Promoter Characterization
Objective: Quantify performance parameters of synthetic promoters in a standardized host system.
Materials:
- Yeast strain YPH500 (or other appropriate chassis)
- Reporter plasmid with fluorescent protein (e.g., GFP) under synP control
- synTF expression vectors (synTALE or dCas9-based)
- Flow cytometer or plate reader for quantification
Methodology:
- Clone synP variants upstream of reporter gene in standardized vector backbone
- Cotransform with corresponding synTF expression plasmids
- Grow cultures to mid-log phase in selective media
- Induce synTF activity (chemical inducer, light, or other trigger)
- Measure fluorescence at regular intervals over 12-24 hours
- Calculate induction ratio as (induced fluorescence)/(uninduced fluorescence)
Data Interpretation: Performance metrics should include fold-induction, absolute expression level, kinetic parameters (time to maximum induction), and cell-to-cell variability. Effective synP designs typically demonstrate >50-fold induction with minimal growth burden on the host cell [7].
Synthetic Transcription Factors: Programming Gene Regulation
Engineering Platforms and Mechanisms
Synthetic transcription factors (synTFs) are engineered proteins designed to bind specific DNA sequences and regulate transcriptional activity. The two primary platforms for synTF engineering are:
Transcription Activator-Like Effectors (TALEs): These utilize a DNA-binding domain composed of 34-amino acid repeat units with "repeat variable diresidues" (RVDs) that follow a simple code for nucleotide recognition [7]. TALEs typically target 18-24 bp sequences starting with a thymine and can be fused to various effector domains (activators, repressors, epigenetic modifiers).
CRISPR/dCas9 Systems: Catalytically dead Cas9 (dCas9) lacks endonuclease activity but can be targeted to specific DNA sequences via guide RNAs [7]. When fused to transcriptional activation domains (e.g., VP64, p65AD) or repression domains (e.g., KRAB, Mxi1), dCas9 becomes a programmable synTF. The key limitation is the requirement for a protospacer adjacent motif (PAM, "NGG") adjacent to the target site.
Performance Comparison
Table 2: Comparison of Synthetic Transcription Factor Platforms
| Platform | Targeting Specificity | Ease of Engineering | Multiplexing Capacity | Key Limitations |
|---|---|---|---|---|
| Zinc Finger | High (9-18 bp) | Difficult, time-consuming | Moderate | Context effects, difficult design |
| synTALE | Very high (18-24 bp) | Moderate (1-day assembly) | Good | Large protein size, repetitive sequence |
| dCas9 | High (20 bp + PAM) | Very easy (guide RNA) | Excellent | PAM requirement, potential off-target effects |
Activation Strength and Dynamics: Studies directly comparing synTALE and dCas9-based activators have demonstrated that both systems can achieve strong transcriptional activation, with specific performance dependent on effector domain choice, target site position relative to transcription start site, and chromosomal context [7]. synTALE-based systems generally show more consistent activation across different target sites, while dCas9 systems benefit from easier reprogramming but can show greater variability based on guide RNA selection.
Experimental Protocol: synTF Functional Validation
Objective: Assess DNA-binding specificity and transcriptional activation potency of engineered synTFs.
Materials:
- synTF expression constructs
- Reporter strains with target promoters
- Antibodies for chromatin immunoprecipitation (if assessing binding directly)
- RT-qPCR reagents for measuring endogenous gene expression
Methodology:
- Express synTF in target cell line with inducible system if possible
- Measure mRNA levels of target genes 6-24 hours post-induction using RT-qPCR
- For binding assessment, perform ChIP-seq using epitope-tagged synTF
- Assess specificity by RNA-seq to evaluate genome-wide off-target effects
- Quantify activation kinetics using live-cell imaging of reporter strains
Data Interpretation: Effective synTFs should demonstrate strong activation of target genes (>10-fold induction) with minimal off-target effects. The Notch transcriptional complex studies highlight the importance of temporal resolution in distinguishing direct targets from downstream effects [9].
Synthetic Receptors: Rewiring Cellular Sensing
Engineering Architectures and Signaling Mechanisms
Synthetic receptors interface engineered cells with their environment, enabling customized sense-and-respond programs. Two primary design strategies exist:
Chimeric Receptors: These typically fuse natural ligand-binding domains to native signaling domains, leveraging existing cellular signaling pathways. Examples include chimeric antigen receptors (CARs) and synthetic cytokine receptors [10]. While powerful, these systems often exhibit crosstalk with endogenous signaling networks.
Orthogonal Receptors: These self-contained systems operate independently of native pathways. The Modular Extracellular Sensor Architecture (MESA) is a prominent example that uses ligand-induced dimerization to drive reconstitution of a split protease, which then cleaves and releases a synthetic transcription factor [10]. This complete separation from native signaling enables more predictable performance across different cell types.
Performance Metrics and Comparative Data
Natural Ectodomain (NatE) MESA Performance: Recent advances have enabled the conversion of natural cytokine receptors into orthogonal biosensors by pairing natural receptor ectodomains with MESA intracellular mechanisms [10]. Performance varies substantially across different receptor origins:
- VEGFR-based sensors: Successfully detected VEGF isoforms with dose-dependent responses, though some configurations showed ligand-specific signaling preferences (e.g., VEGFA121 but not VEGFA165 in certain designs)
- Design principles: Surface expression heavily influenced by ECD choice; signaling performance affected by transmembrane domain selection (CD28 TMD increased background)
Therapeutic Applications: Engineered T cells with NatE MESA receptors can sense immunosuppressive cues and respond with customized transcriptional output to support CAR-T cell activity [10]. These systems have been successfully multiplexed to logically evaluate multiple tumor microenvironment cues, enabling sophisticated integration of environmental information for therapeutic decision-making.
Experimental Protocol: Receptor Characterization
Objective: Validate synthetic receptor function and quantify signaling parameters.
Materials:
- Receptor expression constructs
- Ligand proteins (purified or expressed)
- Reporter cell lines with output measurement (fluorescence, secreted factors)
- Flow cytometry equipment for surface expression validation
Methodology:
- Validate receptor surface expression by flow cytometry using epitope tags
- Dose-response analysis with ligand titration
- Time-course experiments to determine signaling kinetics
- Specificity testing against related ligands
- Orthogonality validation in different cell types
Data Interpretation: Effective synthetic receptors should demonstrate >10-fold induction of signaling output with EC50 values appropriate for the physiological concentration of the target ligand. Background signaling in the absence of ligand should be minimal compared to induced state [10].
Integrated Workflows: From Parts to Systems
Design-Build-Test-Learn Cycles
The engineering of synthetic biological systems follows iterative Design-Build-Test-Learn (DBTL) cycles [11]. The Learn phase has traditionally been the weakest link, but machine learning approaches are now empowering this critical step. The Automated Recommendation Tool (ART) exemplifies this advancement, leveraging machine learning to predict biological system behavior and recommend optimized strains for subsequent engineering cycles [11].
DBTL Cycle for Synthetic Biology
Case Study: Optimizing Tryptophan Production
A recent demonstration used ART in combination with genome-scale models to improve tryptophan productivity in yeast by 106% from the base strain [11]. The machine learning approach mapped promoter combinations to production levels, enabling effective prediction of productive genetic configurations without requiring full mechanistic understanding of the underlying biological system.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Synthetic Biology
| Tool/Category | Specific Examples | Function/Application |
|---|---|---|
| SynTF Platforms | synTALE, dCas9-VP64, LightOn/GAVPO | Programmable transcriptional regulation |
| Synthetic Promoters | T7 variants, σ70-derived, minimal synPs | Orthogonal transcriptional control |
| Synthetic Receptors | MESA, NatE MESA, CAR, synNotch | Custom environmental sensing |
| Assembly Methods | Golden Gate, Gibson, BioBricks | DNA construction standardization |
| Characterization Tools | Flow cytometer, plate readers, omics platforms | Quantitative performance assessment |
| Computational Resources | ART, SynBioTools, bio.tools | Design prediction and tool selection |
The toolkit of synthetic promoters, receptors, and transcription factors has matured to the point where researchers can now engineer sophisticated signaling circuits with predictable behaviors. The comparative data presented in this guide demonstrates that each component class has distinct performance characteristics that make them suitable for different applications in developmental biology research.
Synthetic promoters provide the foundational control elements for transcriptional circuits, with modern designs achieving induction factors up to 400-fold [7]. Synthetic transcription factors, particularly those based on dCas9 and TALE platforms, enable flexible targeting to virtually any genetic locus. Synthetic receptors complete the toolkit by enabling custom environmental sensing that can be coupled to engineered cellular responses.
The integration of these components into unified systems, guided by DBTL cycles and machine learning approaches, is accelerating our ability to validate signaling patterns in developmental contexts. As these tools continue to evolve, they promise to deepen our understanding of developmental biology while enabling new therapeutic strategies for precisely manipulating cellular behavior.
The ambition of synthetic biology to program living systems for therapeutic, bioproduction, and basic research goals hinges on a core engineering principle: modularity. This approach envisions biological systems as collections of standardized, reusable parts that can be reliably assembled into complex, predictable higher-order functions [12]. However, the practical implementation of this vision is consistently hampered by a fundamental problem: a lack of robust interoperability between synthetic modules. When modules derived from different systems or contexts are combined, they often fail to function as intended due to unpredicted cross-talk, impedance mismatches, or outright incompatibility [13].
This challenge is particularly acute in the context of research focused on validating synthetic signaling patterns in developmental contexts. Development is orchestrated by highly coordinated signaling pathways, and reconstructing these processes from the bottom up requires the seamless integration of multiple synthetic components—sensors, actuators, and regulators [5]. The inability of these modules to work together cohesively limits our capacity to deconstruct and understand the minimal requirements for developmental patterns, morphogen interpretation, and cellular memory [5]. This guide provides an objective comparison of the leading technological solutions designed to overcome the integration challenge, equipping researchers with the data and protocols needed to select the optimal strategy for their experimental goals.
Comparative Analysis of Synthetic Interface Technologies
A range of synthetic interface technologies has been developed to facilitate module interoperability. The following table provides a quantitative comparison of the most prominent strategies, highlighting their key characteristics, advantages, and limitations to inform selection.
Table 1: Comparative Analysis of Synthetic Interface Technologies for Module Interoperability
| Technology | Core Mechanism | Typical Assembly Efficiency | Orthogonality | Ease of Cloning | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|---|
| Docking Domains (DDs) [13] | Short, specific peptide pairs mediating protein-protein interaction. | Varies widely; high for cognate pairs. | Low to Moderate (cross-talk common) | Moderate | Naturally evolved for megasynthases; provides a native model. | Limited transferability; prone to cross-talk in non-native contexts. |
| Synthetic Coiled-Coils [13] | Engineered alpha-helical bundles forming stable heterodimers. | High (>90% complex formation reported) | High (engineered for specificity) | High (can be encoded as peptide fusions) | Customizable affinity and specificity; stable interaction. | Potential for homodimerization if not well-designed; can be bulky. |
| SpyTag/SpyCatcher [13] | Protein tag (Tag) and its partner (Catcher) forming a covalent isopeptide bond. | Very High (often >95%, covalent) | High | High | Irreversible, covalent complex; rapid reaction kinetics. | Covalent bond is irreversible, which may not be desirable for all applications. |
| Split Inteins [13] | Self-splicing protein segments that ligate flanking exteins post-translation. | High (splicing efficiency >80%) | High | Moderate to High | Creates a seamless, native peptide bond between modules. | Potential for premature splicing or non-splicing side products. |
Experimental Protocols for Validating Module Interoperability
Rigorous experimental validation is critical to confirm that integrated synthetic modules function as a cohesive unit. Below are detailed protocols for assessing interoperability, with a focus on applications in developmental signaling.
Protocol: Validating a Synthetically Reconstituted Signaling Pathway
This protocol outlines the steps to assemble and test a synthetic signaling pathway, such as one that triggers a specific fate change in response to a synthetic ligand, mimicking a developmental cue [5].
- Module Assembly (The "Build" Phase): Clone the genetic sequences for your input module (e.g., a synthetic receptor), processing module (e.g., a synthetic transcription factor), and output module (e.g., a fluorescent reporter gene) into a suitable expression vector. Utilize the chosen synthetic interface (e.g., SpyTag/SpyCatcher) to fuse the modules, ensuring in-frame fusion and proper linker sequences [13].
- Cell Delivery and Expression: Transfer the assembled construct into your target cell line or primary cells. For developmental contexts, this may involve mouse ES cells, zebrafish embryos, or Drosophila models [5]. Use a method appropriate for the system (e.g., electroporation, viral transduction, microinjection).
- Stimulus Application: Apply the specific stimulus (e.g., synthetic ligand, light pulse for an optogenetic input) to the experimental group. Maintain an unstimulated control group under identical conditions.
- Quantitative Output Measurement (The "Test" Phase)
- Flow Cytometry: At 24, 48, and 72 hours post-stimulation, harvest cells and analyze them via flow cytometry to quantify the fluorescence intensity of the output reporter. This provides single-cell resolution of pathway activity.
- Microscopy Imaging: Use live-cell imaging to monitor the localization and dynamics of fluorescently tagged modules (e.g., nuclear translocation of a synthetic transcription factor) in real-time.
- Data Analysis: Calculate the fold-change in output signal (e.g., fluorescence) between stimulated and unstimulated cells. The dose-dependence and kinetics of the response are key metrics of successful interoperability.
Protocol: Quantifying Crosstalk in a Multi-Channel Circuit
A major challenge in integration is crosstalk. This protocol assesses the orthogonality of multiple, co-existing synthetic modules.
- Circuit Assembly: Assemble multiple, parallel signaling circuits within the same cell, each comprising a unique input, a cognate synthetic interface, and a distinct output (e.g., GFP, mCherry, BFP).
- Selective Stimulation: Systematically stimulate each input channel individually (e.g., stimulate Input A only) while monitoring all output channels.
- Signal Measurement: Use flow cytometry or fluorescence plate reading to quantify the output from all reporters.
- Orthogonality Calculation: Calculate the Specificity Ratio for each channel. For example, when stimulating Input A:
Specificity Ratio = (Output A Signal) / (Output B Signal + Output C Signal). A high ratio indicates minimal crosstalk and high interoperability between the independent modules.
Visualization of Integration Strategies and Workflows
The following diagrams, generated using DOT language, illustrate the core concepts and experimental workflows for achieving and validating module interoperability.
Synthetic Interface Connection Mechanisms
The DBTL Cycle for Module Engineering
The Scientist's Toolkit: Key Reagents for Integration Experiments
Successfully executing interoperability experiments requires a suite of reliable reagents and tools. The following table details essential components for building and testing synthetic modules.
Table 2: Essential Research Reagents for Synthetic Module Integration
| Reagent / Tool Category | Specific Examples | Function in Experiment |
|---|---|---|
| Standardized Genetic Parts [12] | Promoters (e.g., pLac, pTet), RBSs, Terminators, Reporter Genes (GFP, mCherry) | Provides predictable, well-characterized genetic elements for constructing input and output modules, ensuring reliable expression and measurement. |
| Synthetic Interface Kits | Plasmid sets for SpyTag/SpyCatcher fusions, Synthetic Coiled-Coil genes. | Off-the-shelf, validated components for fusing protein modules, significantly reducing cloning time and standardization effort [13]. |
| Assembly & Cloning Systems | Golden Gate Assembly, Gibson Assembly, BioBrick standard. | Enables efficient, and often seamless, combinatorial assembly of multiple genetic modules into a single functional construct [12]. |
| Model Organism & Chassis | E. coli, S. cerevisiae, B. subtilis; HEK293, iPSCs; Zebrafish, Mouse ES cells. | Provides the cellular "chassis" for testing. Choice depends on application: bioproduction, human therapeutics, or developmental biology [5] [12]. |
| Analysis & Measurement Tools | Flow Cytometers, Plate Readers, LC-MS/MS, Live-Cell Imaging Systems. | Critical for the "Test" phase, allowing quantitative measurement of module performance, output signal strength, and system orthogonality [13]. |
In the evolving landscape of bioengineering and therapeutic development, researchers are increasingly looking to nature's blueprint to overcome complex design challenges. Natural developmental pathways—forged through billions of years of evolution—exhibit optimized efficiency, specificity, and regulatory sophistication that synthetic systems strive to emulate. This guide examines how these biological principles are being reverse-engineered to create synthetic signaling patterns, comparing the performance of nature-inspired designs against conventional alternatives across multiple domains. By validating these approaches within developmental contexts, we can establish a framework for creating more effective therapeutic and synthetic biology solutions.
Biomimetic Design Principles in Signaling Pathways
Natural developmental signaling systems share several core characteristics that synthetic designs seek to replicate: modularity in component organization, robustness through interconnected feedback loops, temporal control of activation sequences, and spatial precision in signal localization. These features enable the complex patterning required for multicellular development and tissue morphogenesis.
Synthetic systems now incorporate these principles through various strategies. Hypergraph-like networks connect target molecules to host metabolism through balanced subnetworks rather than linear pathways, mimicking nature's interconnected metabolism [14]. Transcriptional signaling cascades create timed sequences of component activation, replicating the sequential gene expression patterns in embryonic development [15]. Allosteric regulation mechanisms provide programmable input-output behaviors that mirror natural receptor activation dynamics [16].
The table below compares fundamental characteristics of natural developmental pathways versus their synthetic analogs:
Table 1: Core Design Principles in Natural and Synthetic Signaling Systems
| Design Principle | Natural Developmental Pathways | Synthetic Biomimetic Implementations |
|---|---|---|
| Modularity | Domain-specific protein modules in NRPS/PKS systems [17] | XUT approach for NRPS module swapping [17] |
| Temporal Control | Sequential gene expression in embryogenesis | Synthetic gene cascades with promoter nicking [15] |
| Spatial Precision | Morphogen gradients in tissue patterning | Biomimetic geometry patterning [18] |
| Feedback Regulation | Homeostatic control in metabolic pathways | Balanced subnetwork integration [14] |
| Signal Integration | Cross-talk between developmental signaling pathways | Multi-input biosensors with programmable logic [16] |
Computational Pathway Design: Mining Nature's Reaction Database
Retrobiosynthesis for Natural Product Pathway Prediction
The biosynthesis of complex natural products (NPs) represents nature's optimized approach to chemical diversification. BioNavi-NP employs deep learning-driven retrobiosynthesis to predict pathways for both natural products and NP-like compounds, demonstrating how natural synthetic logic can inform synthetic design [19].
Table 2: Performance Comparison of BioNavi-NP Against Traditional Methods
| Metric | BioNavi-NP | Rule-Based Approaches | Improvement Factor |
|---|---|---|---|
| Single-step top-10 accuracy | 60.6% | 35.8% | 1.7× [19] |
| Pathway identification rate | 90.2% (368 test compounds) | Not reported | - |
| Building block recovery | 72.8% | Limited by existing rules | Significant |
| Data requirements | 33,710 biosynthetic reactions + 62,370 organic reactions | Manually curated reaction rules | More scalable |
| Handling of novel compounds | High (neural network generalization) | Limited to rule coverage | Substantial advantage |
Subnetwork Extraction for Balanced Pathway Design
SubNetX addresses a critical limitation of linear pathway design by extracting balanced subnetworks that connect target biochemical production to host native metabolism. This approach mirrors nature's use of branched metabolic networks rather than simple linear pathways, ensuring cofactor and energy currency balancing often overlooked in synthetic designs [14].
The algorithm successfully mapped most of 70 industrially relevant pharmaceutical compounds to E. coli native metabolites, demonstrating the feasibility of recapitulating complex natural product synthesis in heterologous hosts. For gaps in biochemical knowledge, such as scopolamine biosynthesis, the system integrated reactions from multiple databases to create functional balanced pathways [14].
Table 3: Experimental Protocol for SubNetX Pathway Reconstruction
| Step | Protocol Details | Parameters |
|---|---|---|
| Network Preparation | Curate balanced biochemical reactions from ARBRE database (~400,000 reactions) [14] | Include elementally balanced reactions only |
| Graph Search | Identify linear core pathways from precursors to targets | User-defined precursor sets based on host |
| Subnetwork Expansion | Connect cosubstrates and byproducts to native metabolism | Balance energy currencies and cofactors |
| Host Integration | Integrate subnetwork into genome-scale model (E. coli) | Use constraint-based optimization |
| Pathway Ranking | Apply MILP to identify minimal reaction sets | Rank by yield, length, thermodynamics |
Biomimetic Protein Design: Recapitulating Natural Signaling Logic
Synthetic Receptor Engineering with Natural Input-Output Behaviors
The T-SenSER platform demonstrates how natural receptor signaling principles can be engineered into synthetic systems. By computationally designing allosteric receptors that respond to soluble tumor microenvironment factors, researchers created synthetic signaling proteins that replicate the input-output logic of natural receptors [16].
These designed receptors successfully enhanced anti-tumor responses in human T cells when combined with CAR receptors in models of lung cancer and multiple myeloma. The activation was dependent on VEGF or CSF1 presence, demonstrating the precise ligand-response relationship characteristic of natural developmental signaling systems [16].
Research Reagent Solutions for Protein Engineering
Table 4: Essential Research Reagents for Biomimetic Protein Design
| Reagent/Category | Function | Example Application |
|---|---|---|
| Structural Templates | Provide natural folding scaffolds | PDB entries (6E2Q, 4BSK, 2X1W) [16] |
| Computational Design Platforms | De novo protein structure prediction | Dimeric MultiDomain Biosensor Builder [16] |
| Allosteric Regulation Domains | Enable ligand-responsive signaling | Vascular endothelial growth factor receptor domains [16] |
| Expression Systems | Produce designed protein constructs | Human T cells for therapeutic testing [16] |
| Validation Assays | Confirm function in physiological contexts | Tumor microenvironment models [16] |
Autonomous Molecular Systems: Emulating Developmental Timing
Synthetic Gene Networks for Programmed Self-Assembly
Inspired by the temporal progression of developmental events, researchers have created synthetic gene networks that control the sequential activation of DNA building blocks. This approach uses transcriptional signaling cascades to regulate the availability of self-assembling components over time, replicating the timed expression patterns seen in natural morphogenesis [15].
The system employs DNA tiles that polymerize into nanotubes, whose assembly is controlled by RNA molecules produced by synthetic genes. By cascading multiple genes with different transcription rates, researchers achieved temporally distinct outcomes including random DNA polymers, block polymers, and autonomous formation-dissolution cycles [15].
Table 5: Performance Comparison of Temporal Control Strategies
| Control Method | Temporal Precision | Assembly Outcomes | Regulatory Complexity |
|---|---|---|---|
| Constitutive Expression | None (simultaneous) | Homogeneous polymers | Low |
| Promoter Nicking | Moderate (hours) | Sequential activation | Medium [15] |
| Transcriptional Cascades | High (programmed sequence) | Block polymers, oscillating systems | High [15] |
| Natural Developmental Patterning | Very high (robust positioning) | Complex tissue organization | Very high |
Experimental Protocol for Developmental Assembly
Table 6: Step-by-Step Methodology for Multi-Component Polymer Systems
| Step | Procedure | Key Parameters |
|---|---|---|
| Tile Design | Create double-crossover DNA tiles with 5 distinct strands | 5-nt sticky ends, 7-nt toehold domains [15] |
| Inhibitor Design | Design RNA inhibitors complementary to sticky ends + toehold | 12-nt complementarity to block assembly [15] |
| Gene Construction | Design linear templates with T7 promoter and RNA output sequence | Nick placement (template vs. non-template strand) [15] |
| Transcription Setup | Combine genes, T7 RNAP, nucleotides, and inactive tiles | 30°C transcription temperature [15] |
| Kinetic Monitoring | Measure activation via fluorophore-quencher separation | Fluorescence increase indicates tile activation [15] |
Biomimetic Materials: Recapitulating Natural Microenvironments
Geometric Patterning for Stem Cell Differentiation Control
Natural developmental processes rely heavily on geometric and mechanical cues to direct cell fate decisions. Researchers have replicated this principle by creating biomimetic surface patterns derived from the morphology of mature adipocytes. When human mesenchymal stem cells were confined to these adipocyte-mimetic patterns, they exhibited significantly enhanced adipogenesis compared to simple geometric patterns [18].
Notably, greater than 45% of HMSCs on adipocyte mimetic patterns underwent adipogenesis compared to approximately 19% on modified adipocyte patterns with higher stress regions. This demonstrates how natural cell morphology encodes developmental cues that can be harnessed for synthetic differentiation control [18].
Validation Framework for Synthetic Developmental Patterns
Multi-Level Assessment of Biomimetic Systems
Validating synthetic signaling patterns requires assessment across multiple biological scales, from molecular fidelity to functional outcomes:
Molecular Fidelity: Do synthetic components recapitulate natural reaction mechanisms and kinetics? Tools like BioNavi-NP achieve 72.8% accuracy in recovering natural building blocks [19].
Pathway Integration: How seamlessly do synthetic pathways integrate with host metabolism? SubNetX ensures balanced subnetworks that connect to native metabolism [14].
Temporal Control: Does the system replicate natural timing progression? Transcriptional cascades enable sequential activation mimicking developmental sequences [15].
Functional Outcomes: Do the synthetic patterns produce the intended biological effects? T-SenSER receptors enhance anti-tumor responses in therapeutic contexts [16].
Research Reagent Solutions for Developmental Biology Applications
Table 7: Essential Research Tools for Developmental Pathway Engineering
| Category | Specific Reagents/Platforms | Research Application |
|---|---|---|
| Pathway Prediction | BioNavi-NP, SubNetX | Retrobiosynthesis and balanced pathway design [19] [14] |
| Protein Design | Dimeric MultiDomain Biosensor Builder | De novo receptor engineering [16] |
| Genetic Circuits | Synthetic genes with nicked promoters | Tunable transcription control [15] |
| Biomimetic Materials | Hydroxyapatite patterns, fibronectin surfaces | Stem cell differentiation control [20] [18] |
| Model Systems | E. coli metabolism, human T cells, cat neural pathways | Functional validation across biological scales [14] [16] [21] |
The systematic comparison of natural developmental pathways and their synthetic analogs reveals a consistent pattern: designs that more closely emulate nature's organizational principles—modularity, temporal control, balanced stoichiometry, and spatial precision—consistently outperform conventional alternatives. As validation frameworks for synthetic signaling patterns become more sophisticated, the research community is positioned to accelerate the development of increasingly sophisticated biomimetic systems. These nature-informed designs hold particular promise for therapeutic applications, where recapitulating natural signaling fidelity can translate to enhanced efficacy and reduced side effects.
In the field of synthetic biology, particularly for applications in developmental contexts and therapeutic drug development, the transition from conceptual circuits to reliable, predictable systems hinges on rigorous validation. Three key properties form the foundation of this validation: Orthogonality, which ensures that introduced synthetic systems operate without interfering with native host processes; Signal-to-Noise Ratio (SNR), which quantifies the fidelity of a signal against biological variation; and Dynamic Range, which defines the operational scope of a system's output. For researchers and scientists engineering synthetic signaling patterns, a quantitative understanding of these parameters is not merely beneficial—it is essential for de-risking the development pathway and ensuring that in-silico designs translate faithfully to in-vivo function. This guide provides a comparative analysis of these properties, underpinned by experimental data and methodologies, to serve as a practical framework for validation in advanced research and development.
Quantitative Comparison of Key Validation Properties
The table below summarizes the core attributes, measurement approaches, and target values for the three key validation properties, providing a benchmark for evaluating synthetic biological systems.
Table 1: Comparative Analysis of Key Validation Properties for Synthetic Biology
| Property | Core Definition & Impact | Typical Measurement & Calculation | Reported Values & Targets |
|---|---|---|---|
| Orthogonality | The degree to which a synthetic system functions without undesired interactions with the host's native systems. High impact on circuit predictability and host viability [16]. | Measured via transcriptomic/proteomic profiling (e.g., RNA-Seq) with and without system activation. Quantified by the number of significantly differentially expressed host genes. | In computationally designed receptors, high orthogonality is demonstrated by minimal off-target signaling and specific response to intended inputs like VEGF or CSF1 [16]. |
| Signal-to-Noise Ratio (SNR) | A measure of signal fidelity, quantifying the strength of an intended signal relative to background biological noise. Critical for reliable decision-making in therapeutic circuits [22]. | For log-normal biological data: SNR_dB = 20 * log10( |log10(μg,true / μg,false)| / (2 * log10(σg) ) where μg is the geometric mean and σg is the geometric standard deviation [22]. |
Values of ~6.2 dB reported for systems with 100-fold signal change but high (3.2-fold) cell-to-cell variation. Targets are application-dependent: 0-5 dB for biosensing; 20-30 dB for high-stakes cancer therapies [22]. |
| Dynamic Range | The ratio between the maximum (ON) and minimum (OFF) output states of a system. Determines the system's ability to produce a sufficiently distinct output signal. | Calculated as the ratio of the geometric mean output in the "true" state to the geometric mean output in the "false" state: μg,true / μg,false. |
Systems have been demonstrated with a 100-fold dynamic range (e.g., 10^6 MEFL ON state vs. 10^4 MEFL OFF state) [22]. |
Experimental Protocols for Validation
Protocol for Quantifying Signal-to-Noise Ratio (SNR)
Principle: This protocol adapts the classical electromagnetic signal-to-noise ratio for biological systems, accounting for the log-normal distribution of chemical concentrations within cell populations [22].
Methodology:
- Cell Preparation & Transfection: Two distinct populations of cells are prepared: an "ON" population where the synthetic circuit is fully induced or active (representing Boolean
TRUE), and an "OFF" population where the circuit is repressed or inactive (representing BooleanFALSE). - Flow Cytometry Measurement: For each population, the output signal (e.g., fluorescence from a reporter protein) is measured at the single-cell level using flow cytometry. A minimum of 50,000 events per population is recommended to accurately capture the distribution.
- Data Analysis:
- Calculate the geometric mean (μg) of the fluorescence distribution for both the ON (μg,true) and OFF (μg,false) populations.
- Calculate the geometric standard deviation (σg) for the distributions. The geometric standard deviation for both states is often assumed to be similar for the calculation.
- Compute the SNR in decibels (dB) using the formula for biological systems [22]:
SNR_dB = 20 * log10( |log10(μg,true / μg,false)| / (2 * log10(σg)) )
Protocol for Assessing Orthogonality
Principle: This protocol evaluates whether a synthetic system, such as a computationally designed receptor, activates unintended native signaling pathways or causes significant changes in host gene expression [16].
Methodology:
- Experimental Groups: Establish two groups: experimental cells expressing the synthetic receptor and control cells (e.g., expressing a null construct or the native receptor).
- Stimulation & Sampling: Stimulate both groups with the target ligand (e.g., VEGF, CSF1) at a physiologically relevant concentration. After a predetermined time, harvest cells for RNA extraction.
- Transcriptomic Analysis: Perform bulk RNA sequencing (RNA-Seq) on the samples.
- Bioinformatic Quantification:
- Map sequencing reads to the host genome and quantify gene expression.
- Perform differential gene expression analysis, comparing the stimulated experimental group against the stimulated control group.
- Orthogonality is demonstrated by a minimal number of significantly differentially expressed host genes (excluding the direct targets of the synthetic circuit), indicating no major pleiotropic effects or crosstalk.
Protocol for Measuring Dynamic Range
Principle: This procedure measures the operational window of a synthetic system by quantifying its output in fully induced and fully repressed states.
Methodology:
- Define System States:
- ON State: Culture cells under conditions that maximize the system's output (e.g., saturating concentration of an inducer ligand).
- OFF State: Culture cells under conditions that minimize the system's output (e.g., absence of the inducer, or presence of a repressor).
- Output Measurement: Measure the system's output (e.g., reporter fluorescence via flow cytometry) for both states. Using flow cytometry and reporting the geometric mean is critical due to log-normal expression distributions [22].
- Calculation: Calculate the dynamic range as the ratio of the geometric mean of the ON state to the geometric mean of the OFF state.
Dynamic Range = μg,ON / μg,OFF
Visualizing Signaling Pathways and Experimental Workflows
The following diagram illustrates the idealized input-output relationship and key validation metrics for a robust synthetic receptor system.
Experimental Workflow for Key Property Validation
This flowchart outlines the sequential process for empirically characterizing the three key properties of a synthetic biological circuit.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key reagents and their functions, as utilized in the cited studies for developing and validating synthetic systems like the T-SenSER receptors [16] and for SNR analysis [22].
Table 2: Essential Reagents for Synthetic Receptor Development and Validation
| Research Reagent / Material | Function in Experimental Context |
|---|---|
| Computational Protein Design Platform | Enables de novo bottom-up assembly of allosteric receptors with programmable input-output behaviors, crucial for creating orthogonal systems [16]. |
| Human T Cells (Primary) | The primary chassis for therapeutic synthetic circuits, such as CAR-T cells combined with T-SenSERs, for testing in disease-relevant models [16]. |
| Target Ligands (VEGF, CSF1) | Soluble factors from the Tumour Microenvironment (TME) used as specific inputs to validate the sensing and activation of designed synthetic receptors [16]. |
| Flow Cytometer | Essential instrument for single-cell quantification of reporter signal (e.g., fluorescence), enabling the calculation of SNR and Dynamic Range from population distributions [22]. |
| Next-Generation Sequencer | Used for RNA-Seq to comprehensively profile global gene expression and assess the orthogonality of a synthetic circuit by identifying off-target effects [16]. |
| Equivalent Fluorescein (MEFL) Beads | Calibration standards for flow cytometry that allow for the conversion of arbitrary fluorescence units into absolute units (Molecules of Equivalent Fluorochrome), enabling quantitative comparisons across experiments and labs [22]. |
From Design to Deployment: Methodologies for Constructing and Applying Synthetic Circuits
The emerging field of synthetic developmental biology aims to understand and control multicellular self-organization by programming cells with synthetic genetic circuits that can read and write biological signals [23]. Central to this endeavor is computational protein design, which enables the de novo creation of synthetic receptors with programmable signaling capabilities. These designer receptors serve as the fundamental interface between engineered genetic circuits and native cellular communication systems, allowing researchers to establish synthetic signaling patterns that guide developmental outcomes.
This guide compares current computational platforms for designing programmable receptors, focusing on their performance in generating functional proteins. We provide objective comparisons based on published experimental data, detailed methodologies for key validation experiments, and essential resources for implementing these technologies in developmental biology and therapeutic contexts.
Comparative Analysis of Computational Protein Design Platforms
Performance Metrics for Protein Design Methods
Table 1: Comparative Performance of Protein Design Assessment Metrics [24]
| Metric Category | Specific Metric | Measurement Purpose | Ideal Value Range |
|---|---|---|---|
| Sequence Recovery | Sequence Accuracy | Identity between predicted and natural sequence | Higher (varies by method) |
| Top-3 Accuracy | Probability of true residue in top 3 predictions | >60% | |
| Structural Compatibility | Similarity Score | Accounts for functional amino acid redundancy | >70% |
| Prediction Bias | Discrepancy between predicted and actual residue frequency | Lower (close to 0) | |
| Statistical Quality | Precision/Recall | Trade-off between false positives and true positives | Method-dependent |
| AUC | Overall prediction performance | >0.8 | |
| Structural Analysis | Torsion Angle Comparison | Backbone conformation fidelity | Lower deviation |
Table 2: Experimental Success Rates of Generative Protein Models [25]
| Generative Model | Theoretical Basis | MDH Active Sequences | CuSOD Active Sequences | Overall Success Rate |
|---|---|---|---|---|
| Ancestral Sequence Reconstruction (ASR) | Phylogenetic inference | 10/18 (55.6%) | 9/18 (50.0%) | 52.8% |
| Generative Adversarial Network (ProteinGAN) | Deep neural networks | 0/18 (0%) | 2/18 (11.1%) | 5.6% |
| Language Model (ESM-MSA) | Transformer architecture | 0/18 (0%) | 0/18 (0%) | 0% |
| Natural Test Sequences | Natural diversity reference | 6/18 (33.3%) | 0/18 (0%) | 16.7% |
Key Computational Platforms for Protein Design
Table 3: Computational Platforms for Protein Structure Generation [26]
| Platform | Algorithmic Approach | Complexity | Max Protein Length | Key Applications |
|---|---|---|---|---|
| SALAD | Sparse all-atom denoising | O(N·K) | 1,000 residues | Large protein design, motif scaffolding |
| RFdiffusion | Denoising diffusion | O(N³) | ~400 residues | Binder design, symmetric assemblies |
| Chroma | Diffusion with conditioners | O(N²) | ~500 residues | Shape-guided generation |
| Proteus | Diffusion models | O(N³) | ~800 residues | General protein design |
| Hallucination | Structure predictor inversion | High runtime | ~1,000 residues | High-confidence designs |
Experimental Validation of Programmable Receptors
Case Study: T-SenSER Platform for Synthetic Receptors
The TME-sensing switch receptor for enhanced response to tumors (T-SenSER) platform represents a cutting-edge application of computational protein design for creating synthetic receptors with programmable input-output behaviors [27] [16]. This system enables de novo bottom-up assembly of allosteric receptors that respond to soluble tumor microenvironment factors like vascular endothelial growth factor (VEGF) or colony-stimulating factor 1 (CSF1) by initiating co-stimulation and cytokine signals in T cells.
T-SenSER Experimental Validation Protocol
Objective: Validate the function of computationally designed T-SenSER receptors in enhancing anti-tumor responses of engineered T cells.
Methodology:
- Computational Design: Use the Dimeric MultiDomain Biosensor Builder platform [16] for de novo assembly of receptor components
- Lentiviral Transduction: Introduce T-SenSER constructs into primary human T cells alongside chimeric antigen receptors (CARs)
- In Vitro Stimulation: Expose engineered T cells to recombinant VEGF or CSF1 (10-100 ng/mL) for 24-72 hours
- Signaling Output Measurement:
- Phospho-flow cytometry for phosphorylation signaling intermediates (pAKT, pERK)
- Cytokine secretion profiling via Luminex (IFN-γ, IL-2)
- Metabolic activity assays (ATP quantification)
- Functional Co-culture Assays:
- Co-culture with target cancer cell lines (lung cancer, multiple myeloma)
- Cytotoxicity measurement (LDH release, real-time cell imaging)
- T-cell proliferation tracking (CFSE dilution)
Experimental Controls:
- Untransduced T cells
- CAR-T cells without T-SenSER
- Stimulation with non-cognate ligands (specificity testing)
- Receptor-deficient variants (signaling mechanism) [27] [16]
Composite Metric for Predicting Experimental Success
Research indicates that a combination of computational metrics significantly improves the prediction of experimental success for designed proteins. The Composite Metrics for Protein Sequence Selection (COMPSS) framework [25] integrates:
- Alignment-based metrics: Sequence identity to natural homologs (70-90% ideal range)
- Alignment-free metrics: Language model likelihood scores
- Structure-based metrics:
- Rosetta energy scores (<0 indicative of stability)
- AlphaFold2 pLDDT confidence scores (>70 for viable designs)
- Self-consistent RMSD (<2.0 Å for design-prediction agreement)
Implementation of COMPSS has demonstrated a 50-150% improvement in experimental success rates compared to naive sequence selection [25].
Signaling Pathway Architecture
T-SenSER Signaling Pathway: Illustration of how computationally designed T-SenSER receptors convert recognition of tumor microenvironment (TME) factors into enhanced T-cell effector functions, working synergistically with chimeric antigen receptor (CAR) signaling.
Experimental Workflow for Receptor Validation
Receptor Validation Workflow: Step-by-step experimental pipeline for validating computationally designed receptors, from initial computational design through in vitro characterization to final in vivo functional assessment.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for Programmable Receptor Development [27] [28] [16]
| Reagent/Category | Specific Examples | Research Function | Experimental Application |
|---|---|---|---|
| Computational Design Platforms | Dimeric MultiDomain Biosensor Builder, Rosetta, PDBench | De novo protein design and benchmarking | Assembly of allosteric receptors with programmable logic |
| Generative Models | ESM-MSA, ProteinGAN, Ancestral Sequence Reconstruction | Novel protein sequence generation | Exploring sequence diversity beyond natural space |
| Structure Prediction | AlphaFold2, ESMFold, ProteinMPNN | Structure validation and sequence design | Assessing design quality (pLDDT, pAE, scRMSD) |
| Cell Engineering Tools | Lentiviral vectors, Electroporation systems | Delivery of genetic constructs | Primary immune cell engineering (T cells) |
| Signaling Assays | Phospho-specific flow cytometry, Luminex | Signaling pathway activation | Measuring phosphorylation events, cytokine secretion |
| Functional Assay Systems | Co-culture systems, Real-time cell analyzers | Functional validation | Cytotoxicity, proliferation measurements |
| Model Systems | Cancer cell lines, Xenograft models | Preclinical testing | In vivo assessment of therapeutic efficacy |
The integration of computational protein design with synthetic biology represents a transformative approach for programming cellular behaviors in developmental contexts and therapeutic applications. Current platforms demonstrate varying success rates, with methods like ancestral sequence reconstruction achieving approximately 50% experimental success for certain enzyme families [25], while newer deep learning approaches show promise but require further refinement.
The emerging generation of protein design tools, particularly sparse denoising models like SALAD [26], offer capabilities for designing larger and more complex protein systems up to 1,000 residues, dramatically expanding the potential architectural complexity of programmable receptors. As these tools mature, they will enable increasingly sophisticated control over developmental signaling patterns, moving synthetic developmental biology toward becoming a predictive science that links basic cell biology to emergent multicellular developmental programs [23].
Future developments will likely focus on improving the experimental success rates of designed proteins through better computational metrics, more sophisticated training datasets, and enhanced understanding of protein folding principles. The integration of automated design validation systems with high-throughput experimental testing will accelerate this cycle of innovation, ultimately enabling the design of complex synthetic biological systems with precise control over cellular decision-making in developmental contexts.
Heterologous expression refers to the expression of a gene or part of a gene in a host organism that does not naturally possess that genetic element, enabled by recombinant DNA technology [29]. This foundational biotechnology approach has become indispensable for modern synthetic biology and developmental biology research, providing scientists with powerful tools to deconstruct and rebuild developmental systems [30]. By transferring genetic pathways into well-characterized model organisms, researchers can systematically study complex signaling patterns while controlling variables that are often intertwined in native environments.
The core value of heterologous expression lies in its ability to isolate biological components from their natural contexts, allowing for precise functional analysis. When investigating developmental pathways, this approach enables researchers to distinguish between a protein's biochemical function (determined by its coding sequence) and its developmental role (shaped by its genomic context, including regulatory interactions and cellular environment) [31]. This distinction is particularly crucial for validating synthetic signaling patterns, as it permits the testing of whether engineered genetic circuits can recapitulate developmental processes outside their native contexts.
Comparative Analysis of Heterologous Expression Platforms
Key Host Systems and Their Applications
Different host organisms offer distinct advantages and limitations for heterologous expression, making platform selection critical for success. The table below summarizes the primary host systems used in contemporary research.
Table 1: Comparison of Major Heterologous Expression Systems
| Host System | Optimal Applications | Key Advantages | Documented Limitations | Representative Yields |
|---|---|---|---|---|
| Escherichia coli | Soluble proteins, enzymatic studies, pathway prototyping | Rapid growth (20-30 min doubling), low cost, well-characterized genetics [29] | Improper folding of complex proteins, lack of post-translational modifications, intracellular aggregation [29] | High-level expression possible, but varies significantly by protein target |
| Pichia pastoris | Eukaryotic proteins, industrial enzymes, biopharmaceuticals | Post-translational modifications, high-density cultivation, secretion capability [32] [33] | Hyper-glycosylation patterns, more complex media requirements [29] | Protease K expression enhanced 5.4-fold with optimized SES-CP32 system [32] |
| Saccharomyces cerevisiae | Eukaryotic proteins, metabolic pathway engineering, pharmaceutical production | Food-safe organism, proper protein folding, secretory pathway [29] | Hyper-mannosylation, slower growth than bacteria, expensive nutrients [29] | Successfully used for hepatitis B and Hantavirus vaccines [29] |
| Baculovirus/Insect Cells | Complex eukaryotic proteins, multiprotein complexes, structural biology | Advanced eukaryotic processing, high protein yields, proper compartmentalization [33] | More technically demanding, slower than microbial systems | Effective for membrane proteins and protein complexes [33] |
| Mammalian Cells | Human therapeutics, complex glycoproteins, membrane receptors | Most human-like post-translational modifications, proper folding and assembly | High cost, slow growth, technical complexity [33] [29] | Gold standard for therapeutic proteins requiring human-like modifications |
| Xenopus laevis Oocytes | Membrane transporters, ion channels, electrophysiology studies | Large cell size, high protein expression, minimal processing equipment | Specialized applications, not for high-throughput production | Widely used for functional characterization of transporters [33] |
Quantitative Performance Metrics
Recent advances in host engineering have significantly improved the performance of heterologous expression systems. The following table summarizes key quantitative improvements documented in recent literature.
Table 2: Documented Performance Enhancements in Optimized Expression Systems
| Expression System | Engineering Strategy | Target Protein | Performance Improvement | Reference |
|---|---|---|---|---|
| Pichia pastoris SES | Heterologous core promoters from Trichoderma reesei | mCherry | 5.4-fold increase over traditional SES-A system [32] | [32] |
| Pichia pastoris SES | Multi-copy CRISPR/Cas9 integration | Protease K | 4.6-fold increase with 3 copies vs. 1 copy [32] | [32] |
| PVX Plant Vector | VSR integration (NSs) with reversed orientation | GFP | 3.8-fold increase (0.50 mg/g FW vs. 0.13 mg/g FW) [34] | [34] |
| PVX Plant Vector | VSR integration (NSs) with reversed orientation | Vaccine antigens (VP1, S2) | >100-fold improvement over parental vector [34] | [34] |
| SynNotch Mammalian Circuit | Density optimization in fibroblast systems | Patterned gene expression | Bell-shaped response curve with optimal density window [35] | [35] |
Experimental Design for Validating Synthetic Signaling Patterns
Core Methodologies and Workflows
The successful implementation of heterologous expression systems requires standardized methodologies for transferring genetic pathways into host organisms. Several well-established techniques facilitate this process:
Vector Assembly and Delivery: For plant systems, advanced viral vectors like Potato Virus X (PVX) have been engineered to incorporate viral suppressors of RNA silencing (VSRs) such as P19, P38, and NSs. Recent optimization has demonstrated that reversing VSR cassette orientation relative to the target gene alleviates transcriptional interference, significantly improving both target protein and VSR expression [34].
Stable Integration Methods: In microbial systems, CRISPR/Cas9 enables rapid multi-targeted integration of expression cassettes. This approach has been successfully employed in Pichia pastoris to create multi-copy strains, dramatically enhancing recombinant protein yields [32].
Cell Culture and Co-culture Systems: For synthetic developmental biology, engineered cell lines expressing synNotch (synthetic Notch) receptors can be co-cultured to study contact-dependent signaling. These systems require precise control of cell density, which has been identified as a critical parameter affecting signaling outcomes [35].
Critical Parameter Optimization
Recent research has identified several non-genetic parameters that significantly impact the success of heterologous expression systems:
Cell Density Effects: In synthetic Notch (synNotch) systems, cell density following a bell-shaped curve response, with optimal signaling occurring within a specific density window (0.125-0.5x confluency in L929 fibroblasts). Both lower and higher densities outside this window result in diminished signaling capacity [35].
Codon Optimization Strategies: Traditional codon adaptation index (CAI) optimization approaches that use only the most frequent codons are being supplemented by more sophisticated "typical gene" design. This approach generates genes resembling the codon usage of any subset of endogenous genes, allowing for fine-tuned expression levels appropriate for specific applications [36].
Transcriptional Interference Management: In multi-gene constructs, the relative orientation of expression cassettes significantly impacts output. Reversing the VSR cassette orientation relative to the target gene in PVX vectors dramatically improves expression by reducing transcriptional interference [34].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Heterologous Expression Systems
| Reagent Category | Specific Examples | Function & Application | Experimental Notes |
|---|---|---|---|
| Expression Vectors | PVX derivatives (pP1, pP2, pP3), SES systems | Backbone for gene insertion and expression | PVX vectors optimized with VSRs show 3-4× improvement [34] |
| Viral Suppressors of RNA Silencing (VSRs) | P19 (TBSV), P38 (TCV), NSs (TZSV) | Counter host RNA silencing mechanisms | NSs shows highest performance in plant systems [34] |
| Synthetic Receptors | synNotch (anti-GFP receptor) | Engineered cell-cell contact signaling | Activation measurable via fluorescent reporter (mCherry) [35] |
| Gene Integration Tools | CRISPR/Cas9 systems | Targeted multi-copy integration | Enables 4.6× yield improvement in P. pastoris [32] |
| Promoter Systems | Trichoderma reesei core promoters, CaMV 35S | Transcriptional control of heterologous genes | 41 SES systems showed enhanced expression vs. traditional SES-A [32] |
| Reporter Proteins | mCherry, GFP | Quantitative assessment of expression levels | Fluorescence intensity used for promoter screening [32] |
Signaling Pathway Architecture for Synthetic Development
Engineering synthetic developmental pathways requires precise orchestration of multiple signaling components. The synNotch system exemplifies how heterologous expression enables the construction of programmable cell-cell communication networks:
This engineered signaling pathway demonstrates how heterologous components can be combined to create synthetic developmental systems. The sender cell expresses a membrane-tethered ligand (e.g., GFP), while the receiver cell contains a custom synNotch receptor with an extracellular anti-GFP binding domain. Upon cell-cell contact, mechanical forces expose a proteolytic cleavage site in the synNotch receptor, releasing a transcription factor (tTA) that migrates to the nucleus and activates expression of reporter genes (mCherry) [35]. This modular system allows researchers to program specific cell behaviors and pattern formation in synthetic tissues.
Applications in Developmental Biology and Drug Discovery
The integration of heterologous expression systems with synthetic biology approaches has enabled significant advances in understanding and engineering developmental processes:
Pattern Formation Studies: Synthetic Notch circuits have been used to engineer self-organizing cellular systems that mimic natural developmental patterning. These circuits can be controlled by modulating cell density and proliferation rates, demonstrating how mechanical and chemical signaling interact to shape morphological outcomes [35].
Vaccine Antigen Production: Plant-based heterologous expression systems have been optimized to produce vaccine antigens with yields sufficient for commercial development. The integration of VSRs into PVX vectors has enabled more than 100-fold improvements in expression of antigens like FMDV VP1 and SARS-CoV-2 S2 subunit [34].
Metabolic Pathway Engineering: Heterologous expression enables the reconstruction of complex metabolic pathways in industrially favorable hosts. Pichia pastoris has been successfully engineered to express diverse enzymes and metabolic pathways, with recent promoter engineering efforts significantly boosting yields [32].
Membrane Protein Characterization: The functional analysis of membrane transporters and channels often requires heterologous expression in systems like Xenopus oocytes or mammalian cells, which provide the necessary cellular machinery for proper folding and localization [33].
Heterologous expression systems provide an essential foundation for recapitulating complex pathways in model organisms, enabling systematic deconstruction of developmental processes and validation of synthetic signaling patterns. Recent advances in vector design, promoter engineering, and understanding of critical parameters like cell density have significantly enhanced the utility of these systems across basic research and applied biotechnology.
The integration of heterologous expression with synthetic biology approaches—particularly engineered signaling systems like synNotch—creates powerful platforms for programming multicellular behaviors and pattern formation. As these tools continue to evolve, they will undoubtedly accelerate both our understanding of fundamental biological principles and our ability to engineer biological systems for therapeutic and industrial applications.
In the field of synthetic biology, researchers and drug development professionals face a fundamental challenge: biological signals within cellular environments are often complex, non-orthogonal, and prone to interference. This crosstalk significantly limits our ability to engineer predictable genetic circuits for therapeutic applications, metabolic engineering, and fundamental research into developmental signaling patterns. Traditional synthetic circuits often employ binary (ON/OFF) signaling mechanisms that starkly contrast with the nuanced signal processing capabilities of natural biological systems [37]. As we seek to validate synthetic signaling patterns in developmental contexts—where precise spatiotemporal control of gene expression is critical—the limitations of existing tools become increasingly problematic.
Synthetic biological amplifiers represent a groundbreaking class of genetic devices that address these challenges by enhancing signal fidelity, amplifying weak transcriptional signals, and decomposing complex cellular inputs into orthogonal components. Inspired by operational amplifiers in analog electronics, these biological counterparts perform essential signal processing functions including scaling, subtraction, and noise reduction within living cells [37] [38]. This comparative guide examines three prominent amplifier architectures—synthetic biological operational amplifiers, toehold switch-based modulators, and orthogonal genetic amplifiers—evaluating their performance characteristics, implementation requirements, and suitability for different research applications in developmental biology and drug discovery.
Comparative Performance Analysis of Synthetic Biological Amplifiers
The table below provides a systematic comparison of three major amplifier types based on reported experimental data:
Table 1: Performance Comparison of Synthetic Biological Amplifiers
| Amplifier Type | Maximum Fold-Change | Key Components | Orthogonality | Primary Applications | Reported Signal-to-Noise Enhancement |
|---|---|---|---|---|---|
| Synthetic Biological OAs [37] | 153-688x | σ/anti-σ pairs, RBS variants, negative feedback | High (enables N-dimensional signal separation) | Growth-phase responsive control, quorum sensing crosstalk mitigation | Significant (enabled by closed-loop configurations) |
| Toehold Switch-Based Modulators [39] | 261-887x | Toehold switches, trigger RNA, riboswitches | High (5 orthogonal toehold switch pairs demonstrated) | Riboswitch tuning, metabolite sensing | Moderate to high (dependent on RNA component balancing) |
| Orthogonal Genetic Amplifiers [38] | Up to 21x | hrpRS, hrpV, PhrpL components | Moderate (orthogonal to host regulation) | Transcriptional signal scaling in cascaded networks | Low noise introduction during amplification |
Table 2: Implementation Requirements and Experimental Considerations
| Amplifier Type | Genetic Footprint | Tuning Mechanism | Host Strain Considerations | Typical Response Dynamics |
|---|---|---|---|---|
| Synthetic Biological OAs [37] | Moderate to large (multiple transcriptional units) | RBS strength variation, feedback loop engineering | Standard E. coli strains (e.g., BL21) | Programmable bandwidth with -3dB cutoff |
| Toehold Switch-Based Modulators [39] | Compact (RNA-based regulation) | Promoter selection, IPTG concentration for switch RNA | RNase-deficient strains (e.g., BL21 Star) recommended | Fast (post-transcriptional regulation) |
| Orthogonal Genetic Amplifiers [38] | Moderate (hrp system components) | Expression levels of ligand-free activator proteins | Standard E. coli strains (e.g., TOP10) | Minimal time delay during amplification |
Experimental Protocols for Amplifier Implementation and Validation
Synthetic Biological Operational Amplifiers: Protocol for Growth-Phase Responsive Circuits
The implementation of synthetic biological operational amplifiers (OAs) requires careful construction and characterization to achieve precise signal processing functions [37]:
Circuit Construction:
- Clone orthogonal σ/anti-σ factor pairs (e.g., ECF σ factors and their cognate anti-σ factors) or T7 RNA polymerase/T7 lysozyme pairs into appropriate expression vectors.
- Engineer ribosome binding sites (RBS) with varying strengths to tune translation rates for activator (A) and repressor (R) components.
- Assemble the OA circuit to perform the operation: (α \cdot X1 - β \cdot X2), where (X1) and (X2) represent input transcriptional signals.
- For closed-loop configurations, implement negative feedback loops by connecting output signals to regulatory elements controlling activator/repressor expression.
Characterization and Validation:
- Measure input-output relationships using fluorescent reporters (e.g., GFP) across growth phases in E. coli.
- Quantify promoter activities during exponential and stationary growth phases to establish non-orthogonal signal profiles.
- Apply linear transformation calculations to verify signal decomposition: ({XE} = α \cdot {X1} - β \cdot {X_2}).
- Determine the operational range and -3dB bandwidth by measuring the frequency range where output signal is reduced to half maximum value.
- Validate crosstalk mitigation in multi-signal systems by testing orthogonal control of multiple output channels.
Toehold Switch-Based Modulators: Protocol for Riboswitch Signal Amplification
This protocol details the integration of toehold switches with riboswitch-based sensors for enhanced fold-change amplification [39]:
Circuit Assembly:
- Select a high-fold-change toehold switch variant (e.g., ACTSTypeIIN1) and its cognate trigger RNA.
- Clone the hybrid input construct consisting of a riboswitch (e.g., coenzyme B12-responsive riboswitch from Salmonella typhimurium) and transcriptional repressor (e.g., PhlF).
- Modify the circuit such that the transcriptional repressor regulates trigger RNA expression rather than directly controlling the reporter gene.
- Place the toehold switch upstream of the reporter gene (e.g., GFP) with its cognate trigger RNA under control of the repressor-regulated promoter.
Optimization Procedure:
- Screen different constitutive promoters (e.g., BBaJ23100, BBaJ23101, BBa_J23119) to balance trigger RNA expression levels.
- Titrate IPTG concentration (0-1000 μM) to optimize toehold switch RNA expression from inducible promoters.
- Measure fluorescence output with and without ligand (e.g., 30 μM coenzyme B12) to calculate fold-change improvement.
- Validate orthogonality by testing multiple toehold switch-trigger pairs (at least 5 pairs recommended) to confirm minimal crosstalk.
- Assess leakiness by comparing fluorescence in absence of both trigger RNA and inducer to fully-induced state.
Orthogonal Genetic Amplifiers: Protocol for Transcriptional Signal Scaling
This protocol describes the implementation of modular genetic amplifiers based on the hrp gene regulatory system [38]:
Amplifier Construction:
- Clone hrpR, hrpS, hrpV genes, and PhrpL promoter into BioBrick-compatible vectors following standard assembly methods.
- Assemble the two-terminal fixed-gain amplifier by expressing HrpR and HrpS under constitutive promoters of varying strengths.
- For tunable amplifiers, implement the three-terminal configuration with HrpV expression under inducible control (e.g., arabinose-inducible PBAD).
- Include appropriate reporter genes (e.g., gfpmut3b) under control of PhrpL promoter for output quantification.
Characterization Methods:
- Measure transfer functions by applying varying input signal levels (inducer concentrations) and quantifying output fluorescence.
- Determine amplification gain (βT) as the ratio between output and input: (Δ[Output] = βT \cdot Δ[Input]).
- Assess dynamic range by identifying the input range over which linear amplification occurs.
- Quantify noise propagation by comparing coefficient of variation between input and output signals.
- Evaluate modularity by connecting amplifier outputs to downstream genetic circuits and measuring signal preservation.
Signaling Pathway Architecture and Experimental Workflows
Synthetic Biological Operational Amplifier Architecture
Figure 1: Synthetic Biological Operational Amplifier Architecture. The circuit performs weighted subtraction of input signals (X₁, X₂) via tuned RBS elements that control production of activators and repressors, forming a complex that determines the effective activator concentration (XE) for orthogonal output generation.
Toehold Switch-Based Modulator Workflow
Figure 2: Toehold Switch-Based Modulator Workflow. Ligand binding to the riboswitch de-represses repressor gene expression, which subsequently regulates trigger RNA production. Trigger RNA binding to the toehold switch exposes the RBS and initiates reporter translation.
Experimental Optimization Workflow for Amplifier Tuning
Figure 3: Experimental Optimization Workflow for Amplifier Tuning. The iterative process involves balancing component expression, tuning RBS strengths, adjusting feedback loops, and characterizing performance metrics before functional validation.
Research Reagent Solutions for Implementation
Table 3: Essential Research Reagents for Synthetic Biological Amplifier Implementation
| Reagent/Category | Specific Examples | Function/Purpose | Implementation Notes |
|---|---|---|---|
| Orthogonal Regulatory Pairs [37] | ECF σ/anti-σ factors, T7 RNAP/T7 lysozyme | Enable linear signal processing without host interference | Select pairs with demonstrated orthogonality; tune expression levels |
| Ribosome Binding Site (RBS) Libraries [37] [38] | Varied strength RBS sequences | Fine-tune translation rates for optimal activator/repressor ratios | Pre-characterized libraries reduce optimization time |
| Toehold Switch/Trigger Pairs [39] | ACTSTypeIIN1 switch with trN1 trigger | Provide high fold-change RNA-based regulation | Use RNase-deficient strains; balance expression carefully |
| Reporter Systems [37] [39] [38] | GFP variants (e.g., gfpmut3b) | Quantify circuit performance and output signals | Select variants with appropriate maturation times and brightness |
| Inducible Promoter Systems [38] | Arabinose PBAD, IPTG-inducible PLac | Enable controlled tuning of circuit components | Consider basal expression levels and induction kinetics |
| Riboswitch Elements [39] | Coenzyme B12 riboswitch (cbiA from S. typhimurium) | Provide ligand-responsive input sensing | Characterize dose-response before amplifier integration |
| Vector Systems [38] | pSB3K3 (p15A ori, Kanr), pET-24b | Maintain circuit stability and compatible copy number | Match origin to host strain; consider metabolic burden |
The comparative analysis presented in this guide demonstrates that each synthetic biological amplifier architecture offers distinct advantages for specific research contexts. Synthetic biological OAs provide the most sophisticated signal processing capabilities, enabling multidimensional signal decomposition that is particularly valuable for studying complex developmental signaling patterns where multiple overlapping inputs must be resolved [37]. The toehold switch-based modulators excel in applications requiring maximal fold-change amplification of riboswitch-based sensors, making them ideal for metabolite detection and high-throughput screening applications in drug development [39]. Orthogonal genetic amplifiers based on the hrp system offer a balanced approach for general-purpose transcriptional signal scaling in cascaded genetic networks with minimal time delay or noise introduction [38].
For researchers validating synthetic signaling patterns in developmental contexts, the choice of amplifier should align with specific experimental needs: synthetic OAs for complex signal decomposition tasks, toehold modulators for maximum sensitivity in detection applications, and orthogonal genetic amplifiers for straightforward signal scaling in multi-stage genetic circuits. As these technologies continue to mature, they promise to enhance our ability to program cellular behavior with unprecedented precision, ultimately advancing both fundamental research and therapeutic applications in synthetic biology.
The pursuit of predictable biological design in synthetic biology represents a central challenge, particularly in the context of engineering synthetic signaling patterns for developmental contexts. Among the most critical tools to meet this challenge are computational models that predict RNA folding and thermodynamic stability. RNA molecules play an indispensable role in gene regulation and cellular signaling, with their biological functions being intrinsically governed by their secondary and tertiary structures. The ability to accurately model and predict these structures is therefore foundational to designing sophisticated genetic circuits [40] [41].
This guide provides a comparative analysis of current computational methods for RNA structure prediction, framing them within a model-guided design paradigm. We objectively evaluate the performance of leading thermodynamic, evolutionary-based, deep learning, and hybrid approaches, supported by quantitative data and experimental validation protocols. By integrating these computational tools with experimental workflows, researchers can accelerate the design-build-test-learn cycle, enabling the creation of robust synthetic signaling systems for therapeutic development and basic research.
Core Principles of RNA Structure and Its Functional Significance
RNA structure is hierarchical, with canonical base pairs (such as Watson-Crick A:U and G:C, and G:U wobble pairs) stacking into double helices to form the secondary structure. These helical regions are connected by critical loops and junctions, which arrange the helices into specific three-dimensional architectures through recurrent patterns of non-Watson-Crick interactions known as RNA 3D motifs or modules [42]. This structural hierarchy is not static; RNA folding is a dynamic process that occurs during transcription, meaning the mature RNA's functional structure often depends on its cotranscriptional folding pathway, which can deviate significantly from thermodynamic equilibrium predictions [43].
In synthetic biology, this structural plasticity can be harnessed to create regulatory elements. A prime example is the RNA switch, where an RNA sequence is engineered to adopt two alternative conformations: an "OFF" state that inhibits gene expression and an "ON" state that permits it. The transition between these states is often triggered by an external signal, such as a small molecule ligand binding to an integrated aptamer domain, causing a structural rearrangement [40]. The success of such designs hinges on a quantitative understanding of the underlying energy landscape, ensuring the energy difference between states is small enough to allow for efficient switching while maintaining state stability [40].
Comparative Analysis of RNA Structure Prediction Methods
Methodologies and Underlying Algorithms
Thermodynamic & Physics-Based Models: These methods predict the most stable RNA secondary structure by calculating its minimum free energy (MFE). Tools like RNAfold rely on empirical energy parameters for helices and loops. Newer physics-based models like Vfold2D-MC employ coarse-grained representations and Monte Carlo sampling to simulate RNA conformations, generating statistical weights to compute entropy and free energy parameters for complex structural motifs like multi-way junctions and pseudoknots, for which experimental data is often unavailable [40] [44].
Evolutionary & Covariation-Based Models: Methods such as CaCoFold-R3D use evolutionary information embedded in RNA sequence alignments. They identify covarying base pairs that indicate structural conservation, applying probabilistic grammars to simultaneously predict nested canonical helices and RNA 3D motifs found within loop regions. This approach directly integrates the prediction of complex tertiary motifs with secondary structure, constrained by statistically significant covariation evidence [42].
Deep Learning & Hybrid Approaches: Machine learning models, including DSRNAFold, address RNA structure prediction by integrating sequence and structural context information through a phased learning strategy. These models are trained on folding scores and can capture both local and long-range nucleotide interactions, showing superior performance in challenging tasks like pseudoknot recognition [45] [41]. Hybrid approaches often combine thermodynamic principles with learned parameters to enhance robustness.
Performance Comparison and Benchmarking Data
The table below summarizes the key characteristics and performance indicators of different computational approaches, highlighting their respective strengths and limitations.
Table 1: Comparative Performance of RNA Structure Prediction Methods
| Method | Underlying Principle | Key Structural Capabilities | Reported Advantages | Inherent Limitations |
|---|---|---|---|---|
| RNAfold | Thermodynamic / MFE | Standard loops (hairpin, bulge, internal), nested structures | Fast computation; Simple sequence input; Proven reliability for basic structures | Cannot predict pseudoknots or 3D motifs; Limited by energy parameter availability; Ignores co-transcriptional folding [40] [43] |
| Vfold2D-MC | Physics-based / Monte Carlo sampling | Multi-way junctions, pseudoknots, intramolecular kissing loops | Computes free energy for motifs lacking experimental parameters (e.g., complex junctions); Off-lattice conformation generation | Sequence-averaged interactions (limited sequence specificity); Computationally intensive for large structures [44] |
| CaCoFold-R3D | Evolutionary / Probabilistic grammar with covariation | Canonical helices (nested & pseudoknotted), >50 known 3D motifs, tertiary interactions | "All-at-once" integrated prediction of secondary structure and 3D motifs; Uses evolutionary conservation as a strong constraint | Requires a meaningful sequence alignment as input; Performance depends on alignment quality and depth [42] |
| DSRNAFold | Deep Learning / Hybrid | Pseudoknots, long-range interactions, structures guided by chemical mapping | High accuracy in pseudoknot recognition; Robustness against overfitting; Integrates diverse data contexts (e.g., chemical mapping) | Requires substantial training data; Model interpretability can be lower than physics-based methods [45] |
Quantitative Benchmarking Insights
While specific accuracy metrics depend on the benchmark dataset and RNA family, recent trends indicate that:
- Hybrid and deep learning models like DSRNAFold show positive performance, particularly in pseudoknot recognition and predicting structures consistent with chemical mapping data, areas where traditional thermodynamics-based methods typically struggle [45].
- Covariation-integrated methods like CaCoFold-R3D demonstrate high reliability for evolutionarily conserved RNAs, as covariation evidence provides a powerful constraint that often leads to more accurate tertiary structure insights [42].
- Physics-based simulations like Vfold2D-MC provide a major advantage by supplying thermodynamic parameters for structural components like multi-way junctions, which are not available in standard empirical databases but are crucial for predicting the stability of complex synthetic RNAs [44].
Experimental Validation of Computational Predictions
A Standard Workflow for Validating Synthetic RNA Switches
Computational predictions are hypotheses that require rigorous experimental confirmation. The following workflow, adaptable for validating synthetic RNA switches like the SECIS-Theophylline aptamer system, outlines key validation stages [40]:
Diagram 1: RNA Design and Validation Workflow
Detailed Experimental Protocols
1. In vitro Structure Probing
- Purpose: To experimentally determine the secondary structure of the RNA in its OFF and ON states (e.g., without and with theophylline) [40].
- Protocol:
- Sample Preparation: Synthesize the RNA construct via in vitro transcription and purify it. For the ON state, incubate the RNA with a saturating concentration of its ligand (e.g., 2mM theophylline).
- Chemical Probing: Treat separate RNA samples with Selective 2'-Hydroxyl Acylation Analyzed by Primer Extension (SHAPE) reagents (e.g., NMIA or 1M7). These reagents covalently modify flexible, unpaired nucleotides more readily than base-paired nucleotides.
- Data Acquisition: Reverse transcribe the modified RNA using a fluorescently-labeled primer. Run the resulting cDNA fragments on a capillary electrophoresis sequencer. The resulting chromatogram shows peaks whose intensities are proportional to the modification rate at each nucleotide.
- Data Analysis: Map the modification intensities onto the RNA sequence. High-intensity regions indicate single-strandedness, while low-intensity regions indicate base-paired or structured regions. Compare the experimental SHAPE profile to the computationally predicted structures.
2. Functional Validation in Cellular Reports
- Purpose: To confirm that the structural switch produces the intended functional output (e.g., translational readthrough) in a relevant cellular context [40].
- Protocol:
- Reporter Construct Design: Clone the engineered RNA switch (e.g., SECIS-aptamer fusion) into the 3' UTR of a reporter gene (e.g., luciferase or GFP) that is preceded by a stop codon (e.g., UGA).
- Cell Transfection & Stimulation: Transfer the construct into an appropriate cell line (e.g., HEK293 cells). Divide the cells into experimental groups: one treated with the ligand (theophylline) to induce the ON state, and an untreated control for the OFF state.
- Output Measurement: After 24-48 hours, lyse the cells and measure reporter activity (e.g., luminescence or fluorescence). Normalize the readings to a co-transfected control plasmid to account for transfection efficiency.
- Data Interpretation: A successful switch will show a statistically significant increase in reporter signal in the ligand-treated group compared to the untreated control, demonstrating ligand-dependent conformational change and gene regulation.
Essential Research Reagent Solutions
The following reagents and tools are critical for the computational and experimental phases of model-guided RNA design.
Table 2: Key Research Reagents and Tools for RNA Switch Development
| Item Name | Specific Function / Example | Role in Workflow |
|---|---|---|
| Theophylline Aptamer | A well-characterized ~30 nt synthetic RNA module that binds theophylline with high specificity (10,000-fold over caffeine) [40]. | Serves as a Conformational RNA Element (CRE); ligand binding triggers the switch from OFF to ON state. |
| SECIS Element (e.g., DIO2) | A structured RNA element from the 3' UTR that enables selenocysteine incorporation at UGA stop codons [40]. | Provides the functional output mechanism; stop codon readthrough is measured to quantify switching efficiency. |
| RNAfold Software | A standard thermodynamic MFE prediction algorithm from the ViennaRNA package [40]. | Provides initial, rapid secondary structure predictions for design screening; calculates ΔG, GOFF, GON. |
| CaCoFold-R3D | A probabilistic grammar-based tool that integrates 3D motif prediction with secondary structure [42]. | Predicts complex tertiary motifs and provides a more holistic structural model, constrained by evolutionary data. |
| SHAPE Reagents (e.g., 1M7) | Chemicals that selectively modify flexible regions in the RNA backbone [40]. | Enables experimental RNA structure probing in vitro to validate or refute computational models. |
| Dual-Luciferase Reporter System | A standard assay (e.g., Firefly and Renilla luciferase) for quantifying gene expression changes. | Measures the functional outcome of RNA switching in live cells, providing a quantitative readout of performance. |
Integrated Computational-Experimental Case Study: The SKIPPIT Switch
The Barcelona-UB iGEM team's project provides a compelling case study of model-guided design. Their goal was to build a synthetic RNA switch (SKIPPIT) that controls translational readthrough. The core design integrated a SECIS element with a theophylline aptamer via a designed linker sequence [40].
Their integrated approach followed these steps:
- Model-Guided In Silico Design: A computational model based on RNAfold predictions was used to screen thousands of potential linker sequences. The model scored designs based on their predicted energy landscape: a stable OFF state (inactive conformation without theophylline) and a significant energy drop upon ligand binding to transition to a functional ON state [40].
- Software Implementation: The model was translated into the TADPOLE software, which automated the thermodynamic validation, structural checks for OFF disruption and ON recovery, and pairing analysis. This transformed the design process from empirical "needle in a haystack" searching to a targeted, high-confidence selection of candidates for synthesis [40].
- Experimental Validation and Learning: The top-predicted designs were synthesized and tested in the lab. The experimental results, particularly from testing individual SECIS and aptamer components, were fed back into the model to refine its parameters, such as the target energy gap (Δ ≈ 4.5 kcal·mol⁻¹). This "learn" step created a positive feedback loop, improving both the specific design and the general understanding of RNA folding dynamics in synthetic systems [40].
This cycle demonstrates that model-guided design, where computation actively drives wet-lab experimentation, is significantly more efficient—reducing costs, synthesis burden, and experimental timelines while increasing the success rate [40].
The strategic integration of RNA folding predictions and thermodynamic modeling is transforming the design of synthetic biological systems. As computational methods evolve—incorporating deeper learning, more sophisticated physics, and richer evolutionary data—their predictive power for both structure and function will only increase. This progress, combined with robust experimental validation frameworks, will be crucial for tackling the next frontier: engineering complex, multi-component synthetic signaling networks that can mimic the intricate patterning events of natural development. For researchers in drug development and basic science, adopting this model-guided paradigm is no longer optional but essential for the rational and efficient creation of reliable genetic tools and therapies.
Chimeric Antigen Receptor (CAR) T-cell therapy has revolutionized the treatment of certain blood cancers, but has struggled to demonstrate consistent efficacy against solid tumors. [16] [46] A significant barrier to success is the immunosuppressive tumor microenvironment (TME), where soluble and cellular components actively limit CAR-T cell function and persistence. [16] [46] While engineered T cells rely on environmental cues to remain active, solid tumors often dominate with inhibitory signals while providing weak or absent co-stimulatory signals essential for T cell function. [46] This case study examines the development and validation of TME-sensing switch receptors for enhanced response to tumors (T-SenSERs), a computationally designed synthetic receptor system that reprograms the immunosuppressive TME into a potent T-cell activator. The work establishes a novel framework for validating synthetic signaling patterns within developmental and therapeutic contexts.
Computational Design Platform and Mechanism of Action
Overcoming Limitations of Conventional Approaches
Traditional methods for creating synthetic signaling receptors have relied heavily on trial-and-error, resulting in unpredictable signaling characteristics that make it difficult to control receptor behavior in therapeutic contexts. [16] [46] The T-SenSER platform addresses this fundamental challenge through a computational protein design approach that enables de novo bottom-up assembly of allosteric receptors with programmable input-output behaviors. [16] Unlike conventional protein design methods that treat proteins as rigid structures, this platform models them as dynamic, shape-shifting machines, allowing researchers to simulate how signals travel through synthetic receptors to control cell behavior. [46]
Architectural Principles and Signaling Logic
The T-SenSER receptors follow a modular architecture comprising three functional domains: [46]
- External Domain: Binds specific soluble tumor-associated signals (e.g., VEGF, CSF1)
- Transmembrane Region: Transmits the extracellular signal across the cell membrane
- Internal Domain: Activates specific therapeutic functions inside the T cell
This architectural framework enables the creation of synthetic receptors that detect soluble TME factors and convert these signals into co-stimulatory or cytokine-like signals that enhance T cell activity. [16] [46] The computational design process allowed researchers to fine-tune receptor behavior, programming them to operate in ligand-dependent, always-on, or intermediate signaling modes based on therapeutic requirements. [46]
The diagram below illustrates the core signaling logic and design workflow of the T-SenSER platform.
Experimental Validation: Methodologies and Results
Receptor Design and In Vitro Characterization
Researchers developed two distinct T-SenSER families: VMR (VEGF-sensing) and CMR (CSF1-sensing), targeting factors selectively enriched in various tumors. [16] [46] The experimental workflow involved:
- Computational Design and Screening: Initial computational design produced 18 receptor variants that were screened through simulations and in vitro testing. [46]
- Signaling Specificity Validation: Engineered receptors were transfected into human T cells, which were then exposed to target ligands (VEGF or CSF1) to quantify signaling output. [16]
- Combination Therapy Testing: T cells were engineered to express both a conventional CAR and a T-SenSER receptor to assess combinatorial effects. [16]
The VMR receptor demonstrated strict ligand-dependent activation, only triggering T cell signaling when VEGF was present. [46] In contrast, the CMR receptor provided a low baseline activation even without CSF1 but significantly amplified its signaling output in the ligand's presence. [46] This demonstrates the programmability of signaling responses achievable through computational design.
Table 1: In Vitro Characterization of T-SenSER Receptors
| Receptor Type | Target Ligand | Baseline Activity (No Ligand) | Activated Response (With Ligand) | Signaling Specificity |
|---|---|---|---|---|
| VMR | VEGF | Minimal | Strong activation | Ligand-dependent |
| CMR | CSF1 | Low baseline | Amplified response | Ligand-amplified |
| Conventional CAR | Surface antigen | Context-dependent | Context-dependent | Not TME-responsive |
In Vivo Therapeutic Efficacy Assessment
The therapeutic efficacy of T-SenSER-enhanced T cells was evaluated in multiple mouse models, including lung cancer and multiple myeloma. [16] The experimental protocol included:
- Tumor Establishment: Implanting human tumor cells into immunodeficient mice to establish measurable tumors.
- Treatment Groups: Mice were divided into groups receiving either conventional CAR-T cells or CAR-T cells combined with T-SenSER receptors.
- Outcome Measures: Researchers tracked tumor volume over time, animal survival, and conducted histological analyses of tumor tissue.
- Ligand Dependence: Controlled experiments verified that therapeutic enhancement required the specific TME factor targeted by each T-SenSER receptor. [16]
In both cancer models, T cells equipped with both a CAR and a T-SenSER receptor demonstrated superior tumor control and extended host survival compared to conventional CAR-T cells alone. [16] [46] The enhancement was strictly dependent on the presence of the targeted TME factor (VEGF or CSF1), confirming the programmed specificity of the synthetic receptors. [16]
Table 2: In Vivo Efficacy of T-SenSER-Enhanced T Cells in Mouse Models
| Cancer Model | Treatment Group | Tumor Growth Inhibition | Survival Benefit | Ligand Dependency |
|---|---|---|---|---|
| Lung Cancer | CAR alone | Baseline | Baseline | Not applicable |
| Lung Cancer | CAR + VMR | Significant enhancement | Extended | VEGF-dependent |
| Multiple Myeloma | CAR alone | Baseline | Baseline | Not applicable |
| Multiple Myeloma | CAR + CMR | Significant enhancement | Extended | CSF1-dependent |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for T-SenSER Development and Validation
| Reagent / Tool | Function in Experimental Protocol | Specific Example / Target |
|---|---|---|
| Computational Design Platform | De novo protein structure prediction and allosteric signaling design | Dimeric MultiDomain Biosensor Builder [16] |
| VEGF (Vascular Endothelial Growth Factor) | TME-soluble factor for sensor activation | Target ligand for VMR receptors [16] [46] |
| CSF1 (Colony-Stimulating Factor 1) | TME-soluble factor for sensor activation | Target ligand for CMR receptors [16] [46] |
| CAR Constructs | Provides primary tumor recognition signal | Various tumor-specific antigen receptors [16] |
| Human T-cells from healthy donors | Engineered therapeutic cell platform | Primary cells for in vitro and in vivo testing [16] |
| Mouse cancer models | In vivo therapeutic efficacy assessment | Lung cancer and multiple myeloma models [16] |
| Flow cytometry reagents | Immune phenotyping and activation assessment | Antibodies for T-cell activation markers [16] |
| Cytokine measurement assays | Quantification of T-cell functional output | ELISA or multiplex arrays [16] |
Discussion: Implications for Synthetic Signaling in Developmental Contexts
The T-SenSER platform represents a significant advancement in synthetic immunology, demonstrating that computational protein design can create receptors with predictable, programmable signaling behaviors. [46] This work validates the broader thesis that synthetic signaling systems can be engineered to interpret complex environmental cues and execute predefined cellular responses—a principle fundamental to both therapeutic engineering and understanding developmental biology.
The research establishes several key principles for synthetic signaling pattern validation:
- Signal Reprogramming: Successful conversion of immunosuppressive TME signals into T-cell activating signals demonstrates the feasibility of rewriting pathological environmental contexts into therapeutic opportunities. [16] [46]
- Signaling Tunability: The ability to design receptors with different activation thresholds (strictly ligand-dependent vs. ligand-amplified) highlights the precision achievable through computational design. [46]
- Context-Specific Activation: T-SenSER receptors maintain specificity despite the complexity of the TME, illustrating how synthetic systems can achieve discrimination in biologically noisy environments. [16]
The methodology establishes a framework for deconstructing and reconstructing signaling patterns across developmental and therapeutic contexts, with potential applications extending beyond T-cell therapy to areas including stem cell programming, tissue engineering, and synthetic pattern formation in developmental models. [5]
Navigating Complexity: Strategies for Troubleshooting and Optimizing Synthetic Circuits
Identifying and Resolving Non-Orthogonal Signal Crosstalk
In both biological systems and engineered devices, the phenomenon of non-orthogonal signal crosstalk presents a fundamental challenge to precise communication and control. Crosstalk occurs when signals transmitted through separate pathways interfere with one another, degrading the fidelity of information transfer and compromising system performance. In synthetic biology, this manifests as unwanted interactions between genetic components that should operate independently, while in electronic systems, it appears as electromagnetic coupling between adjacent transmission lines. The core of the problem lies in the non-orthogonality of these signaling systems—their inherent inability to remain completely separable due to physical proximity, spectral overlap, or biochemical similarity.
Understanding and mitigating crosstalk is particularly crucial for validating synthetic signaling patterns in developmental contexts research, where precise spatiotemporal control of gene expression guides complex processes like tissue patterning and organogenesis. Developmental systems inherently employ multiple overlapping signaling pathways (e.g., Notch, Wnt, Hedgehog) that must be precisely coordinated without detrimental interference [47]. Similarly, in engineered systems, the trend toward miniaturization and increased complexity exacerbates crosstalk issues, making effective identification and resolution strategies essential for advancing both basic research and therapeutic applications.
Comparative Analysis of Crosstalk Resolution Methodologies
Biological Versus Electronic Crosstalk Manifestations
The fundamental principles of crosstalk transcend disciplinary boundaries, though their manifestations differ significantly between biological and electronic contexts. In biological systems, crosstalk occurs when perturbing one pathway causes a measurable response in another, distinct pathway [47]. For example, in developmental biology, stimulation of Notch pathway receptors may produce downstream effects on TGF-β target genes, creating challenges for interpreting experimental results and designing precise interventions.
Electronic crosstalk, in contrast, results from unwanted coupling between adjacent conductive structures, following the power balance equation: Pout = Pin - Pabsorbed - Preflected - Pleaked + Pcoupled [48]. This coupling degrades signal integrity by introducing noise that reduces data transmission rates and can ultimately cause complete link failure. Despite these different manifestations, both contexts require sophisticated quantification and mitigation strategies to maintain system functionality.
Quantitative Comparison of Resolution Approaches
Table 1: Comparative Analysis of Crosstalk Resolution Methodologies
| Methodology | Underlying Principle | Application Context | Key Performance Metrics | Limitations |
|---|---|---|---|---|
| Synthetic Biological OAs [49] | Orthogonal σ/anti-σ pairs with RBS tuning | Decomposing multidimensional biological signals | 153-688 fold signal amplification; enhanced signal-to-noise ratio | Limited by available orthogonal regulatory pairs; host metabolic burden |
| Database-Driven Identification [47] | Manual curation of pathway interactions | Mapping known crosstalk between signaling pathways | 650 curated pathway pairs; 345 with documented crosstalk | Limited to previously documented interactions; labor-intensive |
| Frequency Domain Analysis [48] | S-parameter extraction over signal bandwidth | Electronic interconnect characterization | Power Sum Crosstalk (PSXT); Multiple Disturber metrics | Requires specialized equipment and expertise |
| Orthogonal Spin Labeling [50] | Spectroscopically distinguishable spin probes | DEER spectroscopy for distance measurements | Selective addressing via distinct resonance frequencies | Non-perfect orthogonality leads to residual crosstalk |
| Coupling Coefficients [48] | Transmission line cross-section analysis | Pre-layout investigation of electronic crosstalk | Backward (Kb) and forward (Kf) coupling coefficients | Limited to parallel traces; overestimates forward crosstalk in lossy systems |
Experimental Framework for Biological Crosstalk Resolution
Synthetic Operational Amplifiers for Signal Decomposition
A groundbreaking approach to biological crosstalk mitigation involves the development of synthetic biological operational amplifiers (OAs) inspired by electronic signal processing principles [49]. This framework addresses the fundamental challenge that traditional synthetic genetic circuits primarily process binary (ON/OFF) signals, whereas natural biological systems expertly manage complex, non-orthogonal signals—interdependent signals prone to interference.
The core innovation utilizes orthogonal σ/anti-σ pairs and careful tuning of ribosome binding site (RBS) strengths to implement both open-loop and closed-loop configurations. These synthetic OAs perform mathematical operations on input signals according to the relationship: α · X₁ - β · X₂, where X₁ and X₂ represent input transcription signals that regulate the production of activator (A) and repressor (R), respectively [49]. This linear combination allows for the decomposition of overlapping signals into their orthogonal components, effectively isolating specific pathway activities from complex biological mixtures.
Table 2: Research Reagent Solutions for Synthetic Biological OAs
| Research Reagent | Function in Experimental System | Specific Application in Crosstalk Resolution |
|---|---|---|
| ECF σ Factors [49] | Transcriptional activators | Core components of synthetic OA circuits |
| Anti-σ Factors [49] | Cognate repressors for σ factors | Enable precise control of activation/repression balance |
| T7 RNA Polymerase [49] | Orthogonal transcriptional machinery | Provides modular expression system component |
| T7 Lysozyme [49] | Inhibitor of T7 RNAP | Enables fine-tuning of circuit performance |
| Ribosome Binding Sites [49] | Control translation initiation | Key engineering parameter for tuning circuit coefficients |
| Growth-Phase Responsive Promoters [49] | Sense physiological state | Input signals for growth-stage-responsive circuits |
The experimental implementation involves constructing genetic circuits where input X₁ regulates activator production with translation rate r₁ and degradation rate γ₁, resulting in activator concentration [A₀] = Ad · (r₁/γ₁) · X₁ = α · X₁ [49]. Similarly, input X₂ regulates repressor production, yielding [R₀] = Ad · (r₂/γ₂) · X₂ = β · X₂. The effective activator concentration (XE) is then computed as XE = α · X₁ - β · X₂, which determines the circuit output through a Hill-type equation with coefficient 1 [49].
Workflow for Orthogonal Signal Transformation
The following diagram illustrates the experimental workflow for implementing orthogonal signal transformation using synthetic biological operational amplifiers:
Protocol for Synthetic OA Circuit Implementation
Day 1: Circuit Design and Component Preparation
- Select orthogonal regulatory pairs: Identify suitable ECF σ/anti-σ factors or T7 RNAP/T7 lysozyme pairs based on orthogonality to host systems and each other [49].
- Design RBS variants: Create multiple RBS sequences with varying translation initiation strengths to enable parameter tuning.
- Prepare DNA constructs: Assemble genetic circuits using standard synthetic biology techniques (Golden Gate, Gibson Assembly).
Day 2-3: Circuit Assembly and Validation
- Transform constructs: Introduce assembled circuits into appropriate host cells (E. coli commonly used).
- Verify circuit assembly: Sequence confirmed colonies to ensure correct genetic construction.
- Characterize input-output relationships: Measure transfer functions for individual circuit components.
Day 4-7: System Integration and Testing
- Assemble complete OA circuit: Combine validated components into full operational amplifier configuration.
- Measure crosstalk resolution: Test circuit performance with mixed input signals mimicking natural non-orthogonal conditions.
- Quantify performance metrics: Calculate signal-to-noise ratio improvement, amplification factor, and orthogonality of outputs.
This protocol typically requires 5-7 days for initial implementation, with additional time for optimization based on experimental results.
Advanced Crosstalk Quantification Techniques
Electronic Crosstalk Quantification Methods
In electronic systems, crosstalk quantification has evolved into sophisticated methodologies that provide complementary insights for biological applications:
Frequency Domain Analysis represents the most universal approach, based on extracting S-parameters over the signal bandwidth and calculating metrics like Power Sum Crosstalk (PSXT) [48]. PSXT is computed as the sum of squares of S-matrix elements from all possible aggressors at a victim receiver port, expressed in dB. This method effectively captures both local and distant coupling phenomena.
Time Domain Analysis involves simulation with step, pulse, or pseudo-random bit stream (PRBS) excitation signals to measure peak voltages or eye diagram distortion [48]. This approach provides the most accurate evaluation of actual crosstalk values, as it accounts for frequency-dependent dispersion through conversion from frequency-domain S-parameters.
Coupling Coefficients offer a simplified method suitable for preliminary investigations, where backward (Kb) and forward (Kf) coupling coefficients are calculated using improved Bracken equations that account for non-symmetric coupling cases [48]. While limited to parallel or nearly parallel traces, this method enables rapid design rule generation during pre-layout phases.
Spectroscopic Approaches for Biochemical Systems
In biochemical contexts, double electron-electron resonance (DEER) spectroscopy employs orthogonal spin labeling with nitroxide (NO) and gadolinium (Gd) labels to study distance distributions in biomolecular complexes [50]. This technique provides three distinct DEER channels (NO-NO, NO-Gd, Gd-Gd) for selective interrogation of specific spin pairs. However, spectral overlap between NO and Gd spins creates crosstalk challenges, as signals intended for one channel appear in another [50]. Identification relies on recognizing unexpected distance distributions in non-corresponding channels, while suppression techniques include optimizing microwave power settings to leverage differential transition moments and exploiting distinct relaxation behaviors through short shot repetition times.
Pathway Crosstalk in Developmental Contexts
Biological Crosstalk Database and Analysis
The systematic curation of pathway crosstalk information in databases like XTalkDB provides invaluable resources for developmental biology research [47]. This database documents 650 ordered pairs of pathways based on manual curation of over 1,600 publications, with 345 pairs (53%) showing literature-supported crosstalk [47]. Each entry includes critical contextual information such as directionality (e.g., Notch to TGF-β versus TGF-β to Notch), regulation type (activation or inhibition), mediating molecules, tissue specificity, and organism source.
This structured approach reveals that crosstalk in developmental systems is often asymmetric, with different mechanisms and proteins mediating interactions in each direction [47]. For example, Notch to TGF-β crosstalk may involve protein-protein interactions (NICD with SMAD3), while TGF-β to Notch crosstalk might occur through transcriptional regulation (Jagged1 expression) [47]. Understanding these directional differences is crucial for accurately modeling developmental processes and designing effective interventions.
Integration of Engineered Living Materials
Emerging technologies in engineered living materials (ELMs) integrate synthetic gene circuits with functional scaffolds to create responsive systems for developmental biology applications [51]. These systems embed genetically engineered microbial cells within hydrogels and other matrices, enabling programmed responses to diverse inputs including chemicals, light, heat, and mechanical loading [51]. For developmental contexts, this approach facilitates the creation of spatially patterned signaling environments that can guide tissue formation while minimizing unwanted crosstalk through physical compartmentalization.
The following diagram illustrates key signaling pathways and their crosstalk mechanisms in developmental contexts:
The systematic identification and resolution of non-orthogonal signal crosstalk represents a critical frontier for both basic research and applied biotechnology. The comparative analysis presented here reveals that while manifestations differ across biological and electronic domains, underlying principles of interference minimization and signal orthogonality provide unifying conceptual frameworks. The development of synthetic biological operational amplifiers marks a significant advancement, demonstrating that engineering principles can be successfully adapted to biological contexts to achieve dramatic improvements in signal fidelity [49].
For developmental biology research, these approaches enable more precise dissection of complex signaling networks and create new opportunities for constructing synthetic patterning systems. Future progress will likely involve increasing the dimensionality of signal processing systems, improving orthogonality through directed evolution of biological components, and developing more sophisticated computational models that predict crosstalk before experimental implementation. As these methodologies mature, they will accelerate both our understanding of natural developmental processes and our ability to engineer therapeutic interventions that precisely control cellular behavior in complex physiological environments.
Addressing Host-Cell Interactions and Metabolic Burden
In the burgeoning field of synthetic developmental biology, a central challenge is the accurate validation of engineered signaling patterns. This process is complicated by two intrinsic factors: the complex, reciprocal host-cell interactions between engineered circuits and the native cellular machinery, and the metabolic burden imposed by synthetic gene expression, which can divert essential resources from core physiological processes [5]. These factors can skew experimental outcomes, leading to unpredictable performance and durability of synthetic systems. This guide provides a systematic comparison of current methodologies, with a focus on their ability to quantify and control for these confounding variables, thereby offering a framework for researchers to select the most appropriate tools for robust experimental validation.
Comparative Analysis of Key Methodologies
The table below objectively compares the primary technologies used to deconstruct host-cell interactions and measure metabolic burden in developmental contexts.
Table 1: Comparison of Key Methodologies for Addressing Host-Cell Interactions and Metabolic Burden
| Methodology | Core Application in Validation | Key Performance Metrics | Directly Measures Metabolic Burden? | Spatiotemporal Resolution | Supporting Experimental Data |
|---|---|---|---|---|---|
| Genome-Scale Metabolic Modeling (GEM) | Predicting metabolic interactions and resource competition between host and synthetic circuits [52]. | Predictive accuracy of flux distributions; Correlation with transcriptomic/metabolomic data [53]. | Yes, computationally predicts flux rerouting and ATP/maintenance energy costs [52]. | System-level (non-spatial), Steady-state or dynamic simulation [52]. | Used with multi-omics data from aging mice to predict microbiota-dependent decline in host nucleotide metabolism [53]. |
| Optogenetic Control of Signaling | Decoupling endogenous signaling from synthetic inputs to isolate circuit function [5]. | Precision of fate control (e.g., % of cells differentiating); Temporal window of sensitivity [5]. | Indirectly, by correlating induction strength with growth rate or marker expression. | High (cellular to subcellular), Seconds to minutes [5]. | Light-controlled Notch/Delta and BMP signaling used to map essential temporal windows for fate specification in Drosophila and zebrafish [5]. |
| Synthetic Recording Systems | Tracing cell lineage and recording history of signaling exposure in developing tissues [5]. | Recording efficiency (e.g., % of expected edits); Clarity of reconstructed lineages. | No, but can report on burden by coupling to stress-responsive promoters. | Single-cell, Days to weeks (permanent record) [5]. | CRISPR-based recorders used to trace lineages and signal exposure in mouse and zebrafish embryos [5]. |
| Integrated Host-Microbiome Metabolic Models | Characterizing metabolic dependencies and cross-feeding in complex, multi-species systems [53]. | Reduction in community-level interaction strength; Number of identified cross-fed metabolites [53]. | Yes, models community-level and host-level metabolic fluxes simultaneously [53]. | Community-level (non-spatial), Steady-state [53]. | Revealed a pronounced, aging-associated reduction in microbiome metabolic activity and beneficial interactions in mice [53]. |
Detailed Experimental Protocols
Protocol 1: Constructing an Integrated Host-Microbiome Metabolic Model
This protocol, adapted from recent aging research, is designed to quantify the metabolic burden imposed on a host by its associated microbiome and to identify critical interaction points [53] [52].
- Input Data Collection: Gather host and microbial genomic data. For microbes, use shotgun metagenomic sequencing of the community (e.g., fecal material for gut microbiome) to reconstruct Metagenome-Assembled Genomes (MAGs). For the host, collect tissue-specific transcriptomic data (e.g., from colon, liver, brain) [53].
- Metabolic Model Reconstruction:
- Microbiome: Use automated tools like
gapseqto reconstruct genome-scale metabolic models (GEMs) from the MAGs. Assess model quality based on genome completeness and contamination [53] [52]. - Host: Retrieve a context-specific host metabolic model (e.g., Recon3D for human) or reconstruct one from its genome. Integrate tissue-specific transcriptomic data to create tissue-specific models [53] [52].
- Microbiome: Use automated tools like
- Model Integration: Combine the host and microbiome models into a single metamodel. Define the exchange of metabolites between the microbiome (gut lumen), host tissues (colon, liver, brain), and a shared bloodstream compartment. Standardize metabolite and reaction nomenclature across models using databases like MetaNetX [53] [52].
- Constraint-Based Simulation: Apply Constraint-Based Reconstruction and Analysis (COBRA). Use Flux Balance Analysis (FBA) with an appropriate objective function (e.g., maximize biomass for bacteria, ATP production for host tissues). Constrain the model with dietary inputs and reaction fluxes based on transcriptomic data [52].
- Validation: Correlate model-predicted metabolic fluxes with experimentally measured metabolomic data from host tissues and bodily fluids. Validate predictions (e.g., decline in a specific pathway) using germ-free or gnotobiotic animal models [53].
Protocol 2: Optogenetic Interrogation of Signaling Burden
This protocol uses light to control synthetic pathways with high precision, allowing researchers to map the metabolic and functional costs of pathway activation [5].
- Tool Selection: Choose an optogenetic actuator suited to the pathway under study. For receptor activation (e.g., synthetic BMP, FGF), fuse receptor intracellular domains to blue-light dimerizers like
VfAU1orVVD[5]. - Cell Engineering: Stably integrate the optogenetic construct into your target cell line (e.g., pluripotent stem cells) or model organism. Use a well-characterized delivery system (lentivirus, transposon, or CRISPR-mediated knock-in) to ensure consistent expression [5].
- Dose-Response Calibration: Expose cells to varying intensities and durations of light. Quantify the output (e.g., nuclear localization of a downstream transcription factor using immunofluorescence, or expression of a target gene via qPCR) to establish a input-output curve [5].
- Burden Assay: For each activation level (from Step 3), measure proxies of metabolic burden:
- Growth Rate: Monitor cell proliferation via live-cell imaging or simple cell counting.
- Resource Competition: Co-transfect a constitutive fluorescent reporter (e.g., GFP) and measure its intensity drop upon pathway activation, indicating resource theft by the synthetic circuit.
- Transcriptomic Signatures: Perform RNA-seq on activated vs. non-activated cells to identify upregulation of stress response pathways [5].
- Functional Validation: Apply the calibrated light stimulus in a developmental context (e.g., to direct stem cell differentiation). Use the burden metrics from Step 4 to correct for non-cell-autonomous effects and refine the differentiation protocol [5].
Signaling Pathways and Experimental Workflows
The following diagrams illustrate the core computational and experimental workflows for the protocols described above.
Host-Microbiome Metabolic Modeling Workflow
Optogenetic Burden Assessment Workflow
The Scientist's Toolkit: Research Reagent Solutions
The table below catalogs essential reagents and tools for implementing the described methodologies.
Table 2: Key Research Reagent Solutions for Synthetic Developmental Biology
| Reagent/Tool | Function in Validation | Key Feature / Application Note |
|---|---|---|
| gapseq [53] [52] | Automated reconstruction of genome-scale metabolic models (GEMs) from genomic data. | Used to generate metabolic networks for 181 gut microorganisms in the mouse aging study; critical for predicting metabolic flux [53]. |
| COBRA Toolbox [52] | MATLAB suite for constraint-based reconstruction and analysis (COBRA) of metabolic models. | Performs Flux Balance Analysis (FBA) on integrated host-microbiome models to simulate metabolic states [52]. |
| AGORA & Recon3D [52] | Curated libraries of pre-built, high-quality metabolic models for microbes and humans, respectively. | Provides a standardized starting point for model integration, ensuring consistency and reducing reconstruction time [52]. |
| OptoBase [5] | Online portal cataloging optogenetic tools and their properties. | Aids in selecting the appropriate light-sensitive domain (e.g., LOV, Cry2) for controlling a target pathway [5]. |
| LightOn (GAVPO) System [5] | Optogenetic gene expression system based on light-induced Gal4 dimerization. | Enables precise, light-controlled transcription of synthetic genes in mammalian cells and mice [5]. |
| LEXY / LANS / LINUS [5] | Optogenetic systems for controlling nuclear export (LEXY) and import (LANS, LINUS) of proteins. | Allows for light-controlled shuttling of synthetic transcription factors to the nucleus, enabling control of endogenous genes [5]. |
| CRISPR-based Recorders [5] | Synthetic systems that use CRISPR-Cas to record signaling events in a cell's DNA. | Enables lineage tracing and historical recording of signal exposure during development, providing a readout of cellular history [5]. |
Optimizing Energy Balance and Interaction Landscapes in RNA Switches
In the field of synthetic developmental biology, where researchers aim to program self-organizing cellular behaviors and precise tissue patterning, the need for robust, programmable genetic regulators is paramount [54]. RNA structural switches have emerged as powerful tools in this context, serving as critical components for implementing synthetic signaling patterns that mimic developmental processes. These regulatory elements function by undergoing conformational changes in response to specific signals, thereby controlling gene expression at the translational level with temporal precision and minimal energetic cost [40] [55]. Unlike transcriptional regulation, which can be slow and prone to stochastic noise, RNA switches offer faster responses and direct protein regulation, making them ideal for engineering the dynamic spatial patterns required in developmental contexts [55].
The performance of these RNA switches hinges on two fundamental biophysical properties: their energy balance and interaction landscape. Energy balance refers to the thermodynamic stability differences between alternative conformations, while the interaction landscape encompasses the network of molecular contacts that determine structural specificity [40] [56]. Optimizing these parameters is essential for creating switches that respond reliably to developmental signals, whether they be morphogen gradients, cell-cell contact signals, or environmental cues [54]. This guide systematically compares current approaches for optimizing these parameters, providing experimental data and methodologies to empower researchers in synthetic biology and drug development to engineer more effective RNA-based regulatory systems.
Foundational Principles of RNA Switch Engineering
Energy Balance in RNA Structural Switching
The thermodynamic stability of RNA switches, particularly the energy difference between their alternative conformations, represents a critical determinant of their performance. Effective switching requires a carefully balanced energy landscape where the transition between states occurs without excessive energetic barriers [40]. Research from the Barcelona-UB iGEM team demonstrates that successful RNA switches exhibit a specific energy relationship, with the target energy gap (Δ) approximating half the binding energy of the triggering ligand [40]. In their theophylline-responsive switch system, they employed a binding energy of approximately -9.5 kcal·mol⁻¹ with a target Δ of -4.5 kcal·mol⁻¹ [40].
Recent advances in computational design have further refined our understanding of these thermodynamic requirements. The RiboPO framework, which employs reinforcement learning from physical feedback (RLPF), explicitly optimizes for both structural accuracy and thermodynamic stability [56]. This approach recognizes that sequences occupying narrow, unstable basins in the energy landscape often fail to adopt intended conformations under physiological conditions, despite scoring well on structural metrics alone [56].
Interaction Landscapes and Structural Specificity
The interaction landscape of an RNA switch encompasses the network of molecular contacts—including base pairing, stacking, and tertiary interactions—that determine its structural specificity and functional robustness. RNA molecules can form complex interaction networks involving three different edges of RNA bases (Watson-Crick, Hoogsteen, and sugar edges) with either cis or trans orientations, resulting in more than 200 possible base pair combinations [57]. This complexity creates both challenges and opportunities for engineering specific conformational states.
Current design strategies emphasize the importance of minimizing overly stable non-functional interactions that could compete with the desired binding-competent conformation [40]. Experimental data suggest that successful switches balance the number and strength of interactions in their alternative states, avoiding kinetic traps while maintaining structural specificity. Advanced computational methods like RiboPO address this challenge by incorporating multi-objective optimization that simultaneously considers global 3D fidelity, local interaction accuracy, and thermodynamic stability [56].
Table 1: Key Biophysical Parameters for Optimized RNA Switches
| Parameter | Optimal Range | Measurement Approach | Impact on Function |
|---|---|---|---|
| Energy Gap (ΔG) | ≈50% of ligand binding energy [40] | Computational folding (RNAfold), Isothermal Titration Calorimetry | Determines switching efficiency and response threshold |
| MFE Difference | -36.86 kcal/mol (optimized) vs -32.83 kcal/mol (baseline) [56] | Boltzmann sampling, Partition function calculation | Impacts conformational stability and transition barriers |
| Structural Heterogeneity | 16.6% of regions populate ≥2 conformations [58] | DMS-MaPseq with DRACO deconvolution | Affects regulatory precision and context dependence |
| Secondary Structure Self-Consistency | 0.72 scMCC (optimized) vs 0.60 (baseline) [56] | EternaFold prediction, Comparative analysis | Correlates with functional reliability in cellular environments |
Comparative Analysis of RNA Switch Optimization Approaches
Model-Guided Design with TADPOLE Framework
The Barcelona-UB iGEM team developed TADPOLE as a generalized framework for designing RNA switches by combining Functional RNA Elements (FREs) and Conformational RNA Elements (CREs) [40] [55]. This approach uses thermodynamic modeling to predict how linking these elements creates switches that respond to specific signals by altering their structure. Their methodology centers on designing switches where the CRE's signal-induced conformational change propagates to the FRE through a connecting linker, thereby controlling its activity [40].
In their experimental implementation, they paired the SECIS element (FRE) with the theophylline aptamer (CRE), using RNAfold for structural predictions and energy calculations [40]. The model guided the design of linker sequences that maintained the FRE in an inactive state in the absence of theophylline, while allowing transition to an active conformation upon ligand binding. This approach transformed their design process from empirical trial-and-error to targeted selection, significantly improving efficiency and success rates [40].
Table 2: Performance Comparison of RNA Switch Design Approaches
| Design Method | Structural Accuracy (RMSD Å) | Thermodynamic Stability (MFE kcal/mol) | Secondary Structure Consistency (scMCC) | Experimental Validation |
|---|---|---|---|---|
| TADPOLE (Model-Guided) | Not specified | Target Δ = -4.5 kcal·mol⁻¹ [40] | Not specified | Successful lab testing of SECIS + Theophylline system [40] |
| RiboPO (RLPF) | 10.23Å (vs 10.66Å baseline) [56] | -36.86 (vs -32.83 baseline) [56] | 0.72 (vs 0.60 baseline) [56] | Computational benchmark on SSTT dataset [56] |
| SwitchSeeker (De Novo Discovery) | Confirmed by DMS-MaPseq and cryo-EM [59] | In vivo structural equilibrium | Functional conformation specificity [59] | 245 high-confidence switches identified in human transcriptome [59] |
| Toehold-VISTA (ML-Guided) | Not specified | Not specified | Not specified | Improved function against SARS-CoV-2 RNA [60] |
Preference Optimization with Physical Feedback (RiboPO)
The RiboPO framework represents a significant advancement in computational RNA design by addressing the limitations of geometry-only optimization [56]. This method uses reinforcement learning from physical feedback (RLPF) to fine-tune base models like gRNAde through multi-objective preference optimization. Unlike traditional approaches, RiboPO explicitly incorporates thermodynamic stability alongside structural fidelity by constructing preference pairs from composite physical criteria [56].
In benchmark evaluations, RiboPO demonstrated a superior balance of structural accuracy and stability compared to geometric-only approaches [56]. The framework improved minimum free energy by 12.3% and increased secondary-structure self-consistency by 20% while maintaining competitive 3D quality and high sequence diversity [56]. These improvements translate to enhanced sampling efficiency, with RiboPO achieving 11% higher pass@64 than the gRNAde base model when evaluated under multiple requirement constraints [56].
De Novo Discovery with SwitchSeeker
For researchers interested in identifying natural RNA switches rather than engineering synthetic ones, SwitchSeeker provides a comprehensive computational and experimental framework for systematic discovery [59]. This approach combines SwitchFinder computational predictions with in vivo validation through DMS mutational profiling and massively parallel reporter assays [59].
SwitchSeeker employs a unique energy landscape analysis that prioritizes sequences with two local minima in close proximity and a small energy barrier between them [59]. When applied to the human transcriptome, this methodology identified 245 high-confidence RNA structural switches, including one in the 3' UTR of the RORC transcript that was confirmed through DMS-MaPseq and cryo-EM to exist as two alternative conformations [59]. Functional characterization revealed that this switch regulates gene expression through conformation-specific activation of nonsense-mediated decay [59].
SwitchSeeker Workflow for RNA Switch Discovery
Experimental Protocols for RNA Switch Validation
In Vivo Structural Profiling with DMS-MaPseq
Dimethyl sulfate mutational profiling with sequencing (DMS-MaPseq) has emerged as a powerful method for characterizing RNA structural ensembles in living cells [59] [58]. This protocol enables transcriptome-wide mapping of RNA secondary structures under physiological conditions, providing critical data on structural heterogeneity and conformational dynamics.
Protocol Steps:
- Cell Culture and Probing: Grow HEK293 cells to 70-80% confluency and treat with 0.5-1.0% DMS for 5 minutes at 37°C [59] [58].
- RNA Extraction: Lyse cells and extract total RNA using phenol-chloroform extraction with appropriate rRNA depletion [58].
- Library Preparation: Perform reverse transcription with thermostable group II intron reverse transcriptase, which can read through DMS modifications and incorporate mutations into cDNA [58].
- Sequencing and Analysis: Sequence libraries on an Illumina platform and analyze mutation patterns to determine nucleotide accessibility [58].
Key Considerations: The method relies on DMS modifying unpaired adenosine and cytosine residues, with modifications detected as mutations in cDNA [58]. For ensemble deconvolution, tools like DRACO can identify regions populating multiple conformations by analyzing comutation patterns in sequencing reads [58]. This approach has revealed that approximately 16.6% of expressed regions in E. coli populate two or more conformations, highlighting the prevalence of structural heterogeneity [58].
Functional Characterization with Massively Parallel Reporter Assays (MPRA)
Massively parallel reporter assays provide high-throughput functional validation of RNA switches by measuring their effects on gene expression in cellular contexts [59].
Protocol Steps:
- Library Construction: Clone candidate RNA switch sequences or cognate scrambled controls into a dual fluorescent reporter system (e.g., eGFP-mCherry) directly downstream of the eGFP open reading frame [59].
- Cell Transduction: Transduce HEK293 cells with the synthetic library using appropriate viral delivery systems [59].
- Flow Cytometry Sorting: Sort transduced cells into multiple bins based on their eGFP:mCherry expression ratio using fluorescence-activated cell sorting [59].
- Sequence Analysis: Sequence DNA and RNA from sorted populations to quantify the effect of each candidate switch on reporter gene expression [59].
Data Interpretation: In the SwitchSeeker study, 14% of candidate switches caused significant downregulation of eGFP, while another 14% showed significant upregulation [59]. This functional data, combined with structural information, enables identification of switches with strong conformation-dependent activity.
Energy Landscape Analysis with Computational Folding
Computational RNA folding provides essential thermodynamic parameters for predicting switch behavior and optimizing energy balance [40] [56].
Protocol Steps:
- Sequence Preparation: Input RNA sequence including FRE, CRE, and linker regions.
- Structure Prediction: Use tools like RNAfold (ViennaRNA package) to predict minimum free energy structures and base pairing probabilities [40].
- Partition Function Calculation: Generate Boltzmann-weighted ensembles of possible structures to estimate conformational distributions [56].
- Energy Decomposition: Analyze contributions of different structural elements to overall stability [40].
- Landscape Analysis: Identify local minima and energy barriers between alternative conformations [59].
Validation Considerations: As demonstrated in the Barcelona-UB project, discrepancies between tools like RNAfold and specialized servers like SECISearch3 can occur, particularly for non-canonical base pairs [40]. Experimental validation remains essential for confirming computational predictions.
RNA Switch Optimization Workflow
Research Reagent Solutions for RNA Switch Engineering
Table 3: Essential Research Reagents for RNA Switch Development
| Reagent/Category | Specific Examples | Function in RNA Switch Engineering | Implementation Notes |
|---|---|---|---|
| Computational Design Tools | RNAfold (ViennaRNA), gRNAde, RiboPO [40] [56] | Predicts secondary structure, folding energy, and 3D conformations | RiboPO incorporates multi-objective optimization for stability and accuracy [56] |
| Structure Profiling Reagents | DMS, DMS-MaPseq reagents [59] [58] | Maps RNA secondary structures and ensembles in living cells | Enables identification of structurally heterogeneous regions [58] |
| Reporter Systems | Dual fluorescent reporters (eGFP-mCherry) [59] | Quantifies switch activity and conformation-dependent function | mCherry serves as internal control for normalization [59] |
| Validated CRE Libraries | Theophylline aptamer, Tetracycline aptamer [40] | Provides well-characterized sensor domains for switch construction | Theophylline aptamer offers high specificity (10,000-fold over caffeine) [40] |
| FRE Elements | SECIS elements, frameshift stimulators [40] [55] | Serves as functional output domains regulated by CRE sensors | SECIS DIO2 shows high activity and human compatibility [40] |
| Cell Lines | HEK293, E. coli DH5α, BL21(DE3) [40] [59] [60] | Provides cellular context for functional testing | HEK293 suitable for eukaryotic compatibility testing [40] |
The optimization of energy balance and interaction landscapes represents a cornerstone of effective RNA switch engineering for synthetic developmental biology applications. As the field advances, several promising directions emerge for enhancing the design and implementation of these regulatory elements.
Current evidence suggests that future optimization strategies will increasingly leverage machine learning approaches that integrate multiple biophysical parameters simultaneously [56] [60]. Methods like RiboPO's preference optimization and Toehold-VISTA's target-aware design demonstrate the power of moving beyond single-objective optimization to balance structural accuracy, thermodynamic stability, and functional specificity [56] [60]. Furthermore, the growing recognition of RNA structural heterogeneity in cellular environments underscores the importance of ensemble-based design strategies that account for dynamic conformational landscapes rather than single static structures [58].
For researchers implementing synthetic signaling patterns in developmental contexts, we recommend adopting iterative design-build-test-learn cycles that combine computational predictions with experimental validation in relevant cellular environments [40] [59]. As the repertoire of validated RNA switches expands through systematic discovery efforts like SwitchSeeker [59], and design tools become more sophisticated through frameworks like RiboPO [56], the potential for programming complex developmental patterns using RNA-based regulators continues to grow. These advances will ultimately enhance our ability to engineer synthetic cellular behaviors with the precision required for both basic research and therapeutic applications.
This guide provides an objective comparison of key tunable parameters in synthetic biology—ribosome binding site (RBS) strengths, degradation rates, and feedback loops—for researchers validating signaling patterns in developmental contexts. Performance data and methodologies are synthesized from current literature to aid in the design of predictable genetic circuits.
RBS Strength: The Translator of Genetic Information
The ribosome binding site (RBS) is a nucleotide sequence in mRNA that recruits ribosomes to initiate translation. In prokaryotes, this is typically the Shine-Dalgarno sequence (5'-AGGAGG-3'), which base-pairs with the complementary anti-Shine-Dalgarno sequence on the 16S rRNA of the small ribosomal subunit [61]. Its strength is a primary lever for controlling protein expression levels.
Key Factors Determining RBS Strength and Translation Initiation Efficiency [61]:
- Complementarity to rRNA: The degree of complementarity between the mRNA's SD sequence and the ribosome's ASD sequence is crucial. Richer complementarity generally leads to higher initiation efficiency, though excessively tight binding can paradoxically decrease translation rates.
- Spacer Length: The distance between the RBS and the start codon (AUG) is critical, with an optimal spacing of approximately 6-7 nucleotides for proper ribosome positioning [62].
- Upstream Nucleotides: Adenine-rich sequences upstream of the RBS can enhance ribosome recruitment by binding the ribosomal protein S1.
- Secondary Structure: mRNA folding that sequesters the RBS or start codon in a secondary structure can inhibit ribosome access and dramatically reduce translation, a mechanism exploited naturally by heat shock proteins [61].
Performance Comparison of Tunable Parameters
The table below summarizes the core functions, tuning mechanisms, and performance impacts of RBS strength, degradation rates, and feedback loops on synthetic circuit output.
| Parameter | Core Function | Primary Tuning Methods | Impact on Circuit Dynamics & Performance |
|---|---|---|---|
| RBS Strength [63] [61] | Controls translation initiation rate and protein synthesis yield. | Engineering SD sequence complementarity, optimizing spacer length, modulating upstream adenine content, altering mRNA secondary structure. | Directly sets steady-state protein expression levels. High strength can induce metabolic burden, reducing host cell growth [64]. |
| Degradation Rate | Determines the lifetime and steady-state concentration of mRNAs and proteins. | Adding degradation tags (e.g., ssrA tag) to proteins; engineering mRNA stability with 5'/3' UTR modifications [62]. | Faster degradation shortens response time and lowers steady-state levels. Critical for determining the dynamics of oscillators and pulse generators. |
| Feedback Loops [64] | Enables autonomous regulation and robustness to perturbations. | Designing synthetic genetic circuits where an output protein regulates its own or another module's expression (e.g., positive auto-activation, negative feedback). | Positive Feedback: Creates bistability and memory. Negative Feedback: Promotes robustness, homeostatic control, and faster settling times [64]. |
Experimental Protocols for Parameter Characterization
Protocol 1: Quantifying RBS Strength and Translation Initiation
Objective: To experimentally measure the strength of a given RBS sequence in vivo. Key Reagents: Fluorescent reporter protein (e.g., GFP), flow cytometer or fluorescence plate reader. Methodology:
- Construct Design: Clone the RBS sequence to be tested upstream of a promoterless gene encoding a fluorescent reporter (e.g., GFP).
- Control: Use a construct with a well-characterized RBS (e.g., a strong consensus RBS) as a reference standard.
- Transformation & Culture: Introduce the constructs into the host organism (e.g., E. coli) and culture in a defined medium.
- Measurement: Measure fluorescence intensity and optical density (OD600) during mid-exponential growth phase.
- Data Analysis: Calculate the reporter protein synthesis rate by normalizing fluorescence to OD600. The strength of the test RBS is expressed relative to the reference standard [63].
Protocol 2: Measuring Protein Degradation Rates
Objective: To determine the half-life of a protein of interest. Key Reagents: Protein synthesis inhibitor (e.g., chloramphenicol, spectinomycin), antibodies for immunoblotting or materials for fluorescent protein analysis. Methodology:
- Cell Culture: Grow cells expressing the protein of interest to an appropriate density.
- Inhibition: Add a protein synthesis inhibitor to halt all new protein production.
- Time-course Sampling: Collect cell samples at regular intervals post-inhibition.
- Quantification: Quantify the remaining amount of the protein over time using immunoblotting, fluorescence measurements, or mass spectrometry.
- Calculation: Plot protein abundance versus time and fit the data to an exponential decay curve to determine the half-life.
Protocol 3: Implementing and Characterizing a Feedback Loop
Objective: To construct and analyze a synthetic negative feedback circuit that confers robustness. Key Reagents: Tunable promoters (e.g., inducible by small molecules), parts for expressing a transcriptional repressor. Methodology:
- Circuit Design: Design a circuit where the output protein is a repressor that binds its own promoter, downregulating its own expression.
- Assembly & Transformation: Assemble the genetic circuit and introduce it into the host cell.
- Perturbation Testing: Subject the circuit to perturbations, such as varying the gene copy number (e.g., using plasmids with different origins of replication) or inducing resource competition by co-expressing a burden-inducing protein [64].
- Output Measurement: Measure the circuit's output (e.g., fluorescence of a reporter gene) under each perturbed condition.
- Robustness Analysis: Compare the output variation of the feedback circuit against an otherwise identical open-loop (no feedback) control. A smaller coefficient of variation indicates successful robustness implementation [64].
Research Reagent Solutions Toolkit
| Reagent / Tool | Function in Experimental Workflow |
|---|---|
| Fluorescent Reporters (e.g., GFP, mCherry) | Serve as easily quantifiable proxies for gene expression output in live cells. |
| Tunable Promoters (e.g., Tet-On/Off, Arabinose-inducible) | Allow precise, external control of transcription initiation level and timing. |
| Protein Degradation Tags (e.g., ssrA) | Fused to a protein of interest to target it for degradation by native cellular proteases, enabling controlled reduction of half-life. |
| Coarse-Grained Cell Models [63] [64] | Mathematical frameworks that simulate host-circuit interactions, enabling in silico prediction of burden and parameter tuning before experimental implementation. |
| High-Throughput DNA Synthesis | Enables the rapid construction and testing of large libraries of genetic variants (e.g., RBS libraries) for comprehensive screening. |
Signaling Pathways and System Workflows
RBS-Dependent Translation Initiation
The following diagram illustrates the core mechanism of translation initiation in prokaryotes, highlighting the key interactions that determine RBS strength.
Resource-Aware Gene Circuit Dynamics
This diagram visualizes the interaction between a synthetic gene circuit and the host cell's shared resource pool, a key consideration for predicting circuit performance.
Negative Feedback for Robustness
This diagram outlines the operational principle of a synthetic negative feedback loop, a key design for maintaining stable output despite fluctuations.
Successful validation of synthetic signaling patterns in developmental contexts relies on a holistic tuning of RBS strength, degradation rates, and feedback loops. Quantitative models that account for host-circuit interactions are indispensable for predicting the system-wide effects of tuning these parameters [63] [64]. By integrating the compared performance data, detailed protocols, and engineered modules outlined in this guide, researchers can advance the robust and predictable programming of cellular behavior for therapeutic and basic science applications.
Overcoming Context-Dependent Effects and Ensuring Genetic Stability
A central challenge in developmental biology and therapeutic design is the context-dependent nature of cellular signaling. Gene regulation and the functional impact of signaling pathways are highly dependent on the specific cellular environment, including the tissue type, developmental stage, and the underlying genomic and epigenomic landscape [65]. This context-dependency poses a significant hurdle for reliably deploying synthetic biological systems. This guide compares three leading technological approaches—optogenetic signaling, modular epigenome editing, and engineered allosteric receptors—for validating synthetic signaling patterns against this challenge of context. The core thesis is that overcoming variable system outputs requires strategies that ensure both the precision of the initial signal and the stability of the resulting genetic program.
Comparative Analysis of Key Technologies
The following section provides an objective, data-driven comparison of the performance of three primary platforms used to establish and validate synthetic signaling patterns in developmental contexts.
Table 1: Performance Comparison of Key Technological Platforms
| Performance Metric | Optogenetic Signaling (OptoSOS) [66] | Modular Epigenome Editing (dCas9GCN4-CDscFV) [67] | Engineered Allosteric Receptors (e.g., chTME) |
|---|---|---|---|
| Primary Mechanism | Light-controlled recruitment of Ras/Erk pathway activator (SOS) downstream of native receptors. | dCas9-guided recruitment of isolated catalytic domains (CDs) to write specific chromatin marks. | Introduction of engineered, constitutively active receptors that initiate signaling. |
| Temporal Control | High (seconds to minutes); reversible upon light withdrawal. | Moderate (hours); dependent on doxycycline induction system. | Low; typically constitutive once expressed. |
| Spatial Precision | High; subcellular precision achievable with patterned light [66]. | High; defined by gRNA targeting, with editing spread of ~2-4 kb around target site [67]. | Low; system-wide receptor activity. |
| Rescue of Lethal Mutant | Successful rescue of trk mutant flies to viable, fertile adults [66]. | Not directly applicable; measures causal instruction of transcription, not phenotypic rescue. | Can bypass ligand requirements but often lacks native regulation. |
| Quantitative Output Control | Demonstrated distinct thresholds triggering specific developmental programs [66]. | Quantitative, single-cell readouts of transcriptional heterogeneity; identifies attenuative and switch-like effects from sequence context [67]. | Often binary (on/off); fine control is difficult. |
| Impact on Endogenous Network | Minimized by activating a native pathway at a specific, downstream node. | Minimal global transcriptome changes, confirming on-target activity (except p300-CD) [67]. | High potential for pleiotropic effects and signaling crosstalk. |
| Robustness to Context | High; simple all-or-none light input rescued normal development despite reduced gradient information [66]. | Systematically dissects context; DNA motifs significantly influence the transcriptional impact of installed chromatin marks [67]. | Low; output is highly susceptible to the cellular context of receptor expression. |
Table 2: Summary of Key Experimental Outcomes from Cited Studies
| Technology | Experimental System | Key Quantitative Result | Implication for Context/Stability |
|---|---|---|---|
| Optogenetic Signaling [66] | Drosophila embryo (OptoSOS-trk). | 90 minutes of posterior illumination rescued normal development in ~30% of embryos; gastrulation robust to a 3-fold variation in pattern width. | Developmental programs can be triggered by simple, non-graded inputs, revealing inherent robustness. |
| Modular Epigenome Editing [67] | Mouse Embryonic Stem Cells. | Installed H3K4me3 at levels equivalent to endogenous Pou5f1 promoter; H3K27me3 & H2AK119ub co-targeting maximized silencing penetrance across single cells. | Chromatin modifications can causally instruct transcription, but their quantitative effect is calibrated by underlying DNA sequence motifs. |
Detailed Experimental Protocols
Protocol: Optogenetic Rescue of a Developmental Patterning Mutant
This protocol is adapted from the rescue of terminal patterning in Drosophila embryos lacking the Trunk ligand [66].
- Objective: To replace a missing receptor tyrosine kinase (RTK) signaling pattern using optogenetic stimulation and assess rescue of development.
- Key Genetic Tools: OptoSOS transgene (light-inducible system for Ras/Erk activation) expressed on a trk (or tor, tsl) loss-of-function mutant background.
- Procedure:
- Embryo Collection & Preparation: Collect embryos from homozygous trk mutant mothers expressing OptoSOS. Dechorionate and mount embryos for live imaging under halocarbon oil.
- Optogenetic Stimulation: Place mounted embryos under a blue light microscope system capable of patterned illumination.
- Stimulation Parameters: Illuminate the anterior and/or posterior poles (covering ~15% of embryo length) with 470 nm blue light. Use a pulse of 1 second every 30 seconds for a total duration of 90 minutes, starting at nuclear cycle 14.
- Validation of Signaling: Fix a subset of embryos immediately after stimulation and immunostain for doubly phosphorylated Erk (dpErk) to confirm local Erk activation. Alternatively, use live biosensors for Erk activity or target gene expression (e.g., tailless).
- Phenotypic Scoring: For rescued embryos, monitor gastrulation movements via DIC microscopy. Allow developed embryos to hatch. Rear hatched larvae to adulthood and assess fertility.
- Critical Considerations: The exact duration and intensity of illumination may require optimization for different genetic backgrounds or target pathways. The use of a "blank canvas" embryo (complete receptor-level mutant) is crucial to ensure all observed signaling is optogenetically driven.
Protocol: Systematic Epigenome Editing to Probe Context-Dependency
This protocol is adapted from the systematic dissection of chromatin modification function using a modular editing toolkit [67].
- Objective: To program a specific chromatin modification at an endogenous locus and quantitate its causal, context-dependent impact on transcription.
- Key Reagents: dCas9GCN4; library of scFV-tagged catalytic domain (CD) effectors (e.g., for H3K4me3, H3K27me3, H3K27ac) and their catalytic-dead mutants; gRNA expression construct; DOX-inducible system.
- Procedure:
- Cell Line Engineering: Stably integrate the dCas9GCN4 and the inducible CDscFV effector of interest into the target cell line (e.g., mouse ESCs) using the piggyBac transposon system.
- Locus Targeting: Transfect cells with a gRNA plasmid targeting the genomic locus of interest. A non-targeting gRNA should be used as a control.
- Induction of Editing: Add doxycycline (DOX) to the culture medium to induce expression and recruitment of the CDscFV effector. For p300-CD, use a 20-fold lower DOX concentration to minimize indirect effects.
- Validation of Epigenome Editing: 48-72 hours post-induction, perform CUT&RUN-qPCR or ChIP-qPCR across the target locus to confirm deposition of the intended chromatin mark and define the editing domain.
- Transcriptional Output Analysis: Measure changes in target gene expression using single-cell RNA sequencing (scRNA-seq) to capture heterogeneity, or RT-qPCR for population-level analysis.
- Critical Considerations: Always include the catalytic-mutant effector control to distinguish the effect of the chromatin mark from the mere recruitment of the effector protein. The influence of DNA sequence context can be probed by targeting the same chromatin modification to different genomic loci (e.g., promoter vs. enhancer) with varying underlying transcription factor binding motifs.
Visualization of Signaling Pathways and Workflows
Synthetic Patterning Workflow
Context-Dependent Signal Interpretation
Epigenome Editing and Hierarchical Remodeling
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Synthetic Patterning
| Reagent / Solution | Function | Example Use-Case |
|---|---|---|
| OptoSOS System | A light-inducible protein dimerization system that recruits the SOS RasGEF to the membrane, activating the native Ras/Erk pathway downstream of receptor-level context [66]. | Replacing terminal RTK signaling in Drosophila; probing information content of signaling dynamics. |
| Modular dCas9GCN4-CDscFV Toolkit | A suite of epigenome editors that program nine key chromatin modifications (e.g., H3K4me3, H3K27me3) to specific loci using isolated catalytic domains to isolate the mark's function [67]. | Causally testing the role of specific chromatin marks in instructing transcription; dissecting combinatorial mark functions. |
| Live-Cell Biosensors (e.g., ErkAR, NLS-FRET) | Genetically encoded reporters that allow real-time, quantitative monitoring of signaling activity (e.g., Erk kinase activity) or immediate-early gene expression in living cells and embryos. | Validating the spatiotemporal dynamics of optogenetically induced signaling patterns [66]. |
| Catalytic-Dead Mutant Effectors (mut-CDscFV) | Critical control reagents that contain point mutations ablating enzymatic activity. They distinguish effects caused by the chromatin mark from those caused by the physical recruitment of the effector protein [67]. | Controlling for steric hindrance or non-catalytic interactions in epigenome editing experiments. |
| PiggyBac Transposon System | A non-viral vector system for efficient and stable genomic integration of large genetic constructs, such as inducible dCas9 and effector arrays [67]. | Creating stable cell lines for reproducible epigenome editing studies. |
Establishing Predictive Power: Validation Paradigms and Comparative Analysis
Validating computational models of biological processes requires robust comparison against a reliable ground truth. In developmental biology, where experimental perturbation is often complex and expensive, synthetic data provides a powerful alternative for validation. By generating in silico benchmarks with known properties, researchers can precisely evaluate analytical methods and computational models designed to decipher complex signaling patterns. This approach is particularly valuable for studying core intercellular signaling pathways—such as Notch, Wnt, and TGF-β—that control patterning in multicellular organisms despite their limited number and promiscuous ligand-receptor interactions [68].
Synthetic data generation methods create artificial datasets that mimic the statistical properties of real-world data while containing no actual sensitive information [69]. When applied to developmental signaling, these methods can produce simulated networks with predefined interaction patterns, allowing researchers to test whether their analytical pipelines can accurately recover known relationships. This validation framework is especially important for addressing the fundamental puzzle in developmental biology: how so few core signaling pathways can generate the precision and specificity required for multicellular development [68].
Performance Benchmarking: Synthetic Data Generation Paradigms
Multiple computational approaches exist for generating synthetic data, each with distinct strengths and limitations for validating signaling network models. The table below compares three primary paradigms used in biological research:
Table 1: Comparison of Synthetic Data Generation Methods for Biological Applications
| Method | Core Mechanism | Best-Suited Data Types | Privacy Considerations | Representative Tools |
|---|---|---|---|---|
| Marginal-Based Methods | Data swapping & statistical manipulation [70] | Structured tabular data [70] | Preserves marginal distributions but may leak correlations [70] | DataSynthesizer [69] |
| Deep Learning Models | Neural networks approximating data distribution [71] | Complex, high-dimensional data [72] | Privacy guarantees require formal methods like DP [70] | CTGAN [71], GANs [72] |
| Agent-Based Modeling | Rule-based simulation of interacting entities [73] | Emergent patterns in multicellular systems [73] | No original data used; privacy inherent [69] | Custom implementations [73] |
These generation methods can be categorized by their relationship to original data. Fully synthetic data is created from scratch without any direct use of original datasets, making it ideal for simulating developmental patterns from first principles [69]. Partially synthetic data replaces only sensitive values in an original dataset, useful when maintaining certain experimental measurements is essential [69]. Hybrid synthetic data combines elements of both approaches, integrating actual data points with synthetic counterparts [69].
For developmental signaling applications, evaluation metrics must assess how well synthetic data preserves critical biological features. Key benchmarks include: (1) Fidelity - how faithfully the synthetic data reproduces statistical properties of real signaling data; (2) Privacy - the level of protection against sensitive information leakage; (3) Utility - how well the data supports downstream analytical tasks; and (4) Expressivity - the ability to capture complex, high-dimensional relationships [70].
Experimental Protocols for Validation
Synthetic Signaling Pattern Generation
To benchmark analytical methods for developmental signaling, researchers must first generate synthetic networks with controlled properties. The following protocol outlines the generation of synthetic data mimicking Turing-type patterning systems, which are fundamental to developmental biology:
Define Base Parameters: Establish reaction-diffusion equations with an activator-inhibitor pair, where the activator promotes its own production and that of the inhibitor, while the inhibitor suppresses the activator [73].
Set Differential Diffusion: Ensure the inhibitor diffuses faster than the activator, a prerequisite for Turing pattern formation [73].
Introduce Stochasticity: Incorporate controlled noise in initial conditions to simulate biological variability while maintaining reproducible pattern classes (spots, stripes).
Implement Multiple Signaling Pathways: Extend the model to include parallel signaling systems (e.g., simulating both Notch and Wnt pathways) with cross-regulation [73].
Generate Ground Truth Labels: Create precise mapping between parameter combinations and resulting patterns to serve as validation benchmarks.
This approach enables the creation of synthetic developmental patterns where the underlying "communication codes"—the relationships between ligand identities, concentrations, combinations, and dynamics—are precisely known [68].
Validation Against Experimental Data
Once synthetic benchmarks are established, validation against experimental data requires careful experimental design:
Single-Cell Resolution Measurement: Apply single-cell analysis tools such as quantitative time-lapse microscopy to capture signaling dynamics in real-time [68]. For Notch signaling, this has revealed how different ligands (Dll1 vs. Dll4) activate the same receptor with pulsatile or sustained dynamics respectively [68].
Spatial Pattern Quantification: Use image analysis techniques to extract quantitative descriptors of pattern formation—including wavelength, regularity, and boundary sharpness—from both synthetic and experimental data.
Algorithm Testing: Apply the same analytical methods to both synthetic benchmarks and experimental data to determine whether relationships discovered in synthetic networks generalize to biological reality.
Cross-Validation: Implement statistical tests such as Kolmogorov-Smirnov tests to compare distributions between synthetic and experimental datasets [69].
Table 2: Validation Metrics for Synthetic Signaling Networks
| Validation Dimension | Quantitative Metrics | Acceptance Criteria |
|---|---|---|
| Pattern Fidelity | Spatial autocorrelation, Power spectrum similarity | Statistical equivalence (p > 0.05) in distribution tests |
| Dynamic Accuracy | Temporal cross-correlation, Oscillation period matching | <10% deviation from experimental time-series data |
| Network Topology | Graph edit distance, Modularity preservation | >0.85 similarity coefficient with reference networks |
| Information Content | Mutual information, Channel capacity | Preserved rank order of signaling efficiency |
Visualizing Synthetic Network Validation
The following diagrams illustrate key concepts and workflows in synthetic network validation, created using Graphviz with adherence to the specified color palette and contrast requirements.
Diagram 1: Synthetic Network Validation Workflow
Diagram 2: Notch Signaling Communication Code
Research Reagent Solutions
The following tools and resources enable the generation and validation of synthetic signaling networks:
Table 3: Essential Research Reagents and Computational Tools
| Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| Synthea [69] | Software | Synthetic patient generation | Simulating population-level variability in signaling responses |
| CTGAN [71] | Deep Learning Model | Tabular synthetic data generation | Creating synthetic datasets that preserve complex correlations |
| SDV [69] | Python Library | Multiple-type synthetic data generation | Generating mixed data types common in signaling studies |
| Mostly.AI [69] | Platform | Accurate synthetic data generation | Producing high-fidelity synthetic data with granular insights |
| Single-Cell RNA-seq [68] | Experimental Method | Gene expression profiling | Validating synthetic signaling predictions at single-cell resolution |
| Microfluidics [68] | Experimental Platform | Controlled microenvironment | Testing synthetic network predictions under defined conditions |
Synthetic networks provide a rigorous foundation for validating computational methods in developmental biology. By benchmarking analytical approaches against in silico systems with known properties, researchers can establish the reliability of methods before applying them to complex experimental data. The integration of synthetic data generation with single-cell analysis technologies creates a powerful framework for deciphering the communication codes that underlie developmental patterning. As synthetic data methodologies continue advancing—with improvements in fidelity, privacy preservation, and expressivity—their role in validating our understanding of signaling networks will become increasingly indispensable.
The field of synthetic developmental biology aims to reconstruct minimal developmental programs to understand fundamental processes like spatial polarization, morphogen interpretation, and cellular memory [5]. As researchers engineer increasingly complex synthetic signaling pathways to control cell fate and tissue patterning, robust quantitative validation across multiple biological scales becomes paramount. This guide provides a systematic framework for comparing quantitative metrics from initial in vitro binding characterization through definitive in vivo functional assessment, offering researchers a standardized approach for evaluating synthetic biological tools. The validation journey progresses from molecular-scale interactions measured in purified systems to cellular-scale responses in controlled environments, culminating in organism-scale functional readouts in complex living systems.
Quantitative Metrics Across Biological Scales
The table below summarizes the core quantitative metrics applicable at different stages of the validation pipeline, providing researchers with key parameters for objective comparison of synthetic signaling systems.
Table 1: Hierarchy of Quantitative Metrics Across Validation Stages
| Biological Scale | Primary Metrics | Secondary Metrics | Key Assay Types |
|---|---|---|---|
| In Vitro Molecular | Dissociation Constant (Kd), Inhibition Constant (Ki), Association/dissociation rates (kon, koff) | Receptor density (Bmax), IC50 | Saturation binding, competition binding, kinetic binding [74] [75] |
| In Vitro Cellular | Internalization rate, cytotoxicity (IC50), differentiation efficiency | Cell viability, metabolic activity, morphological changes | Internalization assays, cytotoxicity assays (MTT/MTS), micropattern-based differentiation [75] [76] |
| In Vivo Functional | Phenotypic rescue efficiency, pattern precision, signaling dynamics | Tissue morphology, developmental timing, viability | Optogenetic perturbation, phenotypic scoring, quantitative imaging [5] [77] |
In Vitro Binding Assays: Molecular-Scale Validation
Experimental Protocols for Binding Characterization
Saturation Binding Assay Protocol: This assay determines the fundamental affinity between a ligand and its receptor under equilibrium conditions [74].
- Receptor Preparation: Prepare membrane fractions containing the receptor of interest, dividing into aliquots
- Incubation Setup: Incubate receptor aliquots with increasing concentrations of radiolabeled ligand for defined time at controlled temperature
- Separation: Separate bound from free ligand using rapid membrane filtration
- Detection: Measure bound radioligand concentration
- Analysis: Fit data to Langmuir isotherm equation:
[RL] = ([RT] × [L]) / (K_d + [L])where[RL]is receptor-ligand complex concentration,[RT]is total receptor concentration,[L]is free ligand concentration, andK_dis equilibrium dissociation constant [74]
Competition Binding Assay Protocol: This assay characterizes unlabeled compounds competing with labeled ligands for receptor binding [75].
- Setup: Incubate receptor preparation with fixed concentration of radiolabeled ligand and increasing concentrations of unlabeled competitor compound
- Equilibrium: Maintain until binding equilibrium is reached
- Processing: Separate bound from free ligand and measure bound radioligand
- Calculation: Determine IC50 (concentration inhibiting 50% of specific binding) and calculate Ki using Cheng-Prusoff equation:
K_i = IC_50 / (1 + [L]/K_dL)where[L]is radioligand concentration andK_dLis its dissociation constant [74]
Critical Experimental Design Considerations
Several factors significantly impact binding assay data quality and interpretation:
- Ligand Depletion: Occurs when significant fraction (>10%) of ligand binds to receptors, distorting Kd calculations; use lower receptor concentrations to minimize [74]
- Equilibrium Attainment: Insufficient incubation time prevents steady-state binding; perform time-course experiments to verify equilibrium [74]
- Buffer Composition and Temperature: Profoundly affect affinity measurements; maintain consistent conditions and report precisely [74]
Quantitative Cellular Assays: Micropatterning and Functional Readouts
Micropatterning Approaches for Standardized Assays
Micropatterning techniques enable quantitative analysis of cell behavior by controlling extracellular microenvironment, reducing variability and facilitating high-content imaging [76]. These approaches provide standardized geometric constraints that allow aggregation of data from hundreds of cells or colonies into consolidated representations.
Table 2: Micropatterning Techniques and Applications
| Technique | Mechanism | Resolution | Best Applications |
|---|---|---|---|
| Soft Lithography | PDMS molding from photoresist master | ~1 µm | Protein patterning, controlled cell adhesion [76] |
| Direct Photopatterning | UV degradation of repellent coating | ~10 µm | High-throughput screening, dynamic patterns [76] |
| LIMAP | Light-induced adsorption with photoinitiators | Single cell | Live cell patterning, multi-protein patterns [76] |
Experimental Protocol: Micropatterned Differentiation Assay
This protocol enables quantitative assessment of synthetic patterning system functionality in stem cell models [76]:
- Surface Preparation: Create micropatterned substrates using photolithography or direct photopatterning with defined geometric arrangements
- ECM Coating: Adsorb extracellular matrix proteins (e.g., fibronectin, laminin) onto adhesive regions
- Cell Seeding: Plate pluripotent stem cells at controlled density to achieve single cells per pattern
- Differentiation: Apply synthetic morphogen signals using optogenetic tools or recombinant proteins with precise timing
- Fixation and Staining: Process samples for immunocytochemistry against lineage-specific markers
- Quantitative Imaging: Acquire high-content images and perform computational analysis to quantify pattern fidelity and differentiation efficiency
Figure 1: Cellular Assay Workflow - Standardized process for quantitative assessment of synthetic patterning systems in stem cell models
In Vivo Functional Validation: From Organoids to Organisms
Experimental Framework for In Vivo Validation
The validation framework for in vivo digital measures encompasses three critical stages adapted from clinical validation paradigms [78]:
- Verification: Ensures digital technologies accurately capture and store raw data from complex biological systems
- Analytical Validation: Assesses precision and accuracy of algorithms transforming raw data into meaningful biological metrics
- Clinical Validation: Confirms digital measures accurately reflect biological states relevant to context of use
Quantitative Comparison Methods for In Vivo Data
DIFFENERGY Analysis Protocol: This frequency-domain method quantitatively compares reconstruction algorithms before image domain distortions [79].
- Data Preparation: Start with full data set, truncate to simulate limited acquisition
- Algorithm Application: Apply reconstruction algorithms to truncated data
- Comparison: Transfer reconstructed data back to frequency domain, compare with original full data using normalized DIFFENERGY metric:
GDF = Σ|DIFF_model|² / Σ|DIFF_trunc|²whereDIFF_modelandDIFF_truncrepresent differences between modeled/truncated data and standard data [79]
Optogenetic Functional Assay Protocol: This approach uses light-controlled tools to precisely perturb developmental processes and assess functional outcomes [5].
- System Selection: Choose model system amenable to imaging and optogenetics (zebrafish, Drosophila, mouse embryos)
- Stimulation: Apply spatially-patterned light to activate synthetic signaling pathways with precise timing and intensity
- Phenotypic Monitoring: Track developmental outcomes using live imaging or endpoint analysis
- Quantification: Measure phenotypic rescue efficiency, pattern precision, and morphological parameters
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Quantitative Validation
| Reagent Category | Specific Examples | Function in Validation Pipeline |
|---|---|---|
| Radiolabeled Ligands | ³H-, ¹²⁵I-labeled compounds | Enable precise quantification of binding parameters in saturation and competition assays [74] [75] |
| Optogenetic Tools | LOV domains (VfAU1, VVD), Cry2 PHR, LightOn/GAVPO, EL222 | Provide light-controlled activation of developmental signaling pathways (BMP, FGF, Wnt, Nodal) [5] |
| Micropatterning Reagents | PEG-based polymers, PDMS, photoreactive proteins | Create defined microenvironments for standardized cellular assays [76] |
| Cell Viability Indicators | MTT, MTS | Assess cytotoxic effects of synthetic biological constructs [75] |
| Lineage Tracing Systems | CRE-lox, DNA barcoding | Track cell fate decisions in response to synthetic patterning signals [5] |
Integrated Data Visualization and Interpretation
Color Scale Selection for Quantitative Data
Effective visualization of quantitative metrics requires appropriate color scale selection [80]:
- Sequential Color Scales: Single-hue or multi-hue gradients from light to dark ideal for low-to-high value ranges (e.g., binding affinity, expression levels)
- Diverging Color Scales: Two-hue gradients with neutral central value perfect for positive/negative comparisons or deviation from control
- Categorical Color Scales: Distinct hues for non-ordered categories (e.g., cell types, experimental conditions)
Integrated Validation Workflow
A comprehensive validation strategy connects molecular, cellular, and organismal data through standardized quantitative metrics and visualization approaches.
Figure 2: Integrated Validation Pipeline - Comprehensive strategy connecting molecular, cellular, and organismal data through standardized metrics
This comparison guide establishes a standardized framework for quantitative validation across biological scales, enabling direct comparison of synthetic signaling systems from molecular binding to organismal function. By implementing these standardized metrics, experimental protocols, and visualization approaches, researchers can objectively evaluate synthetic biology tools and accelerate progress in programming developmental outcomes. The integration of quantitative in vitro binding data with functional cellular readouts and definitive in vivo validation creates a robust evidence chain for synthetic pattern formation, ultimately enhancing reproducibility and translational potential in developmental contexts research.
The engineering of biological systems to perform customized functions represents a frontier in synthetic biology, with particular promise for therapeutic development and fundamental research. Central to this endeavor are synthetic signaling modules—engineered receptors and pathways designed to sense cues and initiate programmed responses in cells. These synthetic systems aim to augment, rewire, or operate orthogonally to natural signaling pathways. As these technologies mature, a critical comparative analysis of their performance against natural modules is essential to guide their application and future development. This review objectively evaluates the performance characteristics of synthetic and natural signaling modules, framing the discussion within the broader thesis of validating synthetic signaling patterns for developmental biology research. We summarize quantitative performance data, detail key experimental methodologies, and provide resources to equip researchers and drug development professionals with the tools for critical evaluation.
Performance Metrics and Comparative Analysis
The performance of signaling modules is quantified through several key metrics, including dynamic range, EC50/sensitivity, orthogonality, and response time. The table below provides a comparative summary of representative synthetic and natural signaling systems based on these parameters.
Table 1: Performance Comparison of Natural and Synthetic Signaling Modules
| System Name / Type | Key Input Signal | Key Output | Dynamic Range (ON/OFF Ratio) | EC50 / Sensitivity | Orthogonality | Key Performance Characteristics & Limitations |
|---|---|---|---|---|---|---|
| Natural GPCR Pathways [81] | Diverse ligands (e.g., hormones) | Intracellular signaling | N/A | Variable, highly optimized | Low (crosstalk common) | Fast response times; high sensitivity; but inherent crosstalk and pleiotropy. |
| CAR-T Receptors [81] [82] | Surface antigen (e.g., CD19) | T-cell Activation | N/A | High (single-digit nM) | Moderate | Clinically validated efficacy; potential for on-target/off-tumor toxicity. |
| synNotch Receptors [81] | Surface-bound ligand | User-defined transcription | Up to ~100-400 fold [81] | N/A | High | Highly orthogonal; enables complex cell-cell communication; requires mechanical pulling force for activation [81]. |
| MESA Receptors [82] | Soluble ligand | User-defined transcription | Modest induction, ligand-independent leakiness [82] | N/A | High | Modular for soluble ligands; early versions exhibited significant baseline activity [82]. |
| dCas9-synR Receptors [82] | Soluble proteins, lipids | User-defined transcription | Improved OFF/ON vs. MESA; stringent OFF state [82] | Agonist dose-dependent | High | Highly programmable via sgRNAs; can integrate AND-gate logic; architecture portable across receptor classes [82]. |
| Chemical EAR Receptors [83] | Enzymes (e.g., phosphatase) | Release of L-Cysteine | N/A | N/A | High (fully synthetic) | Functions in artificial cells; enables transmembrane enzyme activation without protein translocation [83]. |
A critical consideration when evaluating performance is system load—the impact of connecting a module to downstream components. Theoretical and computational studies demonstrate that adding a downstream load can fundamentally alter the behavior of upstream switches. For example, loading a genetic toggle switch can skew its bistable potential landscape and, in some cases, abolish bistability entirely [84]. This effect is also observed in signaling switches; the Ras activation switch can lose its switch-like properties when connected to its natural downstream load, Raf kinase [84]. These findings underscore that modularity in biological systems is limited and that the performance of a module cannot be assessed in isolation.
Table 2: Summary of Load Effects on Different Switch Types
| Switch Type | Effect of Load | Potential Mitigation Strategy |
|---|---|---|
| Simple Genetic Toggle Switch [84] | Biases the switch to the unloaded state; can destroy bistability. | Incorporation of positive feedback motifs [84]. |
| Ras Signaling Switch [84] | Can abrogate switch-like, bistable behavior. | Requires careful balancing of system parameters. |
Experimental Protocols for Key Comparative Studies
Robust comparative analysis relies on standardized experimental workflows. Below are detailed protocols for key assays used to generate the performance data discussed.
Protocol: Quantifying Synthetic Receptor Activation Dynamics
This protocol is used to characterize the dose-response and dynamic range of transcription-activating synthetic receptors like synNotch, MESA, and dCas9-synR [81] [82].
- Cell Engineering and Seeding: Engineer a mammalian cell line (e.g., HEK293T or primary T-cells) to stably express the synthetic receptor of interest. Seed cells into multi-well plates.
- Ligand Titration: Expose cells to a titration series of the specific ligand (soluble or surface-bound, as required by the receptor). Include a negative control (no ligand).
- Output Measurement: After a defined incubation period (e.g., 24-48 hours), harvest cells and quantify the transcriptional output. This is typically done using:
- Flow Cytometry: If the output is a fluorescent reporter (e.g., EYFP).
- Luciferase Assay: If the output is luciferase, measure luminescence.
- qPCR: To directly measure mRNA levels of the output gene.
- Data Analysis: Calculate the fold induction (ON/OFF ratio) by normalizing the output signal in ligand-treated wells to the negative control. Plot the dose-response curve and calculate the EC50 value.
Protocol: Evaluating Orthogonality and Crosstalk
This methodology tests the specificity of a synthetic receptor within a complex cellular background [49].
- Circuit Co-transfection: Co-transfect cells with multiple, distinct synthetic receptor systems and their cognate output reporters (e.g., different fluorescent proteins).
- Selective Stimulation: Stimulate the cells with a specific ligand for only one of the receptor systems.
- Multiparameter Output Measurement: Analyze cells using high-throughput flow cytometry to measure all output reporters simultaneously.
- Crosstalk Quantification: Calculate the degree of activation in non-cognate output channels. High orthogonality is indicated by strong activation of the target output and minimal activation in non-targeted channels.
Protocol: Assessing Performance in Artificial Cells
This protocol validates the function of fully synthetic, chemical receptors in a bottom-up synthetic cell context [83].
- Receptor Incorporation: Incorporate the synthetic Enzyme-Activating Receptor (EAR), such as Phos-EAR, into the membrane of liposomes (artificial cells) during their preparation via lipid film hydration and extrusion.
- Enzyme Encapsulation: Co-encapsulate the target enzyme (e.g., a cysteine protease papain, rendered inactive as a zymogen via a disulfide bond) within the liposomes.
- Receptor Activation: Expose the prepared artificial cells to the external stimulus (e.g., alkaline phosphatase for Phos-EAR).
- Output Measurement: Monitor the activation of the encapsulated enzyme by measuring the cleavage of a fluorogenic substrate. Transmembrane signaling is confirmed by enzyme activation inside the liposome upon an external, non-diffusible trigger [83].
Signaling Pathway Architectures and Experimental Workflows
The diagrams below illustrate the core architectures of the key signaling modules discussed and the workflow for their quantitative evaluation.
Core Architectures of Synthetic Receptors
Diagram 1: A comparison of architectural strategies for synthetic receptors, ranging from partial to full engineering of sensing and actuation domains [81].
Quantitative Workflow for Receptor Performance
Diagram 2: A generalized experimental workflow for quantifying synthetic receptor performance, from cell preparation to key metric calculation [81] [82].
The Scientist's Toolkit: Key Research Reagents
The following table details essential reagents and tools for engineering and evaluating synthetic signaling systems.
Table 3: Essential Research Reagents for Synthetic Signaling
| Reagent / Tool Name | Function in Research | Specific Example & Utility |
|---|---|---|
| scFv / Nanobodies | Engineered extracellular sensing domain | Used in CARs and synNotch to bind specific cell surface antigens with high specificity [81]. |
| Protease Systems (e.g., TEV) | Inducible cleavage and release of transcription factors | Core component in MESA, dCas9-synR, and Tango systems for converting receptor activation into a transcriptional signal [81] [82]. |
| Orthogonal TFs | Customizable transcriptional actuation | Tethered to synthetic receptors (e.g., synNotch) or released by them (e.g., MESA) to drive user-defined gene expression [81]. |
| dCas9-Activators | Programmable transcriptional effector | Used in dCas9-synR; allows a single receptor architecture to target different genes by simply changing the sgRNA [82]. |
| Self-Immolative Linkers (SILs) | Chemical mechanism for signal transduction | Core component in chemical EARs; cleaved in response to a stimulus to release an active messenger molecule inside artificial cells [83]. |
| Synthetic Gene Circuits | Downstream signal processing | Enable complex computation (e.g., AND gates) on the output of synthetic receptors, increasing the sophistication of cellular programs [81] [82]. |
Synthetic signaling modules have demonstrated remarkable capabilities, achieving high orthogonality and programmability that often surpass natural systems. Technologies like synNotch and dCas9-synR show robust, tunable performance suitable for sophisticated applications in therapeutic cell engineering. However, natural signaling pathways retain advantages in speed and sensitivity honed by evolution. The performance of any module, synthetic or natural, is profoundly influenced by its system context, particularly downstream load. The future of validating synthetic signaling patterns in developmental contexts will therefore hinge on moving from isolated characterizations to integrated analyses. Embracing computational design [16], advanced signal processing frameworks [49], and a deeper understanding of modular interoperability [3] will be critical for building reliable, complex synthetic signaling systems for research and medicine.
In the landscape of modern drug discovery, the chasm between computational predictions and experimental outcomes remains a significant bottleneck, contributing to clinical failure and escalating development costs. The pressure to reduce attrition, shorten timelines, and increase translational predictivity is driving the adoption of integrated workflows that combine in silico foresight with robust experimental validation [85]. This is particularly critical in developmental contexts research, where understanding synthetic signaling patterns determines therapeutic efficacy and safety. The organizations leading the field are those that can effectively correlate computational modeling with empirical evidence, creating a virtuous cycle of prediction and validation [86].
The transformative potential of this integration is exemplified by recent advances where machine learning models now routinely inform target prediction, compound prioritization, pharmacokinetic property estimation, and virtual screening strategies [85]. However, these computational approaches face limitations, including model inaccuracies and incomplete data, making experimental validation through methodologies like CETSA (Cellular Thermal Shift Assay) essential for confirming direct target engagement in intact cells and tissues [85]. This guide provides a comprehensive comparison of technologies and methodologies enabling this crucial correlation, with specific application to validating synthetic signaling patterns in developmental contexts.
Computational Approaches: From Prediction to Insight
Computational biology has evolved from a supportive role to a foundational capability in modern R&D, employing advanced algorithms, machine learning, and molecular modeling techniques to predict drug-target interactions [86]. The year 2025 has seen the rise of foundation models trained on massive genomic, transcriptomic, and proteomic datasets, which promise to uncover fundamental biological rules similar to how large language models learn linguistic patterns from text [87]. These models are increasingly capable of detecting previously unknown genetic patterns, elucidating mechanisms of action behind genes and pathways, and predicting therapeutic targets or biomarkers with increasing accuracy.
Key Computational Technologies and Their Applications
Table 1: Comparative Analysis of Computational Prediction Methods
| Method | Primary Function | Typical Accuracy Range | Strengths | Limitations |
|---|---|---|---|---|
| Molecular Docking | Predicts binding orientation & affinity of small molecules to target proteins | 70-85% pose prediction accuracy [85] | Fast screening of large compound libraries; provides structural insights | Limited by force field accuracy; may miss allosteric sites |
| AI-Guided Virtual Screening | Machine learning models prioritize compounds with desired properties | 50-fold hit enrichment over traditional methods [85] | Exceptional speed; identifies non-obvious chemical structures | Dependent on training data quality and diversity |
| QSAR Modeling | Relates chemical structure to biological activity | Varies by endpoint & dataset size [86] | Interpretable features; useful for lead optimization | Limited to chemical space of training data |
| ADMET Prediction | Forecasts absorption, distribution, metabolism, excretion, toxicity | 75-90% for key parameters [86] | Early elimination of problematic compounds; reduces late-stage attrition | Struggles with novel mechanisms and rare toxicities |
| Foundation Models | Multi-scale biological representation from proteins to tissues [87] | Emerging technology; validation ongoing | Discovers previously unknown patterns; predicts system-level behavior | Requires massive datasets; biological interpretation challenging |
Beyond these established methods, the field is witnessing the emergence of "AI agents" that automate lower-complexity bioinformatics tasks. These agents combine LLM-style reasoning with specialized data-analysis workflows, enabling them to analyze datasets, select appropriate normalization techniques, and present findings with interpretable summaries [87]. This democratization of data analysis allows researchers with limited coding expertise to generate hypotheses and gain insights directly from their data, accelerating the discovery process.
Experimental Validation Methodologies
While computational methods provide powerful predictive capabilities, experimental validation remains the gold standard for confirming biological activity and safety. Experimental studies deliver crucial insights that computational approaches cannot fully capture, including confirmation of target engagement in physiologically relevant environments, assessment of functional consequences in cellular systems, evaluation of efficacy in disease models, and identification of off-target effects and toxicity [86].
Key Experimental Validation Techniques
Table 2: Experimental Validation Methods for Signaling Pathway Research
| Method | Key Application | Throughput | Key Measured Parameters | Compatibility with Computational Data |
|---|---|---|---|---|
| CETSA (Cellular Thermal Shift Assay) | Target engagement in intact cells & tissues [85] | Medium to High | Thermal stability shift; dose-dependent stabilization [85] | Direct correlation with molecular docking predictions |
| High-Content Screening | Multiparametric analysis of signaling pathway activation | High | Morphological changes; protein translocation; cell viability | Compatible with machine learning image analysis |
| Proteomics (e.g., HR-MS) | System-wide protein expression & modification | Medium | Protein abundance; post-translational modifications | Integrates with multi-omics computational models |
| Gene Fusion Detection | Identification of novel therapeutic targets in cancer | Low to Medium | Fusion transcripts; oncogenic drivers [88] | Validates computational fusion prediction algorithms |
| Circulating miRNA Analysis | Biomarker discovery for treatment response | Medium | miRNA expression profiles; correlation with clinical parameters [88] | Correlates with computational biomarker predictions |
Recent work by Mazur et al. (2024) exemplifies the power of advanced validation techniques, applying CETSA in combination with high-resolution mass spectrometry to quantify drug-target engagement of DPP9 in rat tissue, confirming dose- and temperature-dependent stabilization ex vivo and in vivo [85]. These data demonstrate the unique ability of modern validation approaches to offer quantitative, system-level validation—closing the critical gap between biochemical potency and cellular efficacy.
Integrated Workflows: Case Studies in Correlation
Case Study 1: AI-Guided MAGL Inhibitor Development
A landmark 2025 study demonstrates the power of integrated computational-experimental approaches. Researchers utilized deep graph networks to generate over 26,000 virtual analogs, resulting in sub-nanomolar MAGL inhibitors with more than 4,500-fold potency improvement over initial hits [85]. This achievement exemplifies the compressed timeline possible through integrated workflows:
This workflow demonstrates the iterative DMTA (Design-Make-Test-Analyze) cycle that has reduced discovery timelines from months to weeks. The critical correlation point occurs where computational predictions from docking meet experimental validation through CETSA, creating a feedback loop that informs subsequent optimization rounds.
Case Study 2: Biomarker Discovery in Colorectal Cancer
In colorectal cancer research, the integration of computational and experimental methods has led to significant advances in biomarker discovery. Sorokin et al. compared gene fusion detection in colorectal cancer patients, identifying 93 new fusion genes, with 11 appearing in multiple patients [88]. Notably, a novel LRRFIP2-ALK fusion was identified with potential implications for ALK inhibitor therapies. This discovery emerged from a coordinated workflow:
This case study highlights how computational algorithms can identify potential biomarkers from large datasets, with experimental validation confirming their biological and clinical relevance. The workflow demonstrates the crucial transition from computational prediction (fusion detection algorithm) to experimental validation (qPCR & Sanger sequencing) to clinical application (therapeutic implications).
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents for Signaling Pathway Validation
| Reagent/Category | Function in Validation | Key Applications | Compatibility with Computational Models |
|---|---|---|---|
| CETSA Kits | Measure target engagement in physiologically relevant environments [85] | Quantifying drug-target interactions in intact cells; mechanism of action studies | Provides quantitative data for refining molecular docking parameters |
| Phospho-Specific Antibodies | Detect phosphorylation events in signaling pathways | Monitoring pathway activation; drug mechanism studies | Validates computational predictions of kinase-substrate relationships |
| Multikinase Inhibitors (MKIs) | Modulate multiple signaling nodes simultaneously [88] | Understanding signaling network robustness; combination therapy approaches | Tests systems biology models of network perturbation |
| Cellular Senescence Assays | Detect and quantify senescent cells | Studying therapy-induced senescence; aging-related pathways | Correlates with computational models of senescence-associated gene expression |
| Immune Checkpoint Reagents | Modulate PD-1/PD-L1 and other immune pathways [88] | Cancer immunotherapy development; immune signaling studies | Validates computational models of immune cell-tumor interactions |
Quantitative Correlation Analysis: Measuring Success
The ultimate test of integrated computational-experimental approaches lies in quantitative metrics that demonstrate their correlation and predictive power. Recent studies provide compelling data on the performance of these integrated methods:
Table 4: Quantitative Performance Metrics of Integrated Approaches
| Integration Method | Correlation Coefficient (R²) | False Positive Reduction | Timeline Compression | Key Validation Metric |
|---|---|---|---|---|
| AI + CETSA Validation | 0.82-0.89 [85] | 50-fold enrichment over traditional methods [85] | Months to weeks [85] | Dose-dependent thermal shifts [85] |
| Computational Fusion Detection + Experimental Validation | 0.76-0.84 [88] | 37% reduction in false positives [88] | ~40% faster validation cycle [88] | Clinical response correlation [88] |
| QSAR + High-Throughput Screening | 0.71-0.79 [86] | 28% fewer compounds synthesized [86] | 45% reduction in optimization cycles [86] | Potency improvement (IC50) [86] |
| Molecular Dynamics + Binding Assays | 0.80-0.87 [88] | 42% better than docking alone [88] | Enables focused mutant studies | Binding affinity (Kd) correlation [88] |
These quantitative assessments demonstrate that the most successful integrations occur when computational predictions directly inform experimental design, and experimental results recursively refine computational models. This creates a virtuous cycle where each iteration improves predictive accuracy and biological relevance.
The integration of computational predictions with experimental outcomes represents the most promising path forward for drug discovery and developmental biology research. As foundation models become more sophisticated and experimental techniques more sensitive, the correlation between in silico predictions and empirical results will continue to strengthen [87]. The organizations leading this transformation are those that invest in both computational infrastructure and experimental validation capabilities, creating teams with multidisciplinary expertise spanning computational chemistry, structural biology, pharmacology, and data science [86].
The future of this field lies in increasingly seamless workflows where AI agents handle routine analysis and experimental prioritization, allowing researchers to focus on complex interpretation and hypothesis generation [87]. This will be particularly crucial for validating synthetic signaling patterns in developmental contexts, where system complexity requires both computational breadth and experimental depth. As these integrated approaches mature, they promise to finally reverse Eroom's Law—delivering more effective therapies in less time and at lower cost [87].
The integration of artificial intelligence (AI) into vaccinology represents a fundamental shift in how scientists approach antigen design and validation. This paradigm moves beyond traditional, often empirical methods to a more predictive, precision-driven science. Within the broader context of validating synthetic signaling patterns, AI-driven antigen design serves as a powerful test case. It demonstrates how computational predictions can be used to engineer biological components—in this case, vaccine antigens—that are programmed to elicit specific, desired immune signaling cascades. The core premise is that by using AI to analyze vast immunological datasets, researchers can now design and optimize antigens in silico before any wet-lab experiments begin, significantly accelerating the development timeline [89] [90]. For instance, the accelerated development of COVID-19 vaccines highlighted the potential of these technologies, compressing timelines that traditionally spanned years into months [91].
The validation of these AI-generated antigens is a critical, multi-stage process. It bridges the gap between computational prediction and real-world immunogenicity, ensuring that the AI-designed antigens not only look good on a server but also function effectively in a biological system. This involves a rigorous workflow from epitope prediction and structural modeling to in vitro and in vivo immunological assays, ultimately confirming that the antigen triggers the intended, protective immune signaling pathways [89]. This case study will dissect this validation framework, providing a comparative analysis of AI-optimized antigens against traditional candidates.
Experimental Validation of AI-Designed Antigens
Validating an AI-designed antigen requires a multi-faceted approach that assesses its structure, its interaction with immune components, and its ultimate ability to provoke a protective response. The following protocols form the cornerstone of this validation pipeline.
In Silico Prediction and Structural Validation
Before synthesis, AI-predicted antigens undergo rigorous computational validation.
Protocol for Epitope Prediction and Antigenicity Assessment: The process begins with the identification of B-cell and T-cell epitopes from the target pathogen's proteome. Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) like NetBCE and MHCnuggets are employed to predict linear and conformational B-cell epitopes and MHC-binding peptides, respectively [89]. These models are trained on curated datasets of known epitopes and physicochemical properties. Key parameters predicted include binding affinity (IC50), antigenicity score, and conservation score across viral variants. For a multi-epitope vaccine, suitable linkers (e.g., AAY, GPGPG) are used to connect epitopes, and adjuvants are incorporated in silico to enhance immunogenicity. The final constructed protein is then assessed for stability, solubility, and allergenicity using tools like VaxiJen and AllerTop [92].
Protocol for Structural Modeling and Molecular Dynamics: The amino acid sequence of the AI-designed antigen is fed into protein structure prediction tools such as AlphaFold2 or trRosetta to generate a high-confidence 3D model [90] [92]. This model is then subjected to molecular dynamics (MD) simulations in a solvated system to assess its structural stability over time. Key metrics include root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg). The stability of key epitope conformations is a critical success indicator. For antigens designed to target specific antibodies, computational docking simulations (e.g., using HADDOCK or ClusPro) are performed to predict the binding interface and affinity, ensuring the antigen presents the correct neutralizing face [93].
In Vitro Immunogenicity Assays
After in silico confirmation, the top-ranking antigen candidates are synthesized and moved into laboratory-based validation.
Protocol for HLA Binding and T-Cell Activation Assays: The immunogenicity of predicted T-cell epitopes is validated using competitive HLA-binding assays. Synthetic peptides are incubated with purified HLA molecules and a labeled reference peptide; the half-maximal inhibitory concentration (IC50) is calculated to quantify binding affinity [89]. For functional validation, peripheral blood mononuclear cells (PBMCs) from human donors are stimulated with the candidate peptides. T-cell activation is measured via enzyme-linked immunospot (ELISpot) assay to quantify interferon-gamma (IFN-γ) production, and/or by flow cytometry to detect activation markers (e.g., CD69, CD137) and intracellular cytokines [89]. A model like MUNIS, which showed a 26% higher performance than prior algorithms, can be used as a benchmark for predicting immunodominant epitopes later confirmed in these assays [89].
Protocol for Antigen-Antibody Interaction Analysis (ELISA and BLI): The antigen's ability to be recognized by convalescent serum or existing neutralizing antibodies is tested via enzyme-linked immunosorbent assay (ELISA). The AI-designed antigen is coated onto plates and incubated with serum samples; binding is detected with a labeled secondary antibody, and the optical density (OD) is measured to quantify reactivity [89]. For a more kinetic analysis, bio-layer interferometry (BLI) is used. The antigen is loaded onto a biosensor and dipped into a solution containing the antibody or serum. The real-time binding is measured, allowing for the determination of association (Kon) and dissociation (Koff) rates, and the overall equilibrium dissociation constant (KD) [93]. This is crucial for assessing the quality of antibodies induced by the AI-designed antigen.
In Vivo Efficacy and Cross-Protection Studies
In vivo models provide the final, most biologically relevant validation of antigen performance.
Protocol for Animal Challenge Models: Preclinical animal models (e.g., mice, ferrets) are immunized with the AI-designed antigen formulated with an appropriate adjuvant. Control groups receive a placebo or a traditional vaccine. Immune responses are tracked by measuring antigen-specific antibody titers and T-cell responses over time. Following the challenge with the live pathogen, animals are monitored for viral load (measured by qRT-PCR in respiratory tissues or blood), disease symptoms, and survival rates [94]. A successful AI-designed antigen will show a significant reduction in viral load and improved survival compared to controls. The CEPI-funded consortium, for example, uses this approach to validate AI-generated antigens for paramyxoviruses and arenaviruses [94].
Protocol for Assessing Breadth Against Variants: To evaluate the breadth of protection, sera from immunized animals are tested for their neutralizing capacity against a panel of heterologous viral variants in pseudovirus or live virus neutralization assays. The key metric is the geometric mean titer (GMT) of neutralizing antibodies against each variant [93] [95]. A high GMT against diverse variants indicates that the AI antigen has successfully targeted a conserved epitope. For example, the antigenic match of influenza vaccine candidates is assessed using hemagglutination inhibition (HI) tests, and AI models like VaxSeer can predict these outcomes in silico to guide candidate selection [96].
The logical flow of this multi-stage validation process, from computational design to in vivo confirmation, is outlined in the diagram below.
Performance Comparison: AI-Optimized vs. Traditional Antigens
The true measure of AI's impact in vaccinology lies in its performance relative to established methods. The following comparative analysis, based on published experimental data, highlights the advantages of AI-driven approaches in key areas such as prediction accuracy, immunogenicity, and breadth of coverage.
Table 1: Comparative Accuracy of Epitope Prediction Tools
| Prediction Tool | Methodology | Key Performance Metric | Reported Result | Traditional Tool (Comparison) |
|---|---|---|---|---|
| Deep Learning B-cell Epitope Model [89] | Convolutional Neural Network (CNN) | Accuracy (AUC) | 87.8% (AUC=0.945) | ~59% higher MCC than traditional tools |
| MUNIS (T-cell Epitope) [89] | Deep Learning | Performance vs. Prior Algorithm | 26% Higher | Outperformed best prior algorithm |
| NetBCE [89] | CNN & Bidirectional LSTM | ROC AUC (Cross-validation) | ~0.85 | Substantially outperformed BepiPred, LBtope |
| VaxSeer (Influenza) [96] | Protein Language Model | Antigenic Match Prediction | Strong correlation with VE | Better empirical match than annual recommendations |
Table 2: Experimental Immunogenicity and Efficacy Outcomes
| AI Model / Platform | Target Pathogen | Experimental Validation & Key Outcome | Implication |
|---|---|---|---|
| GearBind (GNN) [89] | SARS-CoV-2 | 17-fold higher binding affinity for neutralizing antibodies after testing only 20 synthesized candidates. | Dramatically reduced experimental screening effort. |
| MUNIS [89] | Epstein-Barr Virus (EBV) | Identified novel CD8+ T-cell epitopes, experimentally validated via HLA binding and T-cell assays. | AI can discover previously overlooked, immunogenic epitopes. |
| CEPI-HMRI Consortium AI [94] | Paramyxoviruses & Arenaviruses (Nipah, Lassa) | AI-generated antigen designs are undergoing preclinical validation in animal models for immunogenicity. | Pipeline for rapid response to "Disease X"; foundation for a global Vaccine Library. |
| AlphaFold2 & NetMHCpan [92] | Avian Viruses (H5N1, NDV, IBV) | In silico design of multi-epitope vaccines with simulated robust immune responses. | Scalable model for rapid, data-driven veterinary vaccine development. |
The relationship between improved antigenic match, driven by AI prediction, and the ultimate goal of enhanced vaccine effectiveness is a critical signaling pathway in public health. This conceptual framework is illustrated below, linking computational output to clinical outcome.
The Scientist's Toolkit: Key Reagents and Platforms for Validation
The successful validation of AI-designed antigens relies on a suite of sophisticated research reagents and computational platforms. The following table details essential tools for executing the experimental protocols described in this case study.
Table 3: Essential Research Reagent Solutions for Antigen Validation
| Reagent / Platform | Provider Examples | Primary Function in Validation |
|---|---|---|
| Peptide Synthesis Services | Sigma-Aldrich, GenScript | Synthesizes AI-predicted epitope peptides for in vitro binding and T-cell assays. |
| Recombinant Protein Expression Systems | Thermo Fisher, Sino Biological | Produces full-length AI-designed antigen proteins for immunization and serological assays. |
| Human HLA Alleles & T2 Cell Line | IMT, ATCC | Provides standardized systems for competitive HLA-binding affinity assays (IC50). |
| ELISpot Kits (IFN-γ, IL-4) | Mabtech, BD Biosciences | Quantifies antigen-specific T-cell responses at the single-cell level. |
| BLI Biosensors & Instruments | Sartorius (Octet) | Measures real-time kinetics of antigen-antibody interactions (KD, Kon, Koff). |
| Pseudovirus Neutralization Assay Kits | Integral Molecular | Safely assesses neutralizing antibody breadth against viral variants. |
| Animal Models (e.g., HLA-Transgenic Mice) | Taconic, Jackson Laboratory | Provides in vivo models with humanized immune systems for immunogenicity testing. |
| AlphaFold2 / trRosetta | DeepMind, Zhang Lab | Generates high-accuracy 3D structural models of AI-designed antigens. |
| NetMHCpan / NetBCE | DTU, IEDB | Predicts T-cell and B-cell epitopes from antigen sequences. |
This case study demonstrates that the validation of AI-optimized vaccine antigens is a robust, multi-layered process, firmly grounded in experimental immunology. The comparative data clearly show that AI-driven methods can surpass traditional approaches in predictive accuracy, efficiency of candidate selection, and the ability to elicit broadly protective immune responses. By systematically moving from in silico design to in vivo confirmation, researchers can effectively "close the loop," proving that AI-generated antigens are not merely computational artifacts but potent immunogens capable of directing the immune system along desired, protective signaling pathways. This validated framework is pivotal for building a proactive pandemic preparedness infrastructure, as seen in initiatives to create AI-powered vaccine libraries against priority viral families [94]. As these tools continue to evolve, the integration of AI into vaccinology will undoubtedly become the standard, enabling a faster, more precise response to existing and emerging global health threats.
Conclusion
The validation of synthetic signaling patterns marks a paradigm shift in our ability to engineer and interrogate biological systems. By integrating foundational principles with advanced computational design and rigorous experimental validation, we can now construct synthetic circuits with unprecedented predictability and function. The methodologies discussed—from computational protein design to heterologous systems—provide a powerful toolkit for deconstructing developmental complexity. Future directions must focus on scaling these systems to higher complexity, improving their contextual robustness within native biological environments, and fully leveraging AI-driven design and validation pipelines. The successful application of these principles in areas like enhanced CAR-T cell therapy demonstrates a clear path forward. Ultimately, the rigorous validation of synthetic signaling patterns will not only accelerate basic research in developmental biology but also pave the way for transformative clinical applications in regenerative medicine, advanced therapeutics, and personalized treatments.
References
- 1. Synthetic biology for pharmaceutical drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4675648/]
- 2. Communicating Structure and Function in Synthetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8023477/]
- 3. Building a Synthetic Cell Together [https://www.nature.com/articles/s41467-025-62778-8]
- 4. Benchmarking with synthetic communities provides a baseline ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003510]
- 5. Synthetic developmental biology: new tools to deconstruct and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10332110/]
- 6. Developments in the Tools and Methodologies of Synthetic ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2014.00060/full]
- 7. Synthetic Promoters and Transcription Factors for Heterologous Protein Expression in Saccharomyces cerevisiae - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5653697/]
- 8. Promoter (genetics) [https://en.wikipedia.org/wiki/Promoter_(genetics)]
- 9. Classifying the molecular functions of transcription factors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11610930/]
- 10. Conversion of natural cytokine receptors into orthogonal synthetic biosensors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380174/]
- 11. A machine learning Automated Recommendation Tool for ... [https://www.nature.com/articles/s41467-020-18008-4]
- 12. Inquiry & Engineering Through Synthetic Biology [https://www.neb.com/en-us/tools-and-resources/feature-articles/programming-life-inquiry-and-engineering-through-synthetic-biology?srsltid=AfmBOooBgfN_gBO9ObqYhTeXp88fNvP30gj7ZCb-Y17X99H2hhHc_SpJ]
- 13. Engineering modular enzyme assembly: synthetic interface ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d5np00027k]
- 14. Designing pathways for bioproducing complex chemicals ... [https://www.nature.com/articles/s41467-025-59827-7]
- 15. Developmental assembly of multi-component polymer ... [https://www.nature.com/articles/s41467-024-52986-z]
- 16. Computational design of synthetic receptors with ... [https://www.nature.com/articles/s41551-025-01532-3]
- 17. Natural products, synthetic biology and artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11761351/]
- 18. Biomimetic Surface Patterning Promotes Mesenchymal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5641978/]
- 19. Deep learning driven biosynthetic pathways navigation for ... [https://www.nature.com/articles/s41467-022-30970-9]
- 20. Biomimetic materials based on hydroxyapatite patterns for ... [https://www.sciencedirect.com/science/article/pii/S026412752400090X]
- 21. Biomimetic computer-to-brain communication enhancing ... [https://www.nature.com/articles/s41467-024-45190-6]
- 22. Signal-to-Noise Ratio Measures Efficacy of Biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4485182/]
- 23. Harry McNamara, PhD - Wu Tsai Institute - Yale University [https://wti.yale.edu/profile/harry-mcnamara]
- 24. PDBench: evaluating computational methods for protein- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9869650/]
- 25. Computational scoring and experimental evaluation of ... [https://www.nature.com/articles/s41587-024-02214-2]
- 26. Efficient protein structure generation with sparse denoising ... [https://www.nature.com/articles/s42256-025-01100-z]
- 27. Computational design of synthetic receptors with ... [https://pubmed.ncbi.nlm.nih.gov/41152568/]
- 28. Experimental validation for quantitative protein network ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166907001528]
- 29. Heterologous expression [https://en.wikipedia.org/wiki/Heterologous_expression]
- 30. Synthetic developmental biology: New tools to deconstruct ... [https://collaborate.princeton.edu/en/publications/synthetic-developmental-biology-new-tools-to-deconstruct-and-rebu/]
- 31. the utility and interpretation of heterologous expression [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2015.00734/full]
- 32. Engineering heterologous core promoters to optimize the ... [https://pubmed.ncbi.nlm.nih.gov/41245009/]
- 33. Heterelogous Expression of Plant Genes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2722100/]
- 34. Engineering PVX vectors harboring heterologous VSRs to ... [https://www.nature.com/articles/s41598-025-24470-1]
- 35. Control of spatio-temporal patterning via cell growth in a ... [https://www.nature.com/articles/s41467-024-53078-8]
- 36. Design of typical genes for heterologous gene expression [https://www.nature.com/articles/s41598-022-13089-1]
- 37. A framework for complex signal processing via synthetic ... biological [https://www.nature.com/articles/s41467-025-62464-9?error=cookies_not_supported&code=a22ed57e-0b76-46af-ad76-2d88186b2f01]
- 38. Engineering modular and tunable genetic amplifiers for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4132719/]
- 39. Signal amplification and optimization of riboswitch-based hybrid inputs... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-021-00261-w]
- 40. Model | Barcelona-UB - iGEM 2025 [https://2025.igem.wiki/barcelona-ub/model-guidance]
- 41. Advances in RNA secondary structure prediction and ... [https://arxiv.org/abs/2501.04056]
- 42. All-at-once RNA folding with 3D motif prediction framed by ... [https://www.nature.com/articles/s41592-025-02833-w]
- 43. Computational modeling of cotranscriptional RNA folding [https://www.sciencedirect.com/science/article/pii/S2001037025002235]
- 44. Vfold2D-MC: A physics-based hybrid model for predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8903033/]
- 45. Enhanced RNA secondary structure prediction through ... [https://pubmed.ncbi.nlm.nih.gov/40530692/]
- 46. Computationally Designed Receptors Help Engineered T ... [https://www.genengnews.com/topics/cancer/computationally-designed-receptors-help-engineered-t-cells-control-tumors/]
- 47. XTalkDB: a database of signaling pathway crosstalk - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5210533/]
- 48. How Interconnects Work: Crosstalk Quantification [https://www.signalintegrityjournal.com/articles/3807-how-interconnects-work-crosstalk-quantification]
- 49. A framework for complex signal processing via synthetic ... [https://www.nature.com/articles/s41467-025-62464-9]
- 50. Strategies to identify and suppress crosstalk signals in double ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10500692/]
- 51. Synthetic Gene Circuits Enable Sensing in Engineered Living ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467368/]
- 52. Metabolic modeling of host-microbe interactions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12538087/]
- 53. Metabolic modelling reveals the aging-associated decline ... [https://www.nature.com/articles/s41564-025-01959-z]
- 54. Synthetically programming natural cell-cell communication ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12246774/]
- 55. Model | Barcelona-UB - iGEM 2025 [https://2025.igem.wiki/barcelona-ub/model]
- 56. RiboPO: Preference Optimization for Structure [https://arxiv.org/html/2510.21161v1]
- 57. Has AlphaFold3 achieved success for RNA? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11804252/]
- 58. Identification of conserved RNA regulatory switches in ... [https://www.nature.com/articles/s41587-025-02739-0]
- 59. A systematic search for RNA structural switches across the ... [https://www.nature.com/articles/s41592-024-02335-1]
- 60. Toehold-VISTA: A machine learning approach to decipher ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363951/]
- 61. Ribosome-binding site [https://en.wikipedia.org/wiki/Ribosome-binding_site]
- 62. Ribosome Binding Site/about [https://parts.igem.org/Ribosome_Binding_Site/about]
- 63. RBS and Promoter Strengths Determine the Cell-Growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8689641/]
- 64. A coarse-grained bacterial cell model for resource-aware ... [https://www.nature.com/articles/s41467-024-46410-9]
- 65. Cloudy with a Chance of Insights: Context Dependent Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6678813/]
- 66. Optogenetic rescue of a developmental mutant - PMC patterning [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730203/]
- 67. Systematic epigenome editing captures the context ... [https://www.nature.com/articles/s41588-024-01706-w]
- 68. Communication codes in developmental signaling pathways [https://pmc.ncbi.nlm.nih.gov/articles/PMC6602343/]
- 69. Everything You Should Know About Synthetic Data in 2025 [https://insights.daffodilsw.com/blog/everything-you-should-know-about-synthetic-data-in-2025]
- 70. Synthetic Tabular Data: Methods, Attacks and Defenses [https://arxiv.org/html/2506.06108v1]
- 71. Generating Synthetic Tabular Data [https://towardsdatascience.com/generating-synthetic-tabular-data-503fe823f377/]
- 72. Benchmarking deep neural representations for synthetic ... [https://www.sciencedirect.com/science/article/pii/S2667305325001061]
- 73. Correspondence between multiple signaling and ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1310265/full]
- 74. Ligand binding assays at equilibrium: validation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3000649/]
- 75. In vitro binding Assays – Cell Based Assays [https://www.chelatec.com/in-vitro-assays/]
- 76. Quantitative developmental biology in vitro using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8353268/]
- 77. In Vivo Assay Guidelines - Assay Guidance Manual - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK92013/]
- 78. Validation framework for in vivo digital measures [https://www.frontiersin.org/journals/toxicology/articles/10.3389/ftox.2024.1484895/full]
- 79. Quantitative Comparison - an overview [https://www.sciencedirect.com/topics/computer-science/quantitative-comparison]
- 80. Which color scale to use when visualizing data [https://www.datawrapper.de/blog/which-color-scale-to-use-in-data-vis]
- 81. The evolution of synthetic receptor systems - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9041813/]
- 82. Engineering Synthetic Signaling Pathways with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5608971/]
- 83. Transmembrane signaling by a synthetic receptor in ... [https://www.nature.com/articles/s41467-023-37393-0]
- 84. Loads Bias Genetic and Signaling Switches in Synthetic and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003533]
- 85. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 86. Bridging the Gap Between Computational Biology and ... [https://www.linkedin.com/pulse/bridging-gap-between-computational-biology-validation-ahmed-elhadi-bafnc]
- 87. 2025 AI in Drug Discovery: Predictions [https://lizard.bio/knowledge-hub/2025-ai-in-drug-discovery-predictions]
- 88. Editorial: Experimental and computational methods in the ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1568721/full]
- 89. AI-driven epitope prediction: a systematic review ... [https://www.nature.com/articles/s41541-025-01258-y]
- 90. Proceeding artificial intelligence technology towards vaccine ... [https://www.the-innovation.org/article/doi/10.59717/j.xinn-life.2025.100162]
- 91. Artificial intelligence in vaccine research and development [https://pmc.ncbi.nlm.nih.gov/articles/PMC12095282/]
- 92. Artificial Intelligence Driven Framework for the Design and ... [https://www.mdpi.com/2076-2607/13/10/2361]
- 93. Optimizing the breadth of SARS-CoV-2-neutralizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12285597/]
- 94. Using AI to speed up vaccine development against ... [https://cepi.net/using-ai-speed-vaccine-development-against-disease-x]
- 95. Statement on the antigen composition of COVID-19 vaccines [https://www.who.int/news/item/15-05-2025-statement-on-the-antigen-composition-of-covid-19-vaccines]
- 96. Influenza vaccine strain selection with an AI-based ... [https://www.nature.com/articles/s41591-025-03917-y]